

Summary
Genome annotation is, nowadays, performed via automatic pipelines that cannot discriminate between right and wrong annotations. Given their importance in increasing the accuracy of the genome annotations of other organisms, it is critical that the annotations of model organisms reflect the current annotation gold standard. The genome of Bacillus subtilis strain 168 was sequenced twenty years ago. Using a combination of inductive, deductive and abductive reasoning, we present a unique, manually curated annotation, essentially based on experimental data. This reveals how this bacterium lives in a plant niche, while carrying a paleome operating system common to Firmicutes and Tenericutes. Dozens of new genomic objects and an extensive literature survey have been included for the sequence available at the INSDC (AccNum AL009126.3). We also propose an extension to Demerec's nomenclature rules that will help investigators connect to this type of curated annotation via the use of common gene names.
Introduction
With the advent of Next Generation Sequencing (NGS) techniques, sequencing genomes has become routine. While this is of tremendous interest by providing a profusion of sequence data, contributing accurate knowledge coupled to the sequences has become a nightmare (Zallot et al., 2016). The main reason for this dire state of affairs is that automated in silico pipelines draw ‘knowledge’ by inference, relying primarily on protein sequence similarity analysis, with the function tag extracted from databases that basically lack experimental information. Worse, this approach most often uses the majority rule (a function is deemed correct if it is found in the majority of annotations). Several correction systems have been devised to improve this highly inadequate approach, but the fact is that in the absence of a process allowing experimental validation (direct or indirect) of annotations, errors continue to percolate through the system (Gilks et al., 2005). It is therefore of the utmost importance that, for at least some reference genomes, manual curation of sequence data be still maintained on a routine basis (Chang et al., 2016). Unfortunately, there is little or no reward for this type of work despite the fact that individual scientist still develops much of their research and make key discoveries based on knowledge rooted in sequence annotations. Briefly, investigators demand access to knowledge, but they are extremely reluctant to pay for that access in any way. The consequence is that, at the present time, the number of cleanly annotated genomes is vanishingly small (of note are the EcoGene resource for Escherichia coli (Zhou and Rudd, 2013), a recent update for the genome of Pseudomonas putida KT2440 (Belda et al., 2016) and ongoing work on Mycobacterium tuberculosis (Lew et al., 2013)). Bacillus subtilis, strain 168 remains a case in point, and here we present an updated annotation, based on experimental evidence collected for this organism but also from other organisms, that we describe here with the aim of summarizing knowledge about this bacterium as a possible chassis for Synthetic Biology studies.
The genome sequence of B. subtilis 168 was published in 1997 by a consortium mainly formed by European and Japanese laboratories (Harwood and Wipat, 1996; Kunst et al., 1997). At the time, sequencing was very hard work because it primarily rested on cloning fragments of DNA into an E. coli recipient host before sequencing, under conditions where at least 15% of the sequences failed to be cloned. The reason of this unwelcome difficulty was that transcription and translation signals in B. subtilis are unexpectedly efficient in E. coli, resulting in toxic levels of gene expression, particularly of membrane proteins (Frangeul et al., 1999). The situation improved when, late in the project, long‐range PCR became routine. The sequencing of genomes of similar composition remained fairly intractable. This resulted in the genome of B. subtilis being the only Firmicute genome sequence for almost five years, until those of the much smaller genomes of Staphylococcus aureus (Kuroda et al., 2001) and Streptococcus pneumoniae (Hoskins et al., 2001; Tettelin et al., 2001) were published, followed by that of Bacillus anthracis strains of size equivalent to that of B. subtilis (Read et al., 2002).
Being one of the two very first bacterial genomes longer than 4 Mb to be sequenced implied that an appreciable level of errors must have crept in. This was expected because the sequence was obtained in different laboratories, where a variety of experimental protocols was used. It is also likely that mutations occurred even during the cultivation steps that are a prerequisite to sequencing. The genome was therefore entirely re‐sequenced using NGS methods ten years later (Barbe et al., 2009). It can now be expected that, barring the inevitable mutations that appear during propagation in laboratories [see the situation for E. coli (Soupene et al., 2003)], this final sequence corresponds to an exact sequence [International Nucleotide Sequence Database Collaboration (INSDC) AccNum AL009126.2], that does not need to be re‐sequenced. In contrast, sequence annotations inevitably keep changing as the identification of gene function improves almost on a daily basis. Some genes were actually annotation artefacts, while novel genomic objects, in particular untranslated regulatory RNAs, are being discovered on a regular basis. A few years later, it had already been relevant to associate the now exact sequence with an update of the metabolic pathways that were deciphered after analysis of the genome [INSDC AccNum AL009126.3 (Belda et al., 2013)]. Naturally, with the genome sequence available, as well as the new ‘omics’ approaches, discoveries establishing the function of genes previously of unknown function (there was about 2000 of those, half of the genes identified in the first report of the genome sequence) kept accumulating. Here, we report the annotation of the genome sequence at the date of 15 November 2017, twenty years after its initial version, with the inclusion of a large number of newly identified functions (including several unpublished experimentally established functions, Appendix S1) and the discovery of three dozen new genomic objects with experimentally established functions (Table 1). Taking into account the current availability of the sequences of many of its strains, we took the opportunity of the present work to explore again the natural niche of B. subtilis as a species (remembering that because strain 168 is a laboratory strain, it is likely to have lost some of its wild type ecological potential), as well as the nature of the genes that may be considered to characterize the species, focusing on novel entries.
Table 1.
Novel genomic objects introduced in the present annotation of the B. subtilis 168 genome
| Label | Start | Name | Function | References | |
|---|---|---|---|---|---|
| ldRNA | BSU_misc_RNA_3 | 119855 | ldlJ | Ribosomal protein L10 leader mRNA sequence | 26101249 |
| suRNA | BSU_misc_RNA_7 | 486092 | swaO | ATP‐, cyclic di‐AMP‐sensing riboswitch | 25086507, 25086509 |
| CDS | BSU04785 | 528025 | cmpA | Factor allowing degradation of SpoIVA by ClpXP | 26387458 |
| suRNA | BSU_misc_RNA_65 | 532642 | sncO | ICEBs1 mobile element: conserved small untranslated RNA | 20525796, 22505685 |
| suRNA | BSU_misc_RNA_66 | 559610 | sncZ | No identified function: borders undefined | 20525796 |
| suRNA | BSU_misc_RNA_8 | 626446 | aswA | Adenine riboswitch | 25573585 |
| CDS | BSU09958 | 1071402 | sscA | Spore assembly and germination protein | 21670523 |
| CDS | BSU09959 | 1071613 | sscB | Spore assembly and germination protein | 21670523 |
| suRNA | BSU_misc_RNA_67 | 1233405 | roxS | Small regulatory RNA (NO regulated) | 28436820 |
| CDS | BSU12815 | 1348356 | spoIISC | Three component toxin/antitoxin/antitoxin SpoIISABC, antitoxin C | 25039482, 26300872, 27294956 |
| Riboswitch | BSU_misc_RNA_16 | 1376328 | guwA | Guanidinium riboswitch | 28212758 |
| Riboswitch | BSU_misc_RNA_68 | 1395622 | swmG | Magnesium riboswitch (modest affinity) | 28455443 |
| Riboswitch | BSU_misc_RNA_87 | 1410633 | mnrW | Manganese ion riboswitch | 25794618, 25794619 |
| Riboswitch | BSU_misc_RNA_88 | 1457005 | gswA | Riboswitch regulating ptsGHI expression via GlcT binding | 15155854, 22750856 |
| suRNA | BSU_misc_RNA_69 | 1483557 | fsrA | Regulatory RNA controlling iron‐dependent metabolism | 24576839 |
| suRNA | BSU_misc_RNA_70 | 1534070 | srrA | Small regulatory RNA and messenger RNA (arginine metabolism) | 27449348 |
| CDS | BSU14629 | 1534120 | rgpA | Regulator of GapA synthesis | 27449348 |
| CDS | BSU15140 | 1580622 | rsmH | 16S rRNA m4C1402 methyltransferase | 27711192 |
| suRNA | BSU_misc_RNA_89 | 1780554 | surX | sigW‐dependent | 23155385 |
| CDS | BSU17845 | 1916955 | yzzP | No identified function, present in some S. pneumoniae strains | 27144405 |
| CDS | BSU18978 | 2069883 | bsrE | Type I toxin (BsrE/AsrE) | 26940229 |
| suRNA | BSU_misc_RNA_74 | 2070115 | asrE | Small regulatory antitoxin RNA, toxin‐antitoxin type I system (BsrE/AsrE) | 26940229 |
| CDS | BSU19749 | 2146053 | yoyG | Putative toxin of a type I toxin family (sporulation operon) | 20156992, 21670523 |
| fCDS | BSU20049 | 2160397 | nrdFBc | Phage SP beta nucleoside diphosphate reductase minor subunit (C‐terminus) | 23391036 |
| fCDS | BSU20051 | 2161778 | nrdFBn | Phage SP beta nucleoside diphosphate reductase minor subunit (N‐terminus) | 23391036 |
| suRNA | BSU_ncRNA_1 | 2208880 | aimX | Small RNA controlling lysogeny of phage SPbeta | 28099413 |
| CDS | BSU20850 | 2208980 | aimP | Arbitrium lysis /lysogeny regulatory peptide (GMPRGA) | 28099413 |
| CDS | BSU20860 | 2210154 | aimR | Arbitrium peptide sensor regulator | 28099413 |
| asRNA | BSU_misc_RNA_90 | 2219849 | apbT | Antisense RNA of Toxin SpbT | 24576839 |
| CDS | BSU21000 | 2219960 | spbT | Toxin | 24576839 |
| suRNA | BSU_misc_RNA_91 | 2472880 | pswI | Proline T‐box riboswitch upstream of porI | 21233158 |
| suRNA | BSU_misc_RNA_82 | 2773783 | surF | Expressed under sporulation conditions | 25790031 |
| ldRNA | BSU_misc_RNA_43 | 2855915 | ldlU | Ribosomal protein L21 leader mRNA sequence | 27381917 |
| CDS | BSU28475 | 2910746 | lysCB | Beta subunit of aspartokinase II | 1980002 |
| ldRNA | BSU_misc_RNA_47 | 2953550 | ldlT | Ribosomal protein L20 leader mRNA sequence | 23611891 |
| ldRNA | BSU_misc_RNA_93 | 3035589 | ldsD | Ribosomal protein S4 leader mRNA sequence | 23611891 |
| asRNA | BSU_ncRNA_2 | 3335545 | auzJ | Putative antisense RNA for YuzJ putative toxin (toxin I signature) | 20156992, 21670523 |
| suRNA | BSU_misc_RNA_94 | 4169919 | mswM | Manganese riboswitch | 25794618, 25794619 |
Databases for the genome
For many years, the SubtiList database was used by most investigators as the reference database for the B. subtilis 168 sequence (Moszer et al., 2002). It was maintained at the Institut Pasteur until year 2009, when its support was discontinued. In parallel, a mirror with significant modifications (Fang et al., 2005) was established at the HKU‐Pasteur Research Centre Ltd where it was supported by a grant of the Hong Kong government's Innovation and Technology Commission (Biosupport) until 2010. Lack of support from the Institut Pasteur resulted in obsolescence and the Beijing Genome Institute in Shenzhen took over the baton until 2016 via the Microme Genochore microbial support. This resource, MicroSys, which had proposed a database available on tablets (Fig. 1), has since been discontinued without prior notice.
Figure 1.
Based on SubtiList, a draft interface for microbial databases built up for tablets at the BGI.
SubtiWiki
Facing the lack of support for a facility that is of considerable interest for all investigators working with Firmicutes, Jörg Stülke and his colleagues in 2009 decided to create a Wiki site, SubtiWiki, which collates as much as possible information from the literature about the reference B. subtilis genome sequence (Lammers et al., 2010). This resource is now routinely used by the community, providing text‐based access to published information about the genes and proteins of B. subtilis as well as presentations of its metabolic and regulatory pathways (Michna et al., 2016).
The MicroScope/MaGe platform
Annotation of genome sequences must be imbedded in knowledge generated for as many sources of information as possible. Médigue and co‐workers designed an annotation platform, MicroScope/MaGe, meant to make the most of the diverse annotations associated to bacterial genomes by imbedding in the same platform both sequence and annotation data, together with analytical methods designed to explore the data (Medigue et al., 2017; Vallenet et al., 2017). To obtain a cutting‐edge annotation of the genome of B. subtilis 168, we used the MicroScope platform to collect information from as many sources as possible, based on literature and extant databases. This new annotation is now available at the INSDC and at the MicroScope Website (https://www.genoscope.cns.fr/agc/microscope). Since the last update in databanks (January 2013), the annotation of about 96% of the protein‐coding genes (4097 among 4257 CDS) was revised. Furthermore, additional bibliographical references (2097 new publications among a total of 5754) were added and cover approximately 79% of the protein‐coding genes. The annotated sequence is available with the present work as Table S2.
Tentative approaches towards a unified nomenclature
A major challenge facing genome sequence databases is gene nomenclature. Indeed, the first gene names were proposed based on phenotypes [for example in Escherichia coli, related to antibiotic resistance, e.g. ‘ampC’ for ampicillin resistance (Normark and Burman, 1977), or shape, e.g. ‘fts’ genes, yielding filamentation when mutated (Ricard and Hirota, 1973)]. Subsequently, names were chosen following identification of a biological function, often an enzyme function. In parallel, authors liked to propose fancy names to genes (this is well illustrated in the current gene nomenclature of Drosophila melanogaster). Also, genes corresponding to orthologues in different species were often named differently, depending on the inclination of the authors of the first works. Naturally, of course, many enzymes are promiscuous so that the first catalytic activity discovered in one organism could differ from that in another organism, especially when the first identifications were obtained in vitro. This is obviously problematical as browsing knowledge databases using gene names could help investigators to focus rapidly on their genes of interest. It is therefore recommended that the gene names of orthologues should be conserved throughout the tree of life. However, many gene products have more than one function, mediated by interactions with a variety of partners that often differ in different organisms. All this means that a fully consistent nomenclature is unlikely to be reached any time soon. Nevertheless, because having consistent names for common functions each time, a gene that has been correctly annotated would help users immensely and we have tried as much as possible to give identical names to orthologues of E. coli and B. subtilis. This was previously attempted in the GenoChore databases (Fang et al., 2005), where, for example, the ribosomal protein S12 gene would be named rpsL in all genomes, rather than use its original access tag (e.g. in P. putida, PP0449).
To name genes, we used an extended version of Demerec's nomenclature system (Demerec et al., 1968). A gene name is italicized and begins with three low case letters, followed by one or more capitals. In the best systems, there is no numeral in a wild type gene name: numerals are reserved to identifying mutant genes [e.g. relA1 for a common mutation found in laboratory strains of E. coli (Harvey et al., 1988)]. We nevertheless still kept here the numeral 0 in sporulation genes, but we suggest that ‘0’ (spo0A) should soon be replaced by letter ‘O’ (spoOA). When genes are split into several parts, making pseudogenes, their name identifies the relevant part with a low case letter following the standard gene name [e.g. n for the N‐terminus and c for the C‐terminus, as illustrated in pseudogene appA, split into appAn (BSU11381) and appAc (BSU11382) in the present B. subtilis reference laboratory strain]. For genes identified as CDSs of unknown function, we kept the ‘y’ (‘Why’) nomenclature with the Demerec format, until a function could be ascribed to the gene, at which time, the name was changed into a standard gene name, preferably using the name proposed by the authors that identified the function, when it existed. When renaming, we kept the last capital letter of the ‘y’ name in the final name when this did not create duplicated names (e.g. skiX replaces yknX). We finally noticed that investigators often explore the literature and databases using a gene name. It is therefore highly inconvenient when a gene name corresponds to a common English word (e.g. hinT or thiS). We therefore tried as far as possible to avoid such common spelling when creating new names, and we recommend, for future annotations, to try and replace those unwieldy names by new ones (a general possibility is to use the extended Demerec's rule, adding a second letter after the final capitalized letter of the gene name). Because many genes have a variety of names in the literature, synonyms were included in the gene data file, which is indexed using a unique accession number [e.g. thiO, with synonyms yjbR and goxB, AccNum BSU11670 label, codes for a promiscuous glycine oxidase that is involved in the first step of thiamine biosynthesis (Jurgenson et al., 2009)].
Bacillus subtilis in 2017
Experimentally‐rooted database curation is essentially manual, and therefore considerably time‐consuming. Here, data from the literature were systematically collected by exploring PubMed, PubMed Central and SubtiWiki, and browsing the Internet with ‘y’ gene names as keywords as sources of information. In addition, we used a functional analysis approach of the type that is fruitful when trying to construct relevant chassis in synthetic biology [SynBio (Harwood et al., 2013)]. This entails considering cells as computers making computers, with all the relevant prerequisites (Danchin, 2009).
Making inferences using synthetic biology approaches
Function identification can be derived in three major ways (see Fig. 2 for a general scenario used here for genome sequence annotation). First, a bottom‐up approach uses alignments of sequences with proteins of experimentally known function (this is the standard approach). Second, symmetrically, a top‐down complementary approach that follows the trend developed in SynBio studies with emphasis on the machine reading the programme [the ‘chassis’ in the relevant jargon (de Lorenzo and Danchin, 2008)]. It starts from building up a functional partition of the genome into two master functions: 1/functions (hence genes) required for constructing a progeny; 2/functions required for occupying a specific niche and functions used to create specialized devices – cell types or organelles – meant to explore the environment. Finally, an abductive reasoning approach rests on educated guesses that explore the consequences of specific predictions (‘shot in the dark’: facing a forest at night, fire, and if something cries, look for it; if not, try again). An example of this situation is reflected in the discovery of the unexpected pathway allowing the organism to use S‐methyl‐cysteine (SMeC) as a sulphur source. Knowing that dioxygen was involved, the expected pathway was predicted as an oxidation step of the sulphur atom. After a long series of unsuccessful approaches, it was observed that a DefB mutant of strain 168, lacking one of the two amino acid deformylases of strain 168, did not grow on SMeC. This triggered the hypothesis that the methyl‐group was oxidized rather than the sulphur atom. This allowed deciphering of the entire pathway (Chan et al., 2014).
Figure 2.
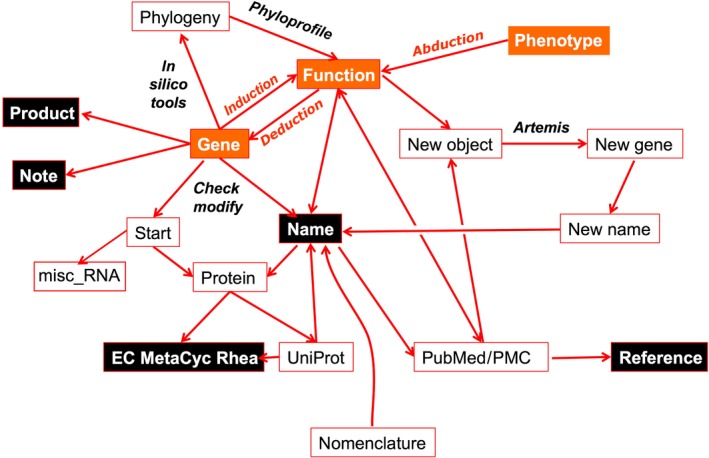
Scenarios for annotation. Annotation combines three approaches: data‐, hypothesis‐ and context‐driven. The first one is based on induction, the second on deduction and the third on abduction, combining functional, phenotypic and sequence data (orange boxes and see text). The outcome of the procedure results in the identification of a gene product, a gene name, participation in metabolic reactions and literature references identified by PubMed identifiers (black boxes). Free text notes are also provided to help understanding the biologically relevant context of each particular gene.
The cell‐as‐a‐computer model splits functions into two major types. Those which run the system [equivalent to the operating system (OS) of a computer] and those which use the cell for specific purposes (equivalent to the applications run by the computer). The former are limited in number. They are those identified in the minimal genomes constructed for SynBio approaches. We named this set the paleome (Acevedo‐Rocha et al., 2013). It comprises the core translation, transcription and replications machineries, together with basic membrane functions involved in waste disposal, basic ion supply, in energy generation and cell wall synthesis, as well as a key set of central metabolism enzymes. Remarkably, exactly as for authentic computers, a few paleome functions are specific to a particular clade, as OS functions may be specific to a particular computer brand. An illustration of such inevitable kludges required to implement an abstract schema into a material world is, in the Firmicutes/Tenericutes clade, the requirement for a protease that splits off the first nine residues of ribosomal protein L27 initially used as a scaffold, after assembly of the ribosome (Danchin and Fang, 2016). In the present annotation, it was found in silico that gene rppA(ysxB) of the B. subtilis genome codes for this function (Wall et al., 2017). As a rule, we further substantiated our predictions, when not directly based on experiments in B. subtilis, by conservation of essential amino acid residues or specific neighbourhoods, provided by synteny or by co‐evolution profile (or both). Another feature of the process of translation has also been revived with the identification of an important role of formylation of the methionine residue loaded on initiator tRNA (Cai et al., 2017). This revives an open question about the apparent redundant role of formylation in translation previously explored in E. coli where an allosteric modulation of the 70S ribosome structure may shift back to initiation in polycistronic operons without a requirement for the dissociation of ribosomal subunits during the translation of contiguous cistrons (Petersen et al., 1976; Yamamoto et al., 2016).
In the domain of replication, the co‐evolution profile of DNA polymerase III alpha subunits (Engelen et al., 2012) contributed fruitfully to the present annotation. It allowed us to identify several important functions specific to the B. subtilis species. In the same way, the degradosome structure of Firmicutes is different from that of Proteobacteria, for example, and quite consistent. In particular, degradation of messenger RNAs involves the combined activities of endonucleases, 3′‐end exonucleases and 5′‐end exonucleases, with a specific set of enzymes that have both activities identified experimentally in B. subtilis RnjA and RnjB. The exact function of the latter will need to be further characterized (Gao et al., 2017) as it seems to be present even in streamlined Tenericutes (Hutchison et al., 2016), while it co‐evolves mainly with genes of unknown functions (Engelen et al., 2012). The set of persistent genes identified in Firmicutes defines the B. subtilis paleome. The function of most of the genes of this basic OS has now been identified. Table S1 summarizes the most recent functional identification of the genes that have long remained without an ascribed function, in parallel with the streamlined paleome functional set.
Strain 168 among other B. subtilis strains
Sequencing genomes has become much simpler and cheaper since the date of publication of the sequence of strain 168. While it remained the only B. subtilis genome available for many years, there has been a significant effort to sequence other strains in the last decade. This resulted in more than 45 completely sequenced genomes by the end of 2016, and around one hundred high‐quality draft genomes are deposited in the NCBI RefSeq database (ftp://ftp.ncbi.nih.gov/genomes/refseq/bacteria/Bacillus_subtilis/). The number of complete genomes is likely to increase further in the near future thanks to long‐read sequencing technologies. The availability of genomes for many strains opens up new avenues for research. Most importantly, it opens up the possibility of using population genomics data to make inferences about the function of genes in the genome and their ecological role. Yet before making such analyses, one must draw a line between authentic genomes of B. subtilis and those of other highly similar species. Currently, and this taxonomic misannotation is unfortunate, several complete genomes labelled as B. subtilis are genetically quite distant from the reference strain, with an average nucleotide identity (ANI) lower than the proposed minimal threshold for defining the species [94% (Konstantinidis et al., 2006; Richter and Rossello‐Mora, 2009)]. While the definition of bacterial species has been based essentially on physiological and biochemical traits, recent works suggest that population genetics and ecological definitions might provide more meaningful definitions of species (Gevers et al., 2005; Ward et al., 2008).
We analysed the diversity of protein‐coding gene repertoires of 36 complete genomes of B. subtilis (selected after using the threshold of ANI > 94% relative to strain 168). The absolute numbers provided by these analyses must be handled with care, as they depend on the methods used and on the homogeneity of the annotations. To ensure that annotations are as homogeneous as possible, we used the re‐annotations of RefSeq (even though, for strain 168, they are not the best ones). The core genome, that is the set of genes present in all strains, was composed of around 2500 genes (Fig. 3A). This value is still slightly decreasing with the increasing number of sequenced genomes, suggesting that it may be even smaller. However, some of the decrease in the core genome with increased sampling may be due to recent deleterious mutations, yet to be purged by natural selection, or to annotation or sequencing errors. Hence, it is more meaningful at this stage to mention that 3291 genes families are present in more than 95% of the strains. These account for around three‐quarters of the genome of strain 168. The pan‐genome, the diversity of different gene families encountered in the set of the 36 genomes in the species, is much larger, reaching ~6250 genes, about 50% more than the gene repertoire of the average genome. Sampling more genomes will certainly increase this number, given the shape of the cumulative curve (Fig. 3A), and as about 1000 gene families are only found in one strain (Fig. 3B). Matching many other bacterial genomes (Touchon et al., 2009; Collins and Higgs, 2012), the majority of gene families are present in either very few, or most genomes of the species.
Figure 3.
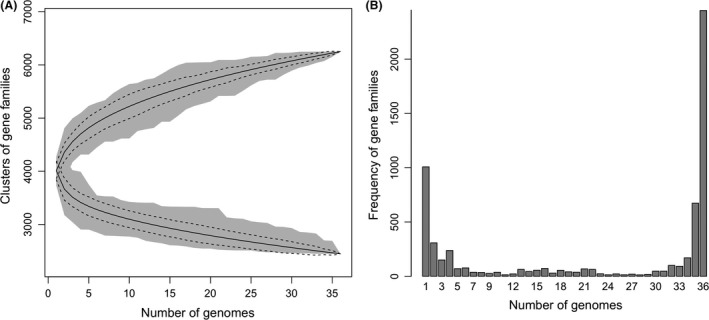
Analysis of protein‐coding genes in 36 complete genomes of B. subtilis.A. The core and pan‐genomes were computed for random samples of increasing size of the 36 genomes. The shaded regions indicate the range of variation of these values.B. The frequency of the presence of each gene family from those that are present in only one strain (peak at 1) to those that are components of the core genome (peak at 36). The identification of the families of core and pan‐genomes followed the methodology of (Touchon et al., 2014).
The genome sequence highlights the B. subtilis ecological niche
This study demonstrates that a core of approximately 2500 genes reflects the gene setup of B. subtilis as a species. It comprises a paleome, described previously, that is essentially shared with that of the minimal Firmicute/Tenericute genome, made of <500 genes (Danchin and Fang, 2016), associated to a complement that defines the minimal niche occupied by this species, its species‐specific cenome – its genes coding for context‐specific functions (Acevedo‐Rocha et al., 2013). Most genes of the paleome have a well‐characterized function. We noticed that several engineer‐type structural functions linked to the building up of the ribosome nanomachine as well as other important RNA structures such as riboswitches are now well understood: RulR(YlxR) codes for molecular ruler (Zhang and Ferre‐D'Amare, 2016), and KtuQ(YlxQ, RpmXA) and KtuS (YbaB, RpmXB) are RNA‐binding proteins specifically associated to kink turns (Huang and Lilley, 2016). Once identified, the association of the functions coded in the cenome match well with the conditions of the first isolation of B. subtilis in the wild, as reported in the Bergey manual (Sneath, 1986); ‘hay, or grass bacillus’ in English, ‘laseczka sienna’ in Polish, ‘kusa no saikin’ in Japanese (B. subtilis enriched from rice straw is used to make the popular soy beans fermented food natto). This also reminds us of the Pasteur/Pouchet controversy about the origin of life (Roll‐Hansen, 1979): Pouchet boiled hay extracts as a way to ‘sterilize’ growth media, and because spores resisted he could antagonize Pasteur. All these experiments point to this bacterium as tightly associated to herbaceous plants, both in the rhizosphere and in the phylloplane. Bacillus subtilis strains have even been found as beneficial endophytic bacteria in a variety of plants (Gond et al., 2015; Ding et al., 2017). A significant number of genes are indeed explicitly involved in direct interaction with plants [e.g. roots (Habib et al., 2017), or leaves (Zeriouh et al., 2014)], either positively or as scavengers of metabolites such as rhamnosides from decaying plants. As a case in point, among many other examples, YfmS, a chemotaxis sensory transducer recognizing a still unknown substrate is involved in the colonization of Arabidopsis thaliana roots (Allard‐Massicotte et al., 2017). Last, the Spo0A protein that controls the fate of cells as vegetative, spores or biofilm‐forming cells, is key to root colonization (Grau et al., 2015). Strain 168, however, is mutated in several genes that would compromise its occupation of this natural biotope: it requires tryptophan for growth because trpC has been inactivated by a frameshift, and, in the same way, it cannot properly colonize roots because of inactivation of gene sfpA for example. However, strain OKB105, which is a derivative with an intact sfpA gene, is able to produce non‐ribosomal peptides and polyketides restoring its authentic plant interaction (Xie et al., 2014).
Finally, and this is remarkable, B. subtilis possesses a blue light receptor, BlrA (formerly YtvA) related to plant phototropins, coupled to a transcriptional complex that monitors the presence of light in the environment. Besides being sensitive to light, this receptor senses the presence of oxygen (Losi et al., 2002). It carries a LOV (light, oxygen and voltage) domain and a STAS (sulphate transporters and antisigma‐factor antagonists) domain and binds FMN (flavin mononucleotide) as a chromophore. Its cycle of activation by light/recovery is also tuned to the environment by features such as hydration (Pennacchietti et al., 2014). Sensing light is a way for cells to tell immediately where they are located. Interestingly, Klebsiella pneumoniae, which, contrary to what its name would suggest, has a plant niche somewhat similar to that of B. subtilis, possesses a putative receptor NifL [involved in oxygen regulation of nitrogenase synthesis, binding flavin nucleotides (Christie et al., 1999)] that has features in common with those of BlrA. Photorhabdus luminescens also codes for a protein, Plu4388, with a domain that may monitor light, perhaps allowing light communication between these photon producing cells. Even Pseudomonas putida PP_4629 protein may be a photoreceptor. In B. subtilis, this is consistent with a plant niche alternating between the phylloplane (light, dioxygen and dry conditions) and the rhizosphere (dark, low oxygen and humidity). In terms of coupling light with environment‐dependent gene expression, this also fits well with another functionally convergent light‐sensing system discovered in E. coli, where the light‐sensitive BluR transcriptional regulator couples the response to light oxygen and temperature [E. coli cycles between a warm dark anaerobic environment and a cool aerobic environment (Tschowri et al., 2012)].
Beside these widespread genes, B. subtilis displays specific features involving cell differentiation, on the one hand via sporulation and motility organelles, or via formation of multicellular entities, biofilms; on the other hand it also encodes phages or phage remnants. The corresponding set of genes that we may name histome (from ἱστος, tissue) comprises an appreciable part of the genome [more than 300 genes for sporulation, and 51 for appendages, flagella (43) and pili (8), often grouped into islands].
The role of phages has also to be revisited. Temperate phages were long considered to be in a dormant state, waking up in specific conditions of the environment. Yet, phage induction is involved in a variety of differentiation processes. In strain 168, the skin element, for example, is removed in the mother cell during the sporulation process, generating the sporulation‐specific sigma factor K (Krogh et al., 1996). In the same way, the spsM gene is interrupted by bacteriophage SPbeta which is excised during the sporulation process using two phage‐encoded proteins, SprA and SprB (Abe et al., 2017). This is now recognized as a new role of lysogeny, named ‘active lysogeny’, that provides yet another account for the presence of bacteriophages within bacteria (Feiner et al., 2015), with B. subtilis as a paradigmatic example.
Novel features extracted for the genome sequence reannotation
In the present release of the B. subtliis 168 reference sequence annotation, we have included some new genomic objects, in particular RNAs, when this was linked to identified functions (we still left aside much of the many transcripts identified via RNAseq sequencing but not explicitly linked to identified functions) and some protein‐coding genes, such as spoIISC coding for the third element of the three‐components toxin/antitoxin/antitoxin SpoIIS sysem [Table 1, (Gabrisko and Barak, 2016)]. We have also experimentally authenticated genes such as the transporters of methylthioribose [MtrA(previously YfnA) for influx and MtrE(B/Y) for efflux, and the ribose transporter, see Appendix S1] and an aminotransferase DapX (previously PatA), required for an essential step in lysine biosynthesis proceeding via an acetylated intermediate, as also does the MetAA intermediate, in contrast to the situation in E. coli [Appendix S1 and see (Bastard et al., 2017)].
We further focused on specific metabolic features that have until recently been overlooked. Bacteria must cope with inevitable errors of metabolism (Danchin, 2017), mediated in particular by a list of expected toxic side reactions (Lerma‐Ortiz et al., 2016). The B. subtilis metabolic setup illustrates variations upon this very general theme. For example, a large variety of organisms use glutathione as a general detoxifying compound. In contrast, in B. subtilis, glutathione is replaced by a counterpart, bacillithiol, that plays most if not all of the roles discovered previously for glutathione (Chandrangsu et al., 2017b). Interestingly, the enzymes that use bacillithiol are often counterparts of enzymes identified elsewhere, but are not true orthologues as they must accommodate a different thiol substrate. A similar situation is observed in Actinomycetes, where mycothiol replaces glutathione (Rawat and Av‐Gay, 2007). This is a very important observation that should be taken into account when considering clusters of orthologues. Metabolic accidents contribute to ageing, in particular via synthesis of dicarbonyls such as methylglyoxal (MGO) or fumarate (Danchin, 2017). The latter reacts with cysteine in proteins or glutathione, forming S‐(2‐succinyl) cysteine inducing senescence in animals (Miglio et al., 2016). MGO results from the action of MGO synthase, MgsA, the function of which is still a matter of speculation (Danchin, 2017). Bacillus subtilis has an arsenal of genes that allows it to cope with this toxic molecule [AkrN(YhdN) aldo/keto reductase specific for NADPH; KhtSTU (YhaSTU) proton/potassium antiporter (Chandrangsu et al., 2013); SufL (YraA) deglycase, a general stress protecting enzyme (Abdallah et al., 2016); GlxB(YurT) methylglyoxalase, lactoylbacillithiol lyase and YvgN promiscuous glyoxal/methylglyoxal reductase, several of them involving bacillithiol directly or indirectly via controlling potassium transport].
Other types of errors result from the presence of mimics of authentic functional metabolites and this must be remedied. As a case in point PgeF (YlmD, EcYfiH), a factor involved in maintaining the composition of the murein peptides complements an E. coli yfiH defect. Lack of PgeF results in the incorporation into the PG sacculi of non‐canonical amino acids, L‐serine or glycine in place of L‐alanine (Parveen and Reddy, 2017). Among widespread sources of metabolic errors, non‐proteinogenic amino acids should be prevented from entering the translation process, and a variety of pathways cope with this situation. A general feature of the metabolic processes that deal with analogues of authentic functional metabolites is similar to that found in a chemist's laboratory: protection (N‐acetylation of the unwanted aminoacid) to prevent hazardous reactions, followed by deprotection at the end of the inactivation pathways (Chan et al., 2014). The large collection of N‐acyl‐transferase genes present in the genome (46 genes) and often with no identified function should be explored for this type of function. Among those are also safeguard systems that protect residues within proteins (usually lysine residues, but also arginine or histidine residues) against spurious modification by reactive metabolic intermediates (Kim et al., 2013). Interestingly, as is commonplace in evolution processes, once a programmed modification exists, it can be recruited for further functions, in particular regulatory functions (Kosono et al., 2015). Coenzymes are prone to accidents: NAD(P)H is hydrated into an analogue that would clog many pathways if it were not converted back to the active form by NnrA(YxkO), a repair enzyme (Petrovova et al., 2014). S‐adenosylmethionine [(S,S)‐AdoMet] may isomerise at the sulfonium atom and the accidental isomer (R,S)‐AdoMet has presumably found a way to remain a methyl‐donor via an homoscysteine methylase using both isomers [a domain in SamT (Lu et al., 2010), and possibly YbgG, similar to S. cerevisiae methyltransferases Mht1 and Sam4 which could also be a much needed AdoMet racemase (Vinci and Clarke, 2010)]
In the same way, while iron is essential in many processes (in particular in respiration), B. subtilis has an interesting preference for manganese [see (Chandrangsu et al., 2017a)], for example with two transporters, a major one MneP(YdfM) and MneS(YeaB) and a minor one (Huang et al., 2016). This may explain why iron is dispensable from a variety of Firmicutes (mostly Lactobacilli (Weinberg, 1997)) and the derived clade of Tenericutes (Danchin and Fang, 2016). Finally, it is important to stress in this update that B. subtilis harbours a new regulator, cyclic diAMP, the main function of which, potassium homoeostasis, has been deciphered by Jörg Stülke and his co‐workers (Gundlach et al., 2017).
Bacillus subtilis exploring its environment
In the previous paragraphs, we have described the behaviour of B. subtilis in its preferred environment as revealed by the present genome annotation update. Several additional features were also revealed during this undertaking. Ecological niches keep changing and bacteria must accommodate to new and often hostile environments, while trying to stick to those environments that evolution has directed them to favour. Three major functions are linked to this situation: overcoming deleterious actions of non‐living and living organisms, escaping to other niches, possibly far away or staying in place. Bacillus subtilis monitors this situation via specific sigma factors (Helmann, 2016) and protein phosphorylation cascades (Schultz, 2016; Pane‐Farre et al., 2017). These will not be further discussed here (except to note that the concept of stress being very ambiguous, as all living organisms suffer multiple transitions, it should probably be avoided to be replaced by the idea of transition management).
Resisting poisons and hostile conditions
Among interesting features, recently identified in the genome is a heteromeric transporter CrcBA CrcBB, allowing resistance to fluoride ions (Ji et al., 2014; Macdonald and Stockbridge, 2017). Indeed, it has been found that fluoride flooding has happened repeatedly (volcanic ashes, local environments and rock weathering) resulting in an average concentration of 625 mg kg−1 in different rock types (Tavener and Clark, 2006) and diffusing into plants. In parallel, GswA, a member of a riboswitch family long of unknown function, has been functionally identified as a result of its ability to bind guanidine (Lilley, 2017), and control expression of a guanidinium exporter, GndCD (YkkCD). A variety of quorum‐sensing systems exists in B. subtilis, with a novel one involving kanosamine, a metabolite that also acts as an antibiotic against a variety of microbes (van Straaten et al., 2013; Tojo et al., 2014; Vetter and Palmer, 2017). Finally, B. subtilis is able to scavenge complex molecules made by other organisms, such as the xenosiderophore schizokinen via the specific transporter SxzYZA (Podkowa et al., 2014).
The plant environment suffers alternating dry and wet conditions and this results in considerable changes in osmotic pressure, monitored by mechanosensing (Belas, 2014). Bacillus subtilis codes for at least five such safety valves (McsC, McsL, McsT, McsY), one specific for sporulation (SpoVAC) that open up upon lethal increase in osmotic pressure. Some of those may also leak out or in antibiotics, leading to constitutive resistance (Song et al., 2013) or sensitivity (Jiafeng et al., 2015).
Moving around
Swimming in liquid media and swarming on surfaces are two major motile behaviours of bacteria. Swimming bacteria use chemotaxis to find nutrients and avoid toxic environments. By contrast, swarming bacteria suppress chemotaxis and self‐organize in a collective motion to explore novel niches while being protected by a mass effect (Harshey and Partridge, 2015). Remarkably, swarming appears to be dependent on a modification of translation factor EF‐P by a 5‐aminopentanol group, as swarming is defective in the absence of EfpI(YmfI) that reduces aminopentanone to aminopentanol (Hummels et al., 2017). In addition, B. subtilis, even when devoid of appendages, is capable of sliding on surfaces (Kovacs et al., 2017), dependent on the presence of surfactin (defective in strain 168, due to pseudogene sfpA) and of exopolysaccharides discussed below, that, interestingly, generate osmotic pressure in the extracellular space (Grau et al., 2015).
Making biofilms
Exploration requires moving around, but when conditions are stably profitable, it is advantageous to find a way to stay around. This is illustrated by yet another case of convergent functional evolution, where many species of bacteria evolved a variety of mechanisms to structure sessile biofilm communities. Bacillus subtilis biofilms display complex architectures that, again, adapt to the plant world, with alternating dry and wet conditions. Cells are encased within a polysaccharide complex made of exopolysaccharides secreted by the bacteria (Hobley et al., 2015). The polyamine spermidine activates matrix synthesis via expression of regulator SlrR (Hobley et al., 2017).
Further exploration of the metabolism of inositol, identified, as expected for catabolism, an NAD‐dependent dehydrogenase IolX. Intriguingly, two dehydrogenases, IolU(YulF) and IolW, associated to a presumably anabolic process because they are NADP‐dependent (Kang et al., 2017), are possibly involved in biofilm formation [inactivation of the counterpart of IolU generates a biofilm defect in Streptococcus mutans (Yoshida and Kuramitsu, 2002)]. FbnA(YloA) is another protein that is likely to be involved in cell adherence to a variety of substrates and belongs to the biofilms’ setup [deficient cells are deficient in biofilm (Rodriguez Ayala et al., 2017)]. Biofilms form highly hydrophobic communities that resist wetting but also solvents and biocides. Hydrophobicity is essentially caused by secreted protein BslA with a small contribution of BslB(YweA), its paralogue (Morris et al., 2017), via production of a leaf/petal‐like hydrophobic behaviour (Werb et al., 2017). In the biofilm, synthesis of BslA is tightly regulated and the resultant protein is secreted into the extracellular environment where it forms a barrier allowing the B. subtilis cells to shelter under a ‘protein raincoat’ (Arnaouteli et al., 2016).
Seeding the earth with a progeny
Plants, which cannot move, nevertheless colonized a considerable area of the Earth. This is because they produce seeds, which carry over their genome using a huge number of processes to escape far from their origin. The same is true for bacteria that make spores, specific structures that can sustain hardships and then germinate when conditions appear to be proper to sustain life. Sporulation, indeed, has been a major research topic for B. subtilis studies, providing models that are used ubiquitously to account for the process in a variety of microbes (Huang and Hull, 2017). The vast majority of our knowledge on B. subtilis sporulation was described in previous updates [in particular with a progressively increasing number of sporulation genes since the early times of genetic analyses (Piggot, 1973)], and we will only point out a recent observation related to this interesting process. Sporulation is costly (it requires the death of a mother cell) and the decision process to choose between other differentiated states of the bacteria is therefore of the utmost importance. Many of the components of the decision‐making machinery have been identified (Decker and Ramamurthi, 2017), most of them converging to protein Spo0A (Dubnau et al., 2016), which appears to be the hub at which the various stages of B. subtilis development are decided. In fact, the exact role of the phosphorylation cascades separating information channels (signal transduction) in cells remains open. As an example, the previously proposed notion that the NAD+/NADH ratio controls the major sporulation kinase KinA activity through the PAS‐A domain of the enzyme has been refuted (Kiehler et al., 2017), opening up again the question of the signals that trigger developmental processes in B. subtilis. These information channels are mediated by histidine kinases that have common properties, but nevertheless can channel information along highly specific pathways (Abriata et al., 2017) avoiding parasitic cross‐talk (Laub, 2016).
Conclusions
Genome annotation is a way to progressively build up a consistent picture of the manner in which living organisms develop in a particular niche. While back in 1991, well before the genome sequence was completed, the acquisition of newly sequenced large genome contigs revealed that half of the putative genes thus identified were unknown both in structure and in function (they were then named elusive, esoteric, conspicuous – EEC – genes by Piotr Slonimski at a meeting in Elounda, in Greece) their role is progressively revealed owing to the hard work of investigators all over the world. Many still remain to be deciphered, and this will often bring about new concepts, such as the CRISPR‐Cas phage immunity system (absent from B. subtilis 168), new structures (such as K‐turn RNA‐binding proteins) or new chemical processes (such as the requirement for a protection/deprotection cycle to cope with close analogues of authentic cell building blocks). We hope that, in addition to the new knowledge that will spread to the community, this type of work will attract young investigators to follow through and take over the helm.
Authors’ contributions
AD organized this work and wrote the bulk of the article, to which all authors contributed. He annotated all genes using the MaGe/Microscope platform, maintained by CM and DV. RB focused on annotation of plant‐related genes and genes involved in secondary metabolism. CRH adapted the text to a large audience of microbiologists. EPCR performed the in silico analyses of strains of B. subtilis related to strain 168 and wrote the corresponding section. AS focused on annotation of sulfur‐related genes and performed experiments to close gaps in metabolic pathways. DV prepared the final annotation table and deposited it at the ENA‐INSDC archive.
Conflict of interest
None declared.
Supporting information
Table S1. The extended Bacillus subtilis paleome
Table S2. Bacillus subtilis 168 annotated genome in the EMBL‐ENA format.
Appendix S1. Experimental identification of methylthioribose transport and a missing step in lysine biosynthesis.
Acknowledgements
This work benefited from the support of AMAbiotics SAS.
Microbial Biotechnology (2018) 11(1), 3–17
Funding information
This work benefited from the support of AMAbiotics SAS.
References
- Abdallah, J. , Mihoub, M. , Gautier, V. , and Richarme, G. (2016) The DJ‐1 superfamily members YhbO and YajL from Escherichia coli repair proteins from glycation by methylglyoxal and glyoxal. Biochem Biophys Res Commun 470: 282–286. [DOI] [PubMed] [Google Scholar]
- Abe, K. , Takamatsu, T. and Sato, T. (2017) Mechanism of bacterial gene rearrangement: SprA‐catalyzed precise DNA recombination and its directionality control by SprB ensure the gene rearrangement and stable expression of spsM during sporulation in Bacillus subtilis . Nucleic Acids Res 45, 6669–6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abriata, L.A. , Albanesi, D. , Dal Peraro, M. , and de Mendoza, D. (2017) Signal sensing and transduction by histidine kinases as unveiled through studies on a temperature sensor. Acc Chem Res 50: 1359–1366. [DOI] [PubMed] [Google Scholar]
- Acevedo‐Rocha, C.G. , Fang, G. , Schmidt, M. , Ussery, D.W. , and Danchin, A. (2013) From essential to persistent genes: a functional approach to constructing synthetic life. Trends Genet 29: 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard‐Massicotte, R. , Tessier, L. , Lecuyer, F. , Lakshmanan, V. , Lucier, J.F. , Garneau, D. , et al (2017) Bacillus subtilis early colonization of Arabidopsis thaliana roots involves multiple chemotaxis receptors. MBio 7, e01664–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaouteli, S. , MacPhee, C.E. , and Stanley‐Wall, N.R. (2016) Just in case it rains: building a hydrophobic biofilm the Bacillus subtilis way. Curr Opin Microbiol 34: 7–12. [DOI] [PubMed] [Google Scholar]
- Barbe, V. , Cruveiller, S. , Kunst, F. , Lenoble, P. , Meurice, G. , Sekowska, A. , et al (2009) From a consortium sequence to a unified sequence: the Bacillus subtilis 168 reference genome a decade later. Microbiology 155: 1758–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastard, K. , Perret, A. , Mariage, A. , Bessonnet, T. , Pinet‐Turpault, A. , Petit, J.L. , et al (2017) Parallel evolution of non‐homologous isofunctional enzymes in methionine biosynthesis. Nat Chem Biol 13: 858–866. [DOI] [PubMed] [Google Scholar]
- Belas, R. (2014) Biofilms, flagella, and mechanosensing of surfaces by bacteria. Trends Microbiol 22: 517–527. [DOI] [PubMed] [Google Scholar]
- Belda, E. , Sekowska, A. , Le Fevre, F. , Morgat, A. , Mornico, D. , Ouzounis, C. , et al (2013) An updated metabolic view of the Bacillus subtilis 168 genome. Microbiology 159: 757–770. [DOI] [PubMed] [Google Scholar]
- Belda, E. , van Heck, R.G. , Jose Lopez‐Sanchez, M. , Cruveiller, S. , Barbe, V. , Fraser, C. , et al (2016) The revisited genome of Pseudomonas putida KT2440 enlightens its value as a robust metabolic chassis. Environ Microbiol 18: 3403–3424. [DOI] [PubMed] [Google Scholar]
- Cai, Y. , Chandrangsu, P. , Gaballa, A. , and Helmann, J.D. (2017) Lack of formylated methionyl‐tRNA has pleiotropic effects on Bacillus subtilis . Microbiology 163: 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, C.M. , Danchin, A. , Marliere, P. , and Sekowska, A. (2014) Paralogous metabolism: S‐alkyl‐cysteine degradation in Bacillus subtilis . Environ Microbiol 16: 101–117. [DOI] [PubMed] [Google Scholar]
- Chandrangsu, P. , Dusi, R. , Hamilton, C.J. , and Helmann, J.D. (2013) Methylglyoxal resistance in Bacillus subtilis: contributions of bacillithiol‐dependent and independent pathways. Mol Microbiol 91: 706–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrangsu, P. , Rensing, C. , and Helmann, J.D. (2017a) Metal homeostasis and resistance in bacteria. Nat Rev Microbiol 15: 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrangsu, P. , Loi, V.V. , Antelmann, H. and Helmann, J.D. (2017b) The role of bacillithiol in Gram‐positive Firmicutes. Antioxid Redox Signal (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, Y.C. , Hu, Z. , Rachlin, J. , Anton, B.P. , Kasif, S. , Roberts, R.J. , and Steffen, M. (2016) COMBREX‐DB: an experiment centered database of protein function: knowledge, predictions and knowledge gaps. Nucleic Acids Res 44: D330–D335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie, J.M. , Salomon, M. , Nozue, K. , Wada, M. , and Briggs, W.R. (1999) LOV (light, oxygen, or voltage) domains of the blue‐light photoreceptor phototropin (nph1): binding sites for the chromophore flavin mononucleotide. Proc Natl Acad Sci USA 96: 8779–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, R.E. , and Higgs, P.G. (2012) Testing the infinitely many genes model for the evolution of the bacterial core genome and pangenome. Mol Biol Evol 29: 3413–3425. [DOI] [PubMed] [Google Scholar]
- Danchin, A. (2009) Bacteria as computers making computers. FEMS Microbiol Rev 33: 3–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchin, A. (2017) Coping with inevitable accidents in metabolism. Microb Biotechnol 10: 57–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchin, A. , and Fang, G. (2016) Unknown unknowns: essential genes in quest for function. Microb Biotechnol 9: 530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker, A.R. , and Ramamurthi, K.S. (2017) Cell death pathway that monitors spore morphogenesis. Trends Microbiol 25: 637–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demerec, M. , Adelberg, E.A. , Clark, A.J. , and Hartman, P.E. (1968) A proposal for a uniform nomenclature in bacterial genetics. J Gen Microbiol 50: 1–14. [DOI] [PubMed] [Google Scholar]
- Ding, T. , Su, B. , Chen, X. , Xie, S. , Gu, S. , Wang, Q. , et al (2017) An endophytic bacterial strain isolated from Eucommia ulmoides inhibits southern corn leaf blight. Front Microbiol 8: 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubnau, E.J. , Carabetta, V.J. , Tanner, A.W. , Miras, M. , Diethmaier, C. , and Dubnau, D. (2016) A protein complex supports the production of Spo0A‐P and plays additional roles for biofilms and the K‐state in Bacillus subtilis . Mol Microbiol 101: 606–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelen, S. , Vallenet, D. , Medigue, C. , and Danchin, A. (2012) Distinct co‐evolution patterns of genes associated to bacterial DNA polymerase III DnaE and PolC. BMC Genom 13: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, G. , Ho, C. , Qiu, Y. , Cubas, V. , Yu, Z. , Cabau, C. , et al (2005) Specialized microbial databases for inductive exploration of microbial genome sequences. BMC Genom 6: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiner, R. , Argov, T. , Rabinovich, L. , Sigal, N. , Borovok, I. , and Herskovits, A.A. (2015) A new perspective on lysogeny: prophages as active regulatory switches of bacteria. Nat Rev Microbiol 13: 641–650. [DOI] [PubMed] [Google Scholar]
- Frangeul, L. , Nelson, K.E. , Buchrieser, C. , Danchin, A. , Glaser, P. , and Kunst, F. (1999) Cloning and assembly strategies in microbial genome projects. Microbiology 145: 2625–2634. [DOI] [PubMed] [Google Scholar]
- Gabrisko, M. and Barak, I. (2016) Evolution of the SpoIISABC toxin‐antitoxin‐antitoxin system in bacilli. Toxins (Basel) 8, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, P. , Pinkston, K.L. , Bourgogne, A. , Murray, B.E. , van Hoof, A. , and Harvey, B.R. (2017) Functional studies of E. faecalis RNase J2 and its role in virulence and fitness. PLoS ONE 12: e0175212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers, D. , Cohan, F.M. , Lawrence, J.G. , Spratt, B.G. , Coenye, T. , Feil, E.J. , et al (2005) Opinion: Re‐evaluating prokaryotic species. Nat Rev Microbiol 3: 733–739. [DOI] [PubMed] [Google Scholar]
- Gilks, W.R. , Audit, B. , de Angelis, D. , Tsoka, S. , and Ouzounis, C.A. (2005) Percolation of annotation errors through hierarchically structured protein sequence databases. Math Biosci 193: 223–234. [DOI] [PubMed] [Google Scholar]
- Gond, S.K. , Bergen, M.S. , Torres, M.S. , and White, J.F. Jr (2015) Endophytic Bacillus spp. produce antifungal lipopeptides and induce host defence gene expression in maize. Microbiol Res 172: 79–87. [DOI] [PubMed] [Google Scholar]
- Grau, R.R. , de Ona, P. , Kunert, M. , Lenini, C. , Gallegos‐Monterrosa, R. , Mhatre, E. , et al (2015) A duo of potassium‐responsive histidine kinases govern the multicellular destiny of Bacillus subtilis . MBio 6: e00581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundlach, J. , Herzberg, C. , Kaever, V. , Gunka, K. , Hoffmann, T. , Weiss, M. , et al (2017) Control of potassium homeostasis is an essential function of the second messenger cyclic di‐AMP in Bacillus subtilis . Sci Signal 10, eaal3011. [DOI] [PubMed] [Google Scholar]
- Habib, C. , Yu, Y. , Gozzi, K. , Ching, C. , Shemesh, M. , and Chai, Y. (2017) Characterization of the regulation of a plant polysaccharide utilization operon and its role in biofilm formation in Bacillus subtilis . PLoS ONE 12: e0179761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harshey, R.M. , and Partridge, J.D. (2015) Shelter in a swarm. J Mol Biol 427: 3683–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey, S. , Hill, C.W. , Squires, C. , and Squires, C.L. (1988) Loss of the spacer loop sequence from the rrnB operon in the Escherichia coli K‐12 subline that bears the relA1 mutation. J Bacteriol 170: 1235–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood, C.R. , and Wipat, A. (1996) Sequencing and functional analysis of the genome of Bacillus subtilis strain 168. FEBS Lett 389: 84–87. [DOI] [PubMed] [Google Scholar]
- Harwood, C.R. , Pohl, S. , Smith, W. , Wipat, A. , Harwood, C. , and Wipat, A. (2013) Bacillus subtilis: model Gram‐positive synthetic biology chassis. Microbial Synthetic Biol 40: 87–117. [Google Scholar]
- Helmann, J.D. (2016) Bacillus subtilis extracytoplasmic function (ECF) sigma factors and defense of the cell envelope. Curr Opin Microbiol 30: 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobley, L. , Harkins, C. , MacPhee, C.E. , and Stanley‐Wall, N.R. (2015) Giving structure to the biofilm matrix: an overview of individual strategies and emerging common themes. FEMS Microbiol Rev 39: 649–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobley, L. , Li, B. , Wood, J.L. , Kim, S.H. , Naidoo, J. , Ferreira, A.S. , et al (2017) Spermidine promotes Bacillus subtilis biofilm formation by activating expression of the matrix regulator slrR . J Biol Chem 292: 12041–12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins, J. , Alborn, W.E. Jr , Arnold, J. , Blaszczak, L.C. , Burgett, S. , DeHoff, B.S. , et al (2001) Genome of the bacterium Streptococcus pneumoniae strain R6. J Bacteriol 183: 5709–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, M. and Hull, C.M. (2017) Sporulation: how to survive on planet Earth (and beyond). Curr Genet 63, 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, L. , and Lilley, D.M.J. (2016) The kink turn, a key architectural element in RNA structure. J Mol Biol 428: 790–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Shin, J.H. , Pinochet‐Barros, A. , Su, T.T. , and Helmann, J.D. (2016) Bacillus subtilis MntR coordinates the transcriptional regulation of manganese uptake and efflux systems. Mol Microbiol 103: 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummels, K.R. , Witzky, A. , Rajkovic, A. , Tollerson, R. 2nd , Jones, L.A. , Ibba, M. and Kearns, D.B. (2017) Carbonyl reduction by YmfI in Bacillus subtilis prevents accumulation of an inhibitory EF‐P modification state. Mol Microbiol 106, 236–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison, C.A. 3rd , Chuang, R.Y. , Noskov, V.N. , Assad‐Garcia, N. , Deerinck, T.J. , Ellisman, M.H. , et al. (2016) Design and synthesis of a minimal bacterial genome. Science 351, aad6253. [DOI] [PubMed] [Google Scholar]
- Ji, C. , Stockbridge, R.B. , and Miller, C. (2014) Bacterial fluoride resistance, Fluc channels, and the weak acid accumulation effect. J Gen Physiol 144: 257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiafeng, L. , Fu, X. , and Chang, Z. (2015) Hypoionic shock treatment enables aminoglycosides antibiotics to eradicate bacterial persisters. Sci Rep 5: 14247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgenson, C.T. , Ealick, S.E. and Begley, T.P. (2009) Biosynthesis of hiamin pyrophosphate. EcoSal Plus 3.6.3.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, D.M. , Tanaka, K. , Takenaka, S. , Ishikawa, S. , and Yoshida, K.I. (2017) Bacillus subtilis iolU encodes an additional NADP+‐dependent scyllo‐inositol dehydrogenase. Biosci Biotechnol Biochem 81: 1026–1032. [DOI] [PubMed] [Google Scholar]
- Kiehler, B. , Haggett, L. and Fujita, M. (2017) The PAS domains of the major sporulation kinase in Bacillus subtilis play a role in tetramer formation that is essential for the autokinase activity. Microbiologyopen 6, e00481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Yu, B.J. , Kim, J.A. , Lee, Y.J. , Choi, S.G. , Kang, S. , and Pan, J.G. (2013) The acetylproteome of Gram‐positive model bacterium Bacillus subtilis . Proteomics 13: 1726–1736. [DOI] [PubMed] [Google Scholar]
- Konstantinidis, K.T. , Ramette, A. , and Tiedje, J.M. (2006) Toward a more robust assessment of intraspecies diversity, using fewer genetic markers. Appl Environ Microbiol 72: 7286–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosono, S. , Tamura, M. , Suzuki, S. , Kawamura, Y. , Yoshida, A. , Nishiyama, M. , and Yoshida, M. (2015) Changes in the acetylome and succinylome of Bacillus subtilis in response to carbon source. PLoS ONE 10: e0131169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs, A.T. , Grau, R. , and Pollitt, E.J.G. (2017) Surfing of bacterial droplets: Bacillus subtilis sliding revisited. Proc Natl Acad Sci USA 114: E8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh, S. , O'Reilly, M. , Nolan, N. , and Devine, K.M. (1996) The phage‐like element PBSX and part of the skin element, which are resident at different locations on the Bacillus subtilis chromosome, are highly homologous. Microbiology 142(Pt 8): 2031–2040. [DOI] [PubMed] [Google Scholar]
- Kunst, F. , Ogasawara, N. , Moszer, I. , Albertini, A.M. , Alloni, G. , Azevedo, V. , et al (1997) The complete genome sequence of the Gram‐positive bacterium Bacillus subtilis . Nature 390: 249–256. [DOI] [PubMed] [Google Scholar]
- Kuroda, M. , Ohta, T. , Uchiyama, I. , Baba, T. , Yuzawa, H. , Kobayashi, I. , et al (2001) Whole genome sequencing of meticillin‐resistant Staphylococcus aureus . Lancet 357: 1225–1240. [DOI] [PubMed] [Google Scholar]
- Lammers, C.R. , Florez, L.A. , Schmeisky, A.G. , Roppel, S.F. , Mader, U. , Hamoen, L. , and Stulke, J. (2010) Connecting parts with processes: SubtiWiki and SubtiPathways integrate gene and pathway annotation for Bacillus subtilis . Microbiology 156: 849–859. [DOI] [PubMed] [Google Scholar]
- Laub, M.T. (2016) Keeping signals straight: how cells process information and make decisions. PLoS Biol 14: e1002519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma‐Ortiz, C. , Jeffryes, J.G. , Cooper, A.J. , Niehaus, T.D. , Thamm, A.M. , Frelin, O. , et al (2016) ‘Nothing of chemistry disappears in biology’: the Top 30 damage‐prone endogenous metabolites. Biochem Soc Trans 44: 961–971. [DOI] [PubMed] [Google Scholar]
- Lew, J.M. , Mao, C. , Shukla, M. , Warren, A. , Will, R. , Kuznetsov, D. , et al (2013) Database resources for the tuberculosis community. Tuberculosis 93: 12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley, D.M. (2017) The guanidine riboswitch‐a poor orphan no longer. Cell Chem Biol 24: 130–131. [DOI] [PubMed] [Google Scholar]
- de Lorenzo, V. , and Danchin, A. (2008) Synthetic biology: discovering new worlds and new words. EMBO Rep 9: 822–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losi, A. , Polverini, E. , Quest, B. , and Gartner, W. (2002) First evidence for phototropin‐related blue‐light receptors in prokaryotes. Biophys J 82: 2627–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, C. , Ding, F. , Chowdhury, A. , Pradhan, V. , Tomsic, J. , Holmes, W.M. , et al (2010) SAM recognition and conformational switching mechanism in the Bacillus subtilis yitJ S box/SAM‐I riboswitch. J Mol Biol 404: 803–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald, C.B. , and Stockbridge, R.B. (2017) A topologically diverse family of fluoride channels. Curr Opin Struct Biol 45: 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medigue, C. , Calteau, A. , Cruveiller, S. , Gachet, M. , Gautreau, G. , Josso, A. , et al (2017) MicroScope‐an integrated resource for community expertise of gene functions and comparative analysis of microbial genomic and metabolic data. Brief Bioinform (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michna, R.H. , Zhu, B. , Mader, U. , and Stulke, J. (2016) SubtiWiki 2.0–an integrated database for the model organism Bacillus subtilis. Nucleic Acids Res 44: D654–D662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miglio, G. , Sabatino, A.D. , Veglia, E. , Giraudo, M.T. , Beccuti, M. , and Cordero, F. (2016) A computational analysis of S‐(2‐succino)cysteine sites in proteins. Biochim Biophys Acta 1864: 211–218. [DOI] [PubMed] [Google Scholar]
- Morris, R.J. , Schor, M. , Gillespie, R.M.C. , Ferreira, A.S. , Baldauf, L. , Earl, C. , et al (2017) Natural variations in the biofilm‐associated protein BslA from the genus Bacillus . Sci Rep 7: 6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moszer, I. , Jones, L.M. , Moreira, S. , Fabry, C. , and Danchin, A. (2002) SubtiList: the reference database for the Bacillus subtilis genome. Nucleic Acids Res 30: 62–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normark, S. , and Burman, L.G. (1977) Resistance of Escherichia coli to penicillins: fine‐structure mapping and dominance of chromosomal beta‐lactamase mutations. J Bacteriol 132: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pane‐Farre, J. , Quin, M.B. , Lewis, R.J. , and Marles‐Wright, J. (2017) Structure and function of the stressosome signalling hub. Subcell Biochem 83: 1–41. [DOI] [PubMed] [Google Scholar]
- Parveen, S. and Reddy, M. (2017) Identification of YfiH (PgeF) as a factor contributing to the maintenance of bacterial peptidoglycan composition. Mol Microbiol 105, 705–720. [DOI] [PubMed] [Google Scholar]
- Pennacchietti, F. , Abbruzzetti, S. , Losi, A. , Mandalari, C. , Bedotti, R. , Viappiani, C. , et al (2014) The dark recovery rate in the photocycle of the bacterial photoreceptor YtvA is affected by the cellular environment and by hydration. PLoS ONE 9: e107489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, H.U. , Danchin, A. , and Grunberg‐Manago, M. (1976) Toward an understanding of the formylation of initiator tRNA methionine in prokaryotic protein synthesis. II. A two‐state model for the 70S ribosome. Biochemistry 15: 1362–1369. [DOI] [PubMed] [Google Scholar]
- Petrovova, M. , Tkadlec, J. , Dvoracek, L. , Streitova, E. , and Licha, I. (2014) NAD(P)H‐hydrate dehydratase‐ a metabolic repair enzyme and its role in Bacillus subtilis stress adaptation. PLoS ONE 9: e112590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piggot, P.J. (1973) Mapping of asporogenous mutations of Bacillus subtilis: a minimum estimate of the number of sporeulation operons. J Bacteriol 114: 1241–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podkowa, K.J. , Briere, L.A. , Heinrichs, D.E. , and Shilton, B.H. (2014) Crystal and solution structure analysis of FhuD2 from Staphylococcus aureus in multiple unliganded conformations and bound to ferrioxamine‐B. Biochemistry 53: 2017–2031. [DOI] [PubMed] [Google Scholar]
- Rawat, M. , and Av‐Gay, Y. (2007) Mycothiol‐dependent proteins in actinomycetes. FEMS Microbiol Rev 31: 278–292. [DOI] [PubMed] [Google Scholar]
- Read, T.D. , Salzberg, S.L. , Pop, M. , Shumway, M. , Umayam, L. , Jiang, L. , et al (2002) Comparative genome sequencing for discovery of novel polymorphisms in Bacillus anthracis. Science 296: 2028–2033. [DOI] [PubMed] [Google Scholar]
- Ricard, M. , and Hirota, Y. (1973) Process of cellular division in Escherichia coli: physiological study on thermosensitive mutants defective in cell division. J Bacteriol 116: 314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter, M. , and Rossello‐Mora, R. (2009) Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA 106: 19126–19131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez Ayala, F. , Bauman, C. , Bartolini, M. , Saball, E. , Salvarrey, M. , Lenini, C. , et al (2017) Transcriptional regulation of adhesive properties of Bacillus subtilis to extracellular matrix proteins through the fibronectin‐binding protein YloA. Mol Microbiol 104: 804–821. [DOI] [PubMed] [Google Scholar]
- Roll‐Hansen, N. (1979) Experimental method and spontaneous generation: the controversy between Pasteur and Pouchet, 1859–64. J Hist Med Allied Sci 34: 273–292. [DOI] [PubMed] [Google Scholar]
- Schultz, D. (2016) Coordination of cell decisions and promotion of phenotypic diversity in B. subtilis via pulsed behavior of the phosphorelay. BioEssays 38: 440–445. [DOI] [PubMed] [Google Scholar]
- Sneath, P.H.A. (1986) Endospore‐forming Gram‐positive rods and cocci In Bergey's Manual of Systematic Bacteriology. Sneath P.H.A., Mair N.S., Sharpe M.E., and Holt J.G. (eds). Baltimore: Williams & Wilkins Co., pp. 1105–1139. [Google Scholar]
- Song, Y. , Rubio, A. , Jayaswal, R.K. , Silverman, J.A. , and Wilkinson, B.J. (2013) Additional routes to Staphylococcus aureus daptomycin resistance as revealed by comparative genome sequencing, transcriptional profiling, and phenotypic studies. PLoS ONE 8: e58469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soupene, E. , van Heeswijk, W.C. , Plumbridge, J. , Stewart, V. , Bertenthal, D. , Lee, H. , et al (2003) Physiological studies of Escherichia coli strain MG1655: growth defects and apparent cross‐regulation of gene expression. J Bacteriol 185: 5611–5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Straaten, K.E. , Ko, J.B. , Jagdhane, R. , Anjum, S. , Palmer, D.R. , and Sanders, D.A. (2013) The structure of NtdA, a sugar aminotransferase involved in the kanosamine biosynthetic pathway in Bacillus subtilis, reveals a new subclass of aminotransferases. J Biol Chem 288: 34121–34130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavener, S.J. , and Clark, J.H. (2006) Fluorine: friend or foe? A green chemist's perspective In Fluorine and the environment: agrochemicals, archaeology, green chemistry and water. Tressaud A. (ed). Amsterdam: Elsevier, pp. 177–202. [Google Scholar]
- Tettelin, H. , Nelson, K.E. , Paulsen, I.T. , Eisen, J.A. , Read, T.D. , Peterson, S. , et al (2001) Complete genome sequence of a virulent isolate of Streptococcus pneumoniae . Science 293: 498–506. [DOI] [PubMed] [Google Scholar]
- Tojo, S. , Kim, J.Y. , Tanaka, Y. , Inaoka, T. , Hiraga, Y. , and Ochi, K. (2014) The mthA mutation conferring low‐level resistance to streptomycin enhances antibiotic production in Bacillus subtilis by increasing the S‐adenosylmethionine pool size. J Bacteriol 196: 1514–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchon, M. , Hoede, C. , Tenaillon, O. , Barbe, V. , Baeriswyl, S. , Bidet, P. , et al (2009) Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet 5: e1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchon, M. , Cury, J. , Yoon, E.J. , Krizova, L. , Cerqueira, G.C. , Murphy, C. , et al (2014) The genomic diversification of the whole Acinetobacter genus: origins, mechanisms, and consequences. Genome Biol Evol 6: 2866–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschowri, N. , Lindenberg, S. , and Hengge, R. (2012) Molecular function and potential evolution of the biofilm‐modulating blue light‐signalling pathway of Escherichia coli . Mol Microbiol 85: 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallenet, D. , Calteau, A. , Cruveiller, S. , Gachet, M. , Lajus, A. , Josso, A. , et al (2017) MicroScope in 2017: an expanding and evolving integrated resource for community expertise of microbial genomes. Nucleic Acids Res 45: D517–D528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter, N.D. , and Palmer, D.R. (2017) Simultaneous measurement of glucose‐6‐phosphate 3‐dehydrogenase (NtdC) catalysis and the nonenzymatic reaction of its product: kinetics and isotope effects on the first step in kanosamine biosynthesis. Biochemistry 56: 2001–2009. [DOI] [PubMed] [Google Scholar]
- Vinci, C.R. , and Clarke, S.G. (2010) Homocysteine methyltransferases Mht1 and Sam4 prevent the accumulation of age‐damaged (R, S)‐AdoMet in the yeast Saccharomyces cerevisiae. J Biol Chem 285: 20526–20531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall, E.A. , Johnson, A.L. , Peterson, D.L. , and Christie, G.E. (2017) Structural modeling and functional analysis of the essential ribosomal processing protease Prp from Staphylococcus aureus . Mol Microbiol 104: 520–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, D.M. , Cohan, F.M. , Bhaya, D. , Heidelberg, J.F. , Kuhl, M. , and Grossman, A. (2008) Genomics, environmental genomics and the issue of microbial species. Heredity (Edinb) 100: 207–219. [DOI] [PubMed] [Google Scholar]
- Weinberg, E.D. (1997) The Lactobacillus anomaly: total iron abstinence. Perspect Biol Med 40: 578–583. [DOI] [PubMed] [Google Scholar]
- Werb, M. , Garcia, C.F. , Bach, N.C. , Grumbein, S. , Sieber, S.A. , Opitz, M. , and Lieleg, O. (2017) Surface topology affects wetting behavior of Bacillus subtilis biofilms. NPJ Biofilms Microbiomes 3: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, S.S. , Wu, H.J. , Zang, H.Y. , Wu, L.M. , Zhu, Q.Q. , and Gao, X.W. (2014) Plant growth promotion by spermidine‐producing Bacillus subtilis OKB105. Mol Plant Microbe Interact 27: 655–663. [DOI] [PubMed] [Google Scholar]
- Yamamoto, H. , Wittek, D. , Gupta, R. , Qin, B. , Ueda, T. , Krause, R. , et al (2016) 70S‐scanning initiation is a novel and frequent initiation mode of ribosomal translation in bacteria. Proc Natl Acad Sci USA 113: E1180–E1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, A. , and Kuramitsu, H.K. (2002) Multiple Streptococcus mutans genes are involved in biofilm formation. Appl Environ Microbiol 68: 6283–6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallot, R. , Harrison, K.J. , Kolaczkowski, B. and de Crecy‐Lagard, V. (2016) Functional annotations of paralogs: a blessing and a curse. Life (Basel) 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeriouh, H. , de Vicente, A. , Perez‐Garcia, A. , and Romero, D. (2014) Surfactin triggers biofilm formation of Bacillus subtilis in melon phylloplane and contributes to the biocontrol activity. Environ Microbiol 16: 2196–2211. [DOI] [PubMed] [Google Scholar]
- Zhang, J. and Ferre‐D'Amare, A.R. (2016) The tRNA elbow in structure, recognition and evolution. Life (Basel) 6, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J. , and Rudd, K.E. (2013) EcoGene 3.0. Nucleic Acids Res 41: D613–D624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The extended Bacillus subtilis paleome
Table S2. Bacillus subtilis 168 annotated genome in the EMBL‐ENA format.
Appendix S1. Experimental identification of methylthioribose transport and a missing step in lysine biosynthesis.