

Abstract
Prostate cancer (PCa) is the most commonly diagnosed malignancy in males, and, in the United States, is the second leading cause of cancer-related death for men older than 40 years. There is a higher incidence of PCa for African Americans (AAs) than for European-Americans (EAs). Investigations related to the incidence of PCa-related health disparities for AAs suggest that there are differences in the genetic makeup of these populations. Other differences are environmentally induced (e.g., diet and lifestyle), and the exposures are different. Men who immigrate from Eastern to Western countries have a higher risk of PCa than men in their native countries. However, the number of immigrants developing PCa is still lower than that of men in Western countries, suggesting that genetic factors are involved in the development of PCa. Altered genetic polymorphisms are associated with PCa progression. Androgens and the androgen receptor (AR) are involved in the development and progression of PCa. For populations with diverse racial/ethnic backgrounds, differences in lifestyle, diet, and biology, including genetic mutations/polymorphisms and levels of androgens and AR, are risk factors for PCa. Here, we provide an immune-biological perspective on PCa in relation to racial/ethnic disparities and identify factors associated with the disproportionate incidence of PCa and its clinical outcomes.
Keywords: Health Disparity, Prostate Cancer, Immunotherapy
1. Introduction
In the United States (US), prostate cancer (PCa) is the most commonly diagnosed malignancy for males and the second leading cause of cancer-related deaths for men older than 40 years [1]. However, there are substantial disparities in the incidence and mortality of this disease among different races/ethnicities and countries. Although the incidence of PCa is declining, the overall PCa-related mortality continues to rise for African-American (AA) men [1]. Relative to European-Americans (EAs), AAs are more susceptible to PCa [2, 3]. There may be various reasons for the disparities in the occurrence and mortality rates between AA and EA men [2]. Relative to other racial/ethnic groups, AA men are often diagnosed with more advanced and aggressive PCa. The different rates of mortality among these populations can be due to environmental factors, diet, genetic makeup, and/or lifestyle. Environmental factors, distributed differentially in various populations, may be associated with prostate carcinogenesis along with genetic mutations, which are, between these populations, present at differing frequencies [4].
Although there have been several investigations of the molecular and biological mechanisms related to PCa racial disparities, the underlying mechanisms/causes remain unclear. Among various populations, environmental factors, health care, genetic makeup, cultural and socioeconomic factors, diet and lifestyle, and awareness and motivation for PCa screening could be reasons for PCa disparities [5]. Further, genetic and molecular influences could be prominent factors for racial disparities in PCa incidence and outcomes [2]. In this report, we consider the immune-biological perspective of PCa racial disparities by presenting information on the associated factors and discussing their relevance in incidence and clinical outcomes.
2. PCa health disparities and population groups
Cancer health disparities are differences in the incidence, prevalence, mortality, or burden of cancer and cancer-related adverse effects that exist among a specific population. Disparities are not limited to cancer; they also include the conditions that lead to the development of cancer and the effects of these conditions on the quality of life and mortality. Factors that contribute to cancer health disparities have an excessive impact on underserved populations. For instance, after a cancer diagnosis, those in underserved populations have poor survival [6–8]. Nevertheless, research on PCa health disparities suggests that, for cancer patients, clinical procedures can improve outcomes [9, 10].
Populations suffering from cancer health disparities can be categorized based on their genetic makeup, age, socioeconomic status, geographic location, gender, and race/ethnicity [11, 12], and differences in the incidence and mortality for PCa can be obtained from the global cancer incidence database [13, 14]. Disparities among racial/ethnic populations are evident. For example, in the US, AAs have the highest overall cancer burden and a higher mortality than the EA population [15, 16]. The incidence and mortality trends for Hispanic and Latino populations in the US resemble those for Latin American nations (Fig. 1).
Fig. 1.
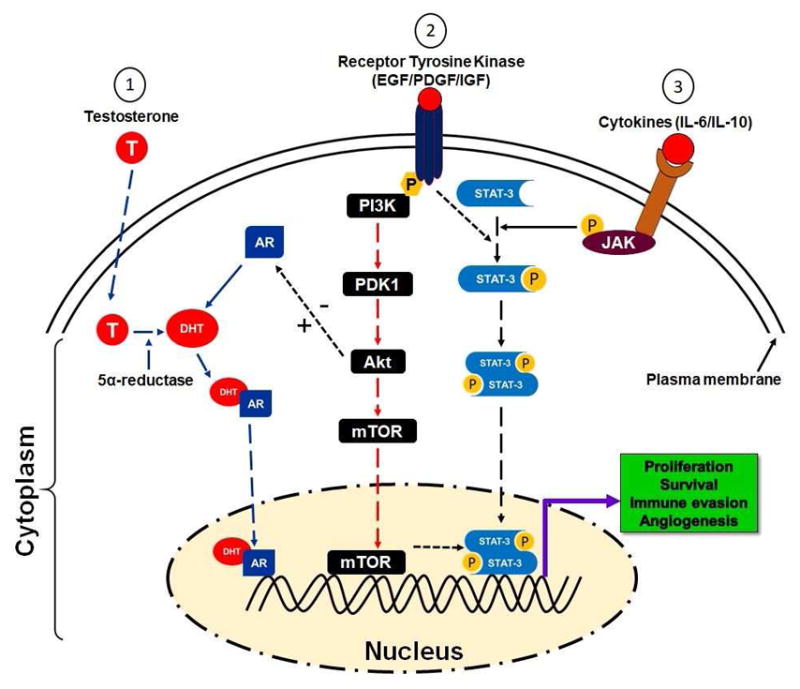
A schematic presentation of different pathways involved in PCa disparity.1. In the cytoplasm, testosterone is reduced to dihydrotestosterone (DHT) in the presence of 5α-reductase and binds to the androgen receptor (AR). The DHT/AR complex migrates to the nucleus and binds to androgen responsive elements (ARE) that further stimulates the activation of gene transcription. 2. After ligand interaction, receptor tyrosine kinase such as EGFR/PDGF/IGF stimulates its phosphorylation and subsequently actives PI3K and Akt pathway which further leads to the activation and migration of mTOR to the nucleus. In the nucleus, activated mTOR induces initiation of gene transcription, which results in proliferation, survival, and angiogenesis. 3. Binding of cytokines such as interleukin 6 (IL-6) to its receptor initiates several cellular events including activation of Janus kinase (JAK)/signal transducer and activating of transcription 3 (STAT3). Activated STAT3 forms homodimers that move to the nucleus and stimulates gene transcription. In brief, activation of; 1. androgen receptor, 2. receptor tyrosine kinase, and 3. Cytokine, induced downstream activation of responsive genes which are critical for PCa cells existence.
In the US, racial/ethnic populations can be defined as self-reported EAs, AAs, Hispanic-Latinos (HLs), Native-Europeans (NEs), and Native-Americans (NAs), as supported by the Surveillance Epidemiology and End Results (SEER) program. However, classification of this population in five race/ethnic groups is more a compromise than an ideal solution. To address the health disparities when characterizing minority populations, social descriptions should also be incorporated [17]. Self-identification of race/ethnicity can be acceptable for some groups but may be inadequate to identify ancestry for other groups [18–20]. Therefore, to determine cancer health disparities, factors such as genetic history, instead of self-identified or self-determined race/ethnicity, should be included [18–20]. Thus, for research on cancer health disparities, procedures have been developed to reduce the potential for confounders and can be used to characterize the ancestry of AAs, EAs, and NAs [12, 20, 21].
Various factors are responsible for PCa health disparities among AA and EA populations [2] [22–37]. These are summarized in a flow diagram (Fig. 1).
3. Genes Involved in PCa tumor biology for different races
The genetic makeup of various population groups is different in regard to PCa disparities. For example, AA men are more susceptible to PCa than EA men, and racial/ethnic genetic abnormalities are associated with high risk of PCa. Genetic alterations possibly associated with PCa disparities are in chromosomal region 8q24 and in the genes, CYP3A4 and EphB2 [38].
3.1 Genetic polymorphisms
Genetic polymorphisms are related to PCa susceptibility and therapeutic outcomes. The CYP3A4 gene, belonging to cytochrome p450 family, is involved in testosterone metabolism and is associated with PCa occurrence and aggressiveness [38–41]. CYP3A4 allele variations among various racial groups are evident for AA men compared to EA men. AA men exhibit higher frequencies of the allele [2]. In contrast, for EA men, a polymorphism of the CYP3A4*1B allele is inversely proportional to the development of cancer. Furthermore, single nucleotide polymorphisms (SNPs) in the 5′ terminal of the CYP3A4 gene lead to a change of adenine to guanine. CYP3A4*1B, and alternatives CYP3A4-V, CYP3A4-392A>G, are associated with a high clinical grade of PCa [11]. The frequency of GG alleles is differentially distributed among populations of various race/ethnicity [12,13]. Furthermore, the distribution of AA, AG, and GG alleles is associated with race/ethnicity, with 92% of EA men having the AA allele, compared to 17% of AA men [38]. The GG homozygous and AG heterozygous allele proportions are 1% and 7% for EA men vs 43% and 39% for AA men [38, 42]. There is an association between the CYP3A4*1B genotype and reoccurrence of PCa, and AA men with the CYP3A4*1B genotype are more susceptible to PCa compared to EA men [38, 43].
Furthermore, AA and EA men show a difference in the frequencies of alleles for CYP3A5 and CYP3A43; for AA men, CYP3A5*1 and CYP3A43*3 a1lele frequencies are higher. For AA men, the occurrence of the genotypes CYP3A4*1B-CYP3A5*1 is related to PCa progression. A pairwise interaction between CYP3A4*1B and CYP3A43*3 suggests a protective effect against PCa progression.
Furthermore, to make progress in dealing with PCa health disparities, we need to understand the role of AR signaling in PCa pathophysiology. AR has two polymorphic repeats of high frequency, CAG (19–20 for AA men) and GGN (21–22 for EA men). For AA men, shorter lengths of CAG repeats relate to PCa disparities [7, 26]. Moreover, allelic variations in chromosome 8q24 are associated with PCa disparities and with the high risk of various epithelial cancers, including PCa. These contribute to the heredity of PCa by altering allele-8 of the microsatellite DG8S737 [44]. For AA and EA men, these associations between 8q24 variants lead to susceptibility to PCa [45]. Genome-wide studies of various populations worldwide reveal that 16 loci on the chromosomal band 8q24 are associated with high risk of PCa [46–48]. Additionally, enhancer elements located on the 8q24 regions that are associated with increased susceptibility to PCa increase MYC promoter activity [49]. Among these genetic variations, SNPs on 8q24 (rs4242382-A, rs6981122, rs7000448, and rs16901896) are associated with a higher risk of PCa for AA men but not for EA men (Fig. 1) [49].
3.2 Gene mutations
Gene mutations also contribute to disparities in PCa among the races. A tumor suppressor gene, EphB2, encodes for expression of EphB2 receptor tyrosine kinase [50–52]. Mutations in this gene are associated with high risk of PCa for AA men relative to EA men. For AA men, the frequency of a germline EphB2 nonsense variant (3055A→T; K1019X) is associated with a familial ancestry of PCa [50–52]. Further, the EphB2 locus is associated with higher risk of PCa for AA men compared to EA men. Thus, a mutation in EphB2 receptor tyrosine kinase suggests that AAs are more prone to PCa than the EA population. Among the races, EphB2 SNPs are associated with PCa risk, with TGen-624 being most relevant. Two SNPs, rs10465543 and rs12090415, show associations with a protective effect. Furthermore, haplotype analysis reveals a low level of linkage equilibrium within the regions, with two blocks being associated with risk for aggressive PCa [51, 52]. The high rate of mutations related to racial disparities in PCa differentiates the AA population from the EA population (Fig. 1).
3.3 MicroRNAs
MicroRNAs (miRNAs) are small, endogenous, non-coding RNA molecules, 18–25 nucleotides long that are involved in the regulation of gene expression. They block mRNA function either by inhibition of translation to the complementary region in the 3′-untranslated regions or by degradation of target mRNAs [2]. By promoting tumor development and resistance to therapy, miRNAs are involved in cancer-related disparities among races/ethnicities [34, 53, 54]. In various tumors, including PCa, altered expression of miRNAs results in tumor growth and progression [34, 53, 54]. For example, miR-26a, miR-30c, miR-30-C1, miR-let7C, miR-1b-1, and paired miRNAs (miR-133a/MCL1, miR-513c/STAT1, miR-96/FOXO3A, and miR-45/ITPR2) are associated with advanced risk of PCa [32, 33]. The expression of miR-26a is higher for AA men with PCa as compared to EA men. For AA men, the expression of miR-26a is proportional to the aggressiveness of PCa compared to the EA men. For those of different races/ethnicities, miR-30c and let7c show differential expression. Additionally, for AA men, 18 of 22 miRNAs are associated with a high risk of PCa [55]. Results of bioinformatics-based analyses show that, in PCa, miRNA-mRNA pairs detect the activation of EGFR-PI3K-AKT signaling and reveal a racial disparity between AA and EA men [56, 57] (Fig. 1).
4. Epigenetic factors in racial disparities
Epigenetic changes are alterations in organisms induced by modifications of gene expression rather than alterations in the genetic makeup. The post-translational modification of histone proteins that change gene expression, along with the expression of gene variants, are associated with the epigenetics of PCa. In various cancers, including PCa, epigenetic markers, such as methylation, acetylation, phosphorylation, sumoylation, and ubiquitination are involved in regulating the expression of genes [58, 59]. The most frequent type of epigenetic modification in PCa is DNA hypermethylation of CpG islands (addition of a methyl group to cytosine-guanine dinucleotides). Such hypermethylation alters gene function by inhibiting transcriptional factors/activators at the target sites [60]. Since the frequency of DNA hypermethylation in normal and malignant cells is race/ethnicity-specific, such changes may contribute to PCa racial disparities [37, 61, 62]. For example, APC, RARβB, SPARC, EDNRB, CD44, RASSF1, CAV1, ANXA2, CDH1, ANKX2, TIMP3, GSTP1, AR, and ARβ2) are more extensively hypermethylated in AA men as compared to EA men. Thus, among AA and EA populations, epigenetic factors are possibly involved in the racial disparities of PCa and may be targeted for cancer therapy (Fig. 1).
5. Tumor biology factors in PCa disparities
As compared to the general population, underserved and minority populations may be discriminated against in clinical trials. It is difficult to establish the efficacy of cancer therapies if the population is not diverse, containing AA, EA, Hispanic/Latino, and Native American patients [4, 63, 64]. Currently, the mortality rates for PCa patients in the US are decreasing for each race/ethnicity; however, low survival rates are persisting among some groups [4]. Before diagnosis, the probability of occurrence of cancer between the races/ethnicities is generally similar [38]. Among men of different race/ethnicity who have detectable PCa, however, AA men have faster growing and more aggressive tumors than EA men [65]. Additional investigations will determine if, for AA men, mechanisms related to the increased proliferation of cells or inhibition of apoptosis lead to the enhanced growth of tumors. Epidermal growth factor receptor (EGFR) signaling is anti-apoptotic and contributes to the activation of other signaling pathways, such as MAPK, PI3K/Akt, and nuclear factor kappa-B (NF-kB). Therefore, it is plausible to target oncogenic molecules that relate to the occurrence of androgen-independent functions. Androgen-independent pathways have been studied in relation to the development of advanced PCa [66]. The findings suggest that, for AA men, receptor-mediated signaling that inhibits apoptosis and promotes cell proliferation and differentiation is more prevalent than for EA men [2, 67].
6. Role of androgen receptor in PCa progression
Androgens are hormones that are essential for normal male sexual characteristics. Signaling of androgen hormones requires an intracellular receptor and a transcription factor, which is involved in the development and progression of PCa. AR, a hormone-dependent transcription factor, belongs to the superfamily of nuclear hormone receptors [68]. When androgens are absent, AR remains in the cytoplasm of stromal and secretory luminal cells. To stabilize its structure and prevent proteolytic cleavage, AR binds to cytosolic heat shock proteins (HSPs) [69, 70]. Androgen stimulates the activation of AR, which is involved in sexual differentiation, development, and maintenance of maleness. Testosterone, an androgen, is derived from cholesterol. Leydig cells of the testes participate in the synthesis of testosterone and are considered its primary source. Adrenal cells synthesize testosterone to a lesser extent and are involved in AR signaling [69, 70]. Testosterone, a hydrophobic hormone, is reduced to 5α-dihydrotestosterone (DHT) by the action of a 5α-reductase [69–72]. Binding of DHT to AR leads to the dissociation from HSPs resulting in homodimerization, phosphorylation, and nuclear translocation of AR [73–76]. In the nucleus, AR binds to androgen-responsive elements of the target genes, resulting in activation of transcription that leads to cell proliferation, differentiation, and secretion. The transcription initiation complex includes co-regulatory proteins, such as steroid receptor coactivator (SRC-1), p160, AR activator (ARA), and CREB binding protein (CBP) (Fig. 2) [77]. Thus, AR signaling is involved in the development and progression of PCa. Apparently, through activation of this pathway, AA men have a higher risk of PCa than EA men.
Fig. 2.
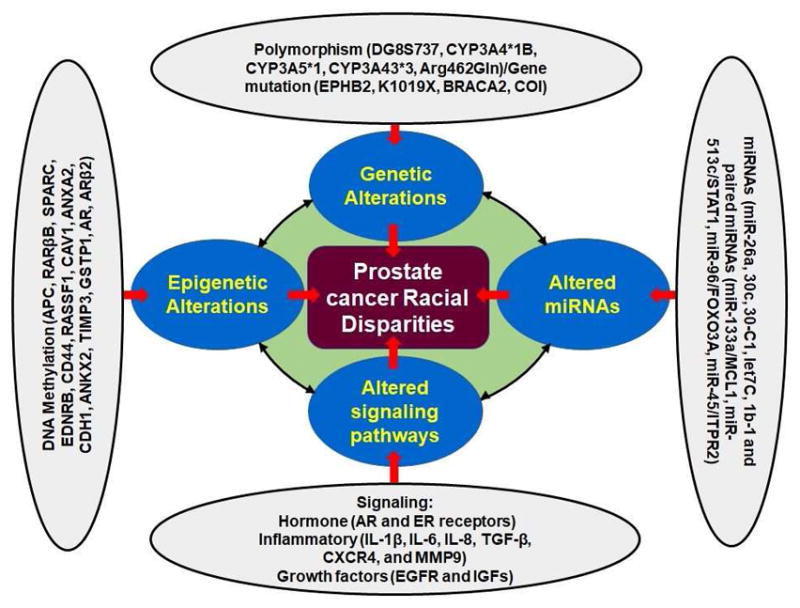
A schematic presentation of the factors associated with PCa disparity. Multiple factors such as genetic polymorphism, gene mutations, overexpression/suppression of microRNAs, epigenetic alteration, and alteration in molecular cell signaling pathways can play a significant role in PCa racial disparity. Defective function in polymorphism associated factors (DG8S737, CYP3A4*1B, CYP3A5*1, CYP3A43*3, Arg462Gln)/Gene mutation (EPHB2, K1019X, BRACA2, COI) may lead to PCa racial disparity. Similarly, overexpression/downregulation of miRNAs (miR-26a, 30c, 30-C1, let7C, 1b-1 and paired miRNAs (miR-133a/MCL1, miR-513c/STAT1, miR-96/FOXO3A, miR-45/ITPR2), alteration in cytokines (IL-1β, IL-6, IL-8, TGF-β, etc.), androgen hormone signaling, and epigenetic factors (APC, RARβB, SPARC, EDNRB, CD44, RASSF1, CAV1, ANXA2, CDH1, ANKX2, TIMP3, GSTP1, AR, ARβ2) may results in PCa disparity.
7. PI3K/Akt/mTOR signaling and drug resistance
In advanced PCa, alterations of components of the PI3K/Akt/mTOR signaling pathway, including mutations in tumor suppressor genes such as PTEN, are more frequent and correlate with high Gleason scores, advanced stages, and resistance to chemotherapy [28, 78]. These alterations result in activation of PI3K/Akt/mTOR signaling and accumulation of abnormalities, such as lower expression of PTEN, a negative regulator of PI3K/Akt/mTOR survival pathways. Additionally, expression of proinflammatory cytokines including IL-1βα, IL-6, IL-8, TNF-α, granulocyte colony-stimulating factor (G-CSF), and vascular endothelial growth factor (VEGF) was also altered [79]. Advanced PCa has high levels of activated Akt [28, 80]. Akt-mediated receptor signaling includes the insulin receptor, epidermal growth factor receptor (EGFR), insulin-like growth factor receptor (IGFR), interleukin-6 receptor, and platelet-derived growth factor receptor (PDGFR), and it likely functions as a cellular sensor for nutrient and growth signals [81, 82]. Through the mammalian target of rapamycin (mTOR) pathway, Akt regulates cell growth, differentiation, apoptosis, and angiogenesis and facilitates translation signals of c-myc, cyclin-D, and vascular endothelial growth factor [80]. In mice, inhibition of PTEN and mTOR activation suppresses the growth of PTEN−/− PCa xenografts by restoring chemotherapeutic sensitivity (Fig. 2) [28], [83, 84]. Thus, accumulated evidence suggests that age, genetic/reproductive history, lifestyle, and mutation in proto-oncogene, are the major risk factor for prostate cancer disparity.
8. EGFR and PDGFR signaling and PCa development
In addition to androgens, prostate growth and function is also regulated by various factors that promote tumor growth and differentiation are potential causes for PCa racial disparities. One of which is the epidermal growth factor and its receptor (EGFR). EGFR signaling pathways are promising targets for treatment of PCa [67]. EGFR belongs to tyrosine kinase family, and mutations in tyrosine kinases, such as EGFR, Bcr-Abl, and c-Kit, result in the development of PCa [85, 86]. EGFR signaling is involved in the androgen-independent progression of PCa, and, for PCa xenografts, inhibitors of EGFR block androgen-dependent and androgen-independent signaling pathways [18, 87, 88]. However, clinical trials of tyrosine kinase inhibitors have produced disappointing results [9, 10, 89].
EGFR is highly expressed in 40–80% of PCa and is more commonly found for AA men as compared to EA men. EGFR expression correlates with Gleason score and androgen-independent progression of PCa [28, 90]. Phase-II studies of the EGFR inhibitor gefitinib demonstrated minimal cytotoxicity but no response as measured by levels of prostate-specific antigen [28, 91]. In PCa, resistance to gefitinib may be associated with hyperactivation of the PI3K/Akt pathway [56, 92].
PCa cells also express high levels of PDGFR, the signaling of which converges with the PI3K/Akt pathway and cooperates in PCa progression. Since targeting the PDGFR pathway by use of a single therapeutic regimen, such as imatinib, may not be effective, combination therapy is being used to target this pathway [56, 92].
Another potential target in the same family is HER2/neu tyrosine kinase, which is upregulated in an androgen-dependent manner, leading to PCa growth and cell survival [93, 94]. Targeting of this kinase by a monoclonal antibody, trastuzumab, showed promising outcomes with little toxicity (Fig. 2).
9. IGF signaling and risk of PCa
Insulin-like growth factor (IGF) and its ligands is an important upstream effector of AKT signaling, and IGF upregulation can promote PCa development suggesting an interrelationship to AKT through its involvement with mitogenesis, anti-apoptosis, and cellular transformation [95]. High expression of IGF-1 and reduced expression of insulin-like growth factor binding protein-3 (IGFBP-3) are associated with a high risk of PCa [96, 97]. Additionally, expression of insulin-like growth factor type-1 receptor (IGF-1R) is higher in PCa, leading to the proliferation of cancer cells and resistance to androgen ablation therapy [96–99]. The IGF pathway can be targeted by monoclonal antibodies (such as IMC-A12 or cixutumumab and CP-751,871 or figitumumab, and fully human IgG1 and IgG2 antibodies) that bind to the extracellular domain of the transmembrane receptor [98, 99]. These molecules inhibit the growth of both androgen-dependent and androgen-independent tumors (Fig. 2).
10. Cytokines and regulation of PCa
The tumor microenvironment consists of various types of immune cells, adipocytes, and blood vessels that secrete soluble and insoluble factors, including cytokines and growth factors. Interleukin-6 (IL-6), a pleiotropic cytokine, is involved in regulation of the cell cycle, apoptosis, cell migration, and angiogenesis. Since, for patients with PCa, the levels of IL-6 in tissues and sera are higher than in controls [100, 101], it is important to know the level of signal transducer and activator of transcription (STAT), mitogen-activated kinases (MAPK), and phosphatidylinositol 3-kinase (PI3K), which are at high levels during IL-6 treatment. Moreover, since the PI3K and MAPK pathways are involved in carcinogenesis, the function of STAT3, which is necessary for IL-6 downstream signaling in normal as well as cancerous prostate tissue, needs to be explored. STAT3 regulates cellular functions in PCa, and its role in these cancers has been explored. Therapeutic approaches, including the use of an antisense oligonucleotide, anti-IL-6 or anti-IL-6R, and CpG-STAT3-specific siRNA have been evaluated, but the results are disappointing [102–104]. For patients with metastatic PCa, IL-6 did not show therapeutic or survival benefits. Moreover, anti-IL-6 agents, such as siltuximab and JAK/STAT3 inhibitors, were not only ineffective but also increased the expression of proliferation markers in PCa (Fig. 2) [105, 106]. Thus, it is likely that an inappropriate tumor microenvironment contributes to increased incidence and unfavorable outcomes for AA men.
11. Tumor antigens: possible option for PCa diagnosis and treatment
Based on serological analyses and discovery of T cells that recognize tumor antigens, new immunotherapeutic regimens to treat various kind of cancers, including PCa, have been developed [107–110]. As described below, there are two types of tumor antigens.
11.1 Defined Antigens
Characteristics of defined antigens that may be useful when they are used for therapeutic interventions are associated with their type, size, and interactions, and their immune responses. These properties suggest that a reasonable target would be the human leukocyte antigen (HLA) molecule. This approach has been used in clinical settings, either as single antigens or in combinations. Combination therapy is an emerging area of research, for combinations of two or more antigens demonstrate favorable outcomes and resolve concerns regarding how antigens escape immune attacks [111–114]. Based on these properties, antigens are categorized in Table 1.
Table 1.
| Antigen | examples |
|---|---|
| Universal antigen | WT1, TERT, Telomerase and survivin |
| Cancer-testis antigens | SUV39H2, MAGE, BAGE, GAGE, TMEM31, NY-ESO-1 |
| Unique antigens | β-catenin, P53, ras, CDK4, CDC27, and α-actinin-4 |
| Differentiation antigens | Tyrosinease, TRPI/gp75, TRP2, gp100, Melan-A/MARTI, gangliosides, PSMA |
| Overexpressed antigens | HER2, WT1, EphA3, EGFR, CD20 |
11.2 Cancer-testis antigens
This group of proteins, normally expressed in the testes of adult males are, in many types of cancer, expressed aberrantly during tumor progression. Cancer-testis antigens are a group of proteins that are restricted to the testis in normal adults but become altered during PCa development [115]. Aberrant expression of cancer testis antigens could be novel biomarkers for PCa disparity. For example, MAGE-1, a testis antigen, induces a CD8+ cytotoxic T cell response, as do antigens from similar families (MAGE, BAGE, and GAGE) [108–110, 116–118]. The best-studied cancer-testis antigens are those of the MAGE-A family and NY-ESO-1, which is found in germ cells and expresses class-I and class-II restricted epitopes [108–110, 116–118]. For patients, restricted expression of these antigens results in fevers and autoimmune dysfunctions (Table 1).
11.3 Unique antigens
This class of antigens shows properties defined by T cell epitopes and exhibits potential immunogenicity. Some of these antigens, derived by the mutation of genes, may have compatible biological properties. For example, antigens that are less susceptible to immuno-selection may maintain or advance tumor growth. Therefore, use of immunogenicity and constitutive expression of tumor antigens could be an approach to design and develop new immunotherapies in this field. Additionally, mutations in proto-oncogenes and tumor suppressor genes, such as CDK4, p53, ras, and b-raf, generate active oncogenes [119]. The chance of mutations in proto-oncogenes such as ras/raf is high. After mutations in these genes, the protein products induce activation of the innate and the cell-mediated arms of the immune system [120–122]. The T cell immune response requires an epitope expression that targets HLA-A*24, the signaling of which leads to activation of cytotoxic T cell-mediated killing of tumor cells [123]. Furthermore, CD8+ cytotoxic T cells recognize if CDK4 has acquired a substitution mutation [124]. However, a mutation in CDC27 results in altered protein trafficking into an endosomal compartment that leads to the presentation of the MHC-II epitope and recognition by CD4+ T cells [125]. Moreover, CD8+ T cells recognize a mutated peptide of α-actinin4 (about 10 amino acids) [125]. Accumulation of α-actinin4 results in actin bundling in the cytoplasm, which enhances cell motility and contributes to metastasis (Table 1) [126]. Although studies summaries significant racial disparities of biological factor, lifestyle, and socio-culture nature affecting PCa survivors, additional research is needed to establish their causes and intervention with respect to unique antigens.
11.4 Differentiation antigens
In addition to other biomarkers, differentiation antigens, protein/lipid/glycoprotein/carbohydrate molecules located on tumors as well as on normal cells with the capacity to evoke an immune response, can be used to study tumor-targeted immunotherapy in relation to restricted expression of HLA types during tumor progression. PCa exhibits the most striking racial disparity, as AA men are at higher risk of being diagnosed and more than two times higher risk of dying of PCa, compared to Caucasian men. For example, tyrosinase is a rate-limiting enzyme for melanin biosynthesis, and the activity of tyrosinase is stabilized by regulation of the expression of tyrosine-related protein-1 (TRP-1) [127]. In melanoma patients, serum IgG that immune-precipitates TRP-1 is recognized by the immune system [128]. Furthermore, since tyrosinase exhibits epitope variations such as TRP-1 and TRP-2, it can be involved in various immune response mechanisms [129, 130]. In mice, TRP-1-based vaccination protects the host in an antibody-dependent manner by providing passive immunity [131, 132]. In addition, melan-A could be a target antigenic protein for cytotoxic T cells; however, further investigations are needed [131, 132]. Gangliosides (GM3, GM2, GD2, and GD3) are a group of differentiation antigens in which GD3 is representative of the related families, MAGE, BAGE, and GAGE. However, NY-ESO-1 is unrelated to those listed in Table 1. In brief, differentiation antigens such as gangliosides are associated with higher risk for Gleason score in PCa disparity, especially in AA patients.
11.5 Overexpressed antigens
Antigens overexpressed on cancer cells are attractive targets for immunotherapy. Proto-oncogenes show homology among the family members (e.g., HER2 shares homology with tyrosine kinase and EGFR) [133–135]. Overexpression of glycoproteins relates to a poor prognosis [107]. Expression of the HER2 protein is commonly upregulated in cancers, including PCa. For breast cancer cells, trastuzumab, a humanized monoclonal antibody, induces cytotoxicity by induction of apoptosis [107]. To activate both arms of the immune system by T cell activation and antibody production, some escape variants have been deleted [107]. This modification of the antibody is effective, in mice, in generating the antitumor immune response [136, 137]. Wilm’s tumor1 (WT1) and ephrin receptor (Eph3) proteins are upregulated and are targets for immunotherapy [138]. For colorectal cancer, EGFR and CD20 are examples of passive immunotherapy (Table 1) [107, 139, 140]. Evidence also suggests that EGFR showed dimerization with HER2 during PCa development [141]. EGF-mediated downstream signaling of EGFR induced transcriptional activation of androgen receptor along with Transforming Growth factor -α which are associated with PCa progression [141]. Additionally, overexpression of HER2 enhances androgen receptor function and hormone-independent growth of PCa cells. Thus, overexpressed antigens are directly associated with PCa disparity.
11.6 Undefined antigens
Undefined/unidentified antigens are present in both allogeneic and autologous vaccines, such as intact cells, cell lysates, total RNA, and heat shock proteins. A benefit of having universal antigens is that the immune system recognizes them due to their large sizes and surfaces. To escape immune surveillance, tumor cells reduce markers on the surface [142]. It seems plausible to target unidentified tumor antigens for cancer immunotherapy because vaccines based on these antigens evoke a strong immune response (Table 1). The previous report suggests that undefined antigens could be used to enhance immune response and may be a potential alternative for PCa therapy [107].
12. T cells and antigen recognition
T cells, a group of lymphocytes produced and processed by the thymus gland, participate in the immune response. Activation of T cells requires turning switches on and off in a coordinated fashion. The T cell receptor (TCR) must engage with its cognate tumor peptide antigen, which, for this purpose, must be bound to a major histocompatibility complex (MHC) molecule expressed on antigen-presenting cells (APCs) [143]. A co-stimulatory signal must be activated; such a pathway is that of the CD28/B7 superfamily. The CD28 receptor, constitutively expressed on the surface of T cells, interacts with the B7 ligands (B7-1 [CD80] and B7-2 [CD86]) on professional APCs [144]. Simultaneously, to prevent hyperactivation of T cells, cytotoxic T lymphocyte-associated protein 4 (CTLA-4), another member of the CD28 family, interact with the B7 family of ligands to inhibit hyperactivation of T cells [144]. It is well known that tumor environment consists several types of immune cells, blood cells, and secreted soluble and insoluble factors such as growth factor and cytokine. Immune dysfunction is the results of continuous secretion of proinflammatory cytokines and other factors. Chronic inflammation and suppressive function of T cell facilitate tumor growth and progression. Thus, T cells are the crucial population that plays a significant role in cancer disparity.
13. Natural killer T (NKT) cells: a possible therapy for PCa
NKT cells share characteristics of the adaptive and innate immune systems. Like conventional T cells, NKT cells have TCRs and antigen specificity, for they recognize lipid antigens more readily than peptide antigens. These cells exhibit limited but rapid response characteristics of the innate immune system [145]. Mucosal-associated invariant T (MAIT) cells and γδ-T cells perform such specialized functions. Like NKT cells, MAIT cells express TCR by use of the Vα19jα33 chain in mice and the Vα7.2jα33 chain in humans and thereby have a regulatory role [146, 147]. MAIT cells depend on gut flora and are not present in germ-free mice [147]. Some γδ-T cells express NK-like markers [148–150]. Previously, these cells were thought to be NKT cells but are now recognized as a distinct subset of T cells [146, 147].
When activated, NKT cells are rapid responders in the innate immune system and are involved in the recruitment other immune cells, such as NK cells and CD4+ and CD8+ T cells of the adaptive immune system [151, 152]. Thus, these cells orchestrate other immune responses. NKT cells produce cytokines of the Th1 (IFN-γ) and Th2 (IL-4 and IL-13) types [153, 154]. By producing IL-17, NK1.1-negative subsets of type I NKT cells contribute to neutrophil recruitment. These cells also produce IL-21, which regulates NKT cells in an autocrine fashion [155]. The presence of cytokines such as IL-4 and IFN-γ allows cells to respond without further activation or gene transcription [156, 157]. Early production of IL-4 by NKT cells induces a Th2-type immune response and production of IgG. However, defective production of IgG in mice is associated with the absence of CD4+ NKT cells, which make IL-4 [158, 159]. NKT cells may not be the only source of IL-4; however, their capacity to respond quickly and subsequent adaptive responses makes their regulatory functions influential in the immune system (Fig 3).
Fig. 3.

Diagrammatic presentation of the role of effector function of NKT cells in immune regulation. Interaction of NKT cells with T cells, macrophages, and dendritic cells results in secretion of several cytokines such as IL-4, IL-10, IL-13, IL-21, IFN-γ which require for the activation of these cells. After activation, T cells divide frequently and secrete cytokines necessary for active immune response. Moreover, B cell interaction with NKT cells resulted in activation of B cells while NKT cell interaction with neutrophil suppresses its effector function.
Like conventional T cells, NKT cells act as a part of the adaptive immune system by recognizing and presenting lipid antigens. The capacity of NKT cells to recognize self-lipid antigens may lead to the development of autoimmune diseases, and their capacity to recognize bacterial lipids leads to an adaptive T cell immune response [160–162]. Thus, NKT cells serve as regulatory cells and potentially as effector cells in the context of autoimmune diseases and resistance to infectious diseases and cancer (Fig. 3). Additionally, PCa tumor environment altered the function of NKT cells by releasing various immunosuppressive factor which results in NKT cells become incompatible to provoke effector immune response. Thus, these cells are associated with PCa disparity.
14. PCa and the biology of immunomodulatory molecules
Cytokines act as a communication bridge for cells of both the adaptive and innate immune systems. They are primary immune modulators and communicators in response to infections and associated immune challenges, and they are involved in various biological processes. As a part of the signaling network, cytokines stimulate and modulate immune system functions and induce their own synthesis and the synthesis of other cytokines. Although cytokines are soluble, some remain cell-bound. Cytokines can be divided into pro-inflammatory (such as IL-1β, IL-6, and TNF-α) and anti-inflammatory cytokines (such as IL-10, TGF-β) [163, 164]. Chemokines are the subpopulation of cytokines that recruit other cells through chemical stimulation [165, 166]. Cells attracted to the chemokines follow a signal toward the source of chemokines such as infected or damaged cells. As signaling molecules, cytokines follow receptor-mediated cell signaling and stimulate intracellular signaling cascades. Moreover, cytokines can be classified based on the distance of source and target cells [165, 166]. Endocrine cytokines pass through the blood before reaching the target cells, paracrine cytokines act near the secreting cells, and autocrine cytokines bind to receptors on the cells that produce them.
B and T lymphocytes maintain the adaptive immune response [167, 168]. Before differentiating into their functional types, Th1, Th2, Treg, and Th17 cells produce cytokines to accomplish specific functions [167, 168]. Proliferation and differentiation of functional types depend on the cytokine-meditated microenvironment and activation of TCR signaling. Differentiated Th1- and Th2-type effector cells determine the nature of the adaptive immune response activated by effector cells [167, 168]. Th1 produces IFN-γ and IL-2, and perhaps TNF-α. The signature cytokines of Th2 cells are IL-4, IL-5, and IL-13. These cells also secrete TNF-α, and some produce IL-9 and modest amounts of IL-2 [167, 168]. An immune regulation requires homeostasis between the activity of Th1 and Th2 cells [169–173]. If one type of cells (e.g., Th1) becomes dominant, another type (Th2) is suppressed. In children, lower numbers of Th1 cells and higher numbers of Th2 cells are associated with autism spectrum disorder (ASD), suggesting that ASD is caused by an imbalance between Th1 and Th2 cytokines [174, 175]. Additionally, peripheral blood mononuclear cells from children with ASD show enhanced activation of Th1 and Th2 cells of the adaptive immune response, with a dominant Th2 and no compensatory increase in the expression of IL-10 [176].
Th1 cells produce IFN-γ, IL-2, and TNF-β; Th2 cells secrete IL-4, IL-5, IL-6, IL-9, and IL-10 [27]. Multiple cells can produce the same type of cytokines. Moreover, more than one type of cell can secrete granulocyte monocyte colony stimulation factor (GM-CSF) and IFN-β [27]. A single cytokine can act on more than one cell type (IL-12 acts on Th1 cells; however, IL-1 acts on T and B cells, macrophages, endothelial cells, fibroblasts, and epithelial cells). Furthermore, almost all interferons act on multiple cell types. Various cytokines act on one type of cell and exhibit a similar effect, called cytokine redundancy. On the other hand, a single cytokine may have multiple effects on a single type of cell. IFN-γ induces antiviral proteins, enhances MHC-I by stimulating NK and IL-12 production, and causes antiproliferative effects [27]. Enhanced expression of IFN-γ and IL-12 lead to greater inflammation, but more TGF-β, IL-4, and IL-10 suppresses inflammation [107, 177]. In brief, besides genetics, epigenetics, signaling of androgens, drug resistance, and tumor antigens, immunomodulatory molecules also play an important role in PCa disparities.
15. HLA class antigen abnormalities in tumors
For PCa, positive outcomes of clinical trials of T cell-based immunotherapy have gained attention in the characterization of the antigen-processing machinery involved in PCa disparities since such immunotherapy is involved in the generation and expression of the trimeric surface antigen complex (HLA-I) on tumor cells. This complex organizes interaction of tumor cells with an HLA-I antigen, and tumor-infiltrating cytotoxic T lymphocytes (CTLs) are recruited through the participation of associated/co-stimulatory molecules. HLA-I presents cytosolic/nuclear proteins or endogenous peptide antigens in the range of 9–11 amino acids long [178, 179]. These peptide antigens pass from the cytosol to the endoplasmic reticulum through transporters associated with antigen processing (TAPs), where they are loaded onto the β2-microglobulin (β-2m)-HLA-I heavy chain complex with the help of the chaperones calnexin, calreticulin, ERp57, and tapasin [180–182]. The trimeric HLA-I-peptide complex moves from the trans-Golgi face to the cell surface for presentation to CD8+ CTL cells [183, 184].
Abnormalities in the expression of antigen-processing machinery are associated with various types of malignancies [185]. These aberrations have clinical significance since they relate to tumor development and progression and poor patient survival. Murine PCa cells express the MHC-I surface antigen; however, lack of detectable LMP2, LMP7, TAP1, and TAP2 transcription leads to tumor development [186]. Expression of IFN-γ induces these four components of antigen-processing machinery and enhances surface expression of MHC-I. The TAP aberration in murine metastatic PCa cells is caused by impaired transcription initiation of TAP1 [186]; however, there is a constitutive expression and IFN-γ-induced transcription of TAP2 HLA-I and expression of HLA-I surface antigen [186–188].
16. Local tumor microenvironments
Differences in the PCa microenvironment contribute to health disparities associated with this disease [189]. The contribution of genetic factors to PCa incidence and mortality for AA men remains unresolved. Various PCa microenvironments may be associated with race/ethnicity [189]. The PCa microenvironment reflects genetic and immune alterations. Genetic abnormalities include chromosomal aberrations such as 8q24, CYP3A4*1B, and EphB2 SNPs. Immune alterations include the low cytotoxic activity of NK cells and CD8+ T cells and high secretion of TGF-β, which inhibits the cytotoxic function of NK cells and CD8+ T cells and induces recruitment and accumulation of Tregs and Th17 lymphocytes, which downregulate anti-tumor immunity [190]. Enhanced levels of TGF-β in prostate tissue are associated with pathologic conditions and, for localized PCa, increase the likelihood of postoperative residual tumors [191]. Additionally, the concentration of TGF-β in PCa metastasis correlates with PCa burden [191]. The prostate immune microenvironment is dynamic, changing over the time and with clinical states, which is associated with a series of phenotypic alterations leading to immunomodulation [192, 193], such as increasing tumor-infiltrating lymphocytes in a prostate bed following androgen-deprivation therapy [194], sensitization of tumor cells to T cell-mediated lysis, and higher expression of PD-L1 and PD-L2 ligands in resistant PCa [195].
17. Conclusions and future perspective
In sum, various factors influence the racial disparities among the AA and EA populations [2, 3, 7, 39, 67]. These include changes in genetic makeup, altered expression of microRNAs, mutations, alterations in signaling, and epigenetic changes. In this review, we have provided an immune-biological perspective on PCa racial disparities by presenting available information on the associated factors and discussing their relevance to the disproportionate incidence of PCa and its clinical outcomes. The highly heterogeneous tumor microenvironment may be a key factor for racial disparities in clinical outcomes for PCa. Thus, the composition of the tumor microenvironment (having various types of immune cells) can differ from person to person because of the genetic makeup of the tumors, their heterogeneity, host genetics and lifestyles, and dietary and environmental factors. Immuno-biological factors may include the type of tumor antigen; the pattern of presentation, recognition, and immune response evoked against those antigens; the heterogeneity of tumors; and host genetics. These data provide a wide and specific perspective of the biological basis of PCa racial disparities and should direct future endeavors in basic, clinical, and translational research. As with all research, availability of resources, funding support, and appropriate tools are necessary to make progress on PCa health disparities. Efforts at the levels of research, the community, clinics, and policymakers are needed to eliminate or at least make irrelevant, race-associated PCa disparities.
At this point, it will also be important to understand the etiological factor such as inherited genotypes, modifiable risk factors, and most importantly, immunobiological factors such as the number and functions of various immune cells (Treg cells and MDSCs) and inflammatory responses. These factors may be the major modulators for PCa disparity.
At this point, it will also be important to understand the etiological factor such as inherited genotypes, modifiable risk factors, and most importantly, immunobiological factors such as the number and functions of various immune cells (Treg cells and MDSCs) and inflammatory responses. These factors may be the major modulators for PCa disparity. Clinically, the chronic inflammation can increase the risk of cancer by recruiting various types of immune cells in the surroundings. These immune cells produced cytokines, chemokines, growth factors, prostaglandins, and reactive oxygen and nitrogen intermediated that may create a hypoxic environment and facilitate tumor growth and progression leading to tumorigenesis; all the way to metastasis progression. Interestingly, in developing tumors, anti- and pro-tumorigenic immune cells co-exist, if the tumor is not rejected, then pro-tumorigenic cells dominates and induced tumor survival pathway(s) such as NF-kB. Thus, investigating immunobiological approach to target PCa disparities would be novel and significant.
Table 2.
| Cytokine | Source | Target | Functions |
|---|---|---|---|
| IL-1 | Mφ, Monocytes, epi- and endothelial cells, fibroblasts, astrocytes | B & T cells, endothelial cells, Hypothalamus, and liver | Co-stimulation, activation, inflammation, fiver, and acute phase reactant |
| IL-2 | NK & T cells | Monocytes, NK, B & T cells | Cell growth/activation |
| IL-3 | T cells | Bone marrow progenitor cells | Cell growth and differential |
| IL-4 | T cells | B & T cells | Th2 differentiation, cell growth/activation, IgE isotyping switching |
| IL-5 | T cells | B cells, eosinophils | Cell growth/activation |
| IL-6 | Mφ, T cells, Fibroblasts | B & T cells, Liver | Co-stimulation, cell growth/activation, acute phase reactants |
| IL-7 | Fibroblasts, Bone marrow stromal cells | Immature lymphoid progenitors | T cell survival, proliferation, homeostasis, B cells development |
| IL-8 | Mφ, epithelial cells, platelets | Neutrophils | Activation chemotaxis |
| IL-10 | Th2 T cells | Mφ and T cells | Block APCs, and cytokine production |
| IL-12 | Mφ and NK cells | T cells | Th1 differentiation |
| IL-15 | Monocytes | NK & T cells | Cell growth/activation, NK cell development, and block apoptosis |
| IL-18 | Mφ | NK, B & T cells | Cell growth/activation, and inflammation |
| IL-21 | NKT & CD4+ T cells | NK, B & T cells | Cell growth/activation, control allergic response and viral infection |
| IL-23 | APCs | DC, NK, & T cells | Chronic inflammation, promotes Th17 cells |
| GM-CSF | T cells, Mφ, endothelial cells, fibroblast s, and mast cells | DC, Mφ, NKT and Bone marrow progenitor cells | ↑ antigen presentation, T cells homeostasis Hematopoitic cells, and growth factor |
| IFN-α | B & T cells, Mφ, endothelial cells, fibroblasts, plasmocytoid DC and Osteoblast cells | Mφ and NK cells | Antiviral and ↑ MHC expression |
| IFN-β | Leucocytes | Anti-proliferative | |
| IFN-γ | NK, NKT & T cells | Mφ, monocyte, endothelial cells | Cell growth activation, ↑ MHC expression |
| TGF-β | T cells and Mφ | T cells | Inhibits cell growth and activation |
| TNF-α | T cells and Mφ | B & T cells, endothelial cells, Hypothalamus, and liver | Co-stimulation, activation, inflammation, fiver, and acute phase reactant |
Highlights.
Role of immunobiological factors for PCa racial disparities are summarized and reviewed.
Advance immunotherapies to improve PCa disparities among AAs and EAs are discussed.
Acknowledgments
19. Funding support:
We are thankful to the National Institutes of Health for support through grants P20CA192976 (MKM) and P20CA192973 (UM) and to the National Science Foundation for grant 1154214 (MKM).
Footnotes
18. Conflict of interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jackson DD, Owens OL, Friedman DB, Hebert JR. An intergenerational approach to prostate cancer education: findings from a pilot project in the southeastern USA. J Cancer Educ. 2014;29:649–656. doi: 10.1007/s13187-014-0618-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhardwaj A, Srivastava SK, Khan MA, Prajapati VK, Singh S, Carter JE, Singh AP. Racial disparities in prostate cancer: a molecular perspective. Front Biosci (Landmark Ed) 2017;22:772–782. doi: 10.2741/4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bock CH, Powell I, Kittles RA, Hsing AW, Carpten J. Racial disparities in prostate cancer incidence, biochemical recurrence, and mortality. Prostate Cancer. 2011;2011:716178. doi: 10.1155/2011/716178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallace TA, Martin DN, Ambs S. Interactions among genes, tumor biology and the environment in cancer health disparities: examining the evidence on a national and global scale. Carcinogenesis. 2011;32:1107–1121. doi: 10.1093/carcin/bgr066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chornokur G, Dalton K, Borysova ME, Kumar NB. Disparities at presentation, diagnosis, treatment, and survival in African American men, affected by prostate cancer. Prostate. 2011;71:985–997. doi: 10.1002/pros.21314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berndt SI, Carter HB, Schoenberg MP, Newschaffer CJ. Disparities in treatment and outcome for renal cell cancer among older black and white patients. J Clin Oncol. 2007;25:3589–3595. doi: 10.1200/JCO.2006.10.0156. [DOI] [PubMed] [Google Scholar]
- 7.Guerrero EG, Marsh JC, Cao D, Shin HC, Andrews C. Gender disparities in utilization and outcome of comprehensive substance abuse treatment among racial/ethnic groups. J Subst Abuse Treat. 2014;46:584–591. doi: 10.1016/j.jsat.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Housri N, Weil RJ, Shalowitz DI, Koniaris LG. Should informed consent for cancer treatment include a discussion about hospital outcome disparities? PLoS Med. 2008;5:e214. doi: 10.1371/journal.pmed.0050214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedman DB, Owens OL, Jackson DD, Johnson KM, Gansauer L, Dickey J, Miller R, Payne J, Bearden JD, Hebert JR. An evaluation of a community-academic-clinical partnership to reduce prostate cancer disparities in the South. J Cancer Educ. 2014;29:80–85. doi: 10.1007/s13187-013-0550-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garg V, Raisch DW, Selig JP, Thompson TA. Health disparities in clinical practice patterns for prostate cancer screening by geographic regions in the United States: a multilevel modeling analysis. Prostate Cancer Prostatic Dis. 2013;16:193–203. doi: 10.1038/pcan.2013.3. [DOI] [PubMed] [Google Scholar]
- 11.Axt J, Murphy AJ, Seeley EH, Martin CA, Taylor C, Pierce J, Caprioli RM, Whiteside M, Lovvorn HN., 3rd Race disparities in Wilms tumor incidence and biology. J Surg Res. 2011;170:112–119. doi: 10.1016/j.jss.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahal BA, Ziehr DR, Aizer AA, Hyatt AS, Lago-Hernandez C, Choueiri TK, Elfiky AA, Hu JC, Sweeney CJ, Beard CJ, D’Amico AV, Martin NE, Kim SP, Lathan CS, Trinh QD, Nguyen PL. Racial disparities in an aging population: the relationship between age and race in the management of African American men with high-risk prostate cancer. J Geriatr Oncol. 2014;5:352–358. doi: 10.1016/j.jgo.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 13.The Science of Global Prostate Cancer Disparities in Black Men Jacksonville, FL, USA. 27–29 August 2010. Abstracts. Infect Agent Cancer. 2011;6(Suppl 1):A1–7. doi: 10.1186/1750-9378-6-S1-A1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rebbeck TR, Haas GP. Temporal trends and racial disparities in global prostate cancer prevalence. Can J Urol. 2014;21:7496–7506. [PMC free article] [PubMed] [Google Scholar]
- 15.Presley CJ, Raldow AC, Cramer LD, Soulos PR, Long JB, Yu JB, Makarov DV, Gross CP. A new approach to understanding racial disparities in prostate cancer treatment. J Geriatr Oncol. 2013;4:1–8. doi: 10.1016/j.jgo.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao H, Tan F, Goovaerts P. Racial and geographic disparities in late-stage prostate cancer diagnosis in Florida. J Health Care Poor Underserved. 2011;22:187–199. doi: 10.1353/hpu.2011.0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Race E G. Genetics Working. The use of racial, ethnic, and ancestral categories in human genetics research. Am J Hum Genet. 2005;77:519–532. doi: 10.1086/491747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holmes L, Jr, Chan W, Jiang Z, Ward D, Essien EJ, Du XL. Impact of androgen deprivation therapy on racial/ethnic disparities in the survival of older men treated for locoregional prostate cancer. Cancer Control. 2009;16:176–185. doi: 10.1177/107327480901600210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahal BA, Aizer AA, Ziehr DR, Hyatt AS, Choueiri TK, Hu JC, Hoffman KE, Sweeney CJ, Beard CJ, D’Amico AV, Martin NE, Kim SP, Trinh QD, Nguyen PL. Racial disparities in prostate cancer-specific mortality in men with low-risk prostate cancer. Clin Genitourin Cancer. 2014;12:e189–195. doi: 10.1016/j.clgc.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Mahal BA, Ziehr DR, Aizer AA, Hyatt AS, Sammon JD, Schmid M, Choueiri TK, Hu JC, Sweeney CJ, Beard CJ, D’Amico AV, Martin NE, Lathan C, Kim SP, Trinh QD, Nguyen PL. Getting back to equal: The influence of insurance status on racial disparities in the treatment of African American men with high-risk prostate cancer. Urol Oncol. 2014;32:1285–1291. doi: 10.1016/j.urolonc.2014.04.014. [DOI] [PubMed] [Google Scholar]
- 21.Printz C. Biological factor may be linked to prostate cancer racial disparities. Cancer. 2014;120:1132–1133. doi: 10.1002/cncr.28692. [DOI] [PubMed] [Google Scholar]
- 22.Singh S, Plaga A, Shukla GC. Racial disparities: disruptive genes in prostate carcinogenesis. Front Biosci (Schol Ed) 2017;9:244–253. doi: 10.2741/S485. [DOI] [PubMed] [Google Scholar]
- 23.Hamano T, Matsui H, Sekine Y, Ohtake N, Nakata S, Suzuki K. Association of SNP rs1447295 and microsatellite marker DG8S737 with familial prostate cancer and high grade disease. J Urol. 2010;184:738–742. doi: 10.1016/j.juro.2010.03.102. [DOI] [PubMed] [Google Scholar]
- 24.Yousef AM, Qosa H, Bulatova N, Abuhaliema A, Almadhoun H, Khayyat G, Olemat M. Effects of Genetic Polymorphism in CYP3A4 and CYP3A5 Genes on Tacrolimus Dose Among Kidney Transplant Recipients. Iran J Kidney Dis. 2016;10:156–163. [PubMed] [Google Scholar]
- 25.Wei B, Xu Z, Ruan J, Zhu M, Jin K, Zhou D, Yan Z, Xuan F, Zhou H, Huang X, Zhang J, Lu P, Shao J. RNASEL Asp541Glu and Arg462Gln polymorphisms in prostate cancer risk: evidences from a meta-analysis. Mol Biol Rep. 2012;39:2347–2353. doi: 10.1007/s11033-011-0985-x. [DOI] [PubMed] [Google Scholar]
- 26.Farrell J, Petrovics G, McLeod DG, Srivastava S. Genetic and molecular differences in prostate carcinogenesis between African American and Caucasian American men. Int J Mol Sci. 2013;14:15510–15531. doi: 10.3390/ijms140815510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee S, Margolin K. Cytokines in cancer immunotherapy. Cancers (Basel) 2011;3:3856–3893. doi: 10.3390/cancers3043856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antonarakis ES, Carducci MA, Eisenberger MA. Novel targeted therapeutics for metastatic castration-resistant prostate cancer. Cancer Lett. 2010;291:1–13. doi: 10.1016/j.canlet.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sfar S, Saad H, Mosbah F, Gabbouj S, Chouchane L. TSP1 and MMP9 genetic variants in sporadic prostate cancer. Cancer Genet Cytogenet. 2007;172:38–44. doi: 10.1016/j.cancergencyto.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 30.Zhang XY, Hong BF, Chen GF, Lu YL, Zhong M. Significance of MMP2 and MMP9 expression in prostate cancer. Zhonghua Nan Ke Xue. 2005;11:359–361. 364. [PubMed] [Google Scholar]
- 31.Erdmann K, Kaulke K, Rieger C, Salomo K, Wirth MP, Fuessel S. MiR-26a and miR-138 block the G1/S transition by targeting the cell cycle regulating network in prostate cancer cells. J Cancer Res Clin Oncol. 2016;142:2249–2261. doi: 10.1007/s00432-016-2222-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kachakova D, Mitkova A, Popov E, Popov I, Vlahova A, Dikov T, Christova S, Mitev V, Slavov C, Kaneva R. Combinations of serum prostate-specific antigen and plasma expression levels of let-7c, miR-30c, miR-141, and miR-375 as potential better diagnostic biomarkers for prostate cancer. DNA Cell Biol. 2015;34:189–200. doi: 10.1089/dna.2014.2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leite KR, Sousa-Canavez JM, Reis ST, Tomiyama AH, Camara-Lopes LH, Sanudo A, Antunes AA, Srougi M. Change in expression of miR-let7c, miR-100, and miR-218 from high grade localized prostate cancer to metastasis. Urol Oncol. 2011;29:265–269. doi: 10.1016/j.urolonc.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 34.Reis ST, Timoszczuk LS, Pontes-Junior J, Viana N, Silva IA, Dip N, Srougi M, Leite KR. The role of micro RNAs let7c, 100 and 218 expression and their target RAS, C-MYC, BUB1, RB, SMARCA5, LAMB3 and Ki-67 in prostate cancer. Clinics (Sao Paulo) 2013;68:652–657. doi: 10.6061/clinics/2013(05)12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang D, Kryvenko ON, Mitrache N, Do KC, Jankowski M, Chitale DA, Trudeau S, Rundle A, Belinsky SA, Rybicki BA. Methylation of the RARB gene increases prostate cancer risk in black Americans. J Urol. 2013;190:317–324. doi: 10.1016/j.juro.2013.01.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu J, Zhang H, Gu J, Lin S, Li J, Lu W, Wang Y, Zhu J. Methylation profiles of thirty four promoter-CpG islands and concordant methylation behaviours of sixteen genes that may contribute to carcinogenesis of astrocytoma. BMC Cancer. 2004;4:65. doi: 10.1186/1471-2407-4-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwabi-Addo B, Wang S, Chung W, Jelinek J, Patierno SR, Wang BD, Andrawis R, Lee NH, Apprey V, Issa JP, Ittmann M. Identification of differentially methylated genes in normal prostate tissues from African American and Caucasian men. Clin Cancer Res. 2010;16:3539–3547. doi: 10.1158/1078-0432.CCR-09-3342. [DOI] [PubMed] [Google Scholar]
- 38.Powell IJ, Bollig-Fischer A. Minireview: the molecular and genomic basis for prostate cancer health disparities. Mol Endocrinol. 2013;27:879–891. doi: 10.1210/me.2013-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Powell IJ, Bock CH, Ruterbusch JJ, Sakr W. Evidence supports a faster growth rate and/or earlier transformation to clinically significant prostate cancer in black than in white American men, and influences racial progression and mortality disparity. J Urol. 2010;183:1792–1796. doi: 10.1016/j.juro.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waxman DJ, Attisano C, Guengerich FP, Lapenson DP. Human liver microsomal steroid metabolism: identification of the major microsomal steroid hormone 6 beta-hydroxylase cytochrome P-450 enzyme. Arch Biochem Biophys. 1988;263:424–436. doi: 10.1016/0003-9861(88)90655-8. [DOI] [PubMed] [Google Scholar]
- 41.Waxman DJ, Lapenson DP, Aoyama T, Gelboin HV, Gonzalez FJ, Korzekwa K. Steroid hormone hydroxylase specificities of eleven cDNA-expressed human cytochrome P450s. Arch Biochem Biophys. 1991;290:160–166. doi: 10.1016/0003-9861(91)90602-f. [DOI] [PubMed] [Google Scholar]
- 42.Powell IJ, Zhou J, Sun Y, Sakr WA, Patel NP, Heilbrun LK, Everson RB. CYP3A4 genetic variant and disease-free survival among white and black men after radical prostatectomy. J Urol. 2004;172:1848–1852. doi: 10.1097/01.ju.0000142779.76603.be. [DOI] [PubMed] [Google Scholar]
- 43.Powell IJ, Banerjee M, Novallo M, Sakr W, Grignon D, Wood DP, Pontes JE. Prostate cancer biochemical recurrence stage for stage is more frequent among African-American than white men with locally advanced but not organ-confined disease. Urology. 2000;55:246–251. doi: 10.1016/s0090-4295(99)00436-7. [DOI] [PubMed] [Google Scholar]
- 44.Helfand BT, Loeb S, Cashy J, Meeks JJ, Thaxton CS, Han M, Catalona WJ. Tumor characteristics of carriers and noncarriers of the deCODE 8q24 prostate cancer susceptibility alleles. J Urol. 2008;179:2197–2201. doi: 10.1016/j.juro.2008.01.110. discussion 2202. [DOI] [PubMed] [Google Scholar]
- 45.Zhao CX, Liu M, Xu Y, Yang K, Wei D, Shi XH, Yang F, Zhang YG, Wang X, Liang SY, Zhao F, Zhang YR, Wang NN, Chen X, Sun L, Zhu XQ, Yuan HP, Zhu L, Yang YG, Tang L, Jiao HY, Huo ZH, Wang JY, Yang Z. 8q24 rs4242382 polymorphism is a risk factor for prostate cancer among multi-ethnic populations: evidence from clinical detection in China and a meta-analysis. Asian Pac J Cancer Prev. 2014;15:8311–8317. doi: 10.7314/apjcp.2014.15.19.8311. [DOI] [PubMed] [Google Scholar]
- 46.Amundadottir LT, Sulem P, Gudmundsson J, Helgason A, Baker A, Agnarsson BA, Sigurdsson A, Benediktsdottir KR, Cazier JB, Sainz J, Jakobsdottir M, Kostic J, Magnusdottir DN, Ghosh S, Agnarsson K, Birgisdottir B, Le Roux L, Olafsdottir A, Blondal T, Andresdottir M, Gretarsdottir OS, Bergthorsson JT, Gudbjartsson D, Gylfason A, Thorleifsson G, Manolescu A, Kristjansson K, Geirsson G, Isaksson H, Douglas J, Johansson JE, Balter K, Wiklund F, Montie JE, Yu X, Suarez BK, Ober C, Cooney KA, Gronberg H, Catalona WJ, Einarsson GV, Barkardottir RB, Gulcher JR, Kong A, Thorsteinsdottir U, Stefansson K. A common variant associated with prostate cancer in European and African populations. Nat Genet. 2006;38:652–658. doi: 10.1038/ng1808. [DOI] [PubMed] [Google Scholar]
- 47.Schumacher FR, Feigelson HS, Cox DG, Haiman CA, Albanes D, Buring J, Calle EE, Chanock SJ, Colditz GA, Diver WR, Dunning AM, Freedman ML, Gaziano JM, Giovannucci E, Hankinson SE, Hayes RB, Henderson BE, Hoover RN, Kaaks R, Key T, Kolonel LN, Kraft P, Le Marchand L, Ma J, Pike MC, Riboli E, Stampfer MJ, Stram DO, Thomas G, Thun MJ, Travis R, Virtamo J, Andriole G, Gelmann E, Willett WC, Hunter DJ. A common 8q24 variant in prostate and breast cancer from a large nested case-control study. Cancer Res. 2007;67:2951–2956. doi: 10.1158/0008-5472.CAN-06-3591. [DOI] [PubMed] [Google Scholar]
- 48.Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, Wang Z, Welch R, Staats BJ, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cancel-Tassin G, Cussenot O, Valeri A, Andriole GL, Gelmann EP, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover R, Hunter DJ, Chanock SJ, Thomas G. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- 49.Sotelo J, Esposito D, Duhagon MA, Banfield K, Mehalko J, Liao H, Stephens RM, Harris TJ, Munroe DJ, Wu X. Long-range enhancers on 8q24 regulate c-Myc. Proc Natl Acad Sci U S A. 2010;107:3001–3005. doi: 10.1073/pnas.0906067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huusko P, Ponciano-Jackson D, Wolf M, Kiefer JA, Azorsa DO, Tuzmen S, Weaver D, Robbins C, Moses T, Allinen M, Hautaniemi S, Chen Y, Elkahloun A, Basik M, Bova GS, Bubendorf L, Lugli A, Sauter G, Schleutker J, Ozcelik H, Elowe S, Pawson T, Trent JM, Carpten JD, Kallioniemi OP, Mousses S. Nonsense-mediated decay microarray analysis identifies mutations of EPHB2 in human prostate cancer. Nat Genet. 2004;36:979–983. doi: 10.1038/ng1408. [DOI] [PubMed] [Google Scholar]
- 51.Kittles RA, Baffoe-Bonnie AB, Moses TY, Robbins CM, Ahaghotu C, Huusko P, Pettaway C, Vijayakumar S, Bennett J, Hoke G, Mason T, Weinrich S, Trent JM, Collins FS, Mousses S, Bailey-Wilson J, Furbert-Harris P, Dunston G, Powell IJ, Carpten JD. A common nonsense mutation in EphB2 is associated with prostate cancer risk in African American men with a positive family history. J Med Genet. 2006;43:507–511. doi: 10.1136/jmg.2005.035790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robbins CM, Hooker S, Kittles RA, Carpten JD. EphB2 SNPs and sporadic prostate cancer risk in African American men. PLoS One. 2011;6:e19494. doi: 10.1371/journal.pone.0019494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhardwaj A, Singh S, Singh AP. MicroRNA-based Cancer Therapeutics: Big Hope from Small RNAs. Mol Cell Pharmacol. 2010;2:213–219. doi: 10.4255/mcpharmacol.10.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang V, Wu W. MicroRNA-based therapeutics for cancer. BioDrugs. 2009;23:15–23. doi: 10.2165/00063030-200923010-00002. [DOI] [PubMed] [Google Scholar]
- 55.Wang BD, Ceniccola K, Yang Q, Andrawis R, Patel V, Ji Y, Rhim J, Olender J, Popratiloff A, Latham P, Lai Y, Patierno SR, Lee NH. Identification and Functional Validation of Reciprocal microRNA-mRNA Pairings in African American Prostate Cancer Disparities. Clin Cancer Res. 2015;21:4970–4984. doi: 10.1158/1078-0432.CCR-14-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Asati V, Mahapatra DK, Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur J Med Chem. 2016;109:314–341. doi: 10.1016/j.ejmech.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 57.Sun P, Wang L, Lu Y, Liu Y, Li L, Yin L, Zhang C, Zhao W, Shen B, Xu W. MicroRNA-195 targets VEGFR2 and has a tumor suppressive role in ACHN cells via PI3K/Akt and Raf/MEK/ERK signaling pathways. Int J Oncol. 2016;49:1155–1163. doi: 10.3892/ijo.2016.3608. [DOI] [PubMed] [Google Scholar]
- 58.Albany C, Alva AS, Aparicio AM, Singal R, Yellapragada S, Sonpavde G, Hahn NM. Epigenetics in prostate cancer. Prostate Cancer. 2011;2011:580318. doi: 10.1155/2011/580318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamashita S. Prostate cancer and epigenetics. Nihon Rinsho. 2016;74(Suppl 3):75–79. [PubMed] [Google Scholar]
- 60.Nelson WG, Yegnasubramanian S, Agoston AT, Bastian PJ, Lee BH, Nakayama M, De Marzo AM. Abnormal DNA methylation, epigenetics, and prostate cancer. Front Biosci. 2007;12:4254–4266. doi: 10.2741/2385. [DOI] [PubMed] [Google Scholar]
- 61.Kim JW, Kim ST, Turner AR, Young T, Smith S, Liu W, Lindberg J, Egevad L, Gronberg H, Isaacs WB, Xu J. Identification of new differentially methylated genes that have potential functional consequences in prostate cancer. PLoS One. 2012;7:e48455. doi: 10.1371/journal.pone.0048455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Woodson K, Hayes R, Wideroff L, Villaruz L, Tangrea J. Hypermethylation of GSTP1, CD44, and E-cadherin genes in prostate cancer among US Blacks and Whites. Prostate. 2003;55:199–205. doi: 10.1002/pros.10236. [DOI] [PubMed] [Google Scholar]
- 63.Chinea FM, Lyapichev K, Epstein JI, Kwon D, Smith PT, Pollack A, Cote RJ, Kryvenko ON. Understanding PSA and its derivatives in prediction of tumor volume: Addressing health disparities in prostate cancer risk stratification. Oncotarget. 2017;8:20802–20812. doi: 10.18632/oncotarget.14903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mullins CD, Onukwugha E, Bikov K, Seal B, Hussain A. Health disparities in staging of SEER-medicare prostate cancer patients in the United States. Urology. 2010;76:566–572. doi: 10.1016/j.urology.2009.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Powell IJ. Epidemiology and pathophysiology of prostate cancer in African-American men. J Urol. 2007;177:444–449. doi: 10.1016/j.juro.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 66.Mimeault M, Batra SK. Recent advances on multiple tumorigenic cascades involved in prostatic cancer progression and targeting therapies. Carcinogenesis. 2006;27:1–22. doi: 10.1093/carcin/bgi229. [DOI] [PubMed] [Google Scholar]
- 67.Shuch B, Mikhail M, Satagopan J, Lee P, Yee H, Chang C, Cordon-Cardo C, Taneja SS, Osman I. Racial disparity of epidermal growth factor receptor expression in prostate cancer. J Clin Oncol. 2004;22:4725–4729. doi: 10.1200/JCO.2004.06.134. [DOI] [PubMed] [Google Scholar]
- 68.Gottlieb B, Beitel LK, Wu J, Elhaji YA, Trifiro M. Nuclear receptors and disease: androgen receptor. Essays Biochem. 2004;40:121–136. doi: 10.1042/bse0400121. [DOI] [PubMed] [Google Scholar]
- 69.Brinkmann AO, Blok LJ, de Ruiter PE, Doesburg P, Steketee K, Berrevoets CA, Trapman J. Mechanisms of androgen receptor activation and function. J Steroid Biochem Mol Biol. 1999;69:307–313. doi: 10.1016/s0960-0760(99)00049-7. [DOI] [PubMed] [Google Scholar]
- 70.Yang YC, Banuelos CA, Mawji NR, Wang J, Kato M, Haile S, McEwan IJ, Plymate S, Sadar MD. Targeting Androgen Receptor Activation Function-1 with EPI to Overcome Resistance Mechanisms in Castration-Resistant Prostate Cancer. Clin Cancer Res. 2016;22:4466–4477. doi: 10.1158/1078-0432.CCR-15-2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bartsch G, Rittmaster RS, Klocker H. Dihydrotestosterone and the concept of 5alpha-reductase inhibition in human benign prostatic hyperplasia. Eur Urol. 2000;37:367–380. doi: 10.1159/000020181. [DOI] [PubMed] [Google Scholar]
- 72.Bartsch G, Rittmaster RS, Klocker H. Dihydrotestosterone and the concept of 5alpha-reductase inhibition in human benign prostatic hyperplasia. World J Urol. 2002;19:413–425. doi: 10.1007/s00345-002-0248-5. [DOI] [PubMed] [Google Scholar]
- 73.Veldscholte J, Berrevoets CA, Zegers ND, van der Kwast TH, Grootegoed JA, Mulder E. Hormone-induced dissociation of the androgen receptor-heat-shock protein complex: use of a new monoclonal antibody to distinguish transformed from nontransformed receptors. Biochemistry. 1992;31:7422–7430. doi: 10.1021/bi00147a029. [DOI] [PubMed] [Google Scholar]
- 74.Gioeli D, Ficarro SB, Kwiek JJ, Aaronson D, Hancock M, Catling AD, White FM, Christian RE, Settlage RE, Shabanowitz J, Hunt DF, Weber MJ. Androgen receptor phosphorylation. Regulation and identification of the phosphorylation sites. J Biol Chem. 2002;277:29304–29314. doi: 10.1074/jbc.M204131200. [DOI] [PubMed] [Google Scholar]
- 75.Larrea F, Chirinos M. The control of gene transcription by steroid hormones: contributions of molecular biology to reproductive medicine. Rev Invest Clin. 2003;55:148–154. [PubMed] [Google Scholar]
- 76.Simons SS, Jr, Oshima H, Szapary D. Higher levels of control: modulation of steroid hormone-regulated gene transcription. Mol Endocrinol. 1992;6:995–1002. doi: 10.1210/mend.6.7.1324423. [DOI] [PubMed] [Google Scholar]
- 77.McEwan IJ. Gene regulation through chromatin remodelling by members of the nuclear receptor superfamily. Biochem Soc Trans. 2000;28:369–373. [PubMed] [Google Scholar]
- 78.McMenamin ME, Soung P, Perera S, Kaplan I, Loda M, Sellers WR. Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res. 1999;59:4291–4296. [PubMed] [Google Scholar]
- 79.Xie S, Chen M, Yan B, He X, Chen X, Li D. Identification of a role for the PI3K/AKT/mTOR signaling pathway in innate immune cells. PLoS One. 2014;9:e94496. doi: 10.1371/journal.pone.0094496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JH, Sawyers CL, Lichtenstein AK. AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. J Biol Chem. 2004;279:2737–2746. doi: 10.1074/jbc.M309999200. [DOI] [PubMed] [Google Scholar]
- 81.Gershtein ES, Scherbakov AM, Shatskaya VA, Kushlinsky NE, Krasil’nikov MA. Phosphatidylinositol 3-kinase/AKT signalling pathway components in human breast cancer: clinicopathological correlations. Anticancer Res. 2007;27:1777–1782. [PubMed] [Google Scholar]
- 82.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 83.Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, Frost P, Gibbons JJ, Wu H, Sawyers CL. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci U S A. 2001;98:10314–10319. doi: 10.1073/pnas.171076798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Podsypanina K, Lee RT, Politis C, Hennessy I, Crane A, Puc J, Neshat M, Wang H, Yang L, Gibbons J, Frost P, Dreisbach V, Blenis J, Gaciong Z, Fisher P, Sawyers C, Hedrick-Ellenson L, Parsons R. An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in Pten+/− mice. Proc Natl Acad Sci U S A. 2001;98:10320–10325. doi: 10.1073/pnas.171060098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shih AJ, Telesco SE, Radhakrishnan R. Analysis of Somatic Mutations in Cancer: Molecular Mechanisms of Activation in the ErbB Family of Receptor Tyrosine Kinases. Cancers (Basel) 2011;3:1195–1231. doi: 10.3390/cancers3011195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tan YH, Krishnaswamy S, Nandi S, Kanteti R, Vora S, Onel K, Hasina R, Lo FY, El-Hashani E, Cervantes G, Robinson M, Hsu HS, Kales SC, Lipkowitz S, Karrison T, Sattler M, Vokes EE, Wang YC, Salgia R. CBL is frequently altered in lung cancers: its relationship to mutations in MET and EGFR tyrosine kinases. PLoS One. 2010;5:e8972. doi: 10.1371/journal.pone.0008972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li J, Mercer E, Gou X, Lu YJ. Ethnical disparities of prostate cancer predisposition: genetic polymorphisms in androgen-related genes. Am J Cancer Res. 2013;3:127–151. [PMC free article] [PubMed] [Google Scholar]
- 88.Wang BD, Yang Q, Ceniccola K, Bianco F, Andrawis R, Jarrett T, Frazier H, Patierno SR, Lee NH. Androgen receptor-target genes in african american prostate cancer disparities. Prostate Cancer. 2013;2013:763569. doi: 10.1155/2013/763569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tyson MD, 2nd, Castle EP. Racial disparities in survival for patients with clinically localized prostate cancer adjusted for treatment effects. Mayo Clin Proc. 2014;89:300–307. doi: 10.1016/j.mayocp.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 90.Syed S. Combination chemotherapy for hormone-refractory prostate carcinoma: progress and pitfalls. Cancer. 2003;98:2088–2090. doi: 10.1002/cncr.11788. [DOI] [PubMed] [Google Scholar]
- 91.Canil CM, Moore MJ, Winquist E, Baetz T, Pollak M, Chi KN, Berry S, Ernst DS, Douglas L, Brundage M, Fisher B, McKenna A, Seymour L. Randomized phase II study of two doses of gefitinib in hormone-refractory prostate cancer: a trial of the National Cancer Institute of Canada-Clinical Trials Group. J Clin Oncol. 2005;23:455–460. doi: 10.1200/JCO.2005.02.129. [DOI] [PubMed] [Google Scholar]
- 92.Butler DE, Marlein C, Walker HF, Frame FM, Mann VM, Simms MS, Davies BR, Collins AT, Maitland NJ. Inhibition of the PI3K/AKT/mTOR pathway activates autophagy and compensatory Ras/Raf/MEK/ERK signalling in prostate cancer. Oncotarget. 2017 doi: 10.18632/oncotarget.18082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gregory CW, Whang YE, McCall W, Fei X, Liu Y, Ponguta LA, French FS, Wilson EM, Earp HS., 3rd Heregulin-induced activation of HER2 and HER3 increases androgen receptor transactivation and CWR-R1 human recurrent prostate cancer cell growth. Clin Cancer Res. 2005;11:1704–1712. doi: 10.1158/1078-0432.CCR-04-1158. [DOI] [PubMed] [Google Scholar]
- 94.Nagasawa J, Mizokami A, Koshida K, Yoshida S, Naito K, Namiki M. Novel HER2 selective tyrosine kinase inhibitor, TAK-165, inhibits bladder, kidney and androgen-independent prostate cancer in vitro and in vivo. Int J Urol. 2006;13:587–592. doi: 10.1111/j.1442-2042.2006.01342.x. [DOI] [PubMed] [Google Scholar]
- 95.da Silva HB, Amaral EP, Nolasco EL, de Victo NC, Atique R, Jank CC, Anschau V, Zerbini LF, Correa RG. Dissecting Major Signaling Pathways throughout the Development of Prostate Cancer. Prostate Cancer. 2013;2013:920612. doi: 10.1155/2013/920612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Renehan AG, Zwahlen M, Minder C, O’Dwyer ST, Shalet SM, Egger M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: systematic review and meta-regression analysis. Lancet. 2004;363:1346–1353. doi: 10.1016/S0140-6736(04)16044-3. [DOI] [PubMed] [Google Scholar]
- 97.Schumacher FR, Cheng I, Freedman ML, Mucci L, Allen NE, Pollak MN, Hayes RB, Stram DO, Canzian F, Henderson BE, Hunter DJ, Virtamo J, Manjer J, Gaziano JM, Kolonel LN, Tjonneland A, Albanes D, Calle EE, Giovannucci E, Crawford ED, Haiman CA, Kraft P, Willett WC, Thun MJ, Le Marchand L, Kaaks R, Feigelson HS, Bueno-de-Mesquita HB, Palli D, Riboli E, Lund E, Amiano P, Andriole G, Dunning AM, Trichopoulos D, Stampfer MJ, Key TJ, Ma J. A comprehensive analysis of common IGF1, IGFBP1 and IGFBP3 genetic variation with prospective IGF-I and IGFBP-3 blood levels and prostate cancer risk among Caucasians. Hum Mol Genet. 2010;19:3089–3101. doi: 10.1093/hmg/ddq210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chan JM, Stampfer MJ, Ma J, Gann P, Gaziano JM, Pollak M, Giovannucci E. Insulin-like growth factor-I (IGF-I) and IGF binding protein-3 as predictors of advanced-stage prostate cancer. J Natl Cancer Inst. 2002;94:1099–1106. doi: 10.1093/jnci/94.14.1099. [DOI] [PubMed] [Google Scholar]
- 99.Lehrer S, Diamond EJ, Droller MJ, Stone NN, Stock RG. Re: Insulin-like growth factor-I (IGF-I) and IGF binding protein-3 as predictors of advanced-stage prostate cancer. J Natl Cancer Inst. 2002;94:1893. doi: 10.1093/jnci/94.24.1893. author reply 1893–1894. [DOI] [PubMed] [Google Scholar]
- 100.Culig Z, Pencik J, Merkel O, Kenner L. Breaking a paradigm: IL-6/STAT3 signaling suppresses metastatic prostate cancer upon ARF expression. Mol Cell Oncol. 2016;3:e1090048. doi: 10.1080/23723556.2015.1090048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Giri D, Ozen M, Ittmann M. Interleukin-6 is an autocrine growth factor in human prostate cancer. Am J Pathol. 2001;159:2159–2165. doi: 10.1016/S0002-9440(10)63067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hossain DM, Pal SK, Moreira D, Duttagupta P, Zhang Q, Won H, Jones J, D’Apuzzo M, Forman S, Kortylewski M. TLR9-Targeted STAT3 Silencing Abrogates Immunosuppressive Activity of Myeloid-Derived Suppressor Cells from Prostate Cancer Patients. Clin Cancer Res. 2015;21:3771–3782. doi: 10.1158/1078-0432.CCR-14-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tawara K, Oxford JT, Jorcyk CL. Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: potential of anti-IL-6 therapies. Cancer Manag Res. 2011;3:177–189. doi: 10.2147/CMR.S18101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wallner L, Dai J, Escara-Wilke J, Zhang J, Yao Z, Lu Y, Trikha M, Nemeth JA, Zaki MH, Keller ET. Inhibition of interleukin-6 with CNTO328, an anti-interleukin-6 monoclonal antibody, inhibits conversion of androgen-dependent prostate cancer to an androgen-independent phenotype in orchiectomized mice. Cancer Res. 2006;66:3087–3095. doi: 10.1158/0008-5472.CAN-05-3447. [DOI] [PubMed] [Google Scholar]
- 105.Pencik J, Schlederer M, Gruber W, Unger C, Walker SM, Chalaris A, Marie IJ, Hassler MR, Javaheri T, Aksoy O, Blayney JK, Prutsch N, Skucha A, Herac M, Kramer OH, Mazal P, Grebien F, Egger G, Poli V, Mikulits W, Eferl R, Esterbauer H, Kennedy R, Fend F, Scharpf M, Braun M, Perner S, Levy DE, Malcolm T, Turner SD, Haitel A, Susani M, Moazzami A, Rose-John S, Aberger F, Merkel O, Moriggl R, Culig Z, Dolznig H, Kenner L. Erratum: STAT3 regulated ARF expression suppresses prostate cancer metastasis. Nat Commun. 2015;6:8802. doi: 10.1038/ncomms8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pencik J, Schlederer M, Gruber W, Unger C, Walker SM, Chalaris A, Marie IJ, Hassler MR, Javaheri T, Aksoy O, Blayney JK, Prutsch N, Skucha A, Herac M, Kramer OH, Mazal P, Grebien F, Egger G, Poli V, Mikulits W, Eferl R, Esterbauer H, Kennedy R, Fend F, Scharpf M, Braun M, Perner S, Levy DE, Malcolm T, Turner SD, Haitel A, Susani M, Moazzami A, Rose-John S, Aberger F, Merkel O, Moriggl R, Culig Z, Dolznig H, Kenner L. STAT3 regulated ARF expression suppresses prostate cancer metastasis. Nat Commun. 2015;6:7736. doi: 10.1038/ncomms8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Srinivasan R, Wolchok JD. Tumor antigens for cancer immunotherapy: therapeutic potential of xenogeneic DNA vaccines. J Transl Med. 2004;2:12. doi: 10.1186/1479-5876-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Boon T, Coulie PG, Van den Eynde B. Tumor antigens recognized by T cells. Immunol Today. 1997;18:267–268. doi: 10.1016/s0167-5699(97)80020-5. [DOI] [PubMed] [Google Scholar]
- 109.Wang RF. Identification of MHC class II-restricted tumor antigens recognized by CD4+ T cells. Methods. 2003;29:227–235. doi: 10.1016/s1046-2023(02)00345-6. [DOI] [PubMed] [Google Scholar]
- 110.Wang RF. Molecular cloning and characterization of MHC class I- and II-restricted tumor antigens recognized by T cells. Curr Protoc Immunol. 2009;(Unit 20):Chapter 20. 10. doi: 10.1002/0471142735.im2010s84. [DOI] [PubMed] [Google Scholar]
- 111.Kochin V, Kanaseki T, Tokita S, Miyamoto S, Shionoya Y, Kikuchi Y, Morooka D, Hirohashi Y, Tsukahara T, Watanabe K, Toji S, Kokai Y, Sato N, Torigoe T. HLA-A24 ligandome analysis of colon and lung cancer cells identifies a novel cancer-testis antigen and a neoantigen that elicits specific and strong CTL responses. Oncoimmunology. 2017;6:e1293214. doi: 10.1080/2162402X.2017.1293214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Garcia-Soto AE, Schreiber T, Strbo N, Ganjei-Azar P, Miao F, Koru-Sengul T, Simpkins F, Nieves-Neira W, Lucci J, 3rd, Podack ER. Cancer-testis antigen expression is shared between epithelial ovarian cancer tumors. Gynecol Oncol. 2017;145:413–419. doi: 10.1016/j.ygyno.2017.03.512. [DOI] [PubMed] [Google Scholar]
- 113.Beppu S, Ito Y, Fujii K, Saida K, Takino H, Masaki A, Murase T, Kusafuka K, Iida Y, Onitsuka T, Yatabe Y, Hanai N, Hasegawa Y, Ijichi K, Murakami S, Inagaki H. Expression of cancer/testis antigens in salivary gland carcinomas with reference to MAGE-A and NY-ESO-1 expression in adenoid cystic carcinoma. Histopathology. 2017 doi: 10.1111/his.13226. [DOI] [PubMed] [Google Scholar]
- 114.Kulkarni P, Uversky VN. Cancer/Testis Antigens: “Smart” Biomarkers for Diagnosis and Prognosis of Prostate and Other Cancers. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18040740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scanlan MJ, Simpson AJ, Old LJ. The cancer/testis genes: review, standardization, and commentary. Cancer Immun. 2004;4:1. [PubMed] [Google Scholar]
- 116.Boel P, Wildmann C, Sensi ML, Brasseur R, Renauld JC, Coulie P, Boon T, van der Bruggen P. BAGE: a new gene encoding an antigen recognized on human melanomas by cytolytic T lymphocytes. Immunity. 1995;2:167–175. doi: 10.1016/s1074-7613(95)80053-0. [DOI] [PubMed] [Google Scholar]
- 117.van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, Knuth A, Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 118.van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde BJ, Knuth A, Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. J Immunol. 2007;178:2617–2621. [PubMed] [Google Scholar]
- 119.Houbiers JG, van der Burg SH, van de Watering LM, Tollenaar RA, Brand A, van de Velde CJ, Melief CJ. Antibodies against p53 are associated with poor prognosis of colorectal cancer. Br J Cancer. 1995;72:637–641. doi: 10.1038/bjc.1995.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cheever MA, Chen W, Disis ML, Takahashi M, Peace DJ. T-cell immunity to oncogenic proteins including mutated ras and chimeric bcr-abl. Ann N Y Acad Sci. 1993;690:101–112. doi: 10.1111/j.1749-6632.1993.tb44000.x. [DOI] [PubMed] [Google Scholar]
- 121.Cheever MA, Disis ML, Bernhard H, Gralow JR, Hand SL, Huseby ES, Qin HL, Takahashi M, Chen W. Immunity to oncogenic proteins. Immunol Rev. 1995;145:33–59. doi: 10.1111/j.1600-065x.1995.tb00076.x. [DOI] [PubMed] [Google Scholar]
- 122.Fossum B, Gedde-Dahl T, 3rd, Breivik J, Eriksen JA, Spurkland A, Thorsby E, Gaudernack G. p21-ras-peptide-specific T-cell responses in a patient with colorectal cancer. CD4+ and CD8+ T cells recognize a peptide corresponding to a common mutation (13Gly-->Asp) Int J Cancer. 1994;56:40–45. doi: 10.1002/ijc.2910560108. [DOI] [PubMed] [Google Scholar]
- 123.Hom SS, Topalian SL, Simonis T, Mancini M, Rosenberg SA. Common expression of melanoma tumor-associated antigens recognized by human tumor infiltrating lymphocytes: analysis by human lymphocyte antigen restriction. J Immunother (1991) 1991;10:153–164. [PubMed] [Google Scholar]
- 124.Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, De Plaen E, Hankeln T, Meyer zum Buschenfelde KH, Beach D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–1284. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- 125.Mami-Chouaib F, Echchakir H, Dorothee G, Vergnon I, Chouaib S. Antitumor cytotoxic T-lymphocyte response in human lung carcinoma: identification of a tumor-associated antigen. Immunol Rev. 2002;188:114–121. doi: 10.1034/j.1600-065x.2002.18810.x. [DOI] [PubMed] [Google Scholar]
- 126.Honda K, Yamada T, Endo R, Ino Y, Gotoh M, Tsuda H, Yamada Y, Chiba H, Hirohashi S. Actinin-4, a novel actin-bundling protein associated with cell motility and cancer invasion. J Cell Biol. 1998;140:1383–1393. doi: 10.1083/jcb.140.6.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Luscombe CJ, French ME, Liu S, Saxby MF, Jones PW, Fryer AA, Strange RC. Prostate cancer risk: associations with ultraviolet radiation, tyrosinase and melanocortin-1 receptor genotypes. Br J Cancer. 2001;85:1504–1509. doi: 10.1054/bjoc.2001.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mattes MJ, Thomson TM, Old LJ, Lloyd KO. A pigmentation-associated, differentiation antigen of human melanoma defined by a precipitating antibody in human serum. Int J Cancer. 1983;32:717–721. doi: 10.1002/ijc.2910320610. [DOI] [PubMed] [Google Scholar]
- 129.Kawakami Y, Robbins PF, Wang X, Tupesis JP, Parkhurst MR, Kang X, Sakaguchi K, Appella E, Rosenberg SA. Identification of new melanoma epitopes on melanosomal proteins recognized by tumor infiltrating T lymphocytes restricted by HLA-A1, -A2, and -A3 alleles. J Immunol. 1998;161:6985–6992. [PubMed] [Google Scholar]
- 130.Wang RF, Appella E, Kawakami Y, Kang X, Rosenberg SA. Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J Exp Med. 1996;184:2207–2216. doi: 10.1084/jem.184.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hara I, Takechi Y, Houghton AN. Implicating a role for immune recognition of self in tumor rejection: passive immunization against the brown locus protein. J Exp Med. 1995;182:1609–1614. doi: 10.1084/jem.182.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Weber LW, Bowne WB, Wolchok JD, Srinivasan R, Qin J, Moroi Y, Clynes R, Song P, Lewis JJ, Houghton AN. Tumor immunity and autoimmunity induced by immunization with homologous DNA. The Journal of clinical investigation. 1998;102:1258–1264. doi: 10.1172/JCI4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bellezza I, Bracarda S, Caserta C, Minelli A. Targeting of EGFR tyrosine kinase by ZD1839 (“Iressa”) in androgen-responsive prostate cancer in vitro. Mol Genet Metab. 2006;88:114–122. doi: 10.1016/j.ymgme.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 134.Festuccia C, Gravina GL, Biordi L, D’Ascenzo S, Dolo V, Ficorella C, Ricevuto E, Tombolini V. Effects of EGFR tyrosine kinase inhibitor erlotinib in prostate cancer cells in vitro. Prostate. 2009;69:1529–1537. doi: 10.1002/pros.20995. [DOI] [PubMed] [Google Scholar]
- 135.Festuccia C, Gravina GL, Muzi P, Millimaggi D, Dolo V, Vicentini C, Bologna M. Akt down-modulation induces apoptosis of human prostate cancer cells and synergizes with EGFR tyrosine kinase inhibitors. Prostate. 2008;68:965–974. doi: 10.1002/pros.20757. [DOI] [PubMed] [Google Scholar]
- 136.Foy TM, Fanger GR, Hand S, Gerard C, Bruck C, Cheever MA. Designing HER2 vaccines. Semin Oncol. 2002;29:53–61. doi: 10.1053/sonc.2002.34056. [DOI] [PubMed] [Google Scholar]
- 137.Marchini C, Kalogris C, Garulli C, Pietrella L, Gabrielli F, Curcio C, Quaglino E, Cavallo F, Amici A. Tailoring DNA Vaccines: Designing Strategies Against HER2-Positive Cancers. Front Oncol. 2013;3:122. doi: 10.3389/fonc.2013.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chiari R, Hames G, Stroobant V, Texier C, Maillere B, Boon T, Coulie PG. Identification of a tumor-specific shared antigen derived from an Eph receptor and presented to CD4 T cells on HLA class II molecules. Cancer Res. 2000;60:4855–4863. [PubMed] [Google Scholar]
- 139.Cohen RB. Epidermal growth factor receptor as a therapeutic target in colorectal cancer. Clin Colorectal Cancer. 2003;2:246–251. doi: 10.3816/CCC.2003.n.006. [DOI] [PubMed] [Google Scholar]
- 140.Scartozzi M, Pierantoni C, Berardi R, Antognoli S, Bearzi I, Cascinu S. Epidermal growth factor receptor: a promising therapeutic target for colorectal cancer. Anal Quant Cytol Histol. 2006;28:61–68. [PubMed] [Google Scholar]
- 141.Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell. 2004;6:517–527. doi: 10.1016/j.ccr.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 142.Wang Z, Cao Y, Albino AP, Zeff RA, Houghton A, Ferrone S. Lack of HLA class I antigen expression by melanoma cells SK-MEL-33 caused by a reading frameshift in beta 2-microglobulin messenger RNA. The Journal of clinical investigation. 1993;91:684–692. doi: 10.1172/JCI116249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kumar S, Deepak P, Kumar S, Jr, Kishore D, Acharya A. Autologous Hsp70 induces antigen specific Th1 immune responses in a murine T-cell lymphoma. Immunol Invest. 2009;38:449–465. doi: 10.1080/08820130902802673. [DOI] [PubMed] [Google Scholar]
- 144.Maus MV, Thomas AK, Leonard DG, Allman D, Addya K, Schlienger K, Riley JL, June CH. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4–1BB. Nat Biotechnol. 2002;20:143–148. doi: 10.1038/nbt0202-143. [DOI] [PubMed] [Google Scholar]
- 145.Kuylenstierna C, Bjorkstrom NK, Andersson SK, Sahlstrom P, Bosnjak L, Paquin-Proulx D, Malmberg KJ, Ljunggren HG, Moll M, Sandberg JK. NKG2D performs two functions in invariant NKT cells: direct TCR-independent activation of NK-like cytolysis and co-stimulation of activation by CD1d. Eur J Immunol. 2011;41:1913–1923. doi: 10.1002/eji.200940278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shimamura M, Huang YY. Presence of a novel subset of NKT cells bearing an invariant V(alpha)19.1-J(alpha)26 TCR alpha chain. FEBS Lett. 2002;516:97–100. doi: 10.1016/s0014-5793(02)02509-7. [DOI] [PubMed] [Google Scholar]
- 147.Treiner E. MAIT lymphocytes, regulators of intestinal immunity? Presse Med. 2003;32:1636–1637. [PubMed] [Google Scholar]
- 148.Arase H, Arase N, Saito T. Fas-mediated cytotoxicity by freshly isolated natural killer cells. J Exp Med. 1995;181:1235–1238. doi: 10.1084/jem.181.3.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Arase H, Ono S, Arase N, Park SY, Wakizaka K, Watanabe H, Ohno H, Saito T. Developmental arrest of NK1.1+ T cell antigen receptor (TCR)-alpha/beta+ T cells and expansion of NK1.1+ TCR-gamma/delta+ T cell development in CD3 zeta-deficient mice. J Exp Med. 1995;182:891–895. doi: 10.1084/jem.182.3.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Carding SR, Egan PJ. Gammadelta T cells: functional plasticity and heterogeneity. Nat Rev Immunol. 2002;2:336–345. doi: 10.1038/nri797. [DOI] [PubMed] [Google Scholar]
- 151.Lin SJ, Chou FJ, Li L, Lin CY, Yeh S, Chang C. Natural killer cells suppress enzalutamide resistance and cell invasion in the castration resistant prostate cancer via targeting the androgen receptor splicing variant 7 (ARv7) Cancer Lett. 2017;398:62–69. doi: 10.1016/j.canlet.2017.03.035. [DOI] [PubMed] [Google Scholar]
- 152.Nowak M, Schmidt-Wolf IG. Natural killer T cells subsets in cancer, functional defects in prostate cancer and implications for immunotherapy. Cancers (Basel) 2011;3:3661–3675. doi: 10.3390/cancers3033661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Baltch AL, Bopp LH, Ritz WJ, Michelsen PB, Yan SB, Um S, Smith RP. Effect of recombinant human activated protein C on the bactericidal activity of human monocytes and modulation of pro-inflammatory cytokines in the presence of antimicrobial agents. J Antimicrob Chemother. 2007;59:1177–1181. doi: 10.1093/jac/dkm080. [DOI] [PubMed] [Google Scholar]
- 154.Rachitskaya AV, Hansen AM, Horai R, Li Z, Villasmil R, Luger D, Nussenblatt RB, Caspi RR. Cutting edge: NKT cells constitutively express IL-23 receptor and RORgammat and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J Immunol. 2008;180:5167–5171. doi: 10.4049/jimmunol.180.8.5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Coquet JM, Kyparissoudis K, Pellicci DG, Besra G, Berzins SP, Smyth MJ, Godfrey DI. IL-21 is produced by NKT cells and modulates NKT cell activation and cytokine production. J Immunol. 2007;178:2827–2834. doi: 10.4049/jimmunol.178.5.2827. [DOI] [PubMed] [Google Scholar]
- 156.Stetson DB, Mohrs M, Reinhardt RL, Baron JL, Wang ZE, Gapin L, Kronenberg M, Locksley RM. Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J Exp Med. 2003;198:1069–1076. doi: 10.1084/jem.20030630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chun T, Page MJ, Gapin L, Matsuda JL, Xu H, Nguyen H, Kang HS, Stanic AK, Joyce S, Koltun WA, Chorney MJ, Kronenberg M, Wang CR. CD1d-expressing dendritic cells but not thymic epithelial cells can mediate negative selection of NKT cells. J Exp Med. 2003;197:907–918. doi: 10.1084/jem.20021366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bradley LM, Yoshimoto K, Swain SL. The cytokines IL-4, IFN-gamma, and IL-12 regulate the development of subsets of memory effector helper T cells in vitro. J Immunol. 1995;155:1713–1724. [PubMed] [Google Scholar]
- 159.Nakanishi K, Yoshimoto T, Chu CC, Matsumoto H, Hase K, Nagai N, Tanaka T, Miyasaka M, Paul WE, Shinka S. IL-2 inhibits IL-4-dependent IgE and IgG1 production in vitro and in vivo. Int Immunol. 1995;7:259–268. doi: 10.1093/intimm/7.2.259. [DOI] [PubMed] [Google Scholar]
- 160.Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 161.Griewank K, Borowski C, Rietdijk S, Wang N, Julien A, Wei DG, Mamchak AA, Terhorst C, Bendelac A. Homotypic interactions mediated by Slamf1 and Slamf6 receptors control NKT cell lineage development. Immunity. 2007;27:751–762. doi: 10.1016/j.immuni.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zullo AJ, Benlagha K, Bendelac A, Taparowsky EJ. Sensitivity of NK1.1-negative NKT cells to transgenic BATF defines a role for activator protein-1 in the expansion and maturation of immature NKT cells in the thymus. J Immunol. 2007;178:58–66. doi: 10.4049/jimmunol.178.1.58. [DOI] [PubMed] [Google Scholar]
- 163.Alvarado A, Bernal L, Ferreira D, Paige C, Romero-Sandoval A. (279) Increase of anti-inflammatory cytokines in human macrophages that overexpress CD163 under different pro-inflammatory conditions. J Pain. 2016;17:S45–S46. [Google Scholar]
- 164.la Torre Fabiola VD, Ralf K, Gabriel B, Victor Ermilo AA, Martha MG, Mirbella CF, Rocio BA. Anti-inflammatory and immunomodulatory effects of Critonia aromatisans leaves: Downregulation of pro-inflammatory cytokines. J Ethnopharmacol. 2016;190:174–182. doi: 10.1016/j.jep.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 165.Salazar N, Castellan M, Shirodkar SS, Lokeshwar BL. Chemokines and chemokine receptors as promoters of prostate cancer growth and progression. Crit Rev Eukaryot Gene Expr. 2013;23:77–91. doi: 10.1615/critreveukaryotgeneexpr.2013006905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wu YM, Robinson DR, Kung HJ. Signal pathways in up-regulation of chemokines by tyrosine kinase MER/NYK in prostate cancer cells. Cancer Res. 2004;64:7311–7320. doi: 10.1158/0008-5472.CAN-04-0972. [DOI] [PubMed] [Google Scholar]
- 167.Cracco CM, Terrone C, Porpiglia F, Scarpa RM. Immune response in prostate cancer. Minerva Urol Nefrol. 2005;57:301–311. [PubMed] [Google Scholar]
- 168.Thara E, Dorff TB, Averia-Suboc M, Luther M, Reed ME, Pinski JK, Quinn DI. Immune response to sipuleucel-T in prostate cancer. Cancers (Basel) 2012;4:420–441. doi: 10.3390/cancers4040420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Perambakam S, Hallmeyer S, Reddy S, Mahmud N, Bressler L, DeChristopher P, Mahmud D, Nunez R, Sosman JA, Peace DJ. Induction of specific T cell immunity in patients with prostate cancer by vaccination with PSA146–154 peptide. Cancer Immunol Immunother. 2006;55:1033–1042. doi: 10.1007/s00262-005-0090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rigamonti N, Bellone M. Prostate cancer, tumor immunity and a renewed sense of optimism in immunotherapy. Cancer Immunol Immunother. 2012;61:453–468. doi: 10.1007/s00262-012-1216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Seton-Rogers S. Prostate cancer: Connecting androgen receptor and immunity. Nat Rev Cancer. 2016;16:273. doi: 10.1038/nrc.2016.46. [DOI] [PubMed] [Google Scholar]
- 172.Simons JW. Prostate cancer immunotherapy: beyond immunity to curability. Cancer Immunol Res. 2014;2:1034–1043. doi: 10.1158/2326-6066.CIR-14-0174. [DOI] [PubMed] [Google Scholar]
- 173.Ueda K, Ohtaguro K, Washida H, Jinno H, Fushimi N, Masegi T. Humoral immunity following double freezing of the prostate of patients with prostatic cancer. Prog Clin Biol Res. 1982;107:815–826. [PubMed] [Google Scholar]
- 174.Suzuki K, Nakazato H, Matsui H, Hasumi M, Shibata Y, Ito K, Fukabori Y, Kurokawa K, Yamanaka H. NK cell-mediated anti-tumor immune response to human prostate cancer cell, PC-3: immunogene therapy using a highly secretable form of interleukin-15 gene transfer. J Leukoc Biol. 2001;69:531–537. [PubMed] [Google Scholar]
- 175.Dell DD, Feleccia M, Hicks L, Longstreth-Papsun E, Politsky S, Trommer C. Care of patients with autism spectrum disorder undergoing surgery for cancer. Oncol Nurs Forum. 2008;35:177–182. doi: 10.1188/08.ONF.177-182. [DOI] [PubMed] [Google Scholar]
- 176.Labarthe MC, Theocharous P, Russell N, Todryk S, Bangma C, Thraves P, Dalgleish AG, Whelan MA. A novel murine model of allogeneic vaccination against prostate cancer. Cancer Immunol Immunother. 2008;57:453–465. doi: 10.1007/s00262-007-0384-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tucker JA, Jochems C, Gulley JL, Schlom J, Tsang KY. Immunotherapy: shifting the balance of cell-mediated immunity and suppression in human prostate cancer. Cancers (Basel) 2012;4:1333–1348. doi: 10.3390/cancers4041333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ayshamgul H, Ma H, Ilyar S, Zhang LW, Abulizi A. Association of defective HLA-I expression with antigen processing machinery and their association with clinicopathological characteristics in Kazak patients with esophageal cancer. Chin Med J (Engl) 2011;124:341–346. [PubMed] [Google Scholar]
- 179.Chen XH, Liu ZC, Zhang G, Wei W, Wang XX, Wang H, Ke HP, Zhang F, Wang HS, Cai SH, Du J. TGF-beta and EGF induced HLA-I downregulation is associated with epithelial-mesenchymal transition (EMT) through upregulation of snail in prostate cancer cells. Mol Immunol. 2015;65:34–42. doi: 10.1016/j.molimm.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 180.Garbi N, Hammerling G, Tanaka S. Interaction of ERp57 and tapasin in the generation of MHC class I-peptide complexes. Curr Opin Immunol. 2007;19:99–105. doi: 10.1016/j.coi.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 181.Harris MR, Lybarger L, Yu YY, Myers NB, Hansen TH. Association of ERp57 with mouse MHC class I molecules is tapasin dependent and mimics that of calreticulin and not calnexin. J Immunol. 2001;166:6686–6692. doi: 10.4049/jimmunol.166.11.6686. [DOI] [PubMed] [Google Scholar]
- 182.Vigneron N, Peaper DR, Leonhardt RM, Cresswell P. Functional significance of tapasin membrane association and disulfide linkage to ERp57 in MHC class I presentation. Eur J Immunol. 2009;39:2371–2376. doi: 10.1002/eji.200939536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Contini P, Zocchi MR, Pierri I, Albarello A, Poggi A. In vivo apoptosis of CD8(+) lymphocytes in acute myeloid leukemia patients: involvement of soluble HLA-I and Fas ligand. Leukemia. 2007;21:253–260. doi: 10.1038/sj.leu.2404494. [DOI] [PubMed] [Google Scholar]
- 184.Poggi A, Contini P, Catellani S, Setti M, Murdaca G, Zocchi MR. Regulation of gammadelta T cell survival by soluble HLA-I: involvement of CD8 and activating killer Ig-like receptors. Eur J Immunol. 2005;35:2670–2678. doi: 10.1002/eji.200526177. [DOI] [PubMed] [Google Scholar]
- 185.Concha-Benavente F, Srivastava R, Ferrone S, Ferris RL. Immunological and clinical significance of HLA class I antigen processing machinery component defects in malignant cells. Oral Oncol. 2016;58:52–58. doi: 10.1016/j.oraloncology.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tertipis N, Haeggblom L, Grun N, Nordfors C, Nasman A, Dalianis T, Ramqvist T. Reduced Expression of the Antigen Processing Machinery Components TAP2, LMP2, and LMP7 in Tonsillar and Base of Tongue Cancer and Implications for Clinical Outcome. Transl Oncol. 2015;8:10–17. doi: 10.1016/j.tranon.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bene L, Bodnar A, Damjanovich S, Vamosi G, Bacso Z, Aradi J, Berta A, Damjanovich J. Membrane topography of HLA I, HLA II, and ICAM-1 is affected by IFN-gamma in lipid rafts of uveal melanomas. Biochem Biophys Res Commun. 2004;322:678–683. doi: 10.1016/j.bbrc.2004.07.171. [DOI] [PubMed] [Google Scholar]
- 188.Poat B, Hazari S, Chandra PK, Gunduz F, Balart LA, Alvarez X, Dash S. SH2 modified STAT1 induces HLA-I expression and improves IFN-gamma signaling in IFN-alpha resistant HCV replicon cells. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013117. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 189.Wallace TA, Prueitt RL, Yi M, Howe TM, Gillespie JW, Yfantis HG, Stephens RM, Caporaso NE, Loffredo CA, Ambs S. Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer Res. 2008;68:927–936. doi: 10.1158/0008-5472.CAN-07-2608. [DOI] [PubMed] [Google Scholar]
- 190.Maia MC, Hansen AR. A comprehensive review of immunotherapies in prostate cancer. Crit Rev Oncol Hematol. 2017;113:292–303. doi: 10.1016/j.critrevonc.2017.02.026. [DOI] [PubMed] [Google Scholar]
- 191.Reis ST, Pontes-Junior J, Antunes AA, Sousa-Canavez JM, Abe DK, Cruz JA, Dall’oglio MF, Crippa A, Passerotti CC, Ribeiro-Filho LA, Viana NI, Srougi M, Leite KR. Tgf-beta1 expression as a biomarker of poor prognosis in prostate cancer. Clinics (Sao Paulo) 2011;66:1143–1147. doi: 10.1590/S1807-59322011000700004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ardiani A, Gameiro SR, Kwilas AR, Donahue RN, Hodge JW. Androgen deprivation therapy sensitizes prostate cancer cells to T-cell killing through androgen receptor dependent modulation of the apoptotic pathway. Oncotarget. 2014;5:9335–9348. doi: 10.18632/oncotarget.2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kwilas AR, Ardiani A, Donahue RN, Aftab DT, Hodge JW. Dual effects of a targeted small-molecule inhibitor (cabozantinib) on immune-mediated killing of tumor cells and immune tumor microenvironment permissiveness when combined with a cancer vaccine. J Transl Med. 2014;12:294. doi: 10.1186/s12967-014-0294-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Thomas BC, Kay JD, Menon S, Vowler SL, Dawson SN, Bucklow LJ, Luxton HJ, Johnston T, Massie CE, Pugh M, Warren AY, Barker P, Burling K, Lynch AG, George A, Burge J, Corcoran M, Stearn S, Lamb AD, Sharma NL, Shaw GL, Neal DE, Whitaker HC. Whole blood mRNA in prostate cancer reveals a four-gene androgen regulated panel. Endocr Relat Cancer. 2016;23:797–812. doi: 10.1530/ERC-16-0287. [DOI] [PubMed] [Google Scholar]
- 195.Bishop JL, Sio A, Angeles A, Roberts ME, Azad AA, Chi KN, Zoubeidi A. PD-L1 is highly expressed in Enzalutamide resistant prostate cancer. Oncotarget. 2015;6:234–242. doi: 10.18632/oncotarget.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]