

Abstract
Adverse drug reactions (ADR) can be broadly categorised as either on-target or off-target. On-target ADRs arise as a direct consequence of the pharmacological properties of the drug and are therefore predictable and dose dependant. On-target ADRs comprise the majority (>80%) of ADRs, relate to the drug’s interaction with its known pharmacological target and are a result of a complex interplay of genetic and ecologic factors. In contrast off-target ADRs, including immune mediated ADRs (IM-ADRs), are due to unintended pharmacological interactions such as inadvertent ligation of host cell receptors or non-pharmacological interactions mediated through an adaptive immune response. IM-ADRs can be classified according to the primary immune cell involved and include B cell-mediated (Gell-Coombs type I-III reactions) and T cell-mediated (Gell-Coombs type IV or delayed hypersensitivity) reactions. IM-ADRs mediated by T cells are associated with phenotypically distinct clinical diagnoses and can vary from a mild delayed rash to a life threatening cutaneous, systemic or organ disease, such as Stephen Johnson syndrome/toxic epidermal necrolysis (SJS/TEN), drug reaction with eosinophilia and systemic symptoms (DRESS) and drug-induced liver disease (DILI). T-cell mediated ADRs are strongly linked to the carriage of particular HLA risk alleles which in the case of abacavir hypersensitivity and HLA-B*57:01 has led to translation into the clinic as a routine screening test. In this review, we will discuss the immunogenetics and pathogenesis of IM-ADRs and how HLA associations inform both pre-drug screening strategies and mechanistic understanding.
Keywords: Abacavir, adverse drug reaction, allopurinol, angioedema, aspirin exacerbated respiratory disease, carbamazepine, human leukocyte antigen, immunological memory, pharmacogenomics
Introduction
Adverse drug reactions (ADRs) are major causes of iatrogenic, potentially preventable patient morbidity and mortality. These reactions have a significant impact on health care systems and are the source of approximately 3–6% of inpatient admissions, comprising 5–10% of inpatient cost. They are estimated to be the fourth most common cause of death1–4. ADRs classified as “on-target” (also known as type A), account for up to 80% of all ADRs, and can be predicted based on the pharmacological activity of the drug. On-target reactions are typically dose dependent and may be compounded by altered pharmacokinetics resulting from comorbidities such as impaired renal or liver function, drug interactions or polymorphisms within drug receptor, transporter or metabolism genes and include reactions such as prolonged bleeding following warfarin therapy.
ADRs arising from “off-target” (also known as type B) interactions account for approximately 20% of all ADRs, however off-target effects may be under-recognized and under-reported. Off-target reactions include those that are directly immune-mediated ADRs (IM-ADRs) and are associated with immunological memory as well as pharmacological drug effects where an interaction of a drug with a receptor can lead to an immunological phenotype (urticaria) but there is no adaptive response. The latter includes interaction of drugs with the mas-related G-protein coupled receptor (MRGPRX2) on mast cell leading to non-IgE mediated mast cell activation5. IM-ADRs encompass several phenotypically distinct clinical entities comprising B-cell (antibody-mediated, Gell Coombs Types I-III) and T-cell (delayed type hypersensitivity, Gell-Coombs Type IV) mediated reactions. IM-ADRs display a range of clinical features including anaphylaxis, angioedema, urticaria, maculopapular exanthema, fever and internal organ involvement (e.g., hepatitis). T-cell mediated - delayed hypersensitivity - reactions present as a variety of clinical phenotypes including severe cutaneous syndromes, such as maculopapular exanthema (MPE), acute generalised exanthema pustulosis (AGEP) and Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN), systemic reactions such as abacavir hypersensitivity syndrome (AHS) and drug reaction with eosinophilia and systemic symptoms (DRESS), or as organ specific manifestations such as drug induced liver injury (DILI) and pancreatitis6,7 (Figure 1).
Figure 1.

Gell and Coombs classification of hypersensitivity reactions. Drugs can elicit all of the defined reaction types, examples are shown in the text boxes at the bottom of the table. These include antibody mediated reactions (Type I-III) and T-cell and cytokine mediated reactions (Type IVa-d). Acute generalised exanthemetous pustulosis (AGEP), polymorphonuclear leukocyte (PMN), cytotoxic T cell (CTL), granulocyte macrophage colony stimulating factor (GM-CSF). Adapted from Pichler, 2007. Drug Hypersensitivity Reactions: Classification and Relationship to T-Cell activation, in Drug Hypersensitivity.
Mechanisms and Specific Immunologically-mediated Adverse Drug Reactions
HLA
Multiple phenotypically distinct T-cell mediated ADRs have been associated with carriage of specific human leukocyte antigen (HLA) risk alleles (Table 1). HLA alleles (Figure 2), and particularly HLA-B which has been prevalently associated with drug-induced IM-ADR, are highly polymorphic with in excess of 8000 class I molecules and just over 3000 class II β-chain variants8. Regions of highest variability map to the peptide binding groves, maximising the diversity of self and pathogen derived peptides that can be presented to T cells. The amino acid sequence of peptides presented by individual HLA class I and class II molecules depends on components of the antigen processing pathway, such as tapasin and the proteasome9, and on the amino acid anchor residues favoured by particular HLA alleles. The binding affinity for these anchor residues is dictated by pockets within the peptide binding groove of the particular HLA allele, designated A, B, C, D, E and F for class I molecules (Figure 2B) and P1, P4, P6 and P9 for class II molecules.
Table 1.
HLA associations for IM-ADR
| Drug | DHR | HLA risk alleles | PPV | NPV | Populations |
|---|---|---|---|---|---|
|
| |||||
| Abacavir | HSS/DIHS | B*57:0158,61,113,114 | 55% | 100% | European, African |
|
| |||||
| Carbamazepine | SJS/TEN | B*15:0270–80 | 3% | 100% in Han Chinese | Han Chinese, Thai, Malaysian, Indian |
|
| |||||
| B*15:11115,116 | Korean, Japanese | ||||
|
| |||||
| B*15:18, B*59:01 and C*07:0481 | Japanese | ||||
|
| |||||
| B*15:21117 | |||||
|
| |||||
| A*31:01116,118–120 | Japanese, northern European, Korean | ||||
|
| |||||
| HSS/DIHS/DRESS | 8.1 AH (HLA A*01:01, Cw*07:01, B*08:01, DRB1*03:01, DQA1*05:01, DQB1*02:01)121 | Caucasians | |||
|
| |||||
| A*31:01122 | 0.89% | 99.98% | Europeans | ||
|
| |||||
| A*31:01122 | 0.59% | 99.97% | Chinese | ||
|
| |||||
| A*31:01116,118–120 | Northern Europeans, Japanese, and Korean | ||||
|
| |||||
| A*11 and B*51 (weak)120 | Japanese | ||||
|
| |||||
| MPE | A*31:01123 | 34.9% | 96.7% | ||
|
| |||||
| Any ADR | A*31:01124 | ||||
|
| |||||
| Allopurinol | SJS/TEN/DIHS/DRESS/MPE | B*58:01 (or B*58 haplotype)85,125–131 | 3% | 100% in Han Chinese | Han Chinese, Thai, European, Italian, Korean |
|
| |||||
| Oxcarbazepine | SJS/TEN | B*15:02 and B*15:18132–134 | 15:02 −0.73% | 15:02 −99.97 | Han Chinese, Taiwanese |
|
| |||||
| Lamotrigine | SJS/TEN | B*15:02 (positive)133 | Han Chinese | ||
|
| |||||
| B*15:02 (no association)135,136 | Han Chinese | ||||
|
| |||||
| Phenytoin | SJS/TEN | B*15:02(weak), Cw*08:01 and DRB1*16:0272,73,137 | Han Chinese | ||
|
| |||||
| DRESS/MPE | B*13:01 (weak) | Han Chinese | |||
| B*5101 (weak)137 | |||||
|
| |||||
| Nevirapine | SJS/TEN | C*04:01138 | Malawian | ||
|
| |||||
| HSS/DIHS/DRESS | DRB1*01:01 & DRB1*01:02 (hepatitis and low CD4+)91,139 | 18% | 96% | Australian, European and South African | |
|
| |||||
| Cw*8 or Cw*8-B*14 haplotype96,140 | Italian and Japanese | ||||
|
| |||||
| Cw*491,141 | Blacks, Asians, Whites, Han Chinese | ||||
|
| |||||
| B*3591 | 16% | 97% | Asian | ||
| B*35:0195 | |||||
| B*35:05142 | |||||
|
| |||||
| Delayed rash | DRB1*01143 | French | |||
|
| |||||
| Cw*0491,93 | African, Asian, European, and Thai | ||||
|
| |||||
| B*35:05142 | Thai | ||||
|
| |||||
| Dapsone | HSS | B*13:01144 | 7.8% | 99.8% | |
|
| |||||
| Efavirenz | Delayed rash | DRB1*01143 | French | ||
|
| |||||
| Sulfamethoxazole | SJS/TEN | B*3885 | European | ||
|
| |||||
| Amoxicillin-clavulanate | DILI | DRB1*15:01 | European | ||
| A*02:01 | |||||
| DQB1*06:02, and rs3135388, a tag SNP of DRB1*15:01-DQB1*06:02 | |||||
| DRB1*07 and HLA-A1 (protective)145–147 | |||||
|
| |||||
| Lumiracoxib | DILI | DRB1*15:01-DQB1*06:02-DRB5*01:01-DQA1*01:02 haplotype148 | International, multi-center | ||
|
| |||||
| Ximelagatran | DILI | DRB1*07 and DQA1*02149 | Swedish | ||
|
| |||||
| Diclofenac | DILI | HLA-A11150 | European | ||
|
| |||||
| Flucloxacilin | DILI | B*57:01 | 0.12% | 99.99% | European |
| DRB1*07:01-DQB1*03:01 151 | |||||
|
| |||||
| Lapatinib | DILI | DRB1*07:01- | International, multi-center | ||
| DQA2*02:01-DQB1*02:02/02:02152 | |||||
|
| |||||
| Methimazole/Carbimazole/Anti-thyroid drugs | Agranulocytosis | HLA-B*38:02 (*5 SNPs)153–155 | 7% | 99.9% | Chinese, Northern Han Chinese |
| HLA-B*27:05(3/5 SNPs)155,156 | *30% | >99% | *European/Northern Han Chinese | ||
| HLA-DRB1*08:03153,155,157 | Chinese, Japanese, Northern Han Chinese | ||||
| Northern Han Chinese | |||||
|
| |||||
| Clozapine | Agranulocytosis/neutropenia | HLA-B*59:01158 | 35.1% | Japanese | |
| HLA-DQB1 (126Q) | European | ||||
| HLA-DQB1*05:02; | |||||
| HLA-B (158T) (HLA-B*39:01, HLA-B*39:06, HLA-B*38:01)159 | |||||
| HLA-DQB1160 | European | ||||
|
| |||||
| Azathioprine | Pancreatitis | HLA-DQA1*02:01; | 9% | European | |
| HLA-DRB1*07:01161 | |||||
|
| |||||
| Statins | Myopathy | HLA-DRB1*11:01162 | European, African | ||
|
| |||||
| Asparaginase | Anaphylaxis | DRB1*07:01163 | European | ||
Figure 2.

The human leukocyte antigen (HLA). A. The HLA genes are amongst the most polymorphic of all human genes and are located on the short arm (p) of human chromosome 6. The class I regions encodes the HLA-A, HLA-C and HLA-B genes whilst the class II regions encode HLA-DR, HLA-DQ and HLA-DP. B. Peptides are presented on the surface of cells in the context of HLA to the T cell receptor (TCR). For class I HLA alleles peptides bind within specific pockets, A, B, C, D, E and F, of the peptide binding groove. The B and F pockets bind the anchor residues, P2 and P9 of each peptide providing binding specificity to a particular HLA molecule. The TCR engages with the CDR3 region of the HLA molecule and appropriate solvent exposed peptide residues.
HLA class I molecules are present on the surface of all nucleated cells and, predominantly, present endogenously processed peptides to CD8 T cells. HLA class II molecules are present on antigen presenting cells such as dendritic cells, macrophages and B cells. Class II molecules present exogenous peptides to CD4 T cells. Typically, class I presented peptides are in the order of 9–11 amino acids in length. As a result of the more open nature of the peptide binding groove, peptides presented by class II molecules are typically in the order of 11–15 amino acids in length. The mechanism by which small drug molecules, typically in the size range of 1–3 amino acids10, stimulate T-cell responses remains incompletely understood, although three non-mutually exclusive models have been proposed to explain this apparent contradiction. These are (1) the hapten/prohapten model, (2) the pharmacological interaction with immune receptors (p-i) model and (3) the altered peptide repertoire model (Figure 3).
Figure 3.

Models of T cell-mediated drug hypersensitivity. (I) In the hapten/prohapten model the drug forms covalent bonds with endogenous peptides or proteins. This modified complex is processed via conventional antigen processing pathways and presented on the surface of cells in the context of HLA. The de novo antigens thus displayed are recognised as foreign by host T cells. (ii) In the p.i model non-modified drug binds directly to immune receptors such as the TCR via non-covalent bonds (dashed line), this response is independent of peptide or antigen processing. (iii) In the altered peptide model drug binds non-covalently within the peptide binding groove thereby altering the chemistry of the antigen binding cleft. This alters the repertoire of peptides capable of binding to a specific allele - creating a pseudo-allogenic HLA molecule - which presents non-tolerised altered self to T cells.
The hapten/prohapten model proposes that drug or drug metabolite binds covalently to a host protein which then undergoes intracellular antigen processing to generate a pool of chemically-modified peptides. When presented in the context of HLA these modified peptides are recognized as foreign by T cells and elicit an immune response11,12. Examples of this model include allergy to penicillin and reactive metabolites of sulfamethoxazole (nitroso-sulfamethoxazole)13,14. The pharmacological interaction with immune receptors (p-i) model postulates that the offending drug binds, non-covalently, to either the T-cell receptor (TCR) or HLA protein in a peptide-independent manner to directly activate T cells. This model has been hypothesized to explain T-cell reactivity that is labile (i.e., reactivity is abrogated by washing drug from the surface of antigen presenting cells) and/or is observed within seconds of drug exposure, a time course too short for intracellular antigen processing15,16. Finally, in the altered peptide repertoire model, the drug occupies a position in the peptide binding groove of the HLA protein changing the structure of the binding cleft and therefore the peptide specificity of the HLA risk allele. The neo-epitopes displayed as a result of altered binding specificity are recognized as foreign by the immune system and therefore elicit a T-cell response17,18.
The T-cell receptor
HLA risk allele restricted T-cell responses have been detected to a range of drugs including HLA-B*57:01 presented abacavir17,19–22, -B*58:01 restricted allopurinol and oxypurinol SJS/TEN and DRESS23,24, -B*15:02 restricted SJS/TEN and -A*31:01 presented carbamazepine MPE≫DRESS≫>SJS/TEN25–27 as well as -B*57:01 restricted flucloxacillin DILI28. Despite the clear role that T cells play in these reactions, the nature of the TCR is poorly defined and the degree of TCR specificity/clonality is likely unique for each drug-HLA combination. Abacavir specific T-cell responses are polyclonal17,19,20 in keeping with the altered peptide model. Oxypurinol specific T-cell lines derived from the blood of patients with allopurinol SJS/TEN appear more restricted and show preferential Vβ TCR use within individual patients. However, public TCRs, those shared across different patients, were not identified in one study29. In contrast, in carbamazepine induced SJS/TEN patients, shared CD8+ T-cell clonotypes bearing a public CDR3 sequence have been identified30. Zhou and colleagues have suggested that carbamazepine may make more intimate contacts with the TCR loops than the HLA molecule31. The carbamazepine data are significant as they suggest for the first time the concomitant involvement of both a specific HLA allotype and a specific TCR clonotype in the pathogenesis of a serious IM-ADR. However, it remains the case that a crystal structure of drug/HLA/TCR complex has yet to been solved for any T-cell mediated IM-ADR.
Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis
SJS and TEN are two of the most severe IM-ADRs with an estimated patient mortality rate over 30% at one year following disease onset32. Cardinal features of SJS/TEN include widespread epidermal necrosis that resembles a severe burn injury and manifests clinically with skin, mucous membrane and eye involvement. SJS/TEN is a single disease with a cohesive immunopathogenesis and is defined by the percentage of body surface area involvement (SJS: 10% BSA affected; SJS/TEN overlap: 10–30% BSA affected; TEN: >30% BSA affected). Internal organ failure and secondary complications such as infection, thrombosis and deconditioning are frequently associated with acute SJS/TEN. Further, the long-term sequelae of this disease, including scarring, blindness and psychiatric illness, are a source of significant disability for survivors. SJS/TEN pathogenesis is characterised by widespread epidermal necrosis and detachment. Early skin lesions are characterized by the epidermis and dermoepidermal infiltration of CD14+CD16+CD11c+HLA-DR+ monocytes33. The pathogenesis is however, driven by cytotoxic CD8+ T cells, NK cells and CD3+CD56+ NK T cells (NKT cells) which are enriched in blister fluid of patients with acute SJS/TEN34–37. Granulysin, a cytotoxic peptide produced by CD8+ T cells, NK and NKT cells, is present in high concentrations in the blister fluid and is the key mediator of epidermal cell death in SJS/TEN38. Serum levels of granulysin associate with the severity of acute SJS/TEN and predict mortality39,40.
Drug Reaction with Eosinophilia and Systemic Symptoms
DRESS, also known as drug induced hypersensitivity syndrome (DIHS), presents as a widespread rash of varying severity, without skin separation or blistering, and is frequently accompanied by fever, internal organ involvement (usually hepatitis) and hematologic abnormalities (often atypical lymphocytes and/or eosinophilia). Diffuse lymphadenopathy, pneumonitis, encephalitis, cardiac failure (myocarditis) and nephritis are variable features of this syndrome, which may mimic a viral illness. Mortality rates in DRESS approximates 10%41. The onset of symptoms typically occurs 2–8 weeks following initiation of the inciting drug and can persist for weeks. Prolonged or recurrent symptoms, sometimes weeks following cessation of the offending drug, as well as late onset autoimmune diseases including thyroiditis, systemic lupus erythematosis and type I diabetes have been described up to four years following disease resolution42. Numerous drugs are associated with the development of DRESS including the allopurinol, antiepileptic medications (carbamazepine, phenytoin, phenobarbital and lamotrigine), beta-lactam antibiotics, NSAIDs, sulfa antimicrobials, other antibiotics such as vancomycin and minocycline and drugs used to treat other infections such as anti-mycobacterial drugs (rifamycins, isoniazid, ethambutol), dapsone and drugs used to treat HIV such as nevirapine, raltegravir and darunavir.
DRESS is associated with expansion of circulating and dermal-infiltrating effector T cells as well as CD4+FoxP3+ regulatory T cells (Treg)43,44. Skin homing CD4+FoxP3+ T cells are postulated to limit the severity of acute disease by suppressing effector T-cell responses45. Reactivation of human herpesviruses, in particular human herpesvirus (HHV)-6, but also Epstein-Barr virus (EBV), HHV-7 and cytomegalovirus (CMV) is universally observed during acute and recovery phase disease. HHV-6 and EBV reactivation has been observed as early as 2–3 weeks after onset of rash and antiviral CD8+ effector T cells are expanded during this phase of disease. Whether viral replication contributes to the events inciting DRESS or is the result of general immune dysfunction, such as breakdown of Treg suppressor function or the up-regulation of the HHV-6 receptor, CD134, on CD4+ T cells, has not been defined44–47. Nevertheless, viral replication and a virus-specific T-cell responses likely contribute to the clinical features of DRESS including prolonged duration, multi-organ involvement and relapsing disease following withdrawal of glucocorticoid steroids.
Drug-induced Liver Disease
DILI is one of the more common causes of primarily single organ IM-ADR and accounts for 10% of all episodes of acute hepatitis and up to 13% of all instances of liver failure in the USA48. DILI can manifest within several days and up to 8 weeks post drug exposure. In some cases where the primary phenotype is severe drug-induced liver disease other features such as skin rash of varying severity have been described. Several drugs have been associated with the development of DILI including drugs withdrawn from the market such as ximelagatran, lumiracoxib, diclofenac, amoxicillin-clavulanate and flucloxacillin (Table 1). Amoxicillin-clavulanate (AC), one of the most heavily prescribed antibiotics, accounts for up to 17% of DILI cases requiring hospitalisation49,50. AC-DILI was first associated with carriage of the class II allele HLA-DRB1*15:0151–53. AC-associated DILI can present as either cholestatic, hepatocellular or mixed, phenotypes. This presentation appears to be subject to ethnicity, with French and Belgian populations experiencing a bias toward a cholestatic presentation. In contrast, Spanish populations presented with an almost equal proportion of cholestatic, hepatocellular or mixed phenotypes54. A later study of Spanish populations indicated that HLA-A*30:02 was associated with hepatocellular liver injury and the class II haplotype DRB1*15:01-DQB1*06:02 was associated with cholestatic or mixed pattern DILI55. Finally, HLA-A*02:01 which is haplotypic with DRB1*1501-DQB1*06:02 is associated with AC-induced DILI in Northwestern Europeans56.
HLA and IM-ADRS: Representative Examples
Abacavir
AHS is an exemplar of T-cell mediated ADR, explaining both the HLA association and the mechanism of T-cell activation. The clinical features of abacavir hypersensitivity are not consistent with DRESS and the AHS is quite unique in its rapid onset and lack of associated eosinophilia and organ involvement paralleled only perhaps by azathioprine hypersensitivity which can present in a similar fashion. Abacavir is a guanosine analogue that is used as part of combination antiretroviral therapy for the treatment of HIV-1 infection. Early use of abacavir was associated with hypersensitivity reactions in 5–8% of patients57. Early reports described that AHS typically manifests within the first 6 weeks of therapy, however patch test positive or immunologically confirm AHS occurs from 1.5 days to 3 weeks following first drug exposure58. AHS is characterized by fever, malaise, gastrointestinal, respiratory symptoms, and/or generalized rash. In 2002, a strong association between carriage of the HLA class I allele, HLA-B*57:01, and AHS was reported59, an association borne out by subsequent studies60,61. Using immunologically defined (patch test positive62) cases as a co-primary clinical endpoint, the PREDICT-1 study demonstrated that screening for, and exclusion of HLA-B*57:01 carriers from abacavir drug exposure could completely eliminate the incidence of true immunologically mediated (patch test positive) AHS. Another case-control study, the SHAPE study confirmed carriage of HLA-B*57:01 as a risk allele for AHS, generalizable across race. The PREDICT-1 study also demonstrated that HLA-B*57:01 carriage provided a 100% negative predictive value (NPV) and a 55% positive predictive value (PPV)63,64 for AHS.
Abacavir shows exquisite specificity for HLA-B*57:01, failing to interact with closely related HLA alleles, HLA-B*57:02, HLA-B*57:03 and HLA-B*58:01, which differ by 2–4 amino acids. Amino acid differences between these alleles locates abacavir binding to the C-terminal end of the peptide binding groove19. The capacity of HLA-B*57:01 to present abacavir requires antigen processing, being dependent on transporter associated with antigen presentation (TAP) and tapasin19, although it does not require the proteasome20. The abacavir binding site on HLA-B*57:01, and the potential mechanism of disease, was defined in 2012 with the simultaneous publication of the crystal structures of HLA-B*57:01 in complex with abacavir and peptide by two independent groups17,65. Abacavir binds non-covalently within the HLA-B*57:01 peptide binding groove at the C, D, E and F pockets (Figure 4). Abacavir interacts directly with the two residues, Asp114 and Ser116, that distinguish HLA-B*57:01 from HLA-B*57:03. This binding alters the F pocket, under the C-terminus of the bound peptide, and induces a change in the binding properties of HLA-B*57:01. The canonical terminal anchor residues for HLA-B*57:01 are large aromatic amino acids such as Tyr or Phe. In the presence of abacavir, peptides with small aliphatic C-terminal residue (Ile, Leu, Val, Ala) are preferentially used as a terminal anchor residue, specificity for the p7 is also altered by the binding of abacavir17,65,66. Consequently, binding of abacavir alters the peptide specificity of HLA-B*57:01 such that 20–45% of the peptides eluted from abacavir-treated HLA-B*57:01 antigen presenting cells are distinct from those recovered from untreated cells17,65,66. These studies defined the altered peptide repertoire model of IM-ADRs and predicts that in the context of drug, numerous novel self-peptides are presented to T cells. These neo-epitopes are not subject to traditional tolerance mechanisms and can activate naïve T cells or stimulate cross reactive pre-formed memory T cells in a manner analogous to graft rejection and graft versus host disease, where T cells are also exposed to novel HLA molecules presenting self-antigens.
Figure 4.
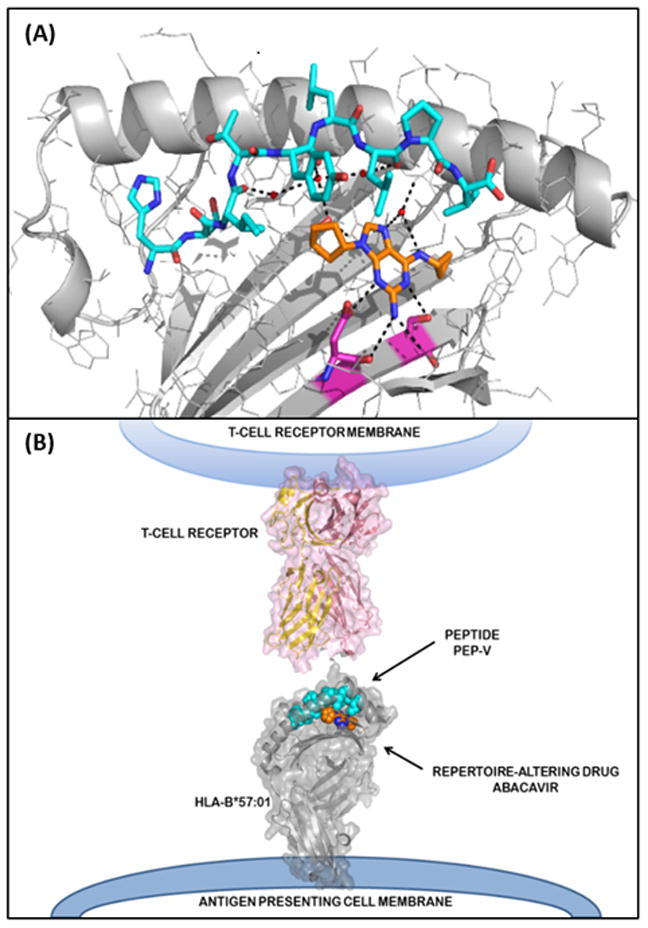
Solved structure of abacavir-peptide-HLA complex. A. Intramolecular contacts within the peptide binding cleft of HLA-B*57:01 and peptide and abacavir. HLA-B*57:01 in grey, synthetic peptide (HSITYLLPV) in cyan. Abacavir is shown as orange for carbon, blue for nitrogen and red for oxygen. Residues that distinguish HLA-B*57:01 from the abacavir insensitive allele, HLA-B*57:03, are shown in magenta for carbon, blue for nitrogen and red for oxygen. Black dashed lines show hydrogen bonds from abacavir to both the peptide and HLA-B*57:01. B. Model of abacavir-peptide-HLA interacting with the TCR. HLA is depicted in grey, peptide in cyan (carbons) and abacavir as orange for carbon and blue for nitrogen. TCR is depicted in pink.
The exact mechanisms driving the pathology seen in AHS are not fully understood. Drug altered peptide binding should generate a vastly different immunopeptidome leading to the generation T cells with multiple specificities in patients with AHS. Abacavir specific CD8+ T cells are present in patients with AHS20,67 and are polyclonal in nature17,19,20. Abacavir specific CD8+ T cell lines can be generated from both memory and naïve precursors21,68, suggesting that abacavir can stimulate cross reactive memory responses as well as promote the generation of de novo responses from naïve T cells. In support of the former proposition, AHS can occur rapidly after administration of the drug, in some instances within 2 days21, well before the generation of de novo responses could occur. Memory responses are also suggested by the rapid and exaggerated clinical responses such as fever and shock seen in AHS patients inadvertently re-challenged with abacavir. Finally, abacavir reactive T cells can be identified in the blood of abacavir-naïve individuals21. The activation threshold for memory T cells is low compared to naïve cells as they do not require second signal. How abacavir leads to the activation of naïve T cells is less clear as these is no obvious danger signal associated with the drug. However a recent study, using supra-physiological concentrations of abacavir suggests that the drug is able to activate the NLRP3 inflammasome following phorbol ester TPA or Toll-like receptor pre-stimulation69. Inflammasomes, a component of the innate immune response, are triggered by pathogen associated patterns and facilitate inflammatory responses by cleaving pro-interleukin 1β to IL-1β. It is possible that naïve T cells are activated via the effects of drug on components of the innate response, such as the NLRP3 inflammasome that creates the initial danger signal, coupled with signals derived from cross reactive memory responses to the drug or response to infectious agents such as HIV.
Carbamazepine
Carbamazepine is anticonvulsant used in the treatment of epilepsy and can lead to the development of MPE, DRESS and SJS/TEN (Table 1). MPE is most strongly associated with the carriage of HLA-A*31:01 Several class I alleles, including HLA-A*31:01 as well as, -A*01:01 and -Cw*07:01, -B*08:01 and class II alleles, DRB1*03:01, DQA1*05:01, DQB1*02:01 have been associated with the development of carbamazepine DRESS. SJS/TEN is associated with carriage of HLA-B*15:02 and HLA-A*31:01 (Table 1). The best characterised of these associations is carriage of HLA-B*15:02 and SJS/TEN. This association was first noted for Han Chinese and later for Thai, Indian and Malaysian and Japanese populations70–82. Other members of the HLA-B75 serotype, HLA-B*15:08, HLA-B*15:11 and HLA-B*15:21 are also associated with carbamazepine SJS/TEN (Table 1). Modelling studies demonstrate that carbamazepine binding to HLA-B*15:02 maps to the B pocket with a likely primary contact at the Arg62 residue on the edge of the cleft, which is a conserved amino acid among HLA B75 serotypes83. Additional contacts at the Asn63, Ile95 and Leu156 residues also likely participate in carbamazepine HLA-B*15:02 interactions, as alteration of these residues results in reduced carbamazepine binding affinity83. Although peptide loading of class I is required, neither drug nor antigen processing is essential for T-cell activation which suggests an alternative mechanism to the altered peptide repertoire of MHC-drug interaction17,83,64.
Allopurinol
Allopurinol is a purine analogue that is used in the treatment of gout and hyperuricemia. Like carbamazepine, allopurinol can cause a range of IM-ADRs (Table 1) including, MPE, DRESS and SJS/TEN. However, unlike carbamazepine, a single HLA risk allele, HLA-B*58:01, is linked to all these phenotypes. The association between allopurinol induced SJS/TEN and HLA-B*58:01 was first reported for the Han Chinese population84 and later in other populations including Europeans85, Thai86 and Japanese82. Carriage of HLA-B*58:01 has a 100% NPV for allopurinol induced SJS/TEN in Han Chinese populations, but only a ~2.7% PPV87. Functional studies indicate that HLA-B*58:01 restricted reactivity is stronger to the metabolite oxypurinol than the parent drug. This and non-covalent interactions between HLA-B*58:01 and oxypurinol are supported by the fact that allopurinol is rapidly metabolised to oxypurinol and patients with renal insufficiency are at higher risk of developing allopurinol SJS/TEN and DRESS and have a poorer prognosis88,89. These later data are consistent with the dose dependency evidenced during the induction of allopurinol and oxypurinol specific T-cell lines24,29.
Similar to carbamazepine, the presentation of allopurinol to T cells does not require-antigen processing. HLA-B*58:01 differs to HLA-B*57:01, which does not present allopurinol, by only 4 amino acids, 45 (Thr/Met), 46 (Glu/Ala), 97 (Arg/Val), and 103 (Leu/Val). Site directed mutagenesis studies suggested that Arg97, between the E and C pocket of HLA-B*58:01, may be a key contact residue for oxypurinol90. These data are consistent with molecular modelling studies which indicate that oxypurinol should make van der Waals interactions with residues surrounding the F pocket and established a hydrogen bond with Arg97 in HLA-B*58:0123. These studies also predict that allopurinol has a lower binding affinity for HLA-B*58:01 due to the lack of a critical oxygen molecule at position six in the pyrimidine ring which affects the hydrogen bond to Arg97. These data are consistent with finding that T-cell responses are skewed toward oxypurinol rather than the parent drug23,29. The putative binding sites of drug and metabolite are not consistent with a p-i model of T cell engagement leading some to suggest that intermittent disassociation of peptide and HLA could allow drug to bind under the peptide without requiring antigen processing23.
Nevirapine
Nevirapine is a non-nucleoside reverse transcriptase inhibitor used in the treatment of HIV-1. NVP hypersensitivity affects approximately 5% of HIV infected individuals who start the drug and encompasses different clinical phenotypes with cutaneous, hepatic or systemic symptoms that include SJS/TEN, DRESS and DILI. The different IM-ADR phenotypes are associated with both shared and specific class I and class II HLA alleles, which have variable distribution and risk across ethnic groups. Cutaneous reactions range in severity from mild rash through to severe diseases with high morbidity and mortality such as SJS/TEN and DRESS. Nevirapine DRESS and SJS/TEN share the same HLA-C*04:01 risk allele in African, Asian and European populations91–93. However the associations of HLA risk alleles with nevirapine DRESS show phenotype and ethnic specific differences with HLA-B*35 a risk allele for DRESS with grade III or IV rash in Asian populations91,94, HLA-DRB1*01:01 and DRB1*01:02 associated with hepatic effects in African, Asian and European populations91 and the HLA-C*08-B*14 haplotype associated with eosinophilia in Caucasians populations95,96.
A recent analysis of cutaneous NVP hypersensitivity across Caucasian, African and Asian patients has shown unique distributions of risk alleles in each ethnic group, and a common F pocket of the HLA-C peptide binding groove and position 156R that are associated with hypersensitivity. The risk HLA-C F pocket and 156R are carried by HLA-C*04:01, as well as HLA-C*05:01 and HLA-C*18:01. An independent association with cutaneous hypersensitivity was demonstrated in a group of class II alleles which share the HLA-DRB1-P4 pocket, as well as NVP HSR protection attributed to a cluster of HLA-B alleles, including HLA-B*15:01, defined by a characteristic peptide binding groove B pocket97. This approach, considering HLA alleles according to specific shared pockets within the peptide binding groove may provide insight into other IM-ADRs in which multiple HLA risk alleles with shared peptide binding specificities are implicated across different ethnic groups.
Translation into Clinical Practice
Mapping of IM-ADR to specific HLA alleles permits the use of pharmocogenomic screening to identify patients are greatest risk for the development of severe drug reactions. However, for all HLA alleles so far identified, even those with NPV as has high as 100%, the PPVs are typically much lower (Table 1). Therefore, where the NPV is 100%, specific HLA risk alleles are necessary but not sufficient for the development of IM-ADR. The utility and safety of pharmocogenomic screening for HLA risk alleles is influenced by the NPV as well as the number needed to treat to prevent one case (NNT). The NNT is a function of PPV, the frequency of the risk allele in the target population and the prevalence of the IM-ADR. Other factors may influence the utility of genetic screening including the cost effectiveness of screening in clinical practice, the severity of the clinical or economic consequences of the disease and the availability of alternative drugs that have a wider safety margin and/or do not require genetic testing98,99. Together these factors determine the cost and number of patients required to be tested to avoid one IM-ADR case and have implications for patients who may unnecessarily be denied optimal treatment, those that carry risk allele, but would not have developed an adverse reaction.
Despite these constraints, screening for risk HLA genes has been successfully applied to the prevention of IM-ADR. The first global screening program for HLA-B*57:01 prior to starting abacavir therapy has successfully eradicated reported cases of AHS in areas where routine HLA-B*57:01 screening has been introduced100,101. The high positive predictive value of HLA-B*57:01 for AHS (55%) has meant that this has been a cost-effective approach. For HLA-B*15:02 driven carbamazepine SJS/TEN, the prevalence of HLA-B*15:02 is highest amongst Asian populations (0.057–0.145 in Han Chinese, 0.085–0.275 in Thais and 0.12–0.157 in Malays) compared with European (0.01–0.02), Japanese (0.002) and Korean populations (0.004). Studies based in Taiwan and Thailand have demonstrated utility and cost-effectiveness of HLA-B*15:02 screening in such populations where the risk allele is most common102,103. Other screening programs currently being implemented or evaluated include HLA-B*58:01 testing prior to allopurinol initiation and CYP2C9*3/HLA-B*15:02/HLA-B*13:01 screening prior to phenytoin prescription in Southeast Asians104–106.
Knowledge gaps and Future Directions
Despite advances in our understanding of the genetic and phenotypic traits that potentiate IM-ADR risk, a series of unanswered questions remain. Chief amongst these are; Although the presence of an HLA risk allele appears to be necessary for the development of a specific IM-ADR, why is the PPV of such risk alleles typically <10%? What drives the exquisite tissue specificity and clinical presentation of many of these reactions? Why do these reactions occur so rapidly in many cases and show evidence of immunological memory?
The variable and for the most part, low PPV associated with specific HLA risk alleles indicates that other mechanisms contribute to the development of IM-ADRs. Some of these will be patient specific variables such as renal and/or liver function or polymorphisms in genes that regulate drug metabolism40,91,107–109. However, many features of the disease may help unravel a more cohesive model of IM-ADR. For some IM-ADRs the first manifestation of disease occurs within 1.5 days of drug exposure21. In addition, drug re-exposure is typically associated with rapid and enhanced toxicity11,57. Taken together these features suggest the involvement of memory T cells. T cells primed via exposure to previously encountered pathogens mature into one of several memory phenotypes. Central memory T cells (TCM) express CD45RO, CCR7 and L-selectin and circulate through lymph nodes via the circulation. Effector memory T cells (TEM) express CD45R0, but do not express CCR7 and L-selectin and are excluded from the lymph node, being found in the peripheral circulation and tissues. Tissue resident memory T cells (TRM) express CD45RO, CD69 and CD103 but not CCR7110. These latter cells are restricted to the tissues and do not recirculate in the peripheral blood. These TRM are poised, ready to activate and proliferate, within tissues known to be affected by IM-ADR. Therefore, it is possible that TRM play a role as key mediators of disease or in the initiation of disease, these cells remain a critical area of study in understanding the pathogenesis of IM-ADRs.
The heterologous immunity model has been proposed as a means of addressing many of the unexplained features of IM-ADR19,111. In this model, pre-formed memory T cells, educated by prior exposure to common pathogens such as HHV, cross recognize the drug-altered self-peptide as foreign and initiates an inappropriate anti-self response. In this model, the tissue specificity is dictated, at least in part, by the location of memory T cells. For instance, skin involvement in SJS/TEN would be mediated by skin TRM, recruited to and resident in the skin following prior infection with pathogens such as herpes simplex type 1 or 2. This may also explain why some patients with risk alleles such as HLA-B*58:01, which predispose to both allopurinol SJS/TEN and DRESS develop one condition over another depending on the specific memory cell population that cross-recognizes drug, the location of this population and the tissue specific repertoire of self-peptides. In an analogous situation, solid organ transplant rejection, it is clear that cross-reactive T cells mediate alloreactivity and in many instances these cross-reactive T cells have cognate specificity HHV112.
Although many questions remain in explaining the nature of T-cell mediated ADRs, the characterisation of clear HLA associations are the critical first step. Well characterised HLA associations for particular IM-ADRs, such as HLA-B*57:01 and AHS or HLA-B*15:02 and SJS/TEN in Asian populations or HLA-B*58:01 and allopurinol SJS/TEN or DRESS continue to provide invaluable models that allow us to explore the unknown factors that contribute to variation in IM-ADR phenotypes and explain susceptibility of certain individuals such as differences in drug metabolism, TCR interactions and contributions from the innate immune system. Taken together, these studies increase our understanding of all ADRs and provide a foundation to explore new drug induced adverse reactions as they arise.
Acknowledgments
Funding Sources:
Dr. Phillips funding is supported through 1P50GM115305-01, 1P30AI110527-01A1, 1 R13AR71267-01, The National Health & Medical Research Association (Australia) and Australian Centre for HIV & Hepatitis Research (ACH2)
Abbreviations used
- ADR
Adverse drug reaction
- AGEP
Acute generalized exanthematous pustulosis
- EBV
Epstein Barr virus
- CMV
Cytomegalovirus
- DILI
Drug-induced liver disease
- DRESS
Drug-reaction with eosinophilia and systemic symptoms
- HHV
Human herpesvirus
- HLA
Human leukocyte antigen
- IM-ADR
Immunologically mediated adverse drug reaction
- MPE
Maculopapular exanthema
- MRGPRX2
Mas-related G protein-coupled receptor
- MHC
Major histocompatibility complex
- NNT
Number needed to treat (to prevent one case)
- NPV
Negative predictive value
- p-i
pharmacological interactions
- PPV
Positive predictive value
- SJS
Stevens-Johnson syndrome
- TAP
transporter associated with antigen presentation
- TCR
T-cell receptor
- TEN
Toxic epidermal necrolysis
- Treg
Regulatory T cells
Footnotes
Conflicts of Interest:
Dr. Phillips is co-Director of IIID Pty Ltd that holds a patent for HLA-B*57:01 testing. The authors have no other competing interests or conflicts of interest to declare.
References
- 1.Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: A meta-analysis of prospective studies. JAMA. 1998;279(15):1200–1205. doi: 10.1001/jama.279.15.1200. [DOI] [PubMed] [Google Scholar]
- 2.Hakkarainen KM, Hedna K, Petzold M, Hägg S. Percentage of Patients with Preventable Adverse Drug Reactions and Preventability of Adverse Drug Reactions – A Meta-Analysis. PLOS ONE. 2012;7(3):e33236. doi: 10.1371/journal.pone.0033236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kongkaew C, Noyce PR, Ashcroft DM. Hospital Admissions Associated with Adverse Drug Reactions: A Systematic Review of Prospective Observational Studies. Annals of Pharmacotherapy. 2008;42(7–8):1017–1025. doi: 10.1345/aph.1L037. [DOI] [PubMed] [Google Scholar]
- 4.Pirmohamed M, James S, Meakin S, et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. BMJ (Clinical research ed) 2004;329(7456):15–19. doi: 10.1136/bmj.329.7456.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McNeil BD, Pundir P, Meeker S, et al. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature. 2015;519(7542):237–241. doi: 10.1038/nature14022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pavlos R, Mallal S, Ostrov D, et al. T Cell-Mediated Hypersensitivity Reactions to Drugs. Annual review of medicine. 2014 doi: 10.1146/annurev-med-050913-022745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White KD, Gaudieri S, Phillips E. HLA and the pharmacogenomics of drug hypersensitivity. In: Padmanabhan S, editor. Handbook of Pharmacogenomics and Stratefied Medicine. Elsevier, Inc; 2014. pp. 437–465. [Google Scholar]
- 8.Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh Steven GE. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Research. 2015;43(Database issue):D423–D431. doi: 10.1093/nar/gku1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blum JS, Wearsch PA, Cresswell P. Pathways of Antigen Processing. Annual review of immunology. 2013;31(1):443–473. doi: 10.1146/annurev-immunol-032712-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Illing PT, Mifsud NA, Purcell AW. Allotype specific interactions of drugs and HLA molecules in hypersensitivity reactions. Curr Opin Immunol. 2016;42:31–40. doi: 10.1016/j.coi.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Pichler W, Yawalkar N, Schmid S, Helbling A. Pathogenesis of drug-induced exanthems. Allergy. 2002;57(10):884–893. doi: 10.1034/j.1398-9995.2002.02161.x. [DOI] [PubMed] [Google Scholar]
- 12.Pichler WJ. Delayed drug hypersensitivity reactions. Annals of internal medicine. 2003;139(8):683–693. doi: 10.7326/0003-4819-139-8-200310210-00012. [DOI] [PubMed] [Google Scholar]
- 13.Naisbitt DJ, Gordon SF, Pirmohamed M, et al. Antigenicity and immunogenicity of sulphamethoxazole: demonstration of metabolism-dependent haptenation and T-cell proliferation in vivo. British journal of pharmacology. 2001;133(2):295–305. doi: 10.1038/sj.bjp.0704074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Padovan E, Mauri-Hellweg D, Pichler WJ, Weltzien HU. T cell recognition of penicillin G: structural features determining antigenic specificity. Eur J Immunol. 1996;26(1):42–48. doi: 10.1002/eji.1830260107. [DOI] [PubMed] [Google Scholar]
- 15.Pichler WJ, Beeler A, Keller M, et al. Pharmacological interaction of drugs with immune receptors: the p-i concept. Allergology international : official journal of the Japanese Society of Allergology. 2006;55(1):17–25. doi: 10.2332/allergolint.55.17. [DOI] [PubMed] [Google Scholar]
- 16.Pichler WJWS. Interaction of small molecules with specific immune receptors: the p-i concept and its consequences. Current Immunology Reviews. 2014;10:7–18. [Google Scholar]
- 17.Illing PT, Vivian JP, Dudek NL, et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature. 2012;486(7404):554–558. doi: 10.1038/nature11147. [DOI] [PubMed] [Google Scholar]
- 18.Ostrov DA, Grant BJ, Pompeu YA, et al. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(25):9959–9964. doi: 10.1073/pnas.1207934109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chessman D, Kostenko L, Lethborg T, et al. Human Leukocyte Antigen Class I-Restricted Activation of CD8+ T Cells Provides the Immunogenetic Basis of a Systemic Drug Hypersensitivity. Immunity. 2008;28(6):822–832. doi: 10.1016/j.immuni.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 20.Adam J, Eriksson KK, Schnyder B, Fontana S, Pichler WJ, Yerly D. Avidity determines T-cell reactivity in abacavir hypersensitivity. European Journal of Immunology. 2012;42(7):1706–1716. doi: 10.1002/eji.201142159. [DOI] [PubMed] [Google Scholar]
- 21.Lucas A, Lucas M, Strhyn A, et al. Abacavir-Reactive Memory T Cells Are Present in Drug Naïve Individuals. PLoS ONE. 2015;10(2):e0117160. doi: 10.1371/journal.pone.0117160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bell CC, Faulkner L, Martinsson K, et al. T-cells from HLA-B*57:01+ human subjects are activated with abacavir through two independent pathways and induce cell death by multiple mechanisms. Chem Res Toxicol. 2013;26(5):759–766. doi: 10.1021/tx400060p. [DOI] [PubMed] [Google Scholar]
- 23.Yun J, Marcaida MJ, Eriksson KK, et al. Oxypurinol Directly and Immediately Activates the Drug-Specific T Cells via the Preferential Use of HLA-B*58:01. The Journal of Immunology. 2014;192(7):2984–2993. doi: 10.4049/jimmunol.1302306. [DOI] [PubMed] [Google Scholar]
- 24.Yun J, Mattsson J, Schnyder K, et al. Allopurinol hypersensitivity is primarily mediated by dose-dependent oxypurinol-specific T cell response. Clin Exp Allergy. 2013;43(11):1246–1255. doi: 10.1111/cea.12184. [DOI] [PubMed] [Google Scholar]
- 25.Ko TM, Chung WH, Wei CY, et al. Shared and restricted T-cell receptor use is crucial for carbamazepine-induced Stevens-Johnson syndrome. The Journal of allergy and clinical immunology. 2011;128(6):1266–1276. e1211. doi: 10.1016/j.jaci.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 26.Farrell J, Lichtenfels M, Sullivan A, et al. Activation of carbamazepine-responsive T-cell clones with metabolically inert halogenated derivatives. Journal of Allergy and Clinical Immunology. 2013;132(2):493–495. doi: 10.1016/j.jaci.2013.02.045. [DOI] [PubMed] [Google Scholar]
- 27.Faulkner L, Gibson A, Sullivan A, et al. Detection of primary T cell responses to drugs and chemicals in HLA-typed volunteers: implications for the prediction of drug immunogenicity. Toxicological sciences : an official journal of the Society of Toxicology. 2016 doi: 10.1093/toxsci/kfw177. [DOI] [PubMed]
- 28.Wuillemin N, Terracciano L, Beltraminelli H, et al. T Cells Infiltrate the Liver and Kill Hepatocytes in HLA-B*57:01-Associated Floxacillin-Induced Liver Injury. The American journal of pathology. 2014;184(6):1677–1682. doi: 10.1016/j.ajpath.2014.02.018. [DOI] [PubMed] [Google Scholar]
- 29.Chung W, Pan RY, Chu MT, et al. Oxypurinol-Specific T Cells Possess Preferential TCR Clonotypes and Express Granulysin in Allopurinol-Induced Severe Cutaneous Adverse Reactions. J Invest Dermatol. 2015;135(9):2237–2248. doi: 10.1038/jid.2015.165. [DOI] [PubMed] [Google Scholar]
- 30.Ko T-M, Chung W-H, Wei C-Y, et al. Shared and restricted T-cell receptor use is crucial for carbamazepine-induced Stevens-Johnson syndrome. Journal of Allergy and Clinical Immunology. 2011;128(6):1266–1276. e1211. doi: 10.1016/j.jaci.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 31.Zhou P, Zhang S, Wang Y, Yang C, Huang J. Structural modeling of HLA-B*1502/peptide/carbamazepine/T-cell receptor complex architecture: implication for the molecular mechanism of carbamazepine-induced Stevens-Johnson syndrome/toxic epidermal necrolysis. Journal of biomolecular structure & dynamics. 2016;34(8):1806–1817. doi: 10.1080/07391102.2015.1092476. [DOI] [PubMed] [Google Scholar]
- 32.Lee HY, Chung WH. Toxic epidermal necrolysis: the year in review. Current opinion in allergy and clinical immunology. 2013;13(4):330–336. doi: 10.1097/ACI.0b013e3283630cc2. [DOI] [PubMed] [Google Scholar]
- 33.Tohyama M, Watanabe H, Murakami S, et al. Possible involvement of CD14+ CD16+ monocyte lineage cells in the epidermal damage of Stevens-Johnson syndrome and toxic epidermal necrolysis. The British journal of dermatology. 2012;166(2):322–330. doi: 10.1111/j.1365-2133.2011.10649.x. [DOI] [PubMed] [Google Scholar]
- 34.Le Cleach L, Delaire S, Boumsell L, et al. Blister fluid T lymphocytes during toxic epidermal necrolysis are functional cytotoxic cells which express human natural killer (NK) inhibitory receptors. Clinical and experimental immunology. 2000;119(1):225–230. doi: 10.1046/j.1365-2249.2000.01119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leyva L, Torres MJ, Posadas S, et al. Anticonvulsant-induced toxic epidermal necrolysis: monitoring the immunologic response. The Journal of allergy and clinical immunology. 2000;105(1 Pt 1):157–165. doi: 10.1016/s0091-6749(00)90191-x. [DOI] [PubMed] [Google Scholar]
- 36.Nassif A, Bensussan A, Dorothee G, et al. Drug specific cytotoxic T-cells in the skin lesions of a patient with toxic epidermal necrolysis. The Journal of investigative dermatology. 2002;118(4):728–733. doi: 10.1046/j.1523-1747.2002.01622.x. [DOI] [PubMed] [Google Scholar]
- 37.Nassif A, Bensussan A, Boumsell L, et al. Toxic epidermal necrolysis: effector cells are drug-specific cytotoxic T cells. The Journal of allergy and clinical immunology. 2004;114(5):1209–1215. doi: 10.1016/j.jaci.2004.07.047. [DOI] [PubMed] [Google Scholar]
- 38.Chung W-H, Hung S-I, Yang J-Y, et al. Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nature medicine. 2008;14(12):1343–1350. doi: 10.1038/nm.1884. [DOI] [PubMed] [Google Scholar]
- 39.Chung WH, Pan RY, Chu MT, et al. Oxypurinol-Specific T Cells Possess Preferential TCR Clonotypes and Express Granulysin in Allopurinol-Induced Severe Cutaneous Adverse Reactions. The Journal of investigative dermatology. 2015;135(9):2237–2248. doi: 10.1038/jid.2015.165. [DOI] [PubMed] [Google Scholar]
- 40.Chung WH, Chang WC, Stocker SL, et al. Insights into the poor prognosis of allopurinol-induced severe cutaneous adverse reactions: the impact of renal insufficiency, high plasma levels of oxypurinol and granulysin. Annals of the rheumatic diseases. 2015;74(12):2157–2164. doi: 10.1136/annrheumdis-2014-205577. [DOI] [PubMed] [Google Scholar]
- 41.Chen Y, Chiu H, Chu C. Drug reaction with eosinophilia and systemic symptoms: A retrospective study of 60 cases. Archives of dermatology. 2010;146(12):1373–1379. doi: 10.1001/archdermatol.2010.198. [DOI] [PubMed] [Google Scholar]
- 42.Shiohara T, Kano Y, Takahashi R, Ishida T, Mizukawa Y. Drug-induced hypersensitivity syndrome: recent advances in the diagnosis, pathogenesis and management. Chemical immunology and allergy. 2012;97:122–138. doi: 10.1159/000335624. [DOI] [PubMed] [Google Scholar]
- 43.Morito H, Ogawa K, Fukumoto T, et al. Increased ratio of FoxP3+ regulatory T cells/CD3+ T cells in skin lesions in drug-induced hypersensitivity syndrome/drug rash with eosinophilia and systemic symptoms. Clinical and experimental dermatology. 2014;39(3):284–291. doi: 10.1111/ced.12246. [DOI] [PubMed] [Google Scholar]
- 44.Takahashi R, Kano Y, Yamazaki Y, Kimishima M, Mizukawa Y, Shiohara T. Defective regulatory T cells in patients with severe drug eruptions: timing of the dysfunction is associated with the pathological phenotype and outcome. Journal of immunology. 2009;182(12):8071–8079. doi: 10.4049/jimmunol.0804002. [DOI] [PubMed] [Google Scholar]
- 45.Shiohara T, Ushigome Y, Kano Y, Takahashi R. Crucial Role of Viral Reactivation in the Development of Severe Drug Eruptions: a Comprehensive Review. Clin Rev Allergy Immunol. 2015;49(2):192–202. doi: 10.1007/s12016-014-8421-3. [DOI] [PubMed] [Google Scholar]
- 46.Miyagawa F, Nakamura Y, Miyashita K, et al. Preferential expression of CD134, an HHV-6 cellular receptor, on CD4T cells in drug-induced hypersensitivity syndrome (DIHS)/drug reaction with eosinophilia and systemic symptoms (DRESS) Journal of dermatological science. 2016;83(2):151–154. doi: 10.1016/j.jdermsci.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 47.Picard D, Janela B, Descamps V, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): a multiorgan antiviral T cell response. Science translational medicine. 2010;2(46):46ra62. doi: 10.1126/scitranslmed.3001116. [DOI] [PubMed] [Google Scholar]
- 48.Ostapowicz G, Fontana RJ, Schiødt FV, et al. REsults of a prospective study of acute liver failure at 17 tertiary care centers in the united states. Annals of internal medicine. 2002;137(12):947–954. doi: 10.7326/0003-4819-137-12-200212170-00007. [DOI] [PubMed] [Google Scholar]
- 49.Andrade RJ, Lucena MI, Fernandez MC, et al. Drug-induced liver injury: an analysis of 461 incidences submitted to the Spanish registry over a 10-year period. Gastroenterology. 2005;129(2):512–521. doi: 10.1016/j.gastro.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 50.Andrade RJ, Lucena MI, Kaplowitz N, et al. Outcome of acute idiosyncratic drug-induced liver injury: Long-term follow-up in a hepatotoxicity registry. Hepatology. 2006;44(6):1581–1588. doi: 10.1002/hep.21424. [DOI] [PubMed] [Google Scholar]
- 51.Hautekeete ML, Horsmans Y, Van Waeyenberge C, et al. HLA association of amoxicillin-clavulanate--induced hepatitis. Gastroenterology. 1999;117(5):1181–1186. doi: 10.1016/s0016-5085(99)70404-x. [DOI] [PubMed] [Google Scholar]
- 52.O’Donohue J, Oien KA, Donaldson P, et al. Co-amoxiclav jaundice: clinical and histological features and HLA class II association. Gut. 2000;47(5):717–720. doi: 10.1136/gut.47.5.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Donaldson PT, Daly AK, Henderson J, et al. Human leucocyte antigen class II genotype in susceptibility and resistance to co-amoxiclav-induced liver injury. J Hepatol. 2010;53(6):1049–1053. doi: 10.1016/j.jhep.2010.05.033. [DOI] [PubMed] [Google Scholar]
- 54.Lucena MI, Andrade RJ, Fernandez MC, et al. Determinants of the clinical expression of amoxicillin-clavulanate hepatotoxicity: a prospective series from Spain. Hepatology. 2006;44(4):850–856. doi: 10.1002/hep.21324. [DOI] [PubMed] [Google Scholar]
- 55.Stephens C, Lopez-Nevot MA, Ruiz-Cabello F, et al. HLA alleles influence the clinical signature of amoxicillin-clavulanate hepatotoxicity. PLoS One. 2013;8(7):e68111. doi: 10.1371/journal.pone.0068111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lucena MI, Molokhia M, Shen Y, et al. Susceptibility to amoxicillin-clavulanate-induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology. 2011;141(1):338–347. doi: 10.1053/j.gastro.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hetherington S, McGuirk S, Powell G, et al. Hypersensitivity reactions during therapy with the nucleoside reverse transcriptase inhibitor abacavir. Clinical Therapeutics. 2001;23(10):1603–1614. doi: 10.1016/s0149-2918(01)80132-6. [DOI] [PubMed] [Google Scholar]
- 58.Mallal S, Phillips E, Carosi G, et al. HLA-B*5701 Screening for Hypersensitivity to Abacavir. New England Journal of Medicine. 2008;358(6):568–579. doi: 10.1056/NEJMoa0706135. [DOI] [PubMed] [Google Scholar]
- 59.Mallal S, Nolan D, Witt C, et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet. 2002;359(9308):727–732. doi: 10.1016/s0140-6736(02)07873-x. [DOI] [PubMed] [Google Scholar]
- 60.Martin AM, Nolan D, Gaudieri S, et al. Predisposition to abacavir hypersensitivity conferred by HLA-B*5701 and a haplotypic Hsp70-Hom variant. Proceedings of the National Academy of Sciences. 2004;101(12):4180–4185. doi: 10.1073/pnas.0307067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hetherington S, Hughes AR, Mosteller M, et al. Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. The Lancet. 2002;359(9312):1121–1122. doi: 10.1016/S0140-6736(02)08158-8. [DOI] [PubMed] [Google Scholar]
- 62.Phillips EJ, Sullivan JR, Knowles SR, Shear NH. Utility of patch testing in patients with hypersensitivity syndromes associated with abacavir. AIDS. 2002;16(16) doi: 10.1097/00002030-200211080-00017. [DOI] [PubMed] [Google Scholar]
- 63.Mallal S, Phillips E, Carosi G, et al. HLA-B*5701 screening for hypersensitivity to abacavir. The New England journal of medicine. 2008;358(6):568–579. doi: 10.1056/NEJMoa0706135. [DOI] [PubMed] [Google Scholar]
- 64.Saag M, Balu R, Phillips E, et al. High sensitivity of human leukocyte antigen-b*5701 as a marker for immunologically confirmed abacavir hypersensitivity in white and black patients. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2008;46(7):1111–1118. doi: 10.1086/529382. [DOI] [PubMed] [Google Scholar]
- 65.Ostrov DA, Grant BJ, Pompeu YA, et al. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proceedings of the National Academy of Sciences. 2012;109(25):9959–9964. doi: 10.1073/pnas.1207934109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Norcross MA, Luo S, Lu L, et al. Abacavir induces loading of novel self-peptides into HLA-B*57: 01: an autoimmune model for HLA-associated drug hypersensitivity. AIDS. 2012;26(11):F21–F29. doi: 10.1097/QAD.0b013e328355fe8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Phillips EJ, Wong GA, Kaul R, et al. Clinical and immunogenetic correlates of abacavir hypersensitivity. AIDS. 2005;19(9):979–981. doi: 10.1097/01.aids.0000171414.99409.fb. [DOI] [PubMed] [Google Scholar]
- 68.Adam J, Wuillemin N, Watkins S, et al. Abacavir Induced T Cell Reactivity from Drug Naïve Individuals Shares Features of Allo-Immune Responses. PLoS ONE. 2014;9(4):e95339. doi: 10.1371/journal.pone.0095339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Toksoy A, Sennefelder H, Adam C, et al. Potent NLRP3 inflammasome activation by the HIV reverse-transcriptase inhibitor abacavir. Journal of Biological Chemistry. 2017 doi: 10.1074/jbc.M116.749473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chung W-H, Hung S-I, Hong H-S, et al. Medical genetics: A marker for Stevens-Johnson syndrome. Nature. 2004;428(6982):486–486. doi: 10.1038/428486a. [DOI] [PubMed] [Google Scholar]
- 71.Hung S-I, Chung W-H, Jee S-H, et al. Genetic susceptibility to carbamazepine-induced cutaneous adverse drug reactions. Pharmacogenetics and Genomics. 2006;16(4):297–306. doi: 10.1097/01.fpc.0000199500.46842.4a. [DOI] [PubMed] [Google Scholar]
- 72.Man CBL, Kwan P, Baum L, et al. Association between HLA-B*1502 Allele and Antiepileptic Drug-Induced Cutaneous Reactions in Han Chinese. Epilepsia. 2007;48(5):1015–1018. doi: 10.1111/j.1528-1167.2007.01022.x. [DOI] [PubMed] [Google Scholar]
- 73.Locharernkul C, Loplumlert J, Limotai C, et al. Carbamazepine and phenytoin induced Stevens-Johnson syndrome is associated with HLA-B*1502 allele in Thai population. Epilepsia. 2008;49(12):2087–2091. doi: 10.1111/j.1528-1167.2008.01719.x. [DOI] [PubMed] [Google Scholar]
- 74.Tassaneeyakul W, Tiamkao S, Jantararoungtong T, et al. Association between HLA-B*1502 and carbamazepine-induced severe cutaneous adverse drug reactions in a Thai population. Epilepsia. 2010;51(5):926–930. doi: 10.1111/j.1528-1167.2010.02533.x. [DOI] [PubMed] [Google Scholar]
- 75.Wu XT, Hu FY, An DM, et al. Association between carbamazepine-induced cutaneous adverse drug reactions and the HLA-B*1502 allele among patients in central China. Epilepsy & Behavior. 2010;19(3):405–408. doi: 10.1016/j.yebeh.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 76.Chang C-C, Too C-L, Murad S, Hussein SH. Association of HLA-B*1502 allele with carbamazepine-induced toxic epidermal necrolysis and Stevens–Johnson syndrome in the multi-ethnic Malaysian population. International journal of dermatology. 2011;50(2):221–224. doi: 10.1111/j.1365-4632.2010.04745.x. [DOI] [PubMed] [Google Scholar]
- 77.Then SM, Rani ZZ, Raymond AA, Ratnaningrum S, Jamal R. Frequency of the HLA-B*1502 allele contributing to carbamazepine-induced hypersensitivity reactions in a cohort of Malaysian epilepsy patients. Asian Pacific Journal of Allergy and Immunology. 2011;29(3):290–293. [PubMed] [Google Scholar]
- 78.Wang Q, Zhou J-q, Zhou L-m, et al. Association between HLA-B*1502 allele and carbamazepine-induced severe cutaneous adverse reactions in Han people of southern China mainland. Seizure. 2011;20(6):446–448. doi: 10.1016/j.seizure.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 79.Zhang Y, Wang J, Zhao L-M, et al. Strong association between HLA-B*1502 and carbamazepine-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in mainland Han Chinese patients. European journal of clinical pharmacology. 2011;67(9):885. doi: 10.1007/s00228-011-1009-4. [DOI] [PubMed] [Google Scholar]
- 80.Kulkantrakorn K, Tassaneeyakul W, Tiamkao S, et al. HLA-B*1502 Strongly Predicts Carbamazepine-Induced Stevens–Johnson Syndrome and Toxic Epidermal Necrolysis in Thai Patients with Neuropathic Pain. Pain Practice. 2012;12(3):202–208. doi: 10.1111/j.1533-2500.2011.00479.x. [DOI] [PubMed] [Google Scholar]
- 81.Ikeda H, Takahashi Y, Yamazaki E, et al. HLA class I markers in Japanese patients with carbamazepine-induced cutaneous adverse reactions. Epilepsia. 2010:51. doi: 10.1111/j.1528-1167.2009.02269.x. [DOI] [PubMed] [Google Scholar]
- 82.Kaniwa N, Saito Y, Aihara M, et al. HLA-B locus in Japanese patients with anti-epileptics and allopurinol-related Stevens-Johnson syndrome and toxic epidermal necrolysis. Pharmacogenomics. 2008:9. doi: 10.2217/14622416.9.11.1617. [DOI] [PubMed] [Google Scholar]
- 83.Wei CY, Chung WH, Huang HW, Chen YT, Hung SI. Direct interaction between HLA-B and carbamazepine activates T cells in patients with Stevens-Johnson syndrome. The Journal of allergy and clinical immunology. 2012;129(6):1562–1569. e1565. doi: 10.1016/j.jaci.2011.12.990. [DOI] [PubMed] [Google Scholar]
- 84.Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci USA. 2005:102. doi: 10.1073/pnas.0409500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lonjou C, Borot N, Sekula P, et al. A European study of HLA-B in Stevens-Johnson syndrome and toxic epidermal necrolysis related to five high-risk drugs. Pharmacogenet Genomics. 2008:18. doi: 10.1097/FPC.0b013e3282f3ef9c. [DOI] [PubMed] [Google Scholar]
- 86.Tassaneeyakul W, Jantararoungtong T, Chen P, et al. Strong association between HLA-B*5801 and allopurinol-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in a Thai population. Pharmacogenet Genomics. 2009:19. doi: 10.1097/FPC.0b013e328330a3b8. [DOI] [PubMed] [Google Scholar]
- 87.Phillips EJ, Mallal SA. Pharmacogenetics of drug hypersensitivity. Pharmacogenomics. 2010;11(7):973–987. doi: 10.2217/pgs.10.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ng CY, Yeh YT, Wang CW, et al. Impact of the HLA-B(*)58:01 Allele and Renal Impairment on Allopurinol-Induced Cutaneous Adverse Reactions. The Journal of investigative dermatology. 2016;136(7):1373–1381. doi: 10.1016/j.jid.2016.02.808. [DOI] [PubMed] [Google Scholar]
- 89.Chung W-H, Chang W-C, Stocker SL, et al. Insights into the poor prognosis of allopurinol-induced severe cutaneous adverse reactions: the impact of renal insufficiency, high plasma levels of oxypurinol and granulysin. Annals of the rheumatic diseases. 2015;74(12):2157–2164. doi: 10.1136/annrheumdis-2014-205577. [DOI] [PubMed] [Google Scholar]
- 90.Lin C-H, Chen J-K, Ko T-M, et al. Immunologic basis for allopurinol-induced severe cutaneous adverse reactions: HLA-B*58:01-restricted activation of drug-specific T cells and molecular interaction. Journal of Allergy and Clinical Immunology. 135(4):1063–1065.e1065. doi: 10.1016/j.jaci.2014.09.041. [DOI] [PubMed] [Google Scholar]
- 91.Yuan J, Guo S, Hall D, et al. Toxicogenomics of nevirapine-associated cutaneous and hepatic adverse events among populations of African, Asian, and European descent. AIDS (London, England) 2011;25(10):1271–1280. doi: 10.1097/QAD.0b013e32834779df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gao S, Gui XE, Liang K, Liu Z, Hu J, Dong B. HLA-Dependent Hypersensitivity Reaction to Nevirapine in Chinese Han HIV-Infected Patients. AIDS Res Hum Retroviruses. 2011 doi: 10.1089/AID.2011.0107. [DOI] [PubMed] [Google Scholar]
- 93.Likanonsakul S, Rattanatham T, Feangvad S, et al. HLA-Cw*04 allele associated with nevirapine-induced rash in HIV-infected Thai patients. AIDS Research and Therapy. 2009;6(1):22. doi: 10.1186/1742-6405-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chantarangsu S, Mushiroda T, Mahasirimongkol S, et al. HLA-B*3505 allele is a strong predictor for nevirapine-induced skin adverse drug reactions in HIV-infected Thai patients. Pharmacogenetics and Genomics. 2009;19(2):139–146. doi: 10.1097/FPC.0b013e32831d0faf. [DOI] [PubMed] [Google Scholar]
- 95.Keane NM, Pavlos RK, McKinnon E, et al. HLA Class I restricted CD8+ and Class II restricted CD4+ T cells are implicated in the pathogenesis of nevirapine hypersensitivity. AIDS. 2014;28(13):1891–1901. doi: 10.1097/QAD.0000000000000345. [DOI] [PubMed] [Google Scholar]
- 96.Littera R, Carcassi C, Masala A, et al. HLA-dependent hypersensitivity to nevirapine in Sardinian HIV patients. AIDS. 2006;20(12) doi: 10.1097/01.aids.0000238408.82947.09. [DOI] [PubMed] [Google Scholar]
- 97.Pavlos R, McKinnon EJ, Ostrov DA, et al. Shared peptide binding of HLA Class I and II alleles associate with cutaneous nevirapine hypersensitivity and identify novel risk alleles. Scientific reports. 2017;7(1):8653. doi: 10.1038/s41598-017-08876-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Veenstra DL. The value of routine pharmacogenomic screening-Are we there yet? A perspective on the costs and benefits of routine screening-shouldn’t everyone have this done? Clinical pharmacology and therapeutics. 2016;99(2):164–166. doi: 10.1002/cpt.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mallal S, Phillips E. Introduction of pharmacogenetic screening to HIV clinical practice: potential benefits and challenges. Eur Infect Dis. 2007 Jun 1;:13–18. [Google Scholar]
- 100.Phillips E, Mallal S. Successful translation of pharmacogenetics into the clinic: the abacavir example. Molecular diagnosis & therapy. 2009;13(1):1–9. doi: 10.1007/BF03256308. [DOI] [PubMed] [Google Scholar]
- 101.Yip VL, Hawcutt DB, Pirmohamed M. Pharmacogenetic Markers of Drug Efficacy and Toxicity. Clinical pharmacology and therapeutics. 2015;98(1):61–70. doi: 10.1002/cpt.135. [DOI] [PubMed] [Google Scholar]
- 102.Chen P, Lin JJ, Lu CS, et al. Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan. The New England journal of medicine. 2011;364(12):1126–1133. doi: 10.1056/NEJMoa1009717. [DOI] [PubMed] [Google Scholar]
- 103.Locharernkul C, Shotelersuk V, Hirankarn N. HLA-B* 1502 screening: time to clinical practice. Epilepsia. 2010;51(5):936–938. doi: 10.1111/j.1528-1167.2010.02549.x. [DOI] [PubMed] [Google Scholar]
- 104.Ko TM, Tsai CY, Chen SY, et al. Use of HLA-B*58:01 genotyping to prevent allopurinol induced severe cutaneous adverse reactions in Taiwan: national prospective cohort study. BMJ (Clinical research ed) 2015;351:h4848. doi: 10.1136/bmj.h4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Saokaew S, Tassaneeyakul W, Maenthaisong R, Chaiyakunapruk N. Cost-effectiveness analysis of HLA-B*5801 testing in preventing allopurinol-induced SJS/TEN in Thai population. Clinical chemistry and laboratory medicine. 2014;9(4):e94294. doi: 10.1371/journal.pone.0094294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Caudle KE, Rettie AE, Whirl-Carrillo M, et al. Clinical pharmacogenetics implementation consortium guidelines for CYP2C9 and HLA-B genotypes and phenytoin dosing. Clinical pharmacology and therapeutics. 2014;96(5):542–548. doi: 10.1038/clpt.2014.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chung WH, Chang WC, Lee YS, et al. Genetic variants associated with phenytoin-related severe cutaneous adverse reactions. JAMA. 2014;312(5):525–534. doi: 10.1001/jama.2014.7859. [DOI] [PubMed] [Google Scholar]
- 108.Tassaneeyakul W, Prabmeechai N, Sukasem C, et al. Associations between HLA class I and cytochrome P450 2C9 genetic polymorphisms and phenytoin-related severe cutaneous adverse reactions in a Thai population. Pharmacogenetics and Genomics. 2016;26(5):225–234. doi: 10.1097/FPC.0000000000000211. [DOI] [PubMed] [Google Scholar]
- 109.Bertrand J, Chou M, Richardson DM, et al. Multiple genetic variants predict steady-state nevirapine clearance in HIV-infected Cambodians. Pharmacogenet Genomics. 2012;22(12):868–876. doi: 10.1097/FPC.0b013e32835a5af2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mueller SN, Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol. 2016;16(2):79–89. doi: 10.1038/nri.2015.3. [DOI] [PubMed] [Google Scholar]
- 111.White KD, Chung W-H, Hung S-I, Mallal S, Phillips EJ. Evolving models of the immunopathogenesis of T cells mediated drug allergy: The role of host, pathogens, and drug response. Journal of Allergy and Clinical Immunology. 2015;136(2):219–234. doi: 10.1016/j.jaci.2015.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Amir AL, D’Orsogna LJ, Roelen DL, et al. Allo-HLA reactivity of virus-specific memory T cells is common. Blood. 2010;115(15):3146–3157. doi: 10.1182/blood-2009-07-234906. [DOI] [PubMed] [Google Scholar]
- 113.Mallal S, Nolan D, Witt C, et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. The Lancet. 2002;359(9308):727–732. doi: 10.1016/s0140-6736(02)07873-x. [DOI] [PubMed] [Google Scholar]
- 114.Saag M, Balu R, Phillips E, et al. High Sensitivity of Human Leukocyte Antigen-B*5701 as a Marker for Immunologically Confirmed Abacavir Hypersensitivity in White and Black Patients. Clinical Infectious Diseases. 2008;46(7):1111–1118. doi: 10.1086/529382. [DOI] [PubMed] [Google Scholar]
- 115.Kaniwa N, Saito Y, Aihara M, et al. HLA-B*1511 is a risk factor for carbamazepine-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in Japanese patients. Epilepsia. 2010;51(12):2461–2465. doi: 10.1111/j.1528-1167.2010.02766.x. [DOI] [PubMed] [Google Scholar]
- 116.Kim S-H, Lee KW, Song W-J, et al. Carbamazepine-induced severe cutaneous adverse reactions and HLA genotypes in Koreans. Epilepsy Research. 2011;97(1–2):190–197. doi: 10.1016/j.eplepsyres.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 117.Jaruthamsophon K, Tipmanee V, Sangiemchoey A, Sukasem C, Limprasert P. HLA-B*15:21 and carbamazepine-induced Stevens-Johnson syndrome: pooled-data and in silico analysis. Scientific reports. 2017;7:45553. doi: 10.1038/srep45553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McCormack M, Alfirevic A, Bourgeois S, et al. HLA-A*3101 and Carbamazepine-Induced Hypersensitivity Reactions in Europeans. New England Journal of Medicine. 2011;364(12):1134–1143. doi: 10.1056/NEJMoa1013297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ozeki T, Mushiroda T, Yowang A, et al. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Human Molecular Genetics. 2011;20(5):1034–1041. doi: 10.1093/hmg/ddq537. [DOI] [PubMed] [Google Scholar]
- 120.Niihara H, Kakamu T, Fujita Y, Kaneko S, Morita E. HLA-A31 strongly associates with carbamazepine-induced adverse drug reactions but not with carbamazepine-induced lymphocyte proliferation in a Japanese population. The Journal of Dermatology. 2012;39(7):594–601. doi: 10.1111/j.1346-8138.2011.01457.x. [DOI] [PubMed] [Google Scholar]
- 121.Alfirevic A, Jorgensen AL, Williamson PR, Chadwick DW, Park BK, Pirmohamed M. HLA-B locus in Caucasian patients with carbamazepine hypersensitivity. Pharmacogenomics. 2006;7(6):813–818. doi: 10.2217/14622416.7.6.813. [DOI] [PubMed] [Google Scholar]
- 122.Genin E, Chen DP, Hung SI, et al. HLA-A*31:01 and different types of carbamazepine-induced severe cutaneous adverse reactions: an international study and meta-analysis. Pharmacogenomics J. 2014;14(3):281–288. doi: 10.1038/tpj.2013.40. [DOI] [PubMed] [Google Scholar]
- 123.Hung SI, Chung WH, Jee SH, et al. Genetic susceptibility to carbamazepine-induced cutaneous adverse drug reactions. Pharmacogenet Genomics. 2006:16. doi: 10.1097/01.fpc.0000199500.46842.4a. [DOI] [PubMed] [Google Scholar]
- 124.Yip VL, Pirmohamed M. The HLA-A*31:01 allele: influence on carbamazepine treatment. Pharmacogenomics and personalized medicine. 2017;10:29–38. doi: 10.2147/PGPM.S108598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tassaneeyakul W, Jantararoungtong T, Chen P, et al. Strong association between HLA-B*5801 and allopurinol-induced Stevens–Johnson syndrome and toxic epidermal necrolysis in a Thai population. Pharmacogenetics and Genomics. 2009;19(9):704–709. doi: 10.1097/FPC.0b013e328330a3b8. [DOI] [PubMed] [Google Scholar]
- 126.Chan SH, Tan T. HLA and allopurinol drug eruption. Dermatologica Sinica. 1989;179(1):32–33. doi: 10.1159/000248097. [DOI] [PubMed] [Google Scholar]
- 127.Hung S-I, Chung W-H, Liou L-B, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(11):4134–4139. doi: 10.1073/pnas.0409500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Génin E, Schumacher M, Roujeau J-C, et al. Genome-wide association study of Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis in Europe. Orphanet Journal of Rare Diseases. 2011;6(1):52. doi: 10.1186/1750-1172-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kang H-R, Jee YK, Kim Y-S, et al. Positive and negative associations of HLA class I alleles with allopurinol-induced SCARs in Koreans. Pharmacogenetics and Genomics. 2011;21(5):303–307. doi: 10.1097/FPC.0b013e32834282b8. [DOI] [PubMed] [Google Scholar]
- 130.Somkrua R, Eickman EE, Saokaew S, Lohitnavy M, Chaiyakunapruk N. Association of HLA-B*5801 allele and allopurinol-induced stevens johnson syndrome and toxic epidermal necrolysis: a systematic review and meta-analysis. BMC Medical Genetics. 2011;12(1):118. doi: 10.1186/1471-2350-12-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sukasem C, Jantararoungtong T, Kuntawong P, et al. HLA-B (*) 58:01 for Allopurinol-Induced Cutaneous Adverse Drug Reactions: Implication for Clinical Interpretation in Thailand. Frontiers in pharmacology. 2016;7:186. doi: 10.3389/fphar.2016.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lin L-C, Lai P-C, Yang S-F, Yang R-C. Oxcarbazepine-induced Stevens-Johnson Syndrome: A Case Report. The Kaohsiung Journal of Medical Sciences. 2009;25(2):82–86. doi: 10.1016/S1607-551X(09)70045-2. [DOI] [PubMed] [Google Scholar]
- 133.Hung SI, Chung WH, Liu ZS, et al. Common risk allele in aromatic antiepileptic-drug induced Stevens-Johnson syndrome and toxic epidermal necrolysis in Han Chinese. Pharmacogenomics. 2010;11(3):349–356. doi: 10.2217/pgs.09.162. [DOI] [PubMed] [Google Scholar]
- 134.Chen CB, Hsiao YH, Wu T, et al. Risk and association of HLA with oxcarbazepine-induced cutaneous adverse reactions in Asians. Neurology. 2017;88(1):78–86. doi: 10.1212/WNL.0000000000003453. [DOI] [PubMed] [Google Scholar]
- 135.An DM, Wu XT, Hu FY, Yan B, Stefan H, Zhou D. Association study of lamotrigine-induced cutaneous adverse reactions and HLA-B*1502 in a Han Chinese population. Epilepsy Res. 2010;92(2–3):226–230. doi: 10.1016/j.eplepsyres.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 136.Shi YW, Min FL, Liu XR, et al. Hla-B alleles and lamotrigine-induced cutaneous adverse drug reactions in the Han Chinese population. Basic Clin Pharmacol Toxicol. 2011;109(1):42–46. doi: 10.1111/j.1742-7843.2011.00681.x. [DOI] [PubMed] [Google Scholar]
- 137.Chung W, Chang W, Lee Y, et al. Genetic variants associated with phenytoin-related severe cutaneous adverse reactions. JAMA. 2014;312(5):525–534. doi: 10.1001/jama.2014.7859. [DOI] [PubMed] [Google Scholar]
- 138.Carr DF, Chaponda M, Jorgensen AL, et al. Association of Human Leukocyte Antigen Alleles and Nevirapine Hypersensitivity in a Malawian HIV-Infected Population. Clinical Infectious Diseases. 2013;56(9):1330–1339. doi: 10.1093/cid/cit021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Martin AM, Nolan D, James I, et al. Predisposition to nevirapine hypersensitivity associated with HLA-DRB1*0101 and abrogated by low CD4 T-cell counts. AIDS. 2005;19(1) doi: 10.1097/00002030-200501030-00014. [DOI] [PubMed] [Google Scholar]
- 140.Gatanaga H, Yazaki H, Tanuma J, et al. HLA-Cw8 primarily associated with hypersensitivity to nevirapine. AIDS. 2007;21(2) doi: 10.1097/QAD.0b013e32801199d9. [DOI] [PubMed] [Google Scholar]
- 141.Gao S, Gui X-e, Liang K, Liu Z, Hu J, Dong B. HLA-Dependent Hypersensitivity Reaction to Nevirapine in Chinese Han HIV-Infected Patients. AIDS Research and Human Retroviruses. 2011;28(6):540–543. doi: 10.1089/AID.2011.0107. [DOI] [PubMed] [Google Scholar]
- 142.Chantarangsu S, Mushiroda T, Mahasirimongkol S, et al. HLA-B*3505 allele is a strong predictor for nevirapine-induced skin adverse drug reactions in HIV-infected Thai patients. Pharmacogenetics and Genomics. 2009;19(2) doi: 10.1097/FPC.0b013e32831d0faf. [DOI] [PubMed] [Google Scholar]
- 143.Vitezica ZG, Milpied B, Lonjou C, et al. HLA-DRB1*01 associated with cutaneous hypersensitivity induced by nevirapine and efavirenz. AIDS. 2008;22(4) doi: 10.1097/QAD.0b013e3282f37812. [DOI] [PubMed] [Google Scholar]
- 144.Zhang F-R, Liu H, Irwanto A, et al. HLA-B*13:01 and the Dapsone Hypersensitivity Syndrome. New England Journal of Medicine. 2013;369(17):1620–1628. doi: 10.1056/NEJMoa1213096. [DOI] [PubMed] [Google Scholar]
- 145.Hautekeete ML, Horsmans Y, van Waeyenberge C, et al. HLA association of amoxicillin-clavulanate–induced hepatitis. Gastroenterology. 1999;117(5):1181–1186. doi: 10.1016/s0016-5085(99)70404-x. [DOI] [PubMed] [Google Scholar]
- 146.O’Donohue J, Oien KA, Donaldson P, et al. Co-amoxiclav jaundice: clinical and histological features and HLA class II association. Gut. 2000;47(5):717–720. doi: 10.1136/gut.47.5.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lucena MI, Molokhia M, Shen Y, et al. Susceptibility to Amoxicillin-Clavulanate-Induced Liver Injury Is Influenced by Multiple HLA Class I and II Alleles. Gastroenterology. 2011;141(1):338–347. doi: 10.1053/j.gastro.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Singer JB, Lewitzky S, Leroy E, et al. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat Genet. 2010;42(8):711–714. doi: 10.1038/ng.632. [DOI] [PubMed] [Google Scholar]
- 149.Kindmark A, Jawaid A, Harbron CG, et al. Genome-wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. Pharmacogenomics J. 2007;8(3):186–195. doi: 10.1038/sj.tpj.6500458. [DOI] [PubMed] [Google Scholar]
- 150.Berson A, Fréneaux E, Larrey D, et al. Possible role of HLA in hepatotoxicity. Journal of Hepatology. 1994;20(3):336–342. doi: 10.1016/s0168-8278(94)80004-9. [DOI] [PubMed] [Google Scholar]
- 151.Daly AK, Donaldson PT, Bhatnagar P, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41(7):816–819. doi: 10.1038/ng.379. [DOI] [PubMed] [Google Scholar]
- 152.Spraggs CF, Budde LR, Briley LP, et al. HLA-DQA1*02:01 Is a Major Risk Factor for Lapatinib-Induced Hepatotoxicity in Women With Advanced Breast Cancer. Journal of Clinical Oncology. 2011;29(6):667–673. doi: 10.1200/JCO.2010.31.3197. [DOI] [PubMed] [Google Scholar]
- 153.Chen P-L, Shih S-R, Wang P-W, et al. Genetic determinants of antithyroid drug-induced agranulocytosis by human leukocyte antigen genotyping and genome-wide association study. Nature Communications. 2015;6:7633. doi: 10.1038/ncomms8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Cheung CL, Sing CW, Tang CSM, et al. HLA-B*38:02:01 predicts carbimazole/methimazole-induced agranulocytosis. Clinical Pharmacology & Therapeutics. 2016;99(5):555–561. doi: 10.1002/cpt.309. [DOI] [PubMed] [Google Scholar]
- 155.He Y, Zheng J, Zhang Q, et al. Association of HLA-B and HLA-DRB1 polymorphisms with antithyroid drug-induced agranulocytosis in a Han population from northern China. Scientific reports. 2017;7(1):11950. doi: 10.1038/s41598-017-12350-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hallberg P, Eriksson N, Ibañez L, et al. Genetic variants associated with antithyroid drug-induced agranulocytosis: a genome-wide association study in a European population. The Lancet Diabetes & Endocrinology. 2016;4(6):507–516. doi: 10.1016/S2213-8587(16)00113-3. [DOI] [PubMed] [Google Scholar]
- 157.Tamai H, Sudo T, Kimura A, et al. Association between the DRB1*08032 histocompatibility antigen and methimazole-induced agranulocytosis in japanese patients with graves disease. Annals of internal medicine. 1996;124(5):490–494. doi: 10.7326/0003-4819-124-5-199603010-00005. [DOI] [PubMed] [Google Scholar]
- 158.Saito T, Ikeda M, Mushiroda T, et al. Pharmacogenomic Study of Clozapine-Induced Agranulocytosis/Granulocytopenia in a Japanese Population. Biological Psychiatry. 2016;80(8):636–642. doi: 10.1016/j.biopsych.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 159.Goldstein JI, Fredrik Jarskog L, Hilliard C, et al. Clozapine-induced agranulocytosis is associated with rare HLA-DQB1 and HLA-B alleles. Nature Communications. 2014;5:4757. doi: 10.1038/ncomms5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Legge SE, Hamshere ML, Ripke S, et al. Genome-wide common and rare variant analysis provides novel insights into clozapine-associated neutropenia. Mol Psychiatry. 2016 doi: 10.1038/mp.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Heap GA, Weedon MN, Bewshea CM, et al. HLA-DQA1-HLA-DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nat Genet. 2014;46(10):1131–1134. doi: 10.1038/ng.3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mammen AL. Statin-Associated Autoimmune Myopathy. New England Journal of Medicine. 2016;374(7):664–669. doi: 10.1056/NEJMra1515161. [DOI] [PubMed] [Google Scholar]
- 163.Fernandez CA, Smith C, Yang W, et al. HLA-DRB1*07:01 is associated with a higher risk of asparaginase allergies. Blood. 2014;124(8):1266–1276. doi: 10.1182/blood-2014-03-563742. [DOI] [PMC free article] [PubMed] [Google Scholar]