

Summary
A range of regulated gene expression systems has been developed for mycobacteria in the last few years to facilitate the study of essential genes, validate novel drug targets and evaluate their vulnerability. Among these, the TetR/Pip‐OFF repressible promoter system was successfully used in several mycobacterial species both in vitro and in vivo. In the first version of the system, the repressible promoter was Pptr, a strong Pip‐repressible promoter of Streptomyces pristinaespiralis, which might hamper effective downregulation of genes with a low basal expression level. Here, we report an enhanced system that allows more effective control of genes expressed at low level. To this end, we subjected Pptr to targeted mutagenesis and produced 16 different promoters with different strength. Three of them, weaker than the wild‐type promoter, were selected and characterized showing that they can indeed improve the performances of TetR/Pip‐OFF repressible system both in vitro and in vivo increasing its stringency. Finally, we used these promoters to construct a series of bacterial biosensors with different sensitivity to DprE1 inhibitors and developed a whole‐cell screening assay to identify inhibitors of this enzyme.
Introduction
Tighty regulated gene expression systems are powerful tools widely used to study essential genes, validate drug targets and evaluate their vulnerability (Schnappinger, 2015). In recent years, several such systems were developed for mycobacteria [reviewed in (Schnappinger and Ehrt, 2014; Choudhary et al., 2016)]. The most widely used are those based on the TetR repressor (Carroll et al., 2005; Ehrt et al., 2005), but recently other systems based on CRISPR interference (Choudhary et al., 2015; Singh et al., 2016), on the theophylline riboswitch (Seeliger et al., 2012) or on dual‐control switches which combine transcriptional regulation and regulated proteolysis (Kim et al., 2011, 2013) were shown to be highly effective.
In previous work, we reported the development of a repressible promoter system (TetR/Pip‐OFF) based on two chromosomally encoded repressors and the tunable promoter Pptr (Boldrin et al., 2010). In this system, entirely integrated in single copy in the mycobacterial genome, the gene encoding the tetracycline‐responsive repressor TetR is constitutively expressed, while the gene coding for the pristinamycin‐responsive repressor Pip is expressed from a TetR‐dependent promoter, and the gene of interest is expressed from Pptr, a strong promoter repressible by Pip. In the absence of ATc, TetR represses pip expression and the target gene is expressed; in the presence of ATc, TetR releases the promoter of the Pip gene resulting in repression of Pptr. The system can be further modulated by the addition of pristinamycin I, which can alleviate Pptr repression by Pip (Boldrin et al., 2010).
Since then, the TetR/Pip‐OFF system has been successfully used by several groups to study essential genes and validate drug targets in vitro and in vivo in different mycobacterial species (Serafini et al., 2009, 2013; Cortes et al., 2011; Di Luca et al., 2012; Mondino et al., 2013; Ventura et al., 2013; Ahmed et al., 2014, 2016; Bazet Lyonnet et al., 2014; Bhowmick et al., 2014; Boldrin et al., 2014; Kolly et al., 2014a,b; Pandey and Rodriguez, 2014; Verma and Chatterji, 2014; Gola et al., 2015; Gupta et al., 2015; Mori et al., 2015; Hu et al., 2016; Degiacomi et al., 2017). This broad experience taught us that the main drawback of this otherwise very succesful system was the strength of the Pptr promoter, which causes overexpression of the gene of interest, when this is physiologically expressed at low level, with resulting accumulation of its product. As a consequence of such accumulation, the effect of gene repression in these cases was often visible only after a number of serial passages of the bacterial culture in the presence of the repressor (ATc). This step was needed to titrate down the protein product. Moreover, we failed to obtain a phenotype in conditional mutants of genes expressed at very low levels (such as transcriptional regulators), probably because even when repressed their residual expression, due to promoter leakage, produced sufficient target protein to effect its physiological function (unpublished data). In this manuscript, we describe the construction and characterization of a series of Pptr promoter mutants of different strength and show that these promoters can be used in the context of the Tet/Pip‐OFF repressible system thus overcoming the problems outlined above.
Results
Promoter mutagenesis
The TetR/Pip‐OFF repressible promoter system has been successfully used to construct several conditional mutants in Mycobacterium tuberculosis (Boldrin et al., 2010). However, the high strength of the Pptr promoter, on which the system is based, may represent a problem when the target gene is physiologically expressed from a weak promoter. To solve this problem, we designed a mutagenic strategy to weaken the promoter by introducing point mutations in the ‐10 or ‐35 consensus sequences or by modifying the length of the spacer region. Using PCR, we obtained eight promoters with mutations in the ‐10 region, six promoters with mutations in the ‐35 region and two promoters where the length of the spacer region was modified (Fig. 1). Mutated and wt promoters were cloned upstream of a promotorless egfp gene in the integrative plasmid pFRA61, containing the TetR/Pip‐OFF system (Boldrin et al., 2010). The resulting plasmids were transformed in both Mycobacterium smegmatis mc2155 and in M. tuberculosis H37Rv (Table S1).
Figure 1.

Mutations introduced in the Pptr promoter. Putative ‐10 and ‐35 are boxed, while the three Pip‐binding regions are underlined. Conserved repeats within the Pip‐binding regions are in capital letters. Mutations are numbered (1–16), and the nucleotides replacing the original are shown below the wt sequence. Mutations 1 and 2 are insertions; the vertical arrow indicates the point where nucleotide(s) was/were inserted. The transcriptional start site is indicated by an arrow.
Characterization of the mutant promoters
To evaluate promoter strength, M. tuberculosis and M. smegmatis strains harbouring the integrative plasmids containing the egfp transcriptional fusions were grown to exponential phase, and their fluorescence was measured using a fluorometer. Results clearly showed that the mutations resulted in promoters with a wide range of strength and that almost all of the new promoters behaved similarly in M. tuberculosis and in M. smegmatis (Fig. 2). Interestingly, mutations in the spacer region and in the ‐35 consensus sequence always caused strong attenuation of the promoter, whereas mutations in the ‐10 consensus sequence resulted in partial attenuation or even an increase in promoter activity (Pptr9, Pptr13, Pptr14). Some promoters showed an activity which was barely over the background. To determine whether their activity was totally abrogated or so low as to be undetectable when present in a single chromosomal copy, some of them were subcloned together with the egfp gene in a replicative plasmid and again introduced in M. tuberculosis. Figure 3 reports the relative fluorescence of the resulting strains, indeed confirming that in these conditions their activity was detectable.
Figure 2.
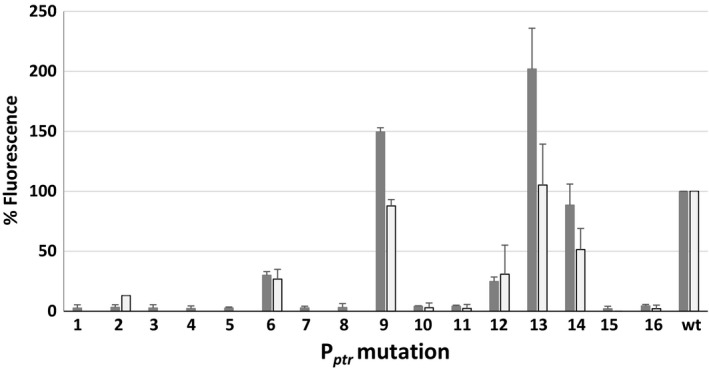
Relative fluorescence of Mycobacterium tuberculosis (dark grey) and M. smegmatis (light grey) strains harbouring integrative plasmids encoding the egfp gene downstream of the different Pptr promoter mutants (1–16). Fluorescence of the strain expressing egfp from wt Pptr was considered as 100%. For unknown reasons, we could not introduce the plasmids containing the Pptr mutations 5 and 8 in M. smegmatis.
Figure 3.
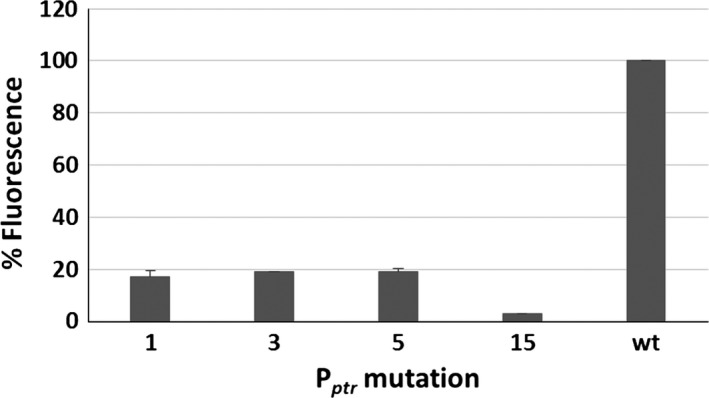
Relative fluorescence of Mycobacterium tuberculosis strains harbouring replicative plasmids encoding the egfp gene downstream Pptr promoters shown to have undetectable activity in Fig. 2. Fluorescence of the strain expressing egfp from wt Pptr was considered as 100%.
Starting from these data, we chose three promoters with different relative strength for further characterization: Pptr11, Pptr12 and Pptr14. To define whether these promoters were still repressible by Pip, the M. tuberculosis strains containing transcriptional fusions to egfp were grown to exponential phase with or without ATc 500 ng ml−1, and fluorescence was measured. As shown in Fig. 4, all of them were still repressible and suitable for use in the TetR/Pip‐OFF system.
Figure 4.
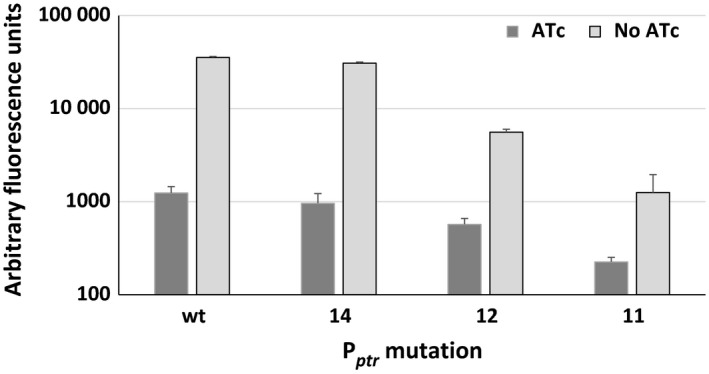
Fluorescence of Mycobacterium tuberculosis strains harbouring integrative plasmids encoding the egfp gene downstream of different Pptr promoter mutants grown with or without ATc.
Construction and characterization of conditional mutants expressing dprE1 from promoters with different strength
Previously, we reported the construction and characterization of a dprE1 conditional mutant based on the TetR/Pip‐OFF system (Kolly et al., 2014a). We demonstrated that dprE1 is an essential gene, as its repression upon exposure to ATc resulted in growth arrest. However, the strain had to be subcultured several times in the presence of ATc to deplete the gene product effectively. This was due to the higher strength of Pptr with respect to the physiological promoter of dprE1, which resulted in its overexpression in the conditional mutant (Kolly et al., 2014a). To rectify this, we constructed three conditional mutants expressing dprE1 from Pptr11, Pptr12, and Pptr14 (TB386, TB387, and TB388 respectively; Table 1). The three conditional mutants were grown in parallel with the original conditional mutant TB110 expressing dprE1 from the wt Pptr in the presence of ATc 500 ng ml−1. As shown in Table 2, the number of passages needed to observe the growth arrest was directly correlated to the strength of the promoter.
Table 1.
dprE1 conditional mutants based on the TetR/Pip‐OFF or the Pip‐ON systems
| Mutation | PlasmidTetR/Pip‐OFF | Strain | Plasmid Pip‐ON | Strain |
|---|---|---|---|---|
| 11 | pFRA184 | TB386 | pFRA212 | TB434 |
| 12 | pFRA186 | TB387 | pFRA213 | TB435 |
| 14 | pFRA188 | TB388 | pFRA214 | TB436 |
Table 2.
Passages in fresh ATc needed to stop the growth of dprE1 conditional mutants
| Strains | Pptr strengtha (%) | Passages in ATc 500 ng ml−1 b |
|---|---|---|
| TB386 | 4 | 3 |
| TB387 | 25 | 4 |
| TB388 | 89 | 7 |
| TB110 | 100 | > 7 |
Referred to Pptr wt which was considered as 100%.
Strains were grown in standing condition with or without ATc. Every 72–96 h, bacteria were diluted into fresh media containing ATc.
The protein encoded by dprE1 is the target of benzothiazinone (BTZ), a new antimycobacterial compound currently under development (Makarov et al., 2009, 2014; Piton et al., 2017). As the level of DprE1 has been shown to correlate with the sensitivity to BTZ, we measured the sensitivity of the new conditional mutants to this molecule: as illustrated in Fig. 5, there was a clear correlation between the level of promoter activity and the MIC to BTZ. The strain expressing dprE1 from Pptr12 (TB387) showed the same MIC for BTZ as H37Rv, suggesting that in these two strains, dprE1 is expressed at a similar level and that Pptr12 and the physiological promoter of dprE1 are similarly active (Table 3). To confirm this hypothesis, the dprE1 natural promoter was cloned upstream of a promoterless egfp gene in the same integrative plasmid used to evaluate the Pptr mutants. The resulting plasmid was introduced into M. tuberculosis to obtain TB419, whose fluorescence was compared with that of the M. tuberculosis strain expressing egfp from Pptr11, Pptr12, or Pptr14. Results in Fig. 6 confirmed that the strength of the dprE1 promoter was comparable to that of Pptr12.
Figure 5.
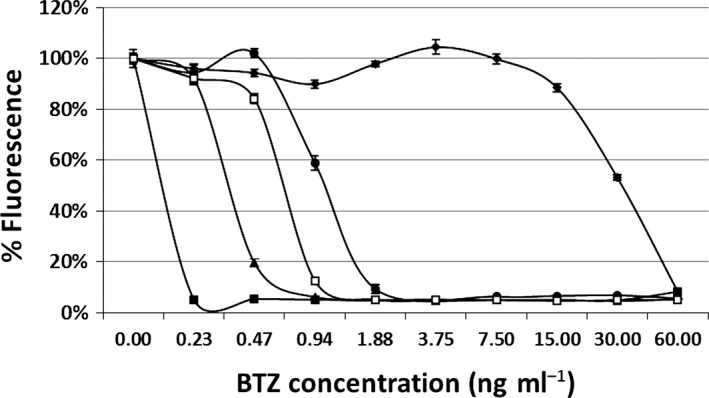
Resazurin microplate assay (REMA) to determine the BTZ sensitivity of Mycobacterium tuberculosis conditional mutants expressing dprE1 from different promoters. Filled squares, TB386 (Pptr11); empty squares, H37Rv; triangles, TB387 (Pptr12); circles, TB388 (Pptr14); diamonds, TB110 (Pptr).
Table 3.
BTZ MIC85 obtained in the experiment shown in Fig. 5 compared with promoter strength
| Strain | Promoter strength (%) | MIC85 (ng ml−1) |
|---|---|---|
| TB386 | 4 | 0.23 |
| TB387 | 25 | 0.94 |
| TB388 | 89 | 1.88 |
| TB110 | 100 | 60 |
Figure 6.
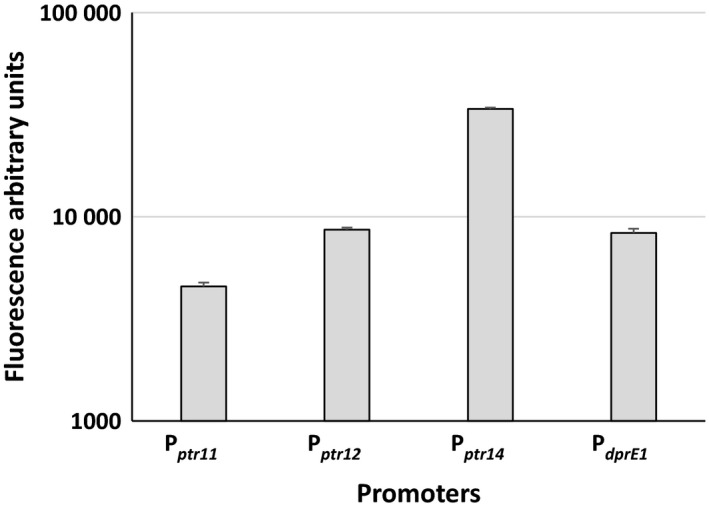
Fluorescence of Mycobacterium tuberculosis strains harbouring integrative plasmids encoding the egfp gene downstream of the different Pptr promoter mutants and of the dprE1 promoter.
Finally, the M. tuberculosis conditional mutants expressing dprE1 from wt Pptr (TB110) or from Pptr11 (TB386) were used to infect two groups of mice, one of which was treated with doxycycline. As visible in Fig. 7, at day 15 post‐infection, mice infected with TB110 showed a higher bacterial burden than those infected with TB386 suggesting that expression of dprE1 from Pptr11 results in suboptimal intracellular DprE1 concentration for in vivo growth. Moreover, 15 days after infection treatment with doxycycline resulted in a 10‐fold decrease in the bacterial burden of TB110 and in TB386 levels decreasing below the limit of detection, thus demonstrating the essentiality of dprE1 during infection.
Figure 7.
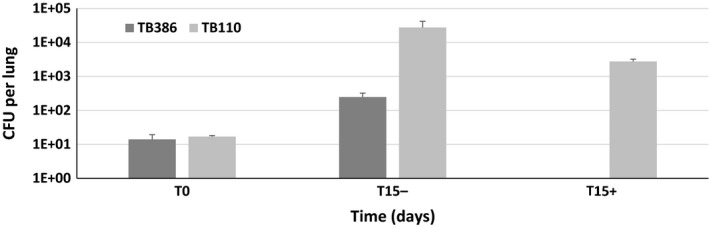
Bacterial burden of TB110 and TB386 in mouse lungs. One group of mice was treated with doxycycline (+) from the beginning of the experiment. Mice were sacrificed at time 0 and 15 days after infection.
Development of a whole‐cell screening assay to identify inhibitors of DprE1
The development of whole‐cell screenings based on bacterial biosensors sensitized to inhibitors of specific targets represents a novel strategy to identify new antibacterial drugs (Hughes and Karlen, 2014; Schnappinger, 2015). As DprE1 has been shown to be a valid and vulnerable target for M. tuberculosis (Makarov et al., 2009), we decided to use our set of promoters to develop an assay to identify molecules able to inhibit its activity. To this end, we constructed a set of dprE1 conditional mutants in which dprE1 was controlled by Pptr11, Pptr12, or Pptr14 under the control of the Pip‐ON inducible system (Forti et al., 2009). These plasmids were then introduced into the conditional dprE1 mutant TB110 by plasmid switching (Pashley and Parish, 2003) obtaining the strains TB434, TB435 and TB436 respectively (Table 1). The resulting strains were pristinamycin‐dependent, as their only functional copy of dprE1 was under transcriptional control of one of the Pptr promoters, which are not active in the absence of pristinamycin due to Pip‐mediated repression (Fig. S1).
Finally, using REMA, we determined the MIC to BTZ of these three strains grown in the presence of different concentrations of pristinamycin. As shown in Table 4 and Fig. 8, the MIC was proportional to the strength of the promoter transcribing dprE1 and to the concentration of pristinamycin with a range going from 0.12 ng ml−1 (TB434, 25 ng ml−1 pristinamycin) to 15 ng ml−1 (TB436, 100 ng ml−1 pristinamycin), while the MIC of H37Rv was 1.88 ng ml−1.
Table 4.
MIC (ng ml−1) to BTZ of H37Rv and different Mycobacterium tuberculosis dprE1 conditional mutants upon the addition of different amounts of pristinamycin
| Strain | Pristinamycin concentration (ng ml−1) | ||||
|---|---|---|---|---|---|
| 0 | 25 | 50 | 75 | 100 | |
| H37Rv | 1.88 | ||||
| TB434 | 0.12 | 0.12 | 0.23 | 0.23 | |
| TB435 | 0.23 | 0.47 | 0.47 | 0.47 | |
| TB436 | 3.75 | 7.5 | 7.5 | 15 | |
Figure 8.
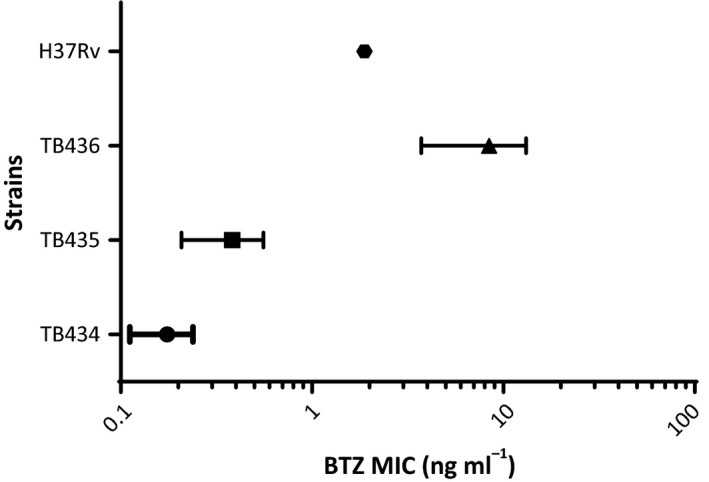
MIC range of different Mycobacterium tuberculosis conditional mutants in the presence of different pristinamycin concentrations.
Discussion
The main aim of this work was to mutagenize the repressible promoter Pptr to increase the tunable range of the TetR/Pip‐OFF repressible system (Boldrin et al., 2010). The pip promoter encloses three Pip operators each containing two directly repeated 4 bp palindromes separated by a 3 bp spacer. Two of these operators overlap the putative ‐10 and ‐35 consensus sequences, while the third is located 10 nucleotides after the ptr transcriptional start site (Fig. 1; Blanc et al., 1995). As our aim was to change Pptr strength without interfering with its regulation by the pristinamycin‐dependent transcriptional repressor Pip, mutations were introduced either before the operators or within the spacers (Fig. 1). Two insertions increased the distance between the putative ‐10 and ‐35 sequences (mutations 1 and 2), and most of the changes in the putative ‐35 sequence (mutations 3–8) resulted in very strong attenuation of transcription. However, mutations in the putative ‐10 sequence showed a wide variation of transcriptional activity: some of them, similarly to mutations in the ‐35 sequence, caused a dramatic decrease in activity (mutations 10, 11, 15 and 16), others (mutations 12 and 14) resulted in decreased, but still appreciable level of activity, or in an increased level of activity (mutations 9 and 13). It should be noted; however, that only mutations of the first and last base of the ‐35 hexamer were designed and tested, given the overlap of this sequence with the first Pip operator (Fig. 1). Mycobacterial promoters usually have well‐conserved ‐10 regions, while ‐35 are less conserved and hardly recognizable, probably because of the high number of sigma factors operating in these bacteria (Bashyam et al., 1996; Newton‐Foot and Gey van Pittius, 2013; Manganelli, 2014). Moreover, several mycobacterial promoters present extended ‐10 regions (containing a TGn sequence at the 5′‐end) which increase their potency, and it was proposed that these promoters do not require the presence of a ‐35 sequence (Kenney and Churchward, 1996). It is not clear if the Pptr promoter has an extended ‐10, as the borders of its ‐10 consensus sequence are not easily recognizable, but a TG sequence is present just before its locus (Fig. 1).
Three mutant promoters with variable strengths were selected, and the possibility for Pip to repress their activity was confirmed. To demonstrate that these promoters could be used to improve the performances of the TetR/Pip‐OFF repressible promoter system, we constructed three conditional dprE1 mutants using these promoters and compared them to the previously characterized dprE1 mutant obtained with wt Pptr. DprE1 is an essential decaprenylphosphoryl D‐ribose epimerase, an enzyme involved in arabinogalactan and lipoarabinomannan biogenesis (Kolly et al., 2014a) and is the target of BTZ, one of the most potent and best characterized candidates in the TB drug development pipeline (Makarov et al., 2009; Brecik et al., 2015). The dprE1 conditional mutant was functional to show the essentiality of this gene, but due to the higher strength of the Pptr promoter in comparison with its physiological promoter, several passages in the presence of ATc were needed to titrate DprE1 down and stop cells growing when cultivated without aeration (Kolly et al., 2014a) and data not shown). Indeed, when this mutant was cultivated in parallel with the new mutants in these conditions, it was clear that the weakest promoters required fewer passages in the presence of ATc to stop bacterial growth. Moreover, this agreed well with the MIC to BTZ of the various mutants which increased with promoter potency (and thus with DprE1 intracellular concentration; Table 3). The MIC of the conditional mutant expressing dprE1 from Pptr12 was equal to that of the wt strain, suggesting a similar potency of these two promoters, which was confirmed when the two promoters were compared (Fig. 6). However, careful inspection of the data obtained by REMA shows that H37Rv is slightly more resistant to BTZ than the conditional mutant (Fig. 5); this may be explained either by the existence of a metabolic burden in the mutant strain due to the presence of the integrated plasmid and the TetR/Pip‐OFF system which might increase its sensitivity, or more interestingly by the possibility that in the presence of a drug inhibiting DprE1, the dprE1 promoter might be induced thereby increasing DprE1 intracellular concentration.
When the original mutant and the mutant in which dprE1 was transcribed from the weakest of the selected promoters (Pptr11) were used to infect mice, we proved that the effect of dprE1 repression was more dramatic in the strain expressing dprE1 at the lower level (more than two log reduction in the number of cfu in 2 weaks vs one log reduction for the other strain). Interestingly, this strain grew more slowly than the other one in the lungs even in the absence of doxycycline treatment. As the two strains grow similarly in vitro (not shown), this suggests that bacteria require more DprE1 expression during infection than during growth in axenic culture, probably for the faster protein turnover in this environment generated by exposure to stress. It is worth noting that as the strength of Pptr11 is lower than that of PdprE1 (Fig. 6), this strain probably contains a suboptimal concentration of DprE1 which might well explain its attenuation even in the absence of doxycyline treatment.
Finally, we wanted to investigate the possibility to use our promoters to develop bacterial biosensors sensitized to inhibitors of specific targets to optimize target‐based whole‐cell screenings. As proof of principle, we used the dprE1 gene. As whole‐cell screenings with M. tuberculosis usually need long incubation times, we reasoned that ATc, which in our experience has a short half‐life, was not the molecule of choice to develop such a system. We therefore used the Pip‐ON inducible system, based on the more stable pristinamycin (Forti et al., 2009). In this system, the pristinamycin‐sensitive transcriptional repressor Pip is constitutively expressed thereby inhibiting transcription at Pptr, but in the presence of pristinamycin, it releases its operators in a dose‐dependent manner and allows transcription. Measurement of MIC to BTZ of the three Pip‐ON‐based conditional mutants in the presence of two different concentrations of pristinamycin clearly showed that each strain was more vulnerable to the drug when the expression of dprE1 was lower due to a lower concentration of pristinamycin. However, the differences due to pristinamycin‐dependent dprE1 expression modulation were smaller than those due to the strength of the different promoters. It is worth noting that the strain with the lower MIC (TB434 grown in the presence of 25 ng ml−1 of pristinamycin) was 16‐fold more sensitive than H37Rv, while the strain with the highest MIC (TB436 grown in the presence of 100 ng ml−1 of pristinamycin) was 8‐fold more resistant than H37Rv (Fig. 8). Consequently, the 128‐fold MIC range, from the most resistant to the most sensitive strain, provides a very sensitive window to screen for active compounds.
In conclusion, the construction and characterization of different Pptr promoter mutants widely expanded the potential of Pip‐based tunable promoter systems, thus increasing the range of genes whose expression can be effectively manipulated for the construction of bacterial biosensors.
Experimental procedures
Strain and culture conditions
The following bacterial strains were used in this study: Escherichia coli Top10 (Invitrogen, Carlsbad, CA, USA), DH5α and HB101 (laboratory stock), M. smegmatis mc2155 (laboratory stock), and M. tuberculosis H37Rv (laboratory stock). E. coli strains were grown at 37°C in Luria‐Bertani (LB) broth or on LB agar plates. Mycobacterial strains were grown at 37°C in Middlebrook 7H9 broth (Difco, Franklin Lakes, NJ, USA) in 150 ml roller bottles with slow rotation (3 rpm), in 10 ml screw‐cap tubes without agitation, or 7H10 agar plates (Difco), supplemented with 0.2% glycerol and 0.05% Tween‐80. For growth of M. tuberculosis, the medium was supplemented with 10% albumin‐dextrose‐sodium chloride complex (ADN; Jacobs et al., 1991). When needed, antibiotics were added to the media at the following concentrations: streptomycin (Sm) 20 μg ml−1, kanamycin (Km) 50 μg ml−1 (E. coli) or 20 μg ml−1 (M. smegmatis and M. tuberculosis), hygromycin (Hyg) 150 μg ml−1 (E. coli) or 50 μg ml−1 (M. smegmatis and M. tuberculosis). Anhydrotetracycline (ATc; Sigma, St. Louis, MO, USA) or pristinamycin (Molcan Corporation, Richmond Hill, ON, Canada) were added when required at the concentrations indicated in the text. Benzothiazinone 043 (BTZ) was dissolved in DMSO at a concentration of 9 μg ml−1. Preparation of electrocompetent cells, electroporation and preparation of mycobacterial genomic DNA were performed as previously described (Maciag et al., 2007).
Plasmid construction and PCR‐mutagenesis
The sequence encoding eGFP was PCR amplified from pMV4‐36 (Cascioferro et al., 2010) using primers RP1335/RP1336 (Table S2) and subcloned in the suicide vector pFRA50 downstream of the repressible promoter Pptr (Boldrin et al., 2010). The Pptr‐egfp fragment was than subcloned in TOPO (Invitrogen). This plasmid was used as a template for PCR‐mutagenesis using upper primers overlapping the Pptr sequence and containing the different point mutations and a lower primer overlapping the final part of egfp (Fig. S2 and Table S2). The amplified mutagenized Pptr‐egfp fragments were cloned in TOPO and, after confirmation of the presence of the correct mutation by sequencing, subcloned in both pFRA61 (integrative plasmid harbouring the TetR/Pip‐OFF system; Kolly et al., 2014a) and in pMV261 (replicative plasmid) (Cooksey et al., 1993). The resulting integrative plasmids were introduced into M. smegmatis mc2155 and M. tuberculosis H37Rv while replicative plasmids were electroporated into M. smegmatis MS212 and M. tuberculosis TB262 (mc2155‐ and H37Rv‐derivatives harbouring the TetR/Pip‐OFF system). Vectors and bacterial strains are listed in Table S1.
Construction of a dprE1‐reporter plasmid
A fragment of 518 bp immediately upstream of dprE1 containing its putative promoter was amplified with primers RP1629/RP1630 using Phusion® High‐Fidelity DNA Polymerase and cloned upstream of a promoterless egfp generating pFRA194. The PdprE1‐egfp fragment was excised from this plasmid and subcloned into the integrative plasmid pFRA61 (Kolly et al., 2014a) obtaining pFRA206. This plasmid was introduced in M. tuberculosis H37Rv to obtain TB419.
Construction of M. tuberculosis dprE1 conditional mutants
The gene encoding DprE1 was amplified using primers RP806/RP807 (Table S2) using Phusion® High‐Fidelity DNA Polymerase (New Englands Biolabs, Ipswich, MA, USA) and cloned into pCR‐Blunt II‐TOPO (Invitrogen). The amplified fragment was than excised and subcloned (i) into three integrative plasmids containing the TetR/Pip‐OFF system and one of the three mutated Pptr (mutations 11, 12, and 14) to obtain pFRA183, pFRA186, and pFRA188 respectively and (ii) into three integrative plasmids containing the Pip‐ON system and one of the three mutated Pptr (mutations 11, 12, and 14) to obtain pFRA212, pFRA213, pFRA214 respectively. These six plasmids were introduced into the dprE1 conditional mutant TB110 (Kolly et al., 2014a) by plasmid switching (Pashley and Parish, 2003) generating TB386, TB387, TB388 (TetR/Pip‐OFF) and TB434, TB435, TB436 (Pip‐ON) respectively (Table 1).
Fluorescence assay
eGFP fluorescence was measured in Nunclon 96‐well Flat Bottom Black Polystyrol FluoroNunc microplates (Thermo Scientific, Carlsbad, CA, USA) to minimize background fluorescence. Bacterial strains were inoculated in 5 ml of 7H9/ADC, supplemented with appropriate antibiotics, and grown to OD540 = 0.5. Cells were harvested by centrifugation (5 min, 10 000 rpm), and pellets were resuspended in a volume of 0.1 ml of Middlebrook 7H9 to obtain a concentration of about 109 cfu ml−1. Then, 108 cfu were added in the microplates, and fluorescence values were measured with Infinite 200Pro microplate reader (Tecan Group, Mannendorf, Switzerland), using excitation and emission wavelengths of 485 and 535 nm respectively. Wells containing only medium or bacterial cultures of M. smegmatis and M. tuberculosis strains not expressing GFP were used as controls to determine bacterial autofluorescence that was used to subtract the background from the measurements. All the experiment were repeated at least twice in triplicate. Data were reported as mean ± standard deviation.
Benzothiazinone susceptibility test
Bacterial strains were grown in 3 ml of Middlebrook 7H9 supplemented with the appropriate antibiotics to OD540 0.2‐0.4. Cultures were diluted to a theoretical OD540 of 0.0005 and 200 μl of the resulting suspension were spotted into each well of the first column of a Nunclon 96‐well Flat Bottom Black Polystyrol FluoroNunc microplates (Thermo Scientific). Starting from the second column, 100 μl of bacteria were added to the other wells of the plate. BTZ (stock concentration = 9 μg ml−1) was added to a final concentration of 60 ng ml−1 in each well of the first column and then serial dilutions (1:1) were made. Plates were incubated 7 days at 37°C, and then 10 μl of AlamarBlue (Invitrogen) was added to each well and incubated at 37°C. The detection was performed after 24 h of incubation using an Infinite 200Pro microplate reader (Tecan Group), with excitation and emission wavelengths of 535 and 590 nm respectively. The lowest drug concentration that resulted in growth inhibition of at least 85% was considered as the MIC. The experiments were repeated twice in triplicate. Data were reported as mean ± standard deviation.
In vivo experiments
Female C57BL/6 mice (Charles River Laboratories, Wilmington, MA, USA) were infected by low‐dose aerosol with the dprE1 conditional knock‐down strains. Mice were fed with doxycycline‐containing food (2000 ppm, Harlan) starting from 5 days before infection. Control groups were fed with regular diet, equal in composition to the doxycycline‐containing food except for the antibiotic. Four mice per group and time point were sacrificed at day 0 and 2 weeks post‐infection. Lung homogenates were plated on 7H10 plates supplemented with ampicillin (100 μg ml−1) and cycloheximide (10 μg ml−1). The experiment was performed once. Data were calculated as mean ± standard deviation. Experimental procedures involving animals were approved by the Swiss Cantonal and Federal Authorities (Authorization number 2658).
Conflict of interest
None of the authors have any potential conflict of interest to declare.
Supporting information
Fig. S1. Pristinamycin‐dependent growth of Pip‐ON based dprE1 conditional mutants.
Fig. S2. Strategy to construct promoter mutants.
Table S1. Mutagenized promoters and corresponding strains.
Table S2. Primers used in this study.
Acknowledgements
This work was supported by the European Community's Seventh Framework Programme under grant agreement 260872.
Microbial Biotechnology (2018) 11(1), 238–247
Funding Information
This work was supported by the European Community's Seventh Framework Programme under grant agreement 260872.
References
- Ahmed, W. , Menon, S. , Godbole, A.A. , Karthik, P.V. , and Nagaraja, V. (2014) Conditional silencing of topoisomerase I gene of Mycobacterium tuberculosis validates its essentiality for cell survival. FEMS Microbiol Lett 353: 116–123. [DOI] [PubMed] [Google Scholar]
- Ahmed, W. , Menon, S. , Karthik, P.V. , and Nagaraja, V. (2016) Autoregulation of topoisomerase I expression by supercoiling sensitive transcription. Nucleic Acids Res 44: 1541–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashyam, M.D. , Kaushal, D. , Dasgupta, S.K. , and Tyagi, A.K. (1996) A study of mycobacterial transcriptional apparatus: identification of novel features in promoter elements. J Bacteriol 178: 4847–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazet Lyonnet, B. , Diacovich, L. , Cabruja, M. , Bardou, F. , Quemard, A. , Gago, G. , and Gramajo, H. (2014) Pleiotropic effect of AccD5 and AccE5 depletion in acyl‐coenzyme A carboxylase activity and in lipid biosynthesis in mycobacteria. PLoS ONE 9: e99853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick, T. , Ghosh, S. , Dixit, K. , Ganesan, V. , Ramagopal, U.A. , Dey, D. , et al (2014) Targeting Mycobacterium tuberculosis nucleoid‐associated protein HU with structure‐based inhibitors. Nat Commun 5: 4124. [DOI] [PubMed] [Google Scholar]
- Blanc, V. , Salah‐Bey, K. , Folcher, M. , and Thompson, C.J. (1995) Molecular characterization and transcriptional analysis of a multidrug resistance gene cloned from the pristinamycin‐producing organism, Streptomyces pristinaespiralis . Mol Microbiol 17: 989–999. [DOI] [PubMed] [Google Scholar]
- Boldrin, F. , Casonato, S. , Dainese, E. , Sala, C. , Dhar, N. , Palu, G. , et al (2010) Development of a repressible mycobacterial promoter system based on two transcriptional repressors. Nucleic Acids Res 38: e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldrin, F. , Ventura, M. , Degiacomi, G. , Ravishankar, S. , Sala, C. , Svetlikova, Z. , et al (2014) The phosphatidyl‐myo‐inositol mannosyltransferase PimA is essential for Mycobacterium tuberculosis growth in vitro and in vivo. J Bacteriol 196: 3441–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecik, M. , Centarova, I. , Mukherjee, R. , Kolly, G.S. , Huszar, S. , Bobovska, A. , et al (2015) DprE1 Is a vulnerable tuberculosis drug target due to its cell wall localization. ACS Chem Biol 10: 1631–1636. [DOI] [PubMed] [Google Scholar]
- Carroll, P. , Muttucumaru, D.G. , and Parish, T. (2005) Use of a tetracycline‐inducible system for conditional expression in Mycobacterium tuberculosis and Mycobacterium smegmatis . Appl Environ Microbiol 71: 3077–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascioferro, A. , Boldrin, F. , Serafini, A. , Provvedi, R. , Palu, G. , and Manganelli, R. (2010) Xer site‐specific recombination, an efficient tool to introduce unmarked deletions into mycobacteria. Appl Environ Microbiol 76: 5312–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary, E. , Thakur, P. , Pareek, M. , and Agarwal, N. (2015) Gene silencing by CRISPR interference in mycobacteria. Nat Commun 6: 6267. [DOI] [PubMed] [Google Scholar]
- Choudhary, E. , Lunge, A. , and Agarwal, N. (2016) Strategies of genome editing in mycobacteria: achievements and challenges. Tuberculosis 98: 132–138. [DOI] [PubMed] [Google Scholar]
- Cooksey, R.C. , Crawford, J.T. , Jacobs, W.R. Jr , and Shinnick, T.M. (1993) A rapid method for screening antimicrobial agents for activities against a strain of Mycobacterium tuberculosis expressing firefly luciferase. Antimicrob Agents Chemother 37: 1348–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes, M. , Singh, A.K. , Reyrat, J.M. , Gaillard, J.L. , Nassif, X. , and Herrmann, J.L. (2011) Conditional gene expression in Mycobacterium abscessus . PLoS ONE 6: e29306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degiacomi, G. , Benjak, A. , Madacki, J. , Boldrin, F. , Provvedi, R. , Palu, G. , et al (2017) Essentiality of mmpL3 and impact of its silencing on Mycobacterium tuberculosis gene expression. Sci Rep 7: 43495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Luca, M. , Bottai, D. , Batoni, G. , Orgeur, M. , Aulicino, A. , Counoupas, C. , et al (2012) The ESX‐5 associated eccB‐EccC locus is essential for Mycobacterium tuberculosis viability. PLoS ONE 7: e52059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt, S. , Guo, X.V. , Hickey, C.M. , Ryou, M. , Monteleone, M. , Riley, L.W. , and Schnappinger, D. (2005) Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res 33: e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forti, F. , Crosta, A. , and Ghisotti, D. (2009) Pristinamycin‐inducible gene regulation in mycobacteria. J Biotechnol 140: 270–277. [DOI] [PubMed] [Google Scholar]
- Gola, S. , Munder, T. , Casonato, S. , Manganelli, R. , and Vicente, M. (2015) The essential role of SepF in mycobacterial division. Mol Microbiol 97: 560–576. [DOI] [PubMed] [Google Scholar]
- Gupta, S. , Banerjee, S.K. , Chatterjee, A. , Sharma, A.K. , Kundu, M. , and Basu, J. (2015) Essential protein SepF of mycobacteria interacts with FtsZ and MurG to regulate cell growth and division. Microbiology 161: 1627–1638. [DOI] [PubMed] [Google Scholar]
- Hu, Y. , Wang, Z. , Feng, L. , Chen, Z. , Mao, C. , Zhu, Y. , and Chen, S. (2016) SigE ‐dependent activation of RbpA controls transcription of the furA‐katG operon in response to oxidative stress in mycobacteria. Mol Microbiol 102: 107–120. [DOI] [PubMed] [Google Scholar]
- Hughes, D. , and Karlen, A. (2014) Discovery and preclinical development of new antibiotics. Ups J Med Sci 119: 162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs, W.R. Jr , Kalpana, G.V. , Cirillo, J.D. , Pascopella, L. , Snapper, S.B. , Udani, R.A. , et al (1991) Genetic systems for mycobacteria. Methods Enzymol 204: 537–555. [DOI] [PubMed] [Google Scholar]
- Kenney, T.J. , and Churchward, G. (1996) Genetic analysis of the Mycobacterium smegmatis rpsL promoter. J Bacteriol 178: 3564–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J.H. , Wei, J.R. , Wallach, J.B. , Robbins, R.S. , Rubin, E.J. , and Schnappinger, D. (2011) Protein inactivation in mycobacteria by controlled proteolysis and its application to deplete the beta subunit of RNA polymerase. Nucleic Acids Res 39: 2210–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J.H. , O'Brien, K.M. , Sharma, R. , Boshoff, H.I. , Rehren, G. , Chakraborty, S. , et al (2013) A genetic strategy to identify targets for the development of drugs that prevent bacterial persistence. Proc Natl Acad Sci USA 110: 19095–19100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolly, G.S. , Boldrin, F. , Sala, C. , Dhar, N. , Hartkoorn, R.C. , Ventura, M. , et al (2014a) Assessing the essentiality of the decaprenyl‐phospho‐d‐arabinofuranose pathway in Mycobacterium tuberculosis using conditional mutants. Mol Microbiol 92: 194–211. [DOI] [PubMed] [Google Scholar]
- Kolly, G.S. , Sala, C. , Vocat, A. , and Cole, S.T. (2014b) Assessing essentiality of transketolase in Mycobacterium tuberculosis using an inducible protein degradation system. FEMS Microbiol Lett 358: 30–35. [DOI] [PubMed] [Google Scholar]
- Maciag, A. , Dainese, E. , Rodriguez, G.M. , Milano, A. , Provvedi, R. , Pasca, M.R. , et al (2007) Global Analysis of the Mycobacterium tuberculosis Zur (FurB) Regulon. J Bacteriol 189: 730–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov, V. , Manina, G. , Mikusova, K. , Mollmann, U. , Ryabova, O. , Saint‐Joanis, B. , et al (2009) Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 324: 801–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov, V. , Lechartier, B. , Zhang, M. , Neres, J. , van der Sar, A.M. , Raadsen, S.A. , et al (2014) Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol Med 6: 372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganelli, R. (2014) Sigma factors: key molecules in Mycobacterium tuberculosis physiology and virulence. Microbiol Spectr 2: MGM2‐0007‐2013. doi: 10.1128/microbiolspec.MGM2‐0007‐2013 [DOI] [PubMed] [Google Scholar]
- Mondino, S. , Gago, G. , and Gramajo, H. (2013) Transcriptional regulation of fatty acid biosynthesis in mycobacteria. Mol Microbiol 89: 372–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori, G. , Chiarelli, L.R. , Esposito, M. , Makarov, V. , Bellinzoni, M. , Hartkoorn, R.C. , et al (2015) Thiophenecarboxamide derivatives activated by EthA kill Mycobacterium tuberculosis by inhibiting the CTP synthetase PyrG. Chem Biol 22: 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton‐Foot, M. , and Gey van Pittius, N.C. (2013) The complex architecture of mycobacterial promoters. Tuberculosis 93: 60–74. [DOI] [PubMed] [Google Scholar]
- Pandey, R. , and Rodriguez, G.M. (2014) IdeR is required for iron homeostasis and virulence in Mycobacterium tuberculosis . Mol Microbiol 91: 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashley, C.A. , and Parish, T. (2003) Efficient switching of mycobacteriophage L5‐based integrating plasmids in Mycobacterium tuberculosis . FEMS Microbiol Lett 229: 211–215. [DOI] [PubMed] [Google Scholar]
- Piton, J. , Foo, C.S. , and Cole, S.T. (2017) Structural studies of Mycobacterium tuberculosis DprE1 interacting with its inhibitors. Drug Discov Today 22: 526–533. [DOI] [PubMed] [Google Scholar]
- Schnappinger, D. (2015) Genetic approaches to facilitate antibacterial drug development. Cold Spring Harb Perspect Med 5: a021139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnappinger, D. , and Ehrt, S. (2014) Regulated expression systems for mycobacteria and their applications. Microbiol Spectr 2: MGM2‐0018‐2013. doi: 10.1128/microbiolspec.MGM2‐0018‐2013 [DOI] [PubMed] [Google Scholar]
- Seeliger, J.C. , Topp, S. , Sogi, K.M. , Previti, M.L. , Gallivan, J.P. , and Bertozzi, C.R. (2012) A riboswitch‐based inducible gene expression system for mycobacteria. PLoS ONE 7: e29266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini, A. , Boldrin, F. , Palu, G. , and Manganelli, R. (2009) Characterization of a Mycobacterium tuberculosis ESX‐3 conditional mutant: essentiality and rescue by iron and zinc. J Bacteriol 191: 6340–6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini, A. , Pisu, D. , Palu, G. , Rodriguez, G.M. , and Manganelli, R. (2013) The ESX‐3 secretion system is necessary for iron and zinc homeostasis in Mycobacterium tuberculosis . PLoS ONE 8: e78351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, A.K. , Carette, X. , Potluri, L.P. , Sharp, J.D. , Xu, R. , Prisic, S. , and Husson, R.N. (2016) Investigating essential gene function in Mycobacterium tuberculosis using an efficient CRISPR interference system. Nucleic Acids Res 44: e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura, M. , Rieck, B. , Boldrin, F. , Degiacomi, G. , Bellinzoni, M. , Barilone, N. , et al (2013) GarA is an essential regulator of metabolism in Mycobacterium tuberculosis . Mol Microbiol 90: 356–366. [DOI] [PubMed] [Google Scholar]
- Verma, A.K. , and Chatterji, D. (2014) Dual role of MsRbpA: transcription activation and rescue of transcription from the inhibitory effect of rifampicin. Microbiology 160: 2018–2029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Pristinamycin‐dependent growth of Pip‐ON based dprE1 conditional mutants.
Fig. S2. Strategy to construct promoter mutants.
Table S1. Mutagenized promoters and corresponding strains.
Table S2. Primers used in this study.