

Summary
Bacterial recombineering typically relies on genomic incorporation of synthetic oligonucleotides as mediated by Escherichia coli λ phage recombinase β – an occurrence largely limited to enterobacterial strains. While a handful of similar recombinases have been documented, recombineering efficiencies usually fall short of expectations for practical use. In this work, we aimed to find an efficient Recβ homologue demonstrating activity in model soil bacterium Pseudomonas putida EM42. To this end, a genus‐wide protein survey was conducted to identify putative recombinase candidates for study. Selected novel proteins were assayed in a standardized test to reveal their ability to introduce the K43T substitution into the rpsL gene of P. putida. An ERF superfamily protein, here termed Rec2, exhibited activity eightfold greater than that of the previous leading recombinase. To bolster these results, we demonstrated Rec2 ability to enter a range of mutations into the pyrF gene of P. putida at similar frequencies. Our results not only confirm the utility of Rec2 as a Recβ functional analogue within the P. putida model system, but also set a complete workflow for deploying recombineering in other bacterial strains/species. Implications range from genome editing of P. putida for metabolic engineering to extended applications within other Pseudomonads – and beyond.
Introduction
In the post‐genomics era, many potential innovations have been hampered by a dearth of rapid, affordable and comprehensive techniques for genetic modification. Fortunately, recombineering is evolving to enable large‐scale genomic editing with a host of applications, ranging from increases in genomic diversity to the optimization of metabolic pathways for biosynthesis (Wang et al., 2009; Gallagher et al., 2014). Recombineering refers to directed genetic recombination between foreign DNA and endogenous homologies during DNA replication, as mediated by phage‐derived recombinases (Yu et al., 2000; Ellis et al., 2001; Marinelli et al., 2012). Modern recombineering technologies originated in Escherichia coli, in which the β, Exo and γ proteins of λ phage were first exploited in tandem to introduce double‐stranded (ds) DNA into the lagging strand of transient replication forks (Murphy, 1998; Yu et al., 2000). Ellis et al. (2001) simplified this method, demonstrating the ability of the λ phage β protein to independently mediate the genomic annealing and recombination of single‐stranded (ss) mutagenic oligonucleotides. Finally, Wang et al. (2009) developed the landmark recombineering protocol known as Multiplex Automated Genomic Engineering (MAGE): a library of synthesized mutagenic ss‐oligonucleotides is introduced into β‐expressing E. coli, allowing for rapid, multisite genomic modification that can be cycled to generate vast spaces of genomic diversity. Since its advent, MAGE recombineering has brought about a multitude of advances, including genome‐wide codon replacement, biosynthesis of lycopene and aromatic amino acids, and comprehensive mutant library generation (Wang et al., 2009; Isaacs et al., 2011; Gallagher et al., 2014; Nyerges et al., 2016). Unfortunately, β‐recombinase (Recβ) recombineering activity is not well‐conserved in bacterial species outside of the Enterobacteriaceae family (Van Kessel and Hatfull, 2008; Binder et al., 2013; Lennen et al., 2016); this limited conservation is thought to stem from specificity of host–phage recombinase interactions (Sun et al., 2015). The apparent species specificity of recombineering systems has spurred a search for functional Recβ analogues in alternative lineages, with limited success in microbes including Salmonella, P. syringae, Corynebacteria, Lactobacilli and Mycobacteria (Van Kessel and Hatfull, 2008; Gerlach et al., 2009; Swingle et al., 2010; Pijkeren and Britton, 2012; Binder et al., 2013; Oh and Pijkeren, 2014). While most reports involve the isolation of a single enzyme, a recent protein survey conducted by Datta et al. (2008) produced several rec genes in E. coli, suggesting the utility of a comprehensive approach to recombinase identification within other microorganisms.
The saprotrophic soil bacterium Pseudomonas putida is known for its tolerance to a variety of environmental stresses, including but not limited to organic solvents and reactive oxygen species (Poblete‐Castro et al., 2012; Nikel et al., 2014; Ramos et al., 2015). P. putida derives this sturdiness from its EDEMP cycle, a series of metabolic pathways that facilitate NAD(P)H production via carbon recycling processes (Nikel et al., 2015). An adaptability to a myriad of harsh conditions has made P. putida a popular focus of biosynthetic studies aimed at industrial biotransformations as well as soil and water bioremediation (Garmendia et al., 2008; Puchałka et al., 2008; Poblete‐Castro et al., 2012; Loeschcke and Thies, 2015). Unfortunately, the collection of genetic tools available in Pseudomonads is limited in scope and, when used in conjunction, is subject to bottlenecks that hinder the generation of a biotechnological chassis (Martínez‐García and de Lorenzo, 2011; Martínez‐García et al., 2011, 2014). Efforts at recombineering in Pseudomonas species have been attempted with limited results (Liang and Liu, 2010; Swingle et al., 2010). One study has isolated a ssDNA annealing recombinase (termed Ssr) demonstrating β‐like activity in P. putida (Aparicio et al., 2016). Although the Ssr protein demonstrates an ability to catalyse P. putida recombineering without off‐target mutagenesis, levels of recombineering are insufficient for large‐scale use.
This study aimed to isolate a recombinase exhibiting activity levels practical for biosynthetic remodelling in P. putida. Using the classical ERF, GP2.5, SAK and SAK4 recombinase families as screening references (Lopes et al., 2010), we employed a protein sequence similarity search to identify enzymes with potential rec capabilities in Pseudomonads genomes. Along with RecTPsy, Recβ and Ssr as controls, we functionally characterized study candidates with a series of standardized assays to assess recombineering activity. This novel biosynthetic workflow allowed us to describe a new set of recombinases demonstrating activity in P. putida. Among these, we identified an ERF superfamily ssDNA binding protein, here termed Rec2, that greatly outperforms current rec standards in P. putida. Our findings support the application of Rec2 towards future recombineering efforts within P. putida, as well as potential extensions of the generated search pipeline and rec panel to alternative Pseudomonads (and other bacteria in general) for industrial, environmental and biomedical purposes.
Results
Sequence search reveals novel protein candidates for recombineering study in P. putida
With its metabolic plasticity and extensive tolerance to environmental stresses, P. putida has recently become the focus of many biosynthetic studies related to industrial catalysis and environmental bioremediation (Loeschcke and Thies, 2015). Unfortunately, recombineering capabilities in this organism are limited by the ongoing search for an analogue of E. coli λ phage Recβ. Thus, we began this work with a comprehensive sequence survey of Pseudomonas sp. genomes for putative functional analogues of the recβ gene. Along with recβ, ssr and recT Psy, we selected ten gene candidates for study in P. putida (rec1 to rec10; Table 1), each of which was Gibson‐assembled into the pSEVA258 vector platform (for more information, see Experimental procedures). A model of the resulting construct – which harboured the promiscuous origin of replication RSF1010 and the 3‐methylbenzoate (3MB)‐inducible, XylS‐dependent expression promoter Pm – is shown in Fig. 1A. Assembled constructs were transformed into P. putida EM42, an industrial reference strain stripped of interference factors known to limit heterologous gene expression (Martínez‐García et al., 2014).
Table 1.
Putative ssDNA binding proteins used in this work
| Name | Organism | Description (Family) | Length (aa) | Locus tag | Accession no. |
|---|---|---|---|---|---|
| Rec1 | P. putida S16 | ERF superfamily single stranded (ss) DNA‐binding protein (ERF) | 253 | PPS_RS12640 | WP_013972384.1 |
| Rec2 | P. putida CSV86 | ERF superfamily ss‐DNA binding protein (ERF) | 267 | CSV86_RS08085 | WP_009397165.1 |
| Rec3 | P. putida KT2440 Prophage 3 | DUF2815 domain‐containing ss‐DNA binding protein (GP2.5) | 227 | PP_2267 | WP_010953240.1 |
| Rec4 | P. sp. KG01 | ss‐DNA binding protein (GP2.5) | 232 | ACR52_RS27 | WP_048731951263.1 |
| Rec5 | P. putida CSV86 | Hypothetical protein (Sak) | 243 | CSV86_RS06065 | WP_009396409.1 |
| Rec6 | P. aeruginosa phage F116 | Hypothetical protein (Sak) | 251 | F116p19 | YP_164283.1 |
| Rec7 | P. aeruginosa AZPAE14939 | Hypothetical protein (Sak) | 254 | NS55_RS18170 | WP_043087219.1 |
| Rec8 | P. putida UASWS0946 | RecT family DNA binding protein (Sak4) | 363 | QV12_RS02185 | WP_043859966.1 |
| Rec9 | P. putida KB9 | DNA binding protein (Sak4) | 314 | A3K88_RS17255 | WP_064314638.1 |
| Rec10 | E. faecalis V583 | RecT family DNA binding protein (Sak4) | 307 | EF2132 | NP_815795.1 |
| RecTPsy | P. syringae B728a | RecT family DNA binding protein (Redβ‐like) | 295 | Psyr_2820 | AAY37_859.1 |
| Recβ | E. coli λ phage | Phage λ ss‐DNA binding protein (Redβ‐like) | 261 | lambdap84 | NP_040617.1 |
| Ssr | P. putida DOT‐T1E | ERF family ss‐DNA binding protein (ERF) | 252 | T1E_1405 | AFO47_260.1 |
Figure 1.
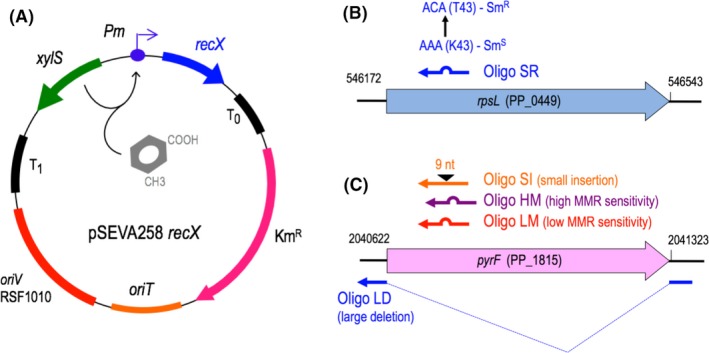
Components of the recombineering testing platform for Pseudomonas putida EM42.
A. Organization of plasmid series pSEVA258‐recX (~8 kb; recX indicates cloned recombinase, e.g. rec1 and rec2) for conditional expression of putative recombinases selected for study in P. putida, image reproduced from Ref. (Aparicio et al., 2016). DNA fragments of all candidate proteins were designed for cloning into the pSEVA258 vector by Gibson assembly, as detailed in Experimental procedures. Proper insertion placed all proteins under control of the 3MB‐inducible Pm promoter. Additional relevant functional segments include the 3MB‐sensitive regulator xylS, flanking transcriptional terminators T1 and T0, origin of transfer (oriT) and replication (oriV), and a kanamycin resistance cassette (KmR) to enable antibiotic selection of pSEVA258‐recX‐transformed cell lines.
B. Designation, location and expected change associated with the SR oligonucleotide targeted to the rpsL gene of P. putida. The gene locus and coordinates are indicated, and the sequence of the SR oligo is listed in Table S1.
C. Depicts analogous information for the pyrF oligonucleotides employed in this work, reproduced directly from Ref. Aparicio et al. (2016).
Design of a rpsL‐based reporter system in P. putida
By inhibiting polypeptide synthesis, streptomycin (Sm) triggers misreads during genomic translation that ultimately lead to bacterial cell death (Ruusala and Kurland, 1984). Several single nucleotide mutations to the rpsL gene, which encodes the ribosomal protein S12, have been characterized as conferring Sm resistance in different bacteria (Funatsu and Wittmann, 1972; Timms et al., 1992; Gregory et al., 2001). This array of modifications make the antibiotic marker a robust tool for genomic mutagenesis in E. coli (Jiang et al., 2013) and P. syringae (Swingle et al., 2010). Given the negligible occurrence rates of spontaneous mutants observed in P. putida (Jatsenko et al., 2010), we chose streptomycin resistance as conferred by oligo‐mediated mutagenesis to the P. putida rpsL gene as a measure of putative recombinase activity. To this end, we designed an rpsL‐directed ss‐oligonucleotide (SR) whose strand invasion would result in the rpsL K43T missense mutation, a P. putida analogue of the K42T mutation in E. coli (Timms et al., 1992). SR was designed to produce a single‐base‐pair mismatch poorly detected by MMR machinery (Babic et al., 1996; Kunkel and Erie, 2005); schemata of the K43T mutation and its targeted disruption to P. putida rpsL appear in Fig. 1B. In keeping with established strand bias and previous study design (Ellis et al., 2001; Costantino and Court, 2003; Aparicio et al., 2016), SR was targeted to the lagging strand of the P. putida rpsL gene and engineered to evade endogenous MMR activity, in order to maximize putative recombinase efficiency. Further information on the design and synthesis of the mutagenic oligonucleotide can be found in the Experimental procedures section.
rpsL‐directed recombineering assay in P. putida EM42 highlights two protein candidates for additional study
To measure the ability of candidate recombinases to direct oligo‐mediated mutagenesis during genomic replication, P. putida EM42/pSEVA258‐recX strains and P. putida EM42 containing the empty vector pSEVA258 were grown to mid‐logarithmic phase and treated with 3MB to induce protein expression. Competent cell mixtures were electroporated with the SR oligonucleotide and allowed to recover overnight before selective plating on LB and LB‐Sm solid agar media. An empty vector strain shocked without the SR oligo was included to measure the frequency of spontaneous Sm resistance in P. putida. Average recombineering frequencies and standard error values for each protein candidate, calculated from absolute colony counts, are shown in Fig. 2. Five to ten SmR clones derived from each experimental condition were isolated, PCR amplified and sequenced at the targeted rpsL region to verify acquisition of the proper K43T mutation.
Figure 2.
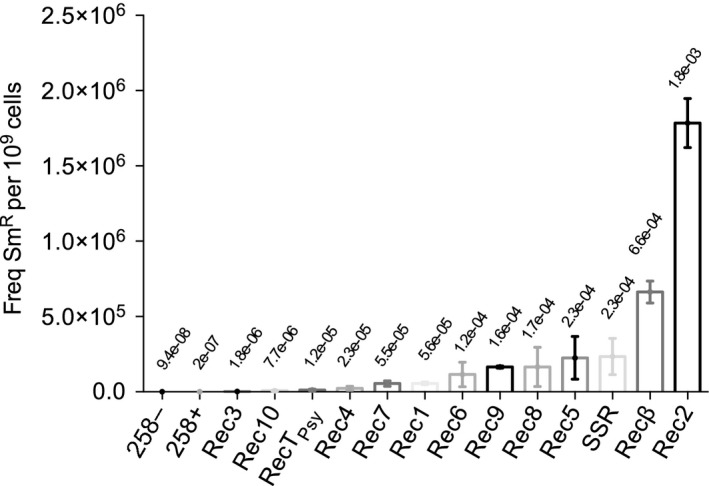
ERF superfamily protein Rec2 exhibits improved recombination frequencies after electroporation with rpsL‐directed mutagenic oligonucleotide SR. Pseudomonas putida EM42 strains containing pSEVA258 and pSEVA258‐recX constructs were induced with 3MB before electroporation with mutagenic oligo SR. After overnight recovery in TB, culture dilutions were plated on LB and LB‐Sm solid media. Colony counts were determined, and correct SR‐mediated changes were checked by amplifying and sequencing the rpsL gene from 5 to 10 SmR clones associated with each experiment (see Experimental procedures for further details). Recombineering efficiency was calculated as the number of SmR mutants per 109 cells. Column graphs of the resulting data were generated with graphpad prism. Column values represent mean recombineering frequencies for each experimental condition; standard error (SE) bars are also shown. Each data set represents at least two independent experimental replicates. Recombineering values are shown on a linear scale to facilitate frequency comparison. Absolute frequency values (mutants per viable cell) are shown above each column. Controls 258+ and 258− indicate SmR colonies arising from P. putida EM42 (pSEVA258) cells electroporated or not, respectively, with the mutagenic oligonucleotide.
We first noted that SmR mutants were detected at low levels in P. putida EM42 (pSEVA258) control strain whether the bacteria were electroporated with the SR oligonucleotide or not. In both cases, absolute recombineering frequencies (RF) were around 10−7 mutants/viable cell (Fig. 2). One hundred percent of resistant colonies arising from the electroporated strain harbored the proper rpsL mutation. This detection of the K43T mutation in the pSEVA258‐containing control strain thus indicates the ability of the SR oligonucleotide to mutate rpsL in the absence of an exogenously expressed recombinase. Similar background levels of recombineering activity in P. putida have been observed previously (Aparicio et al., 2016). In contrast, the SmR clones stemming from cells that were not electroporated with the SR oligonucleotide did not bear the K43T change. These SmR colonies indicate the spontaneous occurrence of Sm resistance mutations, a phenomenon already reported in E. coli (Timms et al., 1992).
Each protein candidate exhibited levels of recombineering activity surpassing experimental controls (RF > 10−7). Sequencing verified the acquired K43T mutation in all colonies originating from protein‐expressing strains. As shown in Fig. 2, allelic exchange rates were diverse, ranging from 1.8 × 10−6 (Rec3) to 1.8 × 10−3 (Rec2). A single recombinase family did not appear to be superior in recombineering activity, although Rec3 and Rec4 of the GP2.5 family produced the lowest recombineering values of proteins derived from the Pseudomonas genus. The lowest efficiency of all proteins tested was detected in Rec3, a recombinase identified in prophage 3 of P. putida KT2440 (Martínez‐García et al., 2015) – a prophage that has been eliminated from the EM42 strain. Rec10, a protein derived from the gram‐positive E. faecalis species, produced relatively efficient recombineering levels (7.7 × 10−6), with RecTPsy from P. syringae performing only slightly better (1.2 × 10−5). Rec6 of P. aeruginosa emerged as a high outlier among the non‐P. putida proteins, producing recombineering frequencies comparable to Ssr and superior to P. putida Rec1. While several recombinases producing superior RF scores came from the P. putida species (Rec2, Rec5, Rec8 and Rec9), average recombineering frequencies of all but one of these enzymes were equal to or lower than those of the current Ssr standard.
Pseudomonas putida EM42 cultures expressing the ERF family ss‐binding protein Rec2 and, surprisingly, the λ phage Recβ protein exhibited respective average recombineering frequencies of 1.8 × 10−3 and 6.6 × 10−4. These were the only values surpassing those of Ssr‐expressing strains (RF = 2.3 × 10−4). With recombineering frequencies from three to eight times greater than those of Ssr, Recβ and Rec2 were selected as promising candidates for further investigation.
Growth curve assays reveal slight toxicity of the Rec2 recombinase
The known toxicity of Ssr – and concomitant associated stress on P. putida growth – led us to wonder whether Rec2 and Recβ might show similar toxicity profiles (Aparicio et al., 2016). Thus, before continuing with recombineering tests, we generated growth profiles for P. putida EM42 strains containing the ssr, recβ and rec2 genes, along with controls, in the presence and in the absence of the 3MB inducer (see Experimental Procedures for details). Results appear in Fig. 3. While all strains exhibited similar growth profiles in the absence of the inducer, strains expressing Ssr and Rec2 were heavily impacted by the introduction of 3MB. Ssr induction resulted in negative P. putida growth rates during the first seven hours of the experimental window, with an average minimum OD600 ~ 0.015 being observed at t = 6.5 h (Fig. 3, Ssr, blue line). The impact of induction on Rec2‐expressing strains was less severe. Strains exhibited lower, but non‐negative early growth rates, with optical densities plateauing at an average OD600 ~ 0.09 at t = 7 h (Fig. 3, Rec2, pink line). These results indicate moderate toxicity in Rec2, as compared to the high capacity of growth inhibition exhibited by Ssr. No growth difference indicating toxicity was detected between 3MB‐induced and non‐induced strains containing recβ (Fig. 3, Recβ, grey and black lines overlap with growth profiles of control strains).
Figure 3.
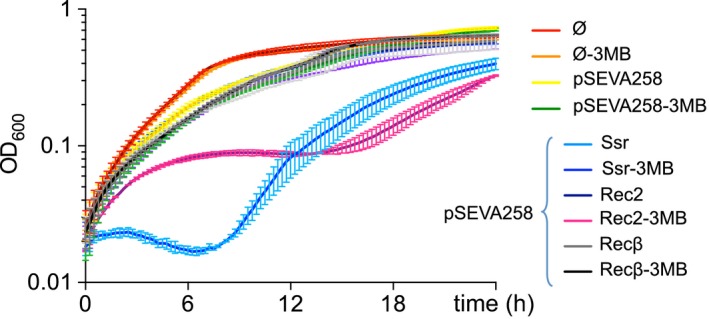
Liquid growth profiles demonstrate slight toxicity of the Rec2 protein. To measure the effects of protein toxicity on cell growth, curves were generated for 3MB‐induced and non‐induced experimental strains using a Spectramax M2e Microplate Reader (Molecular Devices). We prepared overnight LB‐Km cultures of Pseudomonas putida EM42 strains containing pSEVA258‐rec2, pSEVA258‐ssr and pSEVA258‐recβ, as well as EM42/pSEVA258 and EM42 controls. Overnight cultures were back‐diluted in LB‐Km with and without 3MB inducer to an initial OD 600 = 0.05 before being loaded into a 96‐well microtiter plate. Liquid growth profiles of 1 mM 3MB‐induced (t = 0) and non‐induced strains were measured over a 24 h period (interval t = 15 min). Time elapsed (h) is shown on the x‐axis, while optical density at 600 nm (OD 600) is shown on the y‐axis. Strain type is indicated in the figure legend. Individual points represent mean values, and horizontal brackets indicate SE calculated from two independent experimental replicates.
The efficiency of Rec2 is conserved in effecting the E50* mutation in pyrF
Successful genetic remodelling of bacterial chasses using recombineering relies on the capacity to mediate mutagenesis at a range of genetic sites, independent of gene function. Given the efficiency of Rec2 and Recβ in altering the rpsL gene of P. putida, we extended our exploration of recombinase activity to site‐directed editing of an alternative genetic marker: the pyrF gene of P. putida. Briefly, pyrF disruptions in P. putida yield cells both auxotrophic for uracil and resistant to 5FOA, a toxic analogue of uracil (O'Donovan and Neuhard, 1970; Galvao and de Lorenzo, 2005). Scientists have long exploited the dual nature of this gene to isolate edited cell lines in assays of site‐directed mutagenesis (Boeke et al., 1984; Galvao and de Lorenzo, 2005). Thus, following protocols developed for P. putida EM42 in a previous study (Aparicio et al., 2016), we conducted a second round of recombineering experiments with Rec2 and Recβ using the pyrF‐directed LM oligonucleotide (Table S1); the Ssr protein was also tested as a control. The LM oligonucleotide was designed to introduce an A‐G mismatch that replaced the E50 residue of pyrF with a stop codon (Aparicio et al., 2016). The LM and SR mutagenic oligonucleotides are equivalent in nature of mutation and levels of MMR detection. The details of the LM oligonucleotide and its site‐directed mutagenesis of pyrF are depicted in Fig. 1C. A detailed description of pyrF‐directed recombineering procedures, including LM design and synthesis, can be found in the Experimental Procedures section.
Mid‐logarithmic cultures of P. putida EM42 expressing Rec2, Recβ and Ssr were made competent before electroporation with the LM oligonucleotide; after recovery, appropriate dilutions were plated on M9‐citrate solid media supplemented with Ura (viable cells) or Ura/5FOA (allelic changes), and colony counts were corrected by estimating occurrence of spontaneous, non‐pyrF‐related mutants (growing on M9‐citrate). The results of these experiments can be seen in Fig. 4. Rec2 produced an average RF value of 8.6 × 10−4, over seven times that of the Ssr protein (RF = 1.3 × 10−4). Notably, although levels of Rec2 recombineering activity were lowered in pyrF‐directed experiments compared to rpsL, the magnitude of increase in RF relative to Ssr was maintained. Recβ produced the lowest levels of recombinase activity, with an average RF = 1.5 × 10−5. Due to its poor performance under the pyrF reporter system, we decided to exclude Recβ from further recombineering experiments. The discrepancy in Recβ performance between P. putida reporter systems will be addressed in the Discussion.
Figure 4.
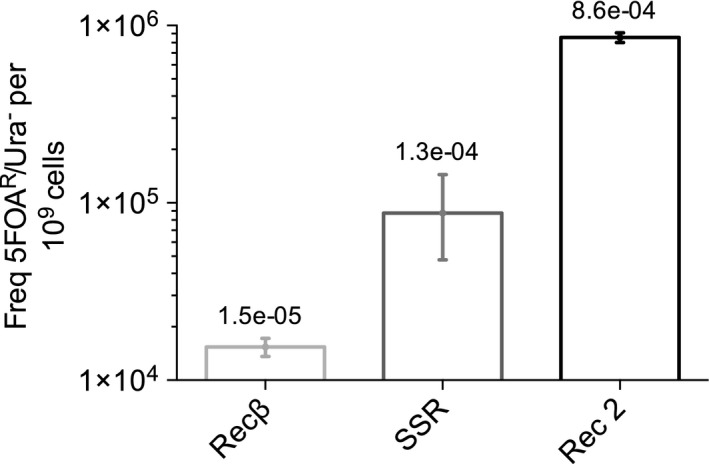
Rec2 maintains recombineering efficiency in entering the LM mutation into the pyrF gene of Pseudomonas putida. P. putida EM42 strains containing pSEVA258‐ssr, pSEVA258‐recβ and pSEVA258‐rec2 were induced with 3MB before electroporation with the LM oligonucleotide (Table S1). After a 6 h recovery period, culture dilutions were plated on selective M9‐Cit‐Ura and M9‐Cit‐Ura‐5FOA solid media. Fifty 5FOAR clones associated with each recombinase were replicated on M9‐Cit and M9‐Cit‐Ura‐5FOA solid media to distinguish authentic pyrF mutants (Ura auxotrophs, no growth on M9‐Cit) from spontaneous 5FOAR colonies (Ura heterotrophs, growth on M9‐Cit). Colony counts were determined and recombineering efficiency calculated (number of authentic pyrF mutants ‐5FOAR/Ura− CFU/109 viable cells). Column graphs of the resulting data were generated with graphpad prism as described in Fig. 2. Each data set represents at least two independent experimental replicates. Recombineering values are shown on a logarithmic scale. Absolute frequency values (mutants per viable cell) are shown above each column.
While Recβ activity was not conserved within the pyrF‐directed system, Rec2 performed at least seven times better than Ssr in both recombineering settings. Further, growth assays indicated diminished toxicity in Rec2 as compared to Ssr in P. putida EM42. These promising features led us to conduct a final round of experiments with the ERF family ssDNA binding protein Rec2, testing its ability to enter a range of mutations into the P. putida genome.
Rec2 demonstrates efficiency in effecting a range of mutations in the pyrF gene
In a final set of recombineering assays, we employed Rec2 to introduce a range of genomic edits to the pyrF gene. As detailed in a previous study (Aparicio et al., 2016), the LD, SI and HM oligonucleotides were designed and coupled with Ssr in P. putida to enter different types of mutations into pyrF: respectively, a complete gene deletion (702‐nt); a short insertion (9 nt) of three stop codons; and a missense mutation of K55 into a stop codon, involving an A‐A mismatch highly detectable by MMR machinery. Thus, we conducted a round of analogous pyrF‐directed recombineering experiments in P. putida with Rec2, testing the Ssr protein in parallel to confirm the robustness of results. The average RF values produced by Rec2 and Ssr using the various pyrF‐directed oligonucleotides are shown in Fig. 5. As in LM‐mediated experiments, Rec2 recombineering assays conducted with SI and HM produced RF values folds greater than those of the Ssr protein. Consistent with the previous study (Aparicio et al., 2016), SI‐mediated experiments involving Rec2 produced the greatest recombineering frequencies, with an average RF = 4.1 × 10−4. HM‐mediated Rec2 experiments produced the lowest average RF = 1.9 × 10−5, likely due to interference by MMR‐repair machinery. LD‐mediated Rec2 experiments yielded intermediate results, with an average RF = 4.7 × 10−5. In comparison, Ssr‐driven recombineering assays produced inferior values for SI and HM oligonucleotides (8.8 × 10−5 and 3.8 × 10−6 respectively) but similar to those with the LD sequence (4.3 × 10−5; see below). In summary, improved recombineering frequencies of Rec2 were upheld across a variety of pyrF‐directed experiments, indicating the utility of this recombinase in effecting a broad range of mutations in the P. putida genome.
Figure 5.
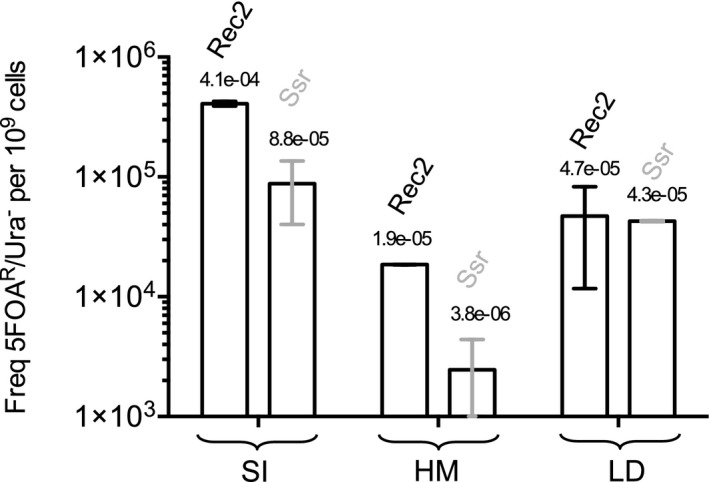
Rec2 is able to enter a range of mutations in Pseudomonas putida. P. putida EM42 strains containing pSEVA258‐ssr and pSEVA258‐rec2 were assayed as explained above for pyrF mutagenesis (see Fig. 4 legend) but using the following pyrF‐directed oligonucleotides (Table S1): SI (introduces a 9‐nt insertion of three stop codons), HM (generates a mismatch highly detectable by MMR, creating a stop codon) and LD (mediates the complete deletion of pyrF). Each data set represents at least two independent experimental replicates. Column graphs of the resulting data were generated with graphpad prism as described in Fig. 2 . Recombineering values are shown on a logarithmic scale.
Discussion
Our comprehensive sequence similarity search for recombinogenic proteins within Pseudomonas and associated bacteriophage genomes allowed us to identify novel recombinases with potential activity in P. putida. We also included in this study three previously documented recombinases: Recβ from E. coli, RecTPsy from P. syringae and EF2132 from E. faecalis (here designated Rec10). Under the rpsL reporter platform, all assayed recs demonstrated some degree of recombineering activity, encompassing three orders of magnitude between the least (Rec3) and the most active proteins (Rec2). While allelic exchange rates varied, all candidates showed high specificity in promoting oligonucleotide‐mediated mutagenesis. Most recs originated from phage‐related genomic regions of the P. putida genus; however, proteins derived from more distant relatives P. aeruginosa (Rec6 and Rec7) and P. syringae (RecTPsy), and even from unrelated species E. coli (Recβ) and E. faecalis (Rec10), demonstrated appreciable activity in P. putida EM42. Further, upon completion of rpsL‐directed assays, no association could be found between recombineering activity and either recombinase family or species proximity to P. putida. Previous recombineering attempts in environmental and pathogenic bacteria have largely relied on proteins originating from the host species or close relatives; this approach has led to successful editing in Mycobacterium tuberculosis (Van Kessel and Hatfull, 2008), Lactobacillus reuteri and Lactococcus lactis (Pijkeren and Britton, 2012), Pseudomonas syringae (Swingle et al., 2010) and Bacillus subtilis (Sun et al., 2015). Our work challenges assumptions of species specificity based on these reports, instead suggesting promiscuity of oligonucleotide‐mediated recombinase activity. Such non‐specific activity has been documented in other bacteria, as in the case of Corynebacterium glutamicum (Binder et al., 2013) and E. coli (Datta et al., 2008). While the first study focused only on RecT of Rac prophage, in the second, authors detected high rates of recombineering activity in E. coli from an array of recombinases originating from both gram‐negative and gram‐positive bacteria (10−4 to 10−1 mutants/viable cell). While such rates of allelic exchange reflect an increase in two orders of magnitude from the numbers we have observed (10−6 to 10−3 mutants/viable cell), we note the use of an E. coli ∆mutS strain devoid of mismatch repair machinery, which could have increased recombineering rates by >100‐fold (Costantino and Court, 2003). In contrast, P. putida EM42 – a streamlined derivative of P. putida KT2440 (Martínez‐García et al., 2014) – contains a fully intact MMR system, making a direct, quantitative study comparison difficult.
In terms of recombineering efficiency, we identified several novel proteins performing at rates approaching the P. putida Ssr standard (Aparicio et al., 2016). P. putida proteins Rec5 (Sak family), Rec8 and Rec9 (Sak4 family) all produced approximate recombineering frequencies of 10−4 mutants/viable cell within the EM42 strain. Interestingly, despite its distant relation to P. putida, Rec6 of P. aeruginosa (Sak4 family) catalysed recombineering at similar rates (10−4 mutants/viable cell). Among recombinases producing poorer results, it should be noted that a low outlier emerged in the only rec gene derived from the P. putida KT2440 genome: rec3 (GP2.5 family, 10−7 mutants/viable cell). With rates of ~10−5 mutants/viable cell, RecTPsy of P. syringae (Redβ‐like family) and Rec10 of E. faecalis (Sak4 family) also fell short of Ssr standards. These results deviate from higher efficiencies reported of each protein in P. syringae (Swingle et al., 2010) and E. coli (Datta et al., 2008) respectively. Only two recombinases outperformed the Ssr protein: ERF family protein Rec2 (P. putida CSV86) and Recβ (E. coli λ phage). Both proteins yielded recombineering values ~10−3 mutants/viable cell, with the former exhibiting rates three times those of the latter. Despite the concept of a broad host spectrum for phage‐derived recombinases supported by this and other studies (Datta et al., 2008), the impressive performance of Recβ in P. putida was unexpected. Recβ has only been shown to catalyze bacterial genomic recombination of foreign ssDNA in enterobacteriaceae (Nyerges et al., 2016). In contrast, it fails to function in species such as P. syringae (Swingle et al., 2010) and C. glutamicum (Binder et al., 2013). Recβ activity in P. putida KT2440 has been documented at ~10−7 mutants/viable cell (Luo et al., 2016), a result equal to, if not lower than, basal recombineering rates in this organism. Notwithstanding our use of an alternative plasmid/reporter system, we take note of Recβ activity reaching ~10−3 mutants/viable cell, a value nearly four orders of magnitude greater than the one reported above.
To check the robustness of the above results, we probed the ability of promising candidates Rec2 and Recβ to direct oligo‐mediated mutagenesis of an alternative reporter gene: pyrF. While the pyrF reporter system facilitates selection of allelic changes resulting in gene disruption, it contains an inherent drawback: Ura‐auxotrophic pyrF knockouts grow slower than wild‐type Ura heterotrophs on LB‐rich media supplemented with uracil (Fig. S1). In the context of recombineering experiments, such a growth differential can lead to bias in strain representation post‐electroporation; over time, quickly dividing wild‐type cells attain higher frequencies in the population, while the global representation of slower‐growing mutants decreases. This trend results in an underestimation of mutant rates that is aggravated by longer recovery times, a phenomenon observed in early trials of pyrF‐directed recombineering (data not shown). To limit this bias, we restricted recovery periods within the pyrF reporter system to 6 h (Aparicio et al., 2016), whereas rpsL reporter assays (unaffected by this trend) were simplified with overnight recovery times. As seen in Fig. 4, Rec2 and Recβ successfully mediated recombination in P. putida under the pyrF reporter system, with Rec2 once again surpassing Recβ. However, despite protocol adaptation, we observed a discrepancy in Recβ performance between the rpsL and pyrF reporter platforms: while the differential recombineering rate was just three times in favour of Rec2 under rpsL, under the pyrF reporter, Rec2 showed sixty times the amount of activity exhibited by Recβ. We argue that the mild toxicity of Rec2 led to slower growth of Rec2‐expressing cultures after electroporation. Rec2 toxicity, for which a mechanism of action is currently unknown, could lead to a lag phase or a higher death rate in Rec2‐expressing cultures during post‐shock recovery. Slower Rec2 culture growth would yield a lower growth differential between wild‐type and mutant cells during this period, as compared to Recβ cultures. As explained above, a greater growth differential in Recβ cultures would aggravate the underestimation of final mutational rates, accounting for diminished Recβ performance under the pyrF reporter system. This said, although Recβ showed noticeable recombineering activities under both experimental conditions, Rec2 alone outperformed Ssr under both reporter platforms within P. putida, making it our sole candidate for further recombineering study.
The main experimental body of this work was conducted by introducing single‐base mutations poorly detected by the MMR system into the rpsL and pyrF genes of P. putida. However, large‐scale genomic‐editing projects often necessitate the introduction of a variety of mutations into the target genome, including insertions, deletions and, depending on the desired modification, changes sensitive to endogenous repair machinery. For this reason, we concluded this study with an assay of Rec2 ability to enter an array of changes into the pyrF gene. As seen in Fig. 5, Rec2 consistently outperformed Ssr in introducing insertions and MMR‐sensitive point mutations to the pyrF gene. However, both proteins produced full pyrF deletions at similar rates. While the reason for this result is unknown, we highlight the difference in toxicity observed between Ssr and Rec2 post‐induction (Fig. 3). The impact of this disparity on growth rate can be appreciated in post‐recovery optical densities, taken six hours after culture back‐dilution to OD600 ~ 0.1: Rec2 produced an average OD600 ~ 0.4, while Ssr produced OD600 values equal to or below 0.1. As mentioned above, higher growth rates exacerbate the underestimation of mutant representation associated with the pyrF reporter platform. Thus, although we cannot exclude interference by Rec2‐specific recombineering factors (e.g. optimal induction time, length of mutagenic oligonucleotides and protein–oligonucleotide interactions), we suspect the relatively worse Rec2 recombineering efficiencies under the pyrF system can be attributed to a greater growth differential between wild‐type and mutant cells during the post‐electroporation recovery period.
In summary, we have isolated an ERF family protein capable of catalyzing mutagenic recombination in P. putida with high efficiency, in some cases surpassing previous standards by an order of magnitude. While Rec2 performs at levels below those of Recβ in E. coli (Wang et al., 2009; Nyerges et al., 2016), we note that the frequencies reported in this study reflect a single cycle of recombineering. We expect that optimized protocols involving several cycles would produce results closer to those seen with Recβ in E. coli. Further, the P. putida EM42 cell factory, while purged of certain endonucleases (Martínez‐García et al., 2014) that could inhibit strand displacement (Mosberg et al., 2012), harbours a fully functional MMR system. As mentioned, MMR machinery is known to interfere with the MAGE platform in various bacterial species, dampening final rates of mutagenesis. For this reason, experimental set‐ups aimed to facilitate deep genome engineering usually rely on permanent or transient suppression of MMR (Wang et al., 2009; Gallagher et al., 2014; Lennen et al., 2016; Nyerges et al., 2016). We expect that implementation of the same approach in P. putida would greatly enhance Rec2 performance, facilitating for projects based on the deployment of high levels of combinatorial genomic diversity.
In practical terms, the suite of recombinases identified in this study has broader implications. With protocols adjusted, Rec2 offers the potential to facilitate auspicious biosynthetic ventures in P. putida ‐ for example, the rewiring of metabolic pathways and implantation of genetic and metabolic circuits in cell factory strains. We note that the array of proteins tested in this study was standardized to the broad‐host‐range plasmid RSF1010. Given the promiscuity of this testing platform, we contend that the broad spectrum of recombinase constructs tested contains viable candidates for future study in environmental bacteria beyond P. putida – in Pseudomonas aeruginosa and other gram‐negative bacteria, for example. Given its remarkable performance in P. putida, Rec2 becomes a particularly promising candidate for future studies in its parent strain. Once perfected, deep genetic programming in Pseudomonas and other environmental bacteria could unlock radical advances in biocatalysis and bioremediation, extending to targeted purification of contaminated soil reserves and biosynthesis of biomedical compounds of interest.
Experimental procedures
Bacterial strains and growth conditions
Table 2 lists the strains and plasmids employed in this work. Experimental strains were grown in liquid Luria Bertani (LB) (10 g l−1 tryptone, 5 g l−1 yeast extract and 5 g l−1 NaCl) or glycerol‐free terrific broth (TB) (24 g l−1 yeast extract, 12 g l−1 tryptone, 9.4 g l−1 K2HPO4, 2 g l−1 KH2PO4) with shaking (170 rpm) at 30°C (P. putida) or 37°C (E. coli). M9 minimal media (Sambrook et al., 1989) was supplemented, when stated, with 0.2% w/v citrate for P. putida growth. Solid media were prepared at the ratios listed above, with 15 g l−1 agar added. When necessary, liquid and solid media were supplemented with 20 μg ml−1 of Uracil (Ura), 50 μg ml−1 of kanamycin (Km), 50 μg ml−1 of chloramphenicol (Cm), 100 μg ml−1 of streptomycin (Sm) or 250 μg ml−1 of 5‐fluoroorotic acid (5‐FOA). During recombineering experiments, 1 mM of 3‐methylbenzoate (3MB) was added to mid‐logarithmic‐phase cultures of P. putida EM42 to induce protein expression for 30 min.
Table 2.
Bacterial strains and plasmids used in this work
| Strain or plasmid | Relevant characteristicsa | Reference |
|---|---|---|
| Strains | ||
| Escherichia coli CC118 | Cloning host; Δ(ara‐leu) araD ΔlacX174 galE galK phoA thiE1 rpsE (SpR) rpoB (RifR) argE(Am) recA1 | Grant et al. (1990) |
| Escherichia coli HB101 |
Helper strain used for conjugation; F− λ− hsdS20(rB− mB−) recA13 leuB6(Am) araC14 Δ(gpt‐proA)62 lacY1 galK2(Oc) xyl‐5 mtl‐1 thiE1 rpsL20(SmR) glnX44(AS) |
Boyer and Roulland‐Dussoix (1969) |
| Pseudomonas putida EM42 |
KT2440 derivative; Δprophage1 Δprophage4 Δprophage3 Δprophage2 ΔTn7 ΔendA‐1 ΔendA‐2 ΔhsdRMS Δflagellum ΔTn4652 |
Martínez‐García et al. (2015) |
| Plasmids | ||
| pRK600 | Helper plasmid used for conjugation; oriV(ColE1), RK2 (mob+ tra+); CmR | Kessler et al. (1992) |
| pSEVA258 | Cloning vector; oriV RSF1010); (xylS‐Pm; standard multiple cloning site; KmR | Silva‐Rocha et al. (2013) |
| pSEVA258‐recX | pSEVA258 derivative bearing X recombinase indicated in Table 2; oriV (RFS1010); cargo [xylS‐Pm → recX], KmR | This work |
Antibiotic markers: Sp, spectinomycin; Rif, rifampicin; Sm, streptomycin; Cm, chloramphenicol; Km, kanamycin.
General DNA manipulation, DataBank Search, Plasmid Construction and Oligonucleotide design
Standard DNA manipulations were conducted according to manufacturer's recommendations and previously established protocol (Sambrook et al., 1989). Isothermal/Gibson assembly was carried out according to procedures outlined in (Gibson et al., 2009; Gibson, 2011). ssDNA binding proteins with potential recombineering activity were selected from the NCBI non‐redundant (nr) protein sequence database: drawing from a recent survey of bacteriophage genomes for homologues to Rad52, Rad51 and Gp2.5 superfamily recombinases (Lopes et al., 2010), homologues from the major recombinase families were identified on Pseudomonas phage genomes. We designated the following proteins belonging to ERF, GP2.5, Sak and Sak4 families as sequence references for a subsequent sequence similarity search: ERF recombinase from P. aeruginosa phage D3 (GenBank no. NP_061548); GP2.5 recombinase from P. putida gh‐1 phage (GenBank no. NP_813756); SAK recombinase from P. aeruginosa phage F116 (GenBank no. YP_164283) and SAK4 recombinase from P. aeruginosa phage 73 (GenBank no. YP_001293439). We conducted a search for sequence similarity using the National Center for Biotechnology Information (NCBI) protein–protein BLAST tool (http://blast.ncbi.nlm.nih.gov). A BlastP search on the NCBI nr protein sequence database against P. putida and related bacteriophage genomes produced seven proteins bearing high sequence similarities to reference recombinases (Recs1, 2, 3, 5, 6, 8 and 9). An equivalent search of all Pseudomonas and related bacteriophage genomes using GP2.5 and SAK phage recombinase references – from Pseudomonas phages gh‐1 and F116 respectively – produced two hypothetical recombinases also included in this study (Recs4, 7). Finally, the EF2132 recombinase RecT of Enterococcus faecalis has been described to facilitate efficient recombineering in a phylogenetically distant gram‐negative host (Datta et al., 2008). Therefore, we also tested EF2132 (here termed Rec10) for recombineering functions in Pseudomonas putida. In total, we selected ten novel protein candidates for investigation. We also incorporated three recombinases previously tested for activity in Pseudomonas. Due to its reported levels of activity in P. syringae (Swingle et al., 2010), we selected RecTPsy, a homologue of the E. coli Rac prophage recombinase RecT. Finally, we included the original E. coli λ phage recombinase Recβ, which has been shown to demonstrate low levels of activity in P. putida KT2440 (Luo et al., 2016). As a control, we added to our study the current leading candidate for recombineering in P. putida, the T1E_1405 protein of P. putida DOT‐T1E (named Ssr). Ssr has been shown to mediate lagging strand invasion and recombination in P. putida at low frequencies with high specificity of mutagenesis (Aparicio et al., 2016). Table 1 characterizes all protein candidates tested in this work. The open reading frames (ORFs) for putative recombinases 1, 2, 4, 5, 6, 7, 8, 9 and 10 were synthesized de novo by GeneCust (GeneCust Europe, Luxembourg) with flanking sequences to allow cloning into the pSEVA258 vector. Following the experimental design outlined in (Aparicio et al., 2016), the 5′ flanking sequence (5′‐TGGAGTCATGACCATGCCTAGGCCGCGGCCGCGCgaattcAGA AGGAGAATATACC‐3′) contained (i) a 40‐bp homology to the sequence upstream of the EcoRI site in pSEVA258 (italics, EcoRI in lowercase), (ii) a ribosome binding site (underlined), and (iii) a 7‐bp spacer. Downstream of the stop codon, the 3′ flanking sequence (5′‐ggatccTCTAGAGTCGACCTGCAGGCATGCAAGCTTGCGG‐3′) contained a 40‐bp homology to the sequence downstream of the BamHI site (lowercase) in pSEVA258. The rec3 gene was PCR amplified directly from the KT2440 chromosome using primers Rec3FW/Rec3REV. The recT Psy and recβ genes were PCR amplified from the pUCP24 (Swingle et al., 2010) and pKD46 (Datsenko and Wanner, 2000) plasmids with primer pairs RecTPsyFW/RecTPsyREV and RecβFW/RecβRV respectively. All primer pairs listed (Table S1) harbored sequence tails containing the same features described for Recs1‐10. In this way, all rec genes were placed in a standard expression platform of pSEVA258 just as the ssr gene (Aparicio et al., 2016). rec genes, either from gene synthesis or from PCR amplification, were Gibson‐assembled into EcoRI/BamHI‐digested pSEVA258 and electroporated into E. coli CC118 (Table 2). Transformed constructs were checked for proper insertion by sequencing with primers 238F and PS2 (Table S1). Constructs were finally introduced into P. putida EM42 via tripartite mating with helper strain HB101 (Table 2), as outlined in Ref. Martínez‐García and de Lorenzo (2012). Plasmid‐containing strains were selected for on M9‐Citrate‐Km solid media. As an additional check, individual clones of P. putida EM42/pSEVA258‐recX (where X is the recombinase specimen 1–10) were replicated on the same media, and plasmid constructs isolated by miniprep were digested with EcoRI/BamHI to verify the transformants. Control strains of P. putida EM42 harboring pSEVA258 and pSEVA258‐ssr were constructed in a previous work (Aparicio et al., 2016). Oligonucleotides used in this work were provided by Sigma‐Aldrich (St Louis, MO, USA) and are characterized in Table S1. pyrF‐directed oligonucleotides employed in genomic recombineering experiments (namely, LM, HM, SI and LD) were described previously in Aparicio et al. (2016). Using the same design criteria applied for the LM oligonucleotide (length, 5′ phosphorothioate protection, folding energy, MMR System sensitivity, etc.), the SR oligonucleotide was modelled after the rpsL gene of P. putida KT2440, which encodes the ribosomal protein S12. SR generates an A‐G mismatch (poorly detected by MMR) that introduces a single nucleotide change (A→C) in codon K43 (AAA‐Lys), resulting in T43 (ACA‐Thr). The K43T missense mutation is analogous to the classical K42T mutation of the rpsL gene of E. coli (Timms et al., 1992; Jiang et al., 2013), which confers resistance to streptomycin.
Assays for recombineering efficiency in P. putida EM42
The ability of recombinase candidates to incorporate foreign ss‐oligonucleotides into endogenous DNA forks occurring during genetic replication (Ogawa and Okazaki, 1980) was first measured through acquisition of Sm resistance by P. putida strains electroporated with rpsL‐targeted SR (Table S1). In rpsL‐directed recombineering experiments, P. putida EM42/pSEVA258‐recX strains were incubated overnight in LB‐Km at 30°C with shaking (170 rpm). A strain transformed with empty vector pSEVA258 was included as an additional control. Overnight strains were back‐diluted to OD600 = 0.1 in 20 ml of fresh LB‐Km and regrown in identical conditions to OD600 = 0.4–0.5. Cultures were supplemented with 1 mM 3MB and incubated for an additional 30 min to induce the expression of Rec proteins. Competent cell preparation was then performed at RT by pelleting cells at 3220 g for 5 minutes, washing successively with 10, 5 and 1 ml of 300 mM sucrose and finally resuspending in 200 μl of the same solution. One hundred microlitres of aliquots of prepared cells was then supplemented with 1 μl of 100 μM SR oligonucleotide stock (~3 μg). Well‐mixed cultures were transferred into 2‐mm gap width cuvettes (Bio‐Rad Laboratories, Hercules, CA, USA) before electroporation at 2.5 kV with a MicropulserTM apparatus (Bio‐Rad Laboratories). P. putida EM42/pSEVA258 control cultures were electroporated with and without oligo SR to determine background recombineering levels. Samples were immediately inoculated in 5 ml of fresh TB and allowed to recover overnight at 30°C with shaking (170 rpm). Recovered cell cultures were plated at appropriate dilutions on LB‐Sm and LB solid media and incubated 24 hours at 30°C. After absolute colony counts were taken, the recombination frequency (RF) was calculated as the ratio between Sm‐resistant cells and viable cells (cells growing on LB), and normalized to 109 viable cells. To ensure accuracy of allelic changes, primers rpsL‐Fw and rpsL‐Rv were used to perform colony PCR on 5‐10 clones derived from each recombinase; the obtained 0.8 Kb fragment (encompassing the rpsL gene) was sequenced with primer rpsL‐Fw (Table S1). Recombineering activity of the most promising protein candidates (RF>2.3 × 10−4) was assayed in a different reporter platform (pyrF) to verify the robustness of rpsL‐targeted recombineering results. pyrF‐targeted recombineering experiments were conducted according to methods described in Ref. (Aparicio et al., 2016). The LM oligonucleotide (Table S1) was used in a first set of experiments to introduce a stop codon into the pyrF gene. Recombineering frequencies were calculated by plating dilutions on M9‐Citrate‐Uracil (viable cells) vs. M9‐Citrate‐Ura‐5‐FOA (allelic changes). In a second set of experiments, the most active Rec protein was characterized, in comparison with Ssr, using the HM, LD and SI oligonucleotides (Table S1). These oligonucleotides each introduce a different genomic change in pyrF as explained above. For all pyrF experiments, recombineering frequency was calculated as the ratio between 5FOAR/Ura‐auxotrophic cells and total viable cells (cells growing on M9‐Cit‐Ura), and normalized to 109 viable cells. To distinguish authentic pyrF‐mutant colonies from spontaneous 5FOAR mutants, fifty 5FOAR clones of each recombinase were replicated on M9‐Cit/M9‐Cit‐Ura‐5FOA plates as outlined in Ref. (Aparicio et al., 2016). Colonies growing on both media were discounted as spontaneous pyrF‐unrelated mutants. Frequencies were multiplied by the ratio between verified pyrF mutants (cells growing on M9‐Cit‐Ura‐FOA only) and total # 5FOAR CFU (cells growing on all media).
Additional experiments
To determine the effect of protein toxicity on strain growth, profiles were generated for 3MB induced and non‐induced experimental strains using a Spectramax M2e Microplate Reader (Molecular Devices, Sunnyvale, CA, USA). Cultures of P. putida EM42 strains harbouring pSEVA258‐rec2, pSEVA258‐ssr and pSEVA258‐recβ, as well as EM42/pSEVA258 and EM42 controls, were grown overnight in LB‐Km‐shaken liquid media at 30°C. Overnight strains were back‐diluted to OD600 = 0.05 on fresh LB‐Km medium before being loaded into a 96‐well microtiter plate. Liquid growth rates of 1 mM 3MB‐induced (t = 0) and non‐induced strains were measured over a 24 h period (interval t = 15 min) following the OD600. Two biological replicates were carried out for each experimental condition.
Conflict of interest
None declared.
Supporting information
Table S1. Oligonucleotides used in this work.
Fig S1. Growth of P. putida EM42∆pyrF.
Acknowledgements
This work was funded by the CAMBIOS Project of the Spanish Ministry of Economy and Competitiveness RTC‐2014‐1777‐3 (MINECO), HELIOS Project of the Spanish Ministry of Economy and Competitiveness BIO 2015‐66960‐C3‐2‐R (MINECO/FEDER). ARISYS (ERC‐2012‐ADG‐322797), EmPowerPutida (EU‐H2020‐BIOTEC‐2014‐2015‐6335536) and FUTURE (704410‐H2020‐MSCA‐IF‐15). Additional support to C.P. was provided by the European Research Council, the Wellcome Trust, GINOP (grants TUMORDNS: GINOP‐2.3.2‐15‐2016‐00020; EVOMER: GINOP‐2.3.2‐15‐2016‐00014) and the Lendület Programme of the Hungarian Academy of Sciences. The support of the Fulbright Foundation to D.E.R. is gratefully acknowledged. Á.N. is the recipient of a PhD Fellowship of the Boehringer Ingelheim Fonds.
Microbial Biotechnology (2018) 11(1), 176–188
Funding information
This work was funded by the CAMBIOS Project of the Spanish Ministry of Economy and Competitiveness RTC‐2014‐1777‐3 (MINECO), HELIOS Project of the Spanish Ministry of Economy and Competitiveness BIO 2015‐66960‐C3‐2‐R (MINECO/FEDER). ARISYS (ERC‐2012‐ADG‐322797), EmPowerPutida (EU‐H2020‐BIOTEC‐2014‐2015‐6335536) and FUTURE (704410‐H2020‐MSCA‐IF‐15). Additional support to C.P. was provided by the European Research Council, the Wellcome Trust, GINOP (grants TUMORDNS: GINOP‐2.3.2‐15‐2016‐00020; EVOMER: GINOP‐2.3.2‐15‐2016‐00014) and the Lendület Programme of the Hungarian Academy of Sciences.
References
- Aparicio, T. , Jensen, S.I. , Nielsen, A.T. , de Lorenzo, V. , and Martínez‐García, E. (2016) The Ssr protein (T1E_1405) from Pseudomonas putida DOT‐T1E enables oligonucleotide‐based recombineering in platform strain P. putida EM42. Biotechnol J 11: 1309–1319. [DOI] [PubMed] [Google Scholar]
- Babic, I. , Andrew, S.E. , and Jirik, F.R. (1996) MutS interaction with mismatch and alkylated base containing DNA molecules detected by optical biosensor. Mutat Res 372: 87–96. [DOI] [PubMed] [Google Scholar]
- Binder, S. , Siedler, S. , Marienhagen, J. , Bott, M. , and Eggeling, L. (2013) Recombineering in Corynebacterium glutamicum combined with optical nanosensors: a general strategy for fast producer strain generation. Nucleic Acids Res 41: 6360–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke, J.D. , LaCroute, F. , and Fink, G.R. (1984) A positive selection for mutants lacking orotidine‐5’‐phosphate decarboxylase activity in yeast: 5‐fluoro‐orotic acid resistance. Mol Gen Genet 197: 345–346. [DOI] [PubMed] [Google Scholar]
- Boyer, H.W. , and Roulland‐Dussoix, D. (1969) A complementation analysis of the restriction and modification of DNA in Escherichia coli . J Mol Biol 41: 459–472. [DOI] [PubMed] [Google Scholar]
- Costantino, N. , and Court, D.L. (2003) Enhanced levels of lambda Red‐mediated recombinants in mismatch repair mutants. Proc Natl Acad Sci USA 100: 15748–15753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko, K.A. , and Wanner, B.L. (2000) One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc Natl Acad Sci USA 97: 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta, S. , Costantino, N. , Zhou, X. , and Court, D.L. (2008) Identification and analysis of recombineering functions from Gram‐negative and Gram‐positive bacteria and their phages. Proc Natl Acad Sci USA 105: 1626–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis, H.M. , Yu, D. , DiTizio, T. , and Court, D.L. (2001) High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single‐stranded oligonucleotides. Proc Natl Acad Sci USA 98: 6742–6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funatsu, G. , and Wittmann, H.G. (1972) Ribosomal proteins. 33: Location of amino‐acid replacements in protein S12 isolated from Escherichia coli mutants resistant to streptomycin. J Mol Biol 68: 547–550. [DOI] [PubMed] [Google Scholar]
- Gallagher, R.R. , Li, Z. , Lewis, A.O. , and Isaacs, F.J. (2014) Rapid editing and evolution of bacterial genomes using libraries of synthetic DNA. Nat Protoc 9: 2301–2316. [DOI] [PubMed] [Google Scholar]
- Galvao, T.C. , and de Lorenzo, V. (2005) Adaptation of the Yeast URA3 selection system to Gram‐negative bacteria and generation of a ∆betCDE Pseudomonas putida strain. Appl Environ Microbiol 71: 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garmendia, J. , de las Heras, A. , Galvão, T.C. and de Lorenzo, V. (2008) Tracing explosives in soil with transcriptional regulators of Pseudomonas putida evolved for responding to nitrotoluenes. Microb Biotechnol 1, 236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach, R.G. , Jäckel, D. , Hölzer, S.U. , and Hensel, M. (2009) Rapid oligonucleotide‐based recombineering of the chromosome of Salmonella enterica . Appl Environ Microbiol 75: 1575–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, D.G. (2011) Enzymatic assembly of overlapping DNA fragments. Methods Enzymol 498: 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, D.G. , Young, L. , Chuang, R.‐Y. , Venter, J.C. , Hutchison, C.A. , and Smith, H.O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345. [DOI] [PubMed] [Google Scholar]
- Grant, S.G.N. , Jessee, J. , Bloom, F.R. , and Hanahan, D. (1990) Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation‐restriction mutants. Proc Natl Acad Sci USA 87: 4645–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory, S.T. , Cate, J.H.D. , and Dahlberg, A.E. (2001) Streptomycin‐resistant and streptomycin‐dependent mutants of the extreme thermophile Thermus thermophilus . J Mol Biol 309: 333–338. [DOI] [PubMed] [Google Scholar]
- Isaacs, F.J. , Carr, P.A. , Wang, H.H. , Lajoie, M.J. , Sterling, B. , Kraal, L. , et al (2011) Precise manipulation of chromosomes in vivo enables genome‐wide codon replacement. Science 333: 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jatsenko, T. , Tover, A. , Tegova, R. , and Kivisaar, M. (2010) Molecular characterization of Rifr mutations in Pseudomonas aeruginosa and Pseudomonas putida . Mutat Res 683: 106–114. [DOI] [PubMed] [Google Scholar]
- Jiang, W. , Bikard, D. , Cox, D. , Zhang, F. , and Marraffini, L.A. (2013) RNA‐guided editing of bacterial genomes using CRISPR‐Cas systems. Nat Biotechnol 31: 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler, B. , Lorenzo, V.D. , and Timmis, K. (1992) A general system to integrate lacZ fusions into the chromosomes of gram‐negative eubacteria: regulation of the Pm promoter of the TOL plasmid studied with all controlling elements in monocopy. Mol Gen Genet 233: 293–301. [DOI] [PubMed] [Google Scholar]
- Kunkel, T.A. , and Erie, D.A. (2005) DNA mismatch repair. Annu Rev Biochem 74: 681–710. [DOI] [PubMed] [Google Scholar]
- Lennen, R.M. , Nilsson Wallin, A.I. , Pedersen, M. , Bonde, M. , Luo, H. , Herrgård, M.J. , and Sommer, M.O.A. (2016) Transient overexpression of DNA adenine methylase enables efficient and mobile genome engineering with reduced off‐target effects. Nucleic Acids Res 44: e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, R. , and Liu, J. (2010) Scarless and sequential gene modification in Pseudomonas using PCR product flanked by short homology regions. BMC Microbiol 10: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeschcke, A. , and Thies, S. (2015) Pseudomonas putida‐a versatile host for the production of natural products. Appl Microbiol Biotechnol 99: 6197–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes, A. , Amarir‐Bouhram, J. , Faure, G. , Petit, M.A. , and Guerois, R. (2010) Detection of novel recombinases in bacteriophage genomes unveils Rad52, Rad51 and Gp2.5 remote homologs. Nucleic Acids Res 38: 3952–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, X. , Yang, Y. , Ling, W. , Zhuang, H. , Li, Q. , and Shang, G. (2016) Pseudomonas putida KT2440 markerless gene deletion using a combination of λ Red recombineering and Cre/loxP site‐specific recombination. FEMS Microbiol Lett 363: fnw014. [DOI] [PubMed] [Google Scholar]
- Marinelli, L.J. , Hatfull, G.F. , and Piuri, M. (2012) Recombineering: a powerful tool for modification of bacteriophage genomes. Bacteriophage 2: 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐García, E. , and de Lorenzo, V. (2011) Engineering multiple genomic deletions in Gram‐negative bacteria: analysis of the multi‐resistant antibiotic profile of Pseudomonas putida KT2440. Environ Microbiol 13: 2702–2716. [DOI] [PubMed] [Google Scholar]
- Martínez‐García, E. , and de Lorenzo, V. (2012) Transposon‐based and plasmid‐based genetic tools for editing genomes of Gram‐negative bacteria. Methods Mol Biol 813: 267–283. [DOI] [PubMed] [Google Scholar]
- Martínez‐García, E. , Calles, B. , Arévalo‐Rodríguez, M. , and de Lorenzo, V. (2011) pBAM1: an all‐synthetic genetic tool for analysis and construction of complex bacterial phenotypes. BMC Microbiol 11: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐García, E. , Nikel, P.I. , Aparicio, T. , and de Lorenzo, V. (2014) Pseudomonas 2.0: genetic upgrading of P. putida KT2440 as an enhanced host for heterologous gene expression. Microb Cell Fact 13: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐García, E. , Jatsenko, T. , Kivisaar, M. , and de Lorenzo, V. (2015) Freeing Pseudomonas putida KT2440 of its proviral load strengthens endurance to environmental stresses. Environ Microbiol 17: 76–90. [DOI] [PubMed] [Google Scholar]
- Mosberg, J.A. , Gregg, C.J. , Lajoie, M.J. , Wang, H.H. , and Church, G.M. (2012) Improving lambda Red renome engineering in Escherichia coli via rational removal of endogenous nucleases. PLoS ONE 7: e44638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, K.C. (1998) Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli . J Bacteriol 180: 2063–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel, P.I. , Martínez‐García, E. , and de Lorenzo, V. (2014) Biotechnological domestication of pseudomonads using synthetic biology. Nat Rev Microbiol 12: 368–379. [DOI] [PubMed] [Google Scholar]
- Nikel, P.I. , Chavarría, M. , Fuhrer, T. , Sauer, U. , and de Lorenzo, V. (2015) Pseudomonas putida KT2440 strain metabolizes glucose through a cycle formed by enzymes of the Entner‐Doudoroff, Embden‐Meyerhof‐Parnas, and pentose phosphate pathways. J Biol Chem 290: 25920–25932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyerges, Á. , Csörgő, B. , Nagy, I. , Bálint, B. , Bihari, P. , Lázár, V. , et al (2016) A highly precise and portable genome engineering method allows comparison of mutational effects across bacterial species. Proc Natl Acad Sci USA 113: 2502–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan, G.A. and Neuhard, J. (1970) Pyrimidine metabolism in microorganisms. Bacteriol Rev 34, 278–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa, T. , and Okazaki, T. (1980) Discontinuous DNA replication. Annu Rev Biochem 49: 421–457. [DOI] [PubMed] [Google Scholar]
- Oh, J.H. , and Pijkeren, J.P. (2014) CRISPR–Cas9‐assisted recombineering in Lactobacillus reuteri . Nucleic Acids Res 42: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijkeren, J.P. , and Britton, R.A. (2012) High efficiency recombineering in lactic acid bacteria. Nucleic Acids Res 40: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poblete‐Castro, I. , Becker, J. , Dohnt, K. , dos Santos, V.M. , and Wittmann, C. (2012) Industrial biotechnology of Pseudomonas putida and related species. Appl Microbiol Biotechnol 93(6): 2279–2290. [DOI] [PubMed] [Google Scholar]
- Puchałka, J. , Oberhardt, M.A. , Godinho, M. , Bielecka, A. , Regenhardt, D. , Timmis, K.N. , et al (2008) Genome‐scale reconstruction and analysis of the Pseudomonas putida KT2440 metabolic network facilitates applications in biotechnology. PLoS Comput Biol 4: e1000210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos, J.‐L. , Sol Cuenca, M. , Molina‐Santiago, C. , Segura, A. , Duque, E. , Gómez‐García, M.R. , et al (2015) Mechanisms of solvent resistance mediated by interplay of cellular factors in Pseudomonas putida. FEMS Microbiol Rev 39: 555–566. [DOI] [PubMed] [Google Scholar]
- Ruusala, T. , and Kurland, C.G. (1984) Streptomycin preferentially perturbs ribosomal proofreading. Mol Gen Genet 198: 100–104. [DOI] [PubMed] [Google Scholar]
- Sambrook, J. , Fritsch, E.F. , and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual. NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Silva‐Rocha, R. , Martínez‐García, E. , Calles, B. , Chavarría, M. , Arce‐Rodríguez, A. , De Las Heras, A. , et al (2013) The Standard European Vector Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res 41: D666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Z. , Deng, A. , Hu, T. , Wu, J. , Sun, Q. , Bai, H. , et al (2015) A high‐efficiency recombineering system with PCR‐based ssDNA in Bacillus subtilis mediated by the native phage recombinase GP35. Appl Microbiol Biotechnol 99: 5151–5162. [DOI] [PubMed] [Google Scholar]
- Swingle, B. , Bao, Z. , Markel, E. , Chambers, A. , and Cartinhour, S. (2010) Recombineering using RecTE from Pseudomonas syringae . Appl Environ Microbiol 76: 4960–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timms, A.R. , Steingrimsdottir, H. , Lehmann, A.R. , and Bridges, B.A. (1992) Mutant sequences in the rpsL gene of Escherichia coli B/r: mechanistic implications for spontaneous and ultraviolet light mutagenesis. Mol Gen Genet 232: 89–96. [DOI] [PubMed] [Google Scholar]
- Van Kessel, J.C. , and Hatfull, G.F. (2008) Efficient point mutagenesis in mycobacteria using single‐stranded DNA recombineering: characterization of antimycobacterial drug targets. Mol Microbiol 67: 1094–1107. [DOI] [PubMed] [Google Scholar]
- Wang, H.H. , Isaacs, F.J. , Carr, P.A. , Sun, Z.Z. , Xu, G. , Forest, C.R. , and Church, G.M. (2009) Programming cells by multiplex genome engineering and accelerated evolution. Nature 460: 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, D. , Ellis, H.M. , Lee, E.C. , Jenkins, N.A. , Copeland, N.G. , and Court, D.L. (2000) An efficient recombination system for chromosome engineering in Escherichia coli . Proc Natl Acad Sci USA 97: 5978–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Oligonucleotides used in this work.
Fig S1. Growth of P. putida EM42∆pyrF.