

Summary
High‐fat diet (HFD) and low‐dose streptozotocin (STZ)‐treated rats provide useful animal model for type II diabetes mellitus. Oxidative stress and advanced glycation end products (AGEs) play a role in the development of diabetic complications. Carnosine (CAR) has anti‐oxidant and anti‐glycating properties. We investigated the effects of CAR on oxidation and glycation products in HFD+STZ rats. Rats were fed with HFD (60% of total calories from fat) for 4 weeks, and then a single dose of STZ (40 mg/kg; i.p.) was applied. Rats with blood glucose levels above 200 mg/dl were fed with HFD until the end of the 12th week. CAR (250 mg/kg body weight; i.p.; five times a week) was administered to the rats for the last four weeks. CAR significantly decreased serum triglyceride (TG) (57.7%), cholesterol (35.6%) levels and hepatic marker enzyme activities of HFD+STZ rats. It significantly reduced serum reactive oxygen species (ROS) (23.7%), AGEs (13.4%) and advanced oxidized protein products (AOPP) (35.9%) and hepatic TG (59%), ROS (26%), malondialdehyde (MDA) (11.5%), protein carbonyl (PC) (19.2%) and AGE (20.2%) levels. Liver steatosis and hepatocyte ballooning were also significantly reduced. However, CAR treatment did not alter serum glucose and blood glycated haemoglobin and hepatic anti‐oxidant enzyme activities/mRNA expressions in HFD+STZ rats. Our results indicate that CAR decreased accumulation of oxidation and glycation products, such as MDA, AGE, AOPP and PC in the serum and liver and ameliorated hepatic dysfunction in HFD+STZ rats. This effect may be related to its anti‐oxidative, anti‐glycating, and anti‐lipogenic potential.
Keywords: advanced glycation end products, Carnosine, diabetes mellitus, oxidative stress
Oxidative stress is accepted to play a major role in the pathophysiology of diabetes and diabetic complications. Several factors such as autoxidation of glucose, formation of advanced glycation end products (AGEs) and activation of the polyol pathway are considered to be responsible for the oxidative stress in DM (Turk 2010; de M Bandeira et al. 2013; Nowotny et al. 2015). Hyperglycaemia‐induced increases in reactive oxygen species (ROS) promote oxidative damage in DNA, lipid and protein and thereby lead to cell injury and death (de M Bandeira et al. 2013). Additionally, AGEs, which are formed through a series of reactions from Schiff bases and Amadori products, play an important role in the formation of diabetic complications (Turk 2010; Nowotny et al. 2015). The interaction between AGEs and their specific receptors (RAGE) induces the activation of oxidative stress and stimulates the production and release of cytokines. Thus, oxidative stress and AGEs play an important role in the development of diabetic complications (Turk 2010; de M Bandeira et al. 2013; Nowotny et al. 2015).
High‐fat diet (HFD) and low‐dose streptozotocin (STZ)‐treated rats provide an animal model for type II diabetes mellitus (T2DM) (Skovso 2014). HFD is known to cause insulin resistance (IR) and hyperinsulinaemia (Öner‐İyidoğan et al. 2013). However, low‐dose STZ results in moderate damage in pancreatic β‐cells. This damage disturbs the balance between IR and hyperinsulinaemia and causes hyperglycaemia (Zheng et al. 2012). Therefore, the HFD+STZ model, which progresses from IR to hypoinsulinaemia and hyperglycaemia, mimics the natural T2DM pathogenesis in humans and is suitable to investigate the pathogenesis of diabetic complications and test the efficiency of anti‐diabetic agents (Zheng et al. 2012; Skovso 2014).
Carnosine (CAR; β‐alanyl‐L‐histidine) is a dipeptide with anti‐oxidant effects. Although it is debatable as to how CAR exerts its anti‐oxidant potential (Decker et al. 2000; Velez et al. 2008), it has been suggested that it is dependent on its ability to inactivate ROS, scavenge free radicals and chelate pro‐oxidant metals. It inhibits lipid peroxidation and protein oxidation and acts as an AGE inhibitor (Hipkiss 2009; Boldyrev et al. 2013). Therefore, the use of CAR has been suggested to be useful in ageing and hepatic, cardiovascular and neuronal disorders because of these properties (Hipkiss 2009; Uysal et al. 2015; Baye et al. 2016). Additionally, some reports demonstrated that CAR treatment was effective in attenuating diabetic complications such as fatty liver, atherosclerosis, retinopathy and nephropathy in STZ‐treated rodents (Lee et al. 2005; Pfister et al. 2011; Riedl et al. 2011; Brown et al. 2014; Peters et al. 2014). However, no in vivo studies have investigated the effect of CAR treatment in HFD+STZ‐induced T2DM rats.
In the current study, the effects of CAR treatment on glycaemia, lipid levels and hepatic function tests, together with glycation and oxidation products, were investigated in the serum and liver of HFD+STZ rats.
Materials and methods
Chemicals
STZ (S0130), CAR (C9625) and other chemicals were obtained from Sigma‐Aldrich (Saint‐Louise, MI, USA).
Animals and diets
Male Wistar rats (3–4 months; 220–240 g body weight) were used for all experiments. Animals were obtained from the Aziz Sancar Experimental Medical Research Institute of Istanbul University. The animals were kept in wire‐bottomed stainless cages (three to four per cage) under a daily cycle of 12‐h light and 12‐h darkness and supplied with food and water ad libitum.
Normal diet and HFD were purchased from Barbaros Denizeri A.Ş. (Kocaeli, Turkey). HFD was composed of 23.5% protein, 27.3% carbohydrate and 34.3% fat with adequate mineral and vitamins.
Ethical approval
The experimental procedure used in this study met the guidelines of the Animal Care and Use Committee of the University of Istanbul (18 February 2014; 2014/18).
Experimental design
Rats were divided into control and experimental groups. The experimental design is shown in Figure 1. Twelve rats in the control group were divided into two separate groups as control and CAR groups. These groups were fed on a normal pellet diet for 12 weeks. CAR (250 mg/kg body weight; i.p.; five times a week) was administered to rats of CAR group in the last 4 weeks. The used dose for CAR was chosen according to our previous study (Kalaz et al. 2014).
Figure 1.

Schematic representation of the experimental procedure followed. Arrows indicate injection moments (CAR, carnosine; HFD, high‐fat diet; STZ, streptozotocin).
Rats in the experimental group (n = 20) were given HFD (60% of total calories from fat) for 4 weeks. After 4 weeks of feeding, these rats were fasted for 12 h and injected with STZ dissolved in 0.1 M cold citrate buffer (pH 4.5) at a dose of 40 mg/kg body weight. Five days after STZ treatment, the tail vein fasting blood glucose was estimated and the rats with persistent hyperglycaemia over 200 mg/dl (n = 16) were considered as diabetic.
Four weeks after STZ injection, the diabetic rats were divided into two groups (n = 8 each): the diabetic control (HFD+STZ group) and CAR‐treated diabetic group (HFD+STZ+CAR group). These groups continued to receive HFD throughout the entire experimental period for 12 weeks. Total food and water intake was recorded daily. CAR (250 mg/kg body weight; i.p.; five times a week) was administered to rats in the last 4 weeks. Additionally, the control and HFD+STZ groups were injected with 0.9% NaCl as vehicle for 4 weeks (five times a week, i.p.).
Samples
At the end of the 12‐week period, animals were fasted overnight and anaesthetized with sodium thiopental (50 mg/kg; i.p.). Blood was collected in dry and anti‐coagulant‐containing tubes by cardiac puncture. Serum and plasma were obtained by centrifugation. The liver was rapidly removed, washed in 0.9% NaCl and kept on ice. A small portion of liver tissue was fixed in 10% buffered formalin for histopathologic analysis. The remaining part of the liver tissue was homogenized in ice‐cold 0.15 M KCl (10%; w/v), and homogenates were centrifuged (600 × g for 10 min at 4°C) to remove unhomogenized tissue fragments and nuclear fraction. This fraction was used for biochemical determinations. In addition, one part of this fraction was recentrifuged at 10,000 × g for 20 min at 4°C to obtain the postmitochondrial fraction which was used for the determination of superoxide dismutase (SOD) and glutathione peroxidase (GSH‐Px) activities.
Determinations in blood and serum/plasma
Glycaemia, serum lipids and hepatic damage markers
Blood HbA1c levels were measured in heparinized blood samples. Levels of serum glucose, cholesterol and triglyceride and activities of alanine aminotransferase (ALT), aspartate aminotransferase (AST) and lactate dehydrogenase (LDH) were determined using a Cobas Integra 800 autoanalyzer (Roche Diagnostics, Mannheim, Germany).
Determination of ROS levels
ROS generation was determined using a fluorometric assay. The fluorescence of 2′,7′‐dichlorofluorescein was determined using a microplate fluorometer and luminometer (Fluoroskan Ascent FL, Thermo Scientific Inc, USA) with an excitation of 485 nm and emission of 538 nm. Results were expressed as relative fluorescence units (RFU) (Wang & Joseph 1999).
Determination of endogenous and induced malondialdehyde (MDA) levels
Endogenous and induced MDA levels in plasma were measured using thiobarbituric acid in accordance with the method of Buege and Aust (1978). Results were calculated using the molar extinction coefficient of the product (1.56 × 10−5 M/cm). For the determination of induced MDA levels, serum was incubated with 100 mM 2,2′‐azobis 2‐amidinopropane (AAPH) for 2 h at 37°C and formation of MDA was also measured spectrophotometrically.
Determination of advanced oxidation products of protein (AOPP) levels
Forty microlitres of plasma and 160 μl of 0.2 M citric acid (test) were pipetted into microplate wells. In addition, reagent blank (190 μl of 0.2 M citric acid and 10 μl of 1.16 M KI) and standards (190 μl of 0–100 μM chloramine‐T in 0.2 M citric acid and 10 μl of 1.16 M KI) were prepared in different wells. After 2 min, absorbances of the test and standards were read at 340 nm against reagent blank. Results were standardized by the formation of triiodide ion upon the oxidation of potassium iodide with chloramine‐T. Results are expressed as μmol/chloramine‐T equivalent (Hanasand et al. 2012).
Determination of AGEs
Plasma was diluted (1:50) with phosphate‐buffered saline (PBS) pH 7.4, and fluorescence intensity was measured (λemission: 440 nm; λexcitation: 350 nm). Fluorescence intensity was expressed in arbitrary units (RFU) (Münch et al. 1997).
Determination of ferric reducing anti‐oxidant power (FRAP)
The FRAP assay, as described by Benzie and Strain (1996), was used to determine anti‐oxidant power in plasma. A ferric‐tripyridyltriazine complex is reduced to the ferrous form by electron‐donating anti‐oxidants present in plasma and monitored by measuring the change in absorbance at 593 nm.
Determinations in the liver
Determinations of cholesterol and triglyceride levels
Hepatic lipids were extracted with chloroform:methanol (2:1). After the extraction and evaporation, lipids were redissolved in ethanol–ether (3:1) as previously reported (Giriş et al. 2014) and hepatic cholesterol and triglyceride levels were assayed using kits provided by Bio‐Science Medical (Madrid, Spain).
Determinations of ROS, AOPP, AGE and FRAP levels
These parameters were measured in liver homogenates as described for serum.
Determination of MDA levels
MDA levels were measured by a thiobarbituric acid assay in accordance with the method of Ohkawa et al. (1979). Results were calculated using the molar extinction coefficient of the product (1.56 × 10−5 M/cm) and expressed as nmol/g tissue.
Determination of protein carbonyl (PC) levels
Oxidative protein damage was measured by the quantification of carbonyl groups based on spectrophotometric detection of the reaction between 2,4‐dinitrophenylhydrazine and PC to form protein hydrazones. Hepatic PC levels were calculated from the maximum absorbance (360 nm) using the molar extinction coefficient of the product (22,000 M/cm). The results were expressed as nmol carbonyl per mg protein (Reznick & Packer 1994).
Determinations of anti‐oxidant parameters
Glutathione (GSH) levels were measured with 5,5‐dithiobis‐(2‐nitrobenzoate) at 412 nm in accordance with the method of Beutler et al. (1963). SOD activity was assayed by its ability to increase riboflavin‐sensitized photo‐oxidation of o‐dianisidine (Mylorie et al. 1986). Catalase (CAT) activity was measured using hydrogen peroxide (H2O2) as substrate (Worthington 1993). The disappearance of H2O2 was followed spectrophotometrically at 240 nm. One unit of CAT was considered the activity of enzyme needed to degrade 1 μmol H2O2 per min at 25°C. Glutathione peroxidase (GSH‐Px) activity was measured using cumene hydroperoxide as substrate (Lawrence & Burk 1976).
Determination of protein levels
Protein levels were determined spectrophotometrically using bicinchoninic acid (Smith et al. 1985).
Determination of mRNA expressions of SOD and GSH‐Px
RNA isolation: mRNA expressions of SOD and GSH‐Px were determined using real‐time quantitative polymerase chain reaction. All reagents for mRNA/cDNA preparation were purchased from Roche Applied Science Diagnostics (Mannheim, Germany). Total RNA was extracted from the liver tissues using a commercially available RNA extraction kit (High Pure PCR Template Preparation Kit) in accordance with the manufacturer's instructions. RNA concentration and purity were detected using UV–vis spectrophotometer (Shimadzu, MD, USA). cDNA was synthesized with a reverse transcription (RT) kit (Transcriptor High Fidelity cDNA Synthesis Kit, Roche Applied Science Diagnostics (Mannheim, Germany)) using 250 ng RNA in a total volume of 20 μl.
Determination of mRNA expressions of SOD and GSH‐Px using ‘real‐time’ quantitative PCR: qPCR was performed using the ‘TaqMan Master kit’ in a lightcycler 2.0 system (Roche Applied Science Diagnostics, Mannheim, Germany). Real‐time ready‐probes were used for SOD (CuZnSOD; #100023905) and GSH‐Px (#100047787). A universal probe library (UPL) probe for glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) (#05046220001) was used as the housekeeping gene. Forty‐five cycles (10 s at 95°C, 30 s at 60°C and 1 s at 72°C) were performed for gene amplification. Each sample was quantified by measuring its fluorescence resonance energy with the light cycler quantification software.
Histopathologic examination
The liver tissue was fixed in a 10% buffered formalin solution, embedded in paraffin, sectioned and stained with haematoxylin and eosin (H&E). Steatosis was scored as 0 = <5% (none), 1 = 5–33% (mild), 2 = 34–66% (moderate) and 3 = >67% (severe). Liver damage was evaluated according to inflammation and hepatocyte ballooning parameters. Lobular inflammation was scored as follows: 0 = no visible cell damage, 1 = focal damage on less than two foci (×200), 2 = focal damage on 2–4 foci (×200) and 3 = focal damage on more than four foci. Hepatocyte ballooning was also scored as follows: 0 = no visible cell damage, 1 = rare hepatocyte ballooning and 2 = extensive hepatocyte ballooning (Goodman 2007).
Statistical analysis
One‐way analysis of variance (anova) followed by Tukey's honestly significant difference post hoc test was used for equal variances. The Kruskal–Wallis test (post hoc Mann–Whitney U‐test) was performed for unequal variances.
Results
Body weight and liver weight
There were no significant differences in final body weights and liver weights between the HFD+STZ and control groups. However, body weight gain during 12 weeks decreased significantly (50.8%) in comparison with the control group. Liver index (liver weight/body weight ×100) increased significantly (22.1%) in the HFD+STZ group. CAR treatment did not alter these parameters in HFD+STZ rats (Table 1).
Table 1.
Effect of carnosine (CAR) treatment on initial and final body weights, body weight gain, liver weight and liver index (liver weight ×100/final body weight) in high‐fat diet and low dose of streptozotocin (HFD+STZ)‐treated rats (Mean ± SD)
| Parameters | Control (n = 6) | CAR (n = 6) | HFD+STZ (n = 8) | HFD+STZ+CAR (n = 8) |
|---|---|---|---|---|
| Initial body weight (g) | 204.7 ± 8.04 | 205.0 ± 8.94 | 211.6 ± 11.1 | 202.4 ± 7.00 |
| Final body weight (g) | 292.5 ± 18.9 | 295.8 ± 17.7 | 254.9 ± 35.7 | 259.2 ± 38.9 |
| Body weight gain (g/12 weeks) | 87.8 ± 15.9 | 90.8 ± 12.0 | 43.2 ± 27.3* | 56.9 ± 32.7 |
| Liver weight (g) | 7.66 ± 0.37 | 7.58 ± 0.30 | 8.02 ± 0.58 | 7.77 ± 0.73 |
| Liver index (%) | 2.62 ± 0.07 | 2.56 ± 0.06 | 3.20 ± 0.49* | 3.06 ± 0.60** |
*P < 0.01; **P < 0.05 as compared to the control group.
Blood HbA1c, serum glucose, triglyceride and cholesterol levels and hepatic damage markers
HFD+STZ treatment caused significant increases in glucose (254.8%), HbA1c (122.2%), triglyceride (889.6%) and cholesterol (115%) levels. Serum ALT (299%), AST (74.2%) and LDH (57.5%) activities also significantly increased in HFD+STZ rats compared with the control group. CAR treatment did not alter glucose and HbA1c levels, whereas triglyceride (57.7%) and cholesterol (35.6%) levels significantly decreased. Serum ALT (37.2%), AST (38.9%) and LDH (20.9%) activities were also observed to be significantly reduced due to CAR treatment (Table 2).
Table 2.
The effect of carnosine (CAR) on serum glucose, blood haemoglobin A1c (HbA1c), triglyceride and cholesterol levels, and alanine aminotransferase (ALT), aspartate aminotransferase (AST) and lactate dehydrogenase (LDH) activities in high‐fat diet and low dose of streptozotocin (HFD+STZ)‐treated rats (Mean ± SD)
| Parameters | Control (n = 6) | CAR (n = 6) | HFD+STZ (n = 8) | HFD+STZ+CAR (n = 8) |
|---|---|---|---|---|
| Glucose(mg/dl) | 105.0 ± 8.94 | 106.2 ± 10.7 | 372.5 ± 33.6** | 353.0 ± 43.5** |
| HbA1c (%) | 2.61 ± 0.11 | 2.63 ± 0.17 | 5.80 ± 0.96** | 5.68 ± 0.86** |
| Triglyceride (mg/dl) | 34.5 ± 13.6 | 36.2 ± 16.0 | 341.4 ± 198.5** | 144.3 ± 35.0** , **** |
| Cholesterol (mg/dl) | 57.5 ± 15.2 | 57.8 ± 13.0 | 123.6 ± 34.0* | 79.6 ± 34.1**** |
| ALT (U/l) | 42.0 ± 9.8 | 41.0 ± 7.53 | 167.7 ± 36.6** | 105.4 ± 37.9* , *** |
| AST (U/l) | 124.8 ± 18.3 | 123.3 ± 15.4 | 217.4 ± 70.8** | 132.7 ± 35.0**** |
| LDH (U/l) | 731.7 ± 115.2 | 716.7 ± 96.5 | 1152.2 ± 192.6* | 911.9 ± 159.0†,*** |
*P < 0.001; **P < 0.01 as compared to the control group.
***P < 0.01; ****P < 0.05 as compared to the HFD+STZ group.
† P < 0.05 as compared to the control group.
Glycation and oxidation products and FRAP levels in plasma
ROS (58.7%), MDA (23.3%), AAPH‐induced MDA (52.4%), AOPP (264.3%) and AGE (32.5%) levels significantly increased, but there was no change in FRAP levels in HFD+STZ rats as compared with the control group. High levels of ROS (23.7%), AAPH‐induced MDA (15.7%), AOPP (35.9%) and AGEs (13.4%) were found to be significantly decreased in HFD+STZ rats due to CAR treatment (Table 3).
Table 3.
The effect of carnosine (CAR) on serum/plasma reactive oxygen species (ROS), endogenous and AAPH‐induced malondialdehyde (i‐MDA), advanced oxidation protein products (AOPP), advanced glycation end products (AGEs) and ferric reducing antioxidant power (FRAP) levels in high‐fat diet and low dose of streptozotocin (HFD+STZ)‐treated rats (Mean± SD)
| Parameters | Control (n = 6) | CAR (n = 6) | HFD+STZ (n = 8) | HFD+STZ+CAR (n = 8) |
|---|---|---|---|---|
| ROS (RFU) | 328.0 ± 33.9 | 330.0 ± 43.8 | 520.5 ± 70.8* | 396.9 ± 82.8**** |
| MDA (μmol/l) | 5.28 ± 0.49 | 5.20 ± 0.52 | 6.51 ± 0.48** | 5.82 ± 1.22 |
| i‐MDA (μmol/l) | 24.6 ± 1.34 | 24.0 ± 2.82 | 37.5 ± 3.93* | 31.6 ± 5.94*** , ***** |
| AOPP (μmol/l) | 171.1 ± 35.5 | 163.3 ± 31.4 | 623.3 ± 169.9** | 399.3 ± 106.8** , **** |
| AGEs (RFU) | 187.8 ± 11.9 | 191.7 ± 19.4 | 248.9 ± 28.5* | 215.5 ± 17.5***** |
| FRAP (μmol/l) | 277.3 ± 49.7 | 288.3 ± 49.1 | 318.4 ± 34.7 | 337.4 ± 56.7 |
*P < 0.001; **P < 0.01; ***P < 0.05 as compared to the control group.
****P < 0.01; *****P < 0.05 as compared to the HFD+STZ group.
Hepatic triglyceride, cholesterol, ROS and MDA levels
Hepatic triglyceride (185.9%), ROS (37.4%) and MDA (32.3%) levels significantly increased following HFD+STZ treatment, but cholesterol levels remained unchanged. CAR treatment significantly decreased high levels of hepatic triglyceride (59%), ROS (26%) and MDA (11.5%) in the liver. However, hepatic cholesterol levels remained unchanged due to CAR treatment (Figure 2).
Figure 2.
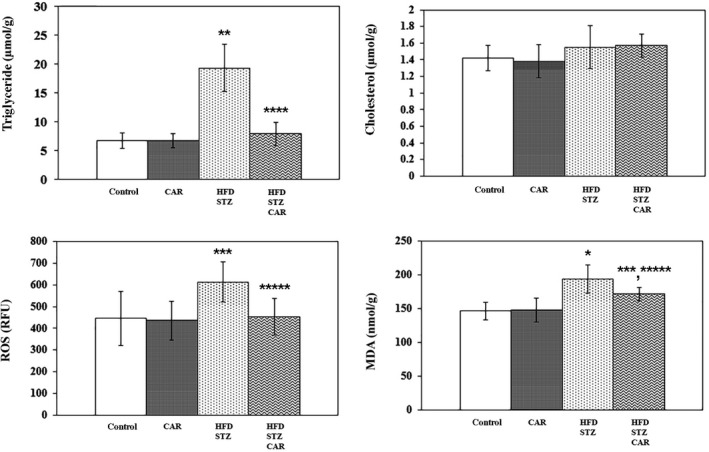
The effect of carnosine (CAR) on hepatic triglyceride, cholesterol, reactive oxygen species (ROS) and malondialdehyde (MDA) levels in high‐fat diet and low dose of streptozotocin (HFD+STZ)‐treated rats (Mean ± SD). *P < 0.001; **P < 0.01; ***P < 0.05 as compared to the control group. ****P < 0.001; *****P < 0.05 as compared to the HFD+STZ group.
Hepatic PC, AOPP and AGE levels
Significant increases in PC (32.7%), AOPP (106.8%) and AGE (137%) levels were found in HFD+STZ rats. PC (19.2%) and AGE (20.2%) levels were diminished by CAR treatment. However, decreases in AOPP levels were not significant (Figure 3).
Figure 3.

The effect of carnosine (CAR) on hepatic protein carbonyl (PC), advanced oxidation protein products (AOPP) and advanced glycation end product (AGE) levels in high‐fat diet and low dose of streptozotocin (HFD+STZ)‐treated rats (Mean ± SD). *P < 0.001; **P < 0.01 as compared to the control group. ***P < 0.001; ****P < 0.05 as compared to the HFD+STZ group.
Hepatic anti‐oxidant parameters and mRNA expressions of SOD and GSH‐Px enzymes
Hepatic FRAP and GSH levels, and SOD and CAT activities did not change in HFD+STZ rats, but GSH‐Px activity decreased significantly (33.4%). There were no changes in anti‐oxidant parameters between HFD+STZ and HFD+STZ+CAR groups (Table 4).
Table 4.
The effect of carnosine (CAR) on hepatic ferric reducing antioxidant power (FRAP) and glutathione (GSH) levels and superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GSH‐Px) activities in high‐fat diet and low dose of streptozotocin (HFD+STZ)‐treated rats (Mean± SD)
| Parameters | Control (n = 6) | CAR (n = 6) | HFD+STZ (n = 8) | HFD+STZ+CAR (n = 8) |
|---|---|---|---|---|
| FRAP (nmol/mg protein) | 53.0 ± 4.42 | 52.3 ± 4.51 | 55.2 ± 3.46 | 58.1 ± 4.03 |
| GSH (μmol/g tissue) | 5.62 ± 0.53 | 5.71 ± 0.48 | 6.18 ± 0.52 | 6.22 ± 0.32 |
| SOD (U/mg protein) | 22.3 ± 5.34 | 22.6 ± 4.22 | 19.3 ± 4.65 | 19.2 ± 3.91 |
| CAT (μmol/mg protein) | 211.0 ± 33.4 | 210.0 ± 25.3 | 234.7 ± 13.1 | 238.0 ± 18.8 |
| GSH‐Px (nmol/min/mg protein) | 534.2 ± 131.3 | 531.3 ± 137.0 | 355.6 ± 24.9* | 341.9 ± 39.9* |
*P < 0.01 as compared to the control group.
mRNA expression of hepatic GSH‐Px significantly decreased (45.8%), but SOD remained unchanged in HFD+STZ rats. These expressions did not alter due to CAR treatment in HFD+STZ rats (Table 5).
Table 5.
The effect of carnosine (CAR) on mRNA expressions of hepatic superoxide dismutase (SOD) and glutathione peroxidase (GSH‐Px) enzymes in high‐fat diet and low dose of streptozotocin (HFD+STZ)‐treated rats
| Parameters | Control | CAR | HFD+STZ | HFD+STZ+CAR |
|---|---|---|---|---|
| SOD | 59.8 ± 4.79 | 56.5 ± 8.73 | 46.7 ± 5.88 | 58.7 ± 12.1 |
| GSH‐Px | 72.3 ± 10.8 | 73.5 ± 11.1 | 39.2 ± 6.31* | 50.2 ± 7.05** |
*P < 0.001; **P < 0.01 as compared to the control group.
Expression levels are relative to expression of glyceraldehyde 3‐phosphate dehydrogenase (Mean± SD; n = 6 each).
Histopathologic observations
Histopathologic scoring was performed using steatosis, lobular inflammation and hepatocyte ballooning scores in H&E staining (Figure 4). Steatosis and hepatocyte ballooning, but not inflammation, scores decreased significantly due to CAR treatment in HFD+STZ rats (Table 6).
Figure 4.
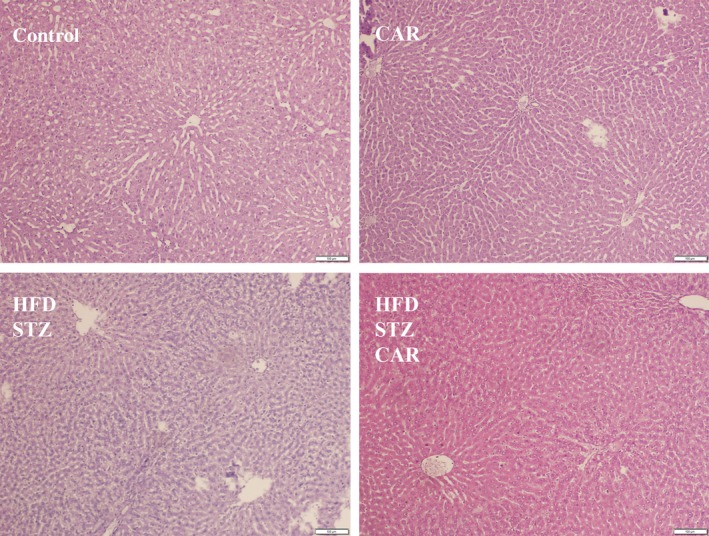
The effect of carnosine (CAR) on liver histopathology in high‐fat diet and low dose of streptozotocin (HFD+STZ)‐treated rats (H&E ×100). [Colour figure can be viewed at wileyonlinelibrary.com]
Table 6.
The effect of carnosine (CAR) on steatosis, inflammation and hepatocyte ballooning scores in the liver of high‐fat diet and low dose of streptozotocin (HFD+STZ)‐treated rats (Mean± SD)
| Parameters | Control (n = 4) | CAR (n = 4) | HFD+STZ (n = 6) | HFD+STZ+CAR (n = 6) |
|---|---|---|---|---|
| Steatosis | 0 | 0 | 1.33 ± 0.52* | 0*** |
| Inflammation | 0 | 0 | 0.83 ± 0.41** | 0.33 ± 0.52 |
| Hepatocyte ballooning | 0 | 0 | 1.50 ± 0.55* | 0.50 ± 0.55**** |
*P < 0.01; **P < 0.05 as compared to the control group.
***P < 0.01; ****P < 0.05 as compared to the HFD+STZ group.
Discussion
HFD+STZ rats show typical symptoms of diabetes such as hyperglycaemia, polyphagia, polydipsia and loss of weight in comparison with control rats (Guo et al. 2014; Silvares et al. 2016). In this study, although no significant changes were detected in body weight due to HFD+STZ treatment, HFD+STZ rats showed a tendency to decrease body weight. Additionally, body weight gain in HFD+STZ rats during the 12 weeks significantly decreased as compared with the controls. Although body weight increases in rats fed on HFD as compared with normal control rats, it has been shown that they show a progressive reduction in body weight gain after injection with STZ. This reduction may be due to increased catabolism of fats and proteins caused by insulin deficiency (Guo et al. 2014).
Metabolic disorders such as dyslipidaemia and liver steatosis are seen in the majority of patients with T2DM. IR may contribute to the development of hepatocyte steatosis in T2DM by disturbing the ability of insulin to suppress lipolysis, resulting in increased circulating free fatty acids and liver lipid accumulation (Tolman et al. 2007). In HFD+STZ rats, dyslipidaemia and hepatic triglyceride accumulation (Li et al. 2015; Zheng et al. 2012) along with histopathologic observations and increased hepatic damage markers in serum (Parveen et al. 2010; Guo et al. 2014; Li et al. 2015; Silvares et al. 2016) were reported by several investigators. Also in the current study, HFD+STZ treatment of rats resulted in hyperglycaemia, dyslipidaemia and hepatic steatosis and inflammation as assessed through biochemical and histopathologic findings.
Mechanisms leading to oxidative stress in DM are not clearly understood yet. However, excessive production of superoxide anion radical (O2 •−) in mitochondrial electron transfer chain, autoxidation of glucose, activation of polyol pathway and activation of protein kinase C (PKC) are being held responsible (King & Loeken 2004; Kawahito et al. 2009; de M Bandeira et al. 2013). Protein glycation, glucose autoxidation, lipid peroxidation and formation of dicarbonyl compounds such as glyoxal and methylglyoxal were observed to increase in hyperglycaemic conditions (Turk 2010; de M Bandeira et al. 2013; Nowotny et al. 2015). As in other complications of diabetes mellitus, oxidative stress and AGEs may also play a significant role in the development of hepatic lesions in DM. Therefore, in the current study, oxidative stress markers and glycation products were investigated in HFD+STZ rats. MDA determination is an indicator of lipid peroxidation, whereas PC and AOPP determinations are indicators of protein oxidation. AOPP, which has some homologies with AGE, also reflects protein glycoxidation (Capeillère‐Blandin et al. 2004). In this study, we found that ROS and lipid and protein oxidation products increased in the liver and serum of HFD+STZ rats. We also aimed to evaluate the anti‐oxidant status of the liver. As is known, GSH is a potent intracellular anti‐oxidant molecule and GSH‐Px, SOD and CAT are major elements of the intracellular anti‐oxidant defence system. Additionally, the FRAP assay is a global marker for anti‐oxidant power. Therefore, GSH and FRAP levels as well as SOD, GSH‐Px and CAT activities and mRNA expressions of SOD and GSH‐Px enzymes were assayed in liver tissue. Hepatic GSH‐Px activity and its mRNA expression decreased, but other anti‐oxidant parameters remained unchanged. Decreases in GSH‐Px activity may cause further increases in oxidative stress in the liver because GSH‐Px plays a primary role in minimizing oxidative damage. Our findings are in accordance with other studies that showed increased oxidative stress in the liver (Parveen et al. 2010; Mahmoud et al. 2012; Silvares et al. 2016) and serum (Mahmoud et al. 2012; Zheng et al. 2012; Govindaraj & Sorimuthu Pillai 2015) in HFD+STZ rats. Additionally, we detected that AGE levels increased in serum and liver of HFD+STZ rats using fluorescence spectroscopy. Although non‐fluorescent AGE cannot be measured by this method, fluorescence spectroscopy is proposed as a valuable method for the determination of AGEs (De la Maza et al. 2012). Some investigators also reported that AGE levels increased in serum (Govindaraj & Sorimuthu Pillai 2015) and liver (Silvares et al. 2016) of HFD+STZ rats.
CAR treatment has gained increasing importance in the prevention of diabetes and its chronic complications (Hipkiss 2009; Baye et al. 2016). Human studies have shown that serum and urine CAR concentrations depend on carnosinase activity and that there is an association between diabetic nephropathy and the polymorphism of carnosinase gene, which encodes an enzyme that metabolizes CAR (Janssen et al. 2005; Freedman et al. 2007). However, in rodents, carnosinase expression has been shown only in kidney tissue (Teufel et al. 2003) and it is absent in serum and other tissues of these animals. Therefore, it has been shown that CAR intake causes significant increases in its levels in plasma and tissues of rats (Hipkiss 2009; Boldyrev et al. 2013). Although no in vivo studies have investigated the effect of CAR treatment in HFD+STZ rats, some investigators have demonstrated that CAR treatment attenuated the diabetic complications in STZ‐treated rodents, but there were some conflicting results in these studies. CAR treatment protected diabetic kidneys from apoptosis and podocyte loss (Riedl et al. 2011), prevented retinal vascular damage (Pfister et al. 2011) and suppressed atherosclerotic plaque instability (Brown et al. 2014) without affecting blood glucose or HbA1c levels. Contrarily, CAR alleviated diabetic nephropathy and cataract formation by decreasing blood HbA1c levels, and serum carboxyl‐methyl lysine levels (Peters et al. 2014). Serum glucose, plasma cytokines and hepatic lipid and lipid peroxide levels were also found to be decreased together with increases in hepatic catalase and GSH‐Px activities due to CAR treatment (1 g/l in drinking water for 4 weeks) in STZ diabetic mice (Lee et al. 2005). Conflicting results about the effect of CAR on glucose metabolism are interesting. A possible reason for this may be the dosage of CAR treatment. In their study, Yamano et al. (2001) induced hyperglycaemia by intracranial injection of 2‐deoxy‐D‐glucose (2‐DG) and examined the effect of CAR under these circumstances. They found that CAR caused a suppression of the hyperglycaemia probably through the regulation of autonomic nerves. Interestingly, a lower or higher dose than the effective dose did not inhibit the 2‐DG hyperglycaemia.
In the current study, the effects of CAR on oxidation and glycation products were investigated for the first time in the serum and the liver of HFD+STZ‐treated rats. The dose of CAR use did not alter serum glucose and HbA1c levels, but it decreased serum lipid levels and hepatic damage markers in HFD+STZ rats. This treatment also led to significant decreases in hepatic triglyceride levels and ameliorated histopathologic findings in the liver of HFD+STZ rats. Similarly, CAR treatment was found to improve insulin resistance, dyslipidaemia, hepatic dysfunction and liver steatosis grade in non‐diabetic mice fed on HFD (60% of calories derived from fat) for 8 weeks (Mong et al. 2011). This effect was related to diminished activities and mRNA expressions of some lipogenic enzymes such as malic enzyme, fatty acid synthase (FAS) and 3‐hydroxy‐3‐methylglutaryl coenzyme A (HMG‐CoA) reductase. HMG‐CoA reductase is a key enzyme in cholesterol biosynthesis and both malic enzyme and FAS are involved in fatty acid and triglyceride synthesis. Furthermore, CAR was reported to be able to suppress HFD‐induced expression of sterol regulatory element‐binding proteins (SREBP)‐1c and SREBP‐2, which are modulators of FAS and HMG‐CoA reductase, respectively, in both liver and adipose tissues (Mong et al. 2011). In the current study, CAR treatment was also observed to decrease ROS, MDA, PC, AOPP and AGE levels without any change in hepatic anti‐oxidant parameters or mRNA expressions of SOD and GSH‐Px enzymes. Similar decreases in lipid and protein oxidation products and AGE levels were also detected in serum of HFD+STZ rats due to CAR treatment.
CAR has anti‐oxidant, anti‐inflammatory, anti‐ischaemic, anti‐glycating, transglycating and chelating roles, and is a protector of biomolecules against oxidative/carbonyl stress (Hipkiss 2009; Boldyrev et al. 2013; Baye et al. 2016; Hipkiss et al. 2016). It detoxifies reactive dicarbonyl compounds such as 4‐hydroxynonenal and forms adducts with them (Aldini et al. 2005). It also reacts directly with PC groups and produces PC‐CAR adducts or carnosinylated proteins and inhibits oxidative conversion of glycated proteins to AGEs (Hipkiss et al. 2001, 2016). These properties of CAR are important for inactivation/removal of damaged proteins. In addition, CAR was found to have anti‐lipogenic effects (Mong et al. 2011). The efficiency of CAR treatment on glycation and oxidation products observed in the current study may be related to its multifunctional properties.
In conclusion, our results indicate that CAR treatment may be useful in the prevention of the accumulation of oxidation and glycation products in the serum and liver, and the amelioration of hepatic dysfunction in HFD+STZ‐induced diabetic rats.
Conflict of interest
Authors declare no conflict of interest.
Funding
Research Fund of Istanbul University (project Number: YÖP: 43042; UDP‐53865).
Acknowledgements
The authors wish to thank David F. Chapman BSc. for editing the English of the final version of the manuscript. This study was supported by the Research Fund of Istanbul University (project Number: YÖP: 43042; UDP‐53865).
References
- Aldini G., Facino R.M., Beretta G. & Carini M. (2005) Carnosine and related dipeptides as quenchers of reactive carbonyl species: from structural studies to therapeutic perspectives. BioFactors 24, 77–87. [DOI] [PubMed] [Google Scholar]
- Baye E., Ukropcova B., Ukropec J., Hipkiss A., Aldini G. & de Courten B. (2016) Physiological and therapeutic effects of carnosine on cardiometabolic risk and disease. Amino Acids 48, 1131–1149. [DOI] [PubMed] [Google Scholar]
- Benzie I.F.F. & Strain J.J. (1996) The ferric reducing ability of plasma (FRAP) as a measure of ‘antioxidant power’’: the FRAP assay. Anal. Biochem. 239, 70–76. [DOI] [PubMed] [Google Scholar]
- Beutler E., Duron O. & Kelly B.M. (1963) Improved method for the determination of blood glutathione. J. Lab. Clin. Med. 61, 882–888. [PubMed] [Google Scholar]
- Boldyrev A.A., Aldini G. & Derave W. (2013) Physiology and pathophysiology of carnosine. Physiol. Rev. 93, 1803–1845. [DOI] [PubMed] [Google Scholar]
- Brown B.E., Kim C.H., Torpy F.R., et al (2014) Supplementation with carnosine decreases plasma triglycerides and modulates atherosclerotic plaque composition in diabetic apoE(‐/‐) mice. Atherosclerosis 232, 403–409. [DOI] [PubMed] [Google Scholar]
- Buege J.A. & Aust S.D. (1978) Microsomal lipid peroxidation. Methods Enzymol. 52, 302–310. [DOI] [PubMed] [Google Scholar]
- Capeillère‐Blandin C., Gausson V., Descamps‐Latscha B. & Witko‐Sarsat V. (2004) Biochemical and spectrophotometric significance of advanced oxidized protein products. Biochim. Biophys. Acta 1689, 91–102. [DOI] [PubMed] [Google Scholar]
- De la Maza M.P., Garrido F., Escalante N., et al (2012) Fluorescent advanced glycation end‐products (ages) detected by spectro‐photofluorimetry, as a screening tool to detect diabetic microvascular complications. J. Diabetes Mellitus 2, 221–226. [Google Scholar]
- Decker E.A., Livisay S.A. & Zhou S. (2000) A re‐evaluation of the antioxidant activity of purified carnosine. Biochemistry (Mosc) 65, 766–770. [PubMed] [Google Scholar]
- Freedman B.L., Hicks P.J., Sale M.M., et al (2007) A leucine repeat in the carnosinase gene CNDP1 is associated with diabetic end‐stage renal disease in European Americans. Nephrol. Dial. Transplant. 22, 1131–1135. [DOI] [PubMed] [Google Scholar]
- Giriş M., Doğru‐Abbasoğlu S., Kumral A., Olgaç V., Koçak‐Toker N. & Uysal M. (2014) Effect of carnosine alone or combined with α‐tocopherol on hepatic steatosis and oxidative stress in fructose‐induced insulin resistant rats. J. Physiol. Biochem. 70, 385–395. [DOI] [PubMed] [Google Scholar]
- Goodman Z.D. (2007) Grading and staging systems for inflammation and fibrosis in chronic liver diseases. J. Hepatol. 47, 598–607. [DOI] [PubMed] [Google Scholar]
- Govindaraj J. & Sorimuthu Pillai S. (2015) Rosmarinic acid modulates the antioxidant status and protects pancreatic tissues from glucolipotoxicity mediated oxidative stress in high‐fat diet: streptozotocin‐induced diabetic rats. Mol. Cell. Biochem. 404, 143–159. [DOI] [PubMed] [Google Scholar]
- Guo C., Zhang C., Li L., Wang Z., Xiao W. & Yang Z. (2014) Hypoglycemic and hypolipidemic effects of oxymatrine in high‐fat diet and streptozotocin‐induced diabetic rats. Phytomedicine 21, 807–814. [DOI] [PubMed] [Google Scholar]
- Hanasand M., Omdal R., Norheim K.B., Gøransson L.G., Brede C. & Jonsson G. (2012) Improved detection of advanced oxidation protein products in plasma. Clin. Chim. Acta 413, 901–906. [DOI] [PubMed] [Google Scholar]
- Hipkiss A.R. (2009) Carnosine and its possible roles in nutrition and health. Adv. Food Nutr. Res. 57, 87–154. [DOI] [PubMed] [Google Scholar]
- Hipkiss A.R., Brownson C. & Carrier M.J. (2001) Carnosine, the anti‐ageing, antioxidant dipeptide, may react with carbonyl groups. Mech. Ageing Dev. 122, 1431–1445. [DOI] [PubMed] [Google Scholar]
- Hipkiss A.R., Baye E. & de Courten B. (2016) Carnosine and the processes of ageing. Maturitas 93, 28–33. [DOI] [PubMed] [Google Scholar]
- Janssen B., Hohenadel D., Brinkkoetter P., et al (2005) Carnosine as a protective factor in diabetic nephropathy: association with a leucine repeat of the carnosinase gene CDNP1. Diabetes 54, 2320–2327. [DOI] [PubMed] [Google Scholar]
- Kalaz E.B., Çoban J., Aydın A.F., et al (2014) Carnosine and taurine treatments decreased oxidative stress and tissue damage induced by D‐galactose in rat liver. J. Physiol. Biochem. 70, 15–25. [DOI] [PubMed] [Google Scholar]
- Kawahito S., Kitahata H. & Oshita S. (2009) Problems associated with glucose toxicity: Role of hyperglycemia‐induced oxidative stress. World J. Gastroenterol. 15, 4137–4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King G.L. & Loeken M.R. (2004) Hyperglycemia‐induced oxidative stress in diabetic complications. Histochem. Cell Biol. 122, 333–338. [DOI] [PubMed] [Google Scholar]
- Lawrence R.A. & Burk R.F. (1976) Glutathione peroxidase activity in selenium–deficient rat liver. Biochem. Biophys. Res. Commun. 71, 952–958. [DOI] [PubMed] [Google Scholar]
- Lee Y.T., Hsu C.C., Lin M.H., Liu K.S. & Yin M.C. (2005) Histidine and carnosine delay diabetic deterioration in mice and protect human low density lipoprotein against oxidation and glycation. Eur. J. Pharmacol. 513, 145–150. [DOI] [PubMed] [Google Scholar]
- Li Y.G., Ji D.F., Zhong S., Lin T.B. & Lv Z.Q. (2015) Hypoglycemic effect of deoxynojirimycin‐polysaccharide on high fat diet and streptozotocin‐induced diabetic mice via regulation of hepatic glucose metabolism. Chem. Biol. Interact. 225, 70–79. [DOI] [PubMed] [Google Scholar]
- de M Bandeira S., da Fonseca L.J., da S Guedes G., Rabelo L.A., Goulart M.O., Vasconcelos S.M. (2013) Oxidative stress as an underlying contributor in the development of chronic complications in diabetes mellitus. Int. J. Mol. Sci. 14, 3265–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud A.M., Ashour M.B., Abdel‐Moneim A. & Ahmed O.M. (2012) Hesperidin and naringin attenuate hyperglycemia‐mediated oxidative stress and proinflammatory cytokine production in high fat fed/streptozotocin‐induced type 2 diabetic rats. J. Diabetes Complications 26, 483–490. [DOI] [PubMed] [Google Scholar]
- Mong M.C., Chao C.Y. & Yin M.C. (2011) Histidine and carnosine alleviated hepatic steatosis in mice consumed high saturated fat diet. Eur. J. Pharmacol. 653, 82–88. [DOI] [PubMed] [Google Scholar]
- Münch G., Keis R., Wessels A., et al (1997) Determination of advanced glycation end products in serum by fluorescence spectroscopy and competitive ELISA. Eur. J. Clin. Chem. Clin. Biochem. 35, 669–677. [DOI] [PubMed] [Google Scholar]
- Mylorie A.A., Collins H., Umbles C. & Kyle J. (1986) Erythrocyte superoxide dismutase activity and other parameters of copper status in rats ingesting lead acetate. Toxicol. Appl. Pharmacol. 82, 512–520. [DOI] [PubMed] [Google Scholar]
- Nowotny K., Jung T., Höhn A., Weber D. & Grune T. (2015) Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 5, 194–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkawa H., Ohishi N. & Yagi K. (1979) Assay for lipid peroxidation in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 95, 351–358. [DOI] [PubMed] [Google Scholar]
- Öner‐İyidoğan Y., Koçak H., Seyidhanoğlu M., et al (2013) Curcumin prevents liver fat accumulation and serum fetuin‐A increase in rats fed a high‐fat diet. J. Physiol. Biochem. 69, 677–686. [DOI] [PubMed] [Google Scholar]
- Parveen K., Khan M.R., Mujeeb M. & Siddiqui W.A. (2010) Protective effects of Pycnogenol on hyperglycemia‐induced oxidative damage in the liver of type 2 diabetic rats. Chem. Biol. Interact. 186, 219–227. [DOI] [PubMed] [Google Scholar]
- Peters V., Riedl E., Braunagel M., et al (2014) Carnosine treatment in combination with ACE inhibition in diabetic rats. Regul. Pept. 194–195, 36–40. [DOI] [PubMed] [Google Scholar]
- Pfister F., Riedl E., Wang Q., et al (2011) Oral carnosine supplementation prevents vascular damage in experimental diabetic retinopathy. Cell. Physiol. Biochem. 28, 125–136. [DOI] [PubMed] [Google Scholar]
- Reznick A.Z. & Packer L. (1994) Oxidative damage to proteins: spectrophotometric method for carbonyl assay. Methods Enzymol. 233, 357–363. [DOI] [PubMed] [Google Scholar]
- Riedl E., Pfister F., Braunagel M., et al (2011) Carnosine prevents apoptosis of glomerular cells and podocyte loss in STZ diabetic rats. Cell. Physiol. Biochem. 28, 279–288. [DOI] [PubMed] [Google Scholar]
- Silvares R.R., Pereira E.N., Flores E.E., et al (2016) Combined therapy with metformin and insulin attenuates systemic and hepatic alterations in a model of high‐fat diet‐/streptozotocin‐induced diabetes. Int. J. Exp. Pathol. 97, 266–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovso S. (2014) Modeling type 2 diabetes in rats using high fat diet and streptozotocin. J. Diabetes Invest. 5, 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P.K., Krohn R.I., Hermanson G.T., et al (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85. [DOI] [PubMed] [Google Scholar]
- Teufel M., Saudek V., Ledig J.P., et al (2003) Sequence identification and characterization of human carnosinase and a closely related non‐specific dipeptidase. J. Biol. Chem. 278, 6521–6531. [DOI] [PubMed] [Google Scholar]
- Tolman K.G., Fonseca V., Dalpiaz A. & Tan M.H. (2007) Spectrum of liver disease in type 2 diabetes and management of patients with diabetes and liver disease. Diabetes Care 30, 734–743. [DOI] [PubMed] [Google Scholar]
- Turk Z. (2010) Glycotoxines, carbonyl stress and relevance to diabetes and its complications. Physiol. Res. 59, 147–156. [DOI] [PubMed] [Google Scholar]
- Uysal M., Koçak‐Toker N., Doğru‐Abbasoğlu S. (2015) Carnosine protection against liver injury In: Food and Nutritional Component in Focus No. 8, Imidazole Peptides: Chemistry, Analysis, Function and Effects, pp. 510–527 (ed Preedy V.R.), Cambridge, UK: The Royal Society of Chemistry. [Google Scholar]
- Velez S., Nair N.G. & Reddy V.P. (2008) Transition metal ion binding studies of carnosine and histidine: biologically relevant antioxidants. Colloids Surf. B. Biointerfaces 66, 291–294. [DOI] [PubMed] [Google Scholar]
- Wang H. & Joseph J.A. (1999) Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic. Biol. Med. 27, 612–616. [DOI] [PubMed] [Google Scholar]
- Worthington V. (1993) Catalase In: Worthington Enzyme Manual: enzymes and related biochemical, pp. 77–80 (ed Worthington V.), New Jersey: Worthington Biochem. Corp. [Google Scholar]
- Yamano T., Nijima A., Iimori S., Tsuruoka N., Kiso Y. & Nagai K. (2001) Effects of L‐carnosine on the hyperglycemia caused by intracranial injection of 2‐deoxy‐D‐glucose in rats. Neurosci. Lett. 313, 78–82. [DOI] [PubMed] [Google Scholar]
- Zheng T., Shu G., Yang Z., Mo S., Zhao Y. & Mei Z. (2012) Antidiabetic effect of total saponins from Entada phaseoloides (L.) Merr. in type 2 diabetic rats. J. Ethnopharmacol. 139, 814–821. [DOI] [PubMed] [Google Scholar]