

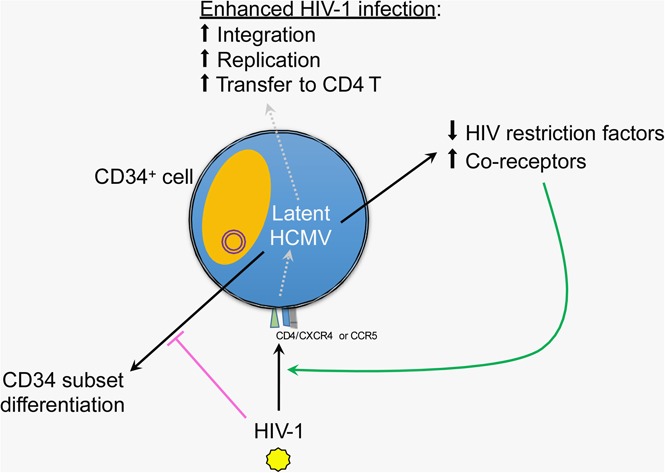
Key Points
HCMV latency modulates host CD34+ cells in favoring HIV-1 infection.
Latent HCMV upregulates HIV entry coreceptors and downregulates HIV restriction factors in CD34+ cells.
Abstract
Individuals who have been preinfected by human cytomegalovirus (HCMV) are more prone to AIDS disease progression after subsequent HIV-1 infection but the underlying mechanism remains elusive. HCMV is a ubiquitous DNA virus that commonly establishes lifelong latent infection in CD34+ progenitor cells, where latency-specific HCMV genes may modulate host restriction to HIV-1 infection. To test this hypothesis, we studied progenitor cells that are known to resist replicative HIV-1 infection because of the intrinsic expression of host restriction factors. Interestingly, in primary CD34+ cells undergoing latent HCMV infection, an enhanced level of HIV-1 proviral DNA and replication was observed as measured by digital polymerase chain reaction, quantitative polymerase chain reaction, and Gag expression, and confirmed using dual-reporter pseudovirus encoding X4- or R5-tropic envelope and T-cell transfer. This phenomenon may be partially explained by the upregulation of HIV-1 entry coreceptors, including chemokine receptors CXCR4 and CCR5, but not of the primary receptor CD4. Furthermore, latent HCMV infection downregulated the expression of HIV-1 restriction factors SAMHD1, APOBEC3G, tetherin, and Mx2 in CD34+ progenitor cells, which may confer to enhanced HIV-1 infection. However, this enhancement was abrogated when ultraviolet-inactivated HCMV was used for comparison, suggesting that expression of latent HCMV genes is essential for this effect. Importantly, HCMV gB and HIV-1 p24 can be detected in the same cell by immunofluorescence and flow cytometry; therefore, the establishment of HCMV latency in CD34+ cells likely leads to host cell gene modulation that favors HIV-1 infection.
Visual Abstract
Introduction
Human cytomegalovirus (HCMV) is a ubiquitous DNA virus that is prevalent in 50% to 100% of human populations. It establishes a natural lifelong latent infection in CD34+ hematopoietic progenitor cells, where it remains asymptomatic in the immunocompetent host. Latent HCMV infection is defined by (1) the absence of productive or lytic infection gene expression, (2) no new infectious virus being produced, (3) latency-associated gene expression, and (4) being capable of reactivation to revert to productive infection. During latency, HCMV is quiescent, with limited gene expression of a unique subset of ∼30 viral genes,1,2 in contrast to ∼200 genes being expressed in a cascade during productive infection. These latency-associated genes include UL111.5A, LUNA, and UL138 and can regulate host cell responses, such as downregulating major histocompatibility complex (MHC) class II molecules, while inducing host interleukin-10 (IL-10), chemokine (C-C motif) ligand 8 (CCL8), and the multidrug resistance-associated protein-1 (MRP1).3-8 These HCMV latent genes are important for facilitating establishment/maintenance of latency in which host cell mechanisms are modulated. However, only few latent genes have known functions.
Recent studies showed that infection of CD34+ progenitor cells by HIV-1 may serve as a latent reservoir.9,10 Latent HIV-1 exists in the proviral state in multiple hematopoietic progenitor cell subsets that is regulated by NF-κB activation.10 As with other myeloid-lineage cells, however, CD34+ progenitor cells exhibit restriction to HIV-1 infection11; thus, it may be difficult for HIV-1 to undergo successful replication or establish latent infection in these cells. In fact, the study by Carter et al estimated that 0.4% of CD34+ cells harbor latent HIV-1 in patient’s bone marrow.9 Myeloid-lineage cells resist HIV-1 replication due to the constitutive high expression of restriction factors, including APOBEC3G, SAMHD1, tetherin, and Mx2. These factors can act on different stages of HIV-1 replication such as viral RNA synthesis (APOBEC3G),12-18 reverse transcription (SAMHD1),19-22 viral release (tetherin),23-26 and integration (Mx2).27 However, HIV-1 and HIV-2 encode different proteins (Vif, Vpx, and Vpu) to counteract these restriction factors, but whether they are regulated in CD34+ cells has not been studied before.
The interplay between HCMV and HIV-1 in CD34+ cells has not been investigated to date. Previous studies showed that HIV-1–infected individuals who were HCMV-seropositive progressed faster to AIDS from an average of 19 months compared with 49.5 months for HCMV-seronegative individuals.28-30 Although the mechanism remains elusive, this finding suggests that HCMV infection could modulate the host in favor for HIV/AIDS. In a human cervical tissue explant model, HCMV and HIV-1 appears to coinfect macrophages, although the outcome remains unknown.31,32 Thus, we hypothesized that latent HCMV infection could modulate CD34+ progenitor cells and result in enhanced infection by HIV-1. To test this hypothesis, we first established a primary CD34+ cell culture model by expanding peripheral blood (PB)-derived CD34+ cells. HCMV can successfully establish latent infection in these cells with no viral release, but is capable of reactivation when triggered by coculture with permissive fibroblasts. Importantly, CD34+ cells latently infected with HCMV had significantly decreased expression of HIV-1 restriction factors SAMHD1, APOBEC3G, tetherin, and Mx2, and upregulated HIV-1 coreceptors. As a result, HIV-1 infection of CD34+ cells with latent HCMV had increased replication as detected by real-time quantitative polymerase chain reaction (qPCR), digital PCR, and HIV Gag p24 protein levels with both X4- and R5-tropic viruses and dual-reporter pseudoviruses. On the contrary, HIV-1 infection did not seem to induce HCMV reactivation in these cells. Taken together, we propose that HCMV latency can influence HIV-1 pathogenesis in CD34+ progenitor cells.
Materials and methods
Viruses
HCMV strains Towne and Merlin were purchased from ATCC and propagated as described previously2 in MRC5 human lung fibroblast cell line (ATCC). HIV-1 live strains NL4-3 (X4-tropic) and JR-FL (R5-tropic) were propagated in PHA/IL-2 activated healthy human PB mononuclear cells (PBMCs) as described previously.1 Viral concentrations for HCMV and live HIV-1 were determined by plaque assay on MRC5 cells and p24 enzyme-linked immunosorbent assay, respectively. HCMV inactivation was performed by placing 1 mL of virus inoculum in a 6-well plate and exposed to 20 minutes of ultraviolet (UV) light (30 W, 20-cm distance) in a sterile tissue culture cabinet.
PB-CD34 cell culture model and viral infection
CD34+ cells were isolated using the Diamond CD34 Isolation kit (Miltenyi Biotec) from PBMCs generated by Ficoll-Paque PREMIUM (GE Healthcare) density centrifugation of healthy donor buffy coats (Hong Kong Red Cross). Ethics approval was received from Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (#UW13-476). Freshly isolated CD34+ cells were cultured in StemPro-34 serum-free medium (GIBCO) supplemented with 25 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF), 100 ng/mL stem cell factor, and 50 ng/mL IL-3 in 96-well culture plates. Every 3 to 4 days, 50% media change occurred and cell counts were performed over time. Cells expanded up to 30 days of culture (named PB-CD34 cells) were used for viral infections. HCMV infection occurred at multiplicity of infection = 3 for 3 hours before cells were rigorously washed 3 times with phosphate-buffered saline (PBS) as described previously.2 Live HIV-1 infection or pseudovirus infection at 50 and 20 ng p24, respectively, occurred at 2 × 106 cells/mL in a 24-well plate for 6 hours before cells were washed 3 times with PBS. After washing, cells were cultured in StemPro-34 media without cytokines. Plaque assay was performed using MRC5 cell monolayer at 70% to 80% confluence, where culture supernatants, infected PB-CD34 cells, or viral inoculum were added for 3 hours before washed off and replaced with fresh media and monitored for cytopathic effect (CPE) for <20 days. The HIV-1 transfer experiment was performed by coculture of mock or infected PB-CD34 cells at day 5 postinfection (PI) (washed 3 times with excess PBS) at a ratio of 1:5 to autologous naïve CD4+ T cells, which was negatively isolated by naïve CD4 T-cell microbeads (Miltenyi Biotec) in RPMI supplemented with 10% fetal bovine serum without stimulation. Transwell was used to prevent cell–cell interaction in the same setup. Immunostaining for CD34, CD4, CD25, and intracellular p24 expression was performed on the cocultured cells at 48 hours PI and analyzed by flow cytometry. Reactivation of infected PB-CD34 cells was performed by adding GM-CSF (100 ng/mL) and tumor necrosis factor-α (TNF-α; 2.5 ng/mL) to the culture cells for 14 hours,10 before being assessed by immunofluorescence staining using anti-gB (Novus Biologicals) and anti-p24 (DAKO) primary antibodies and anti-mouse immunoglobulin G2a (IgG2a) Alexa Fluor 488 and anti-mouse IgG1 Alexa Fluor 568 (Invitrogen), respectively. Images were acquired using Carl Zeiss LSM 700 confocal microscope using the 40× oil lens and ZEN software and analyzed by ImageJ software (http://imagej.nih.gov/ij).
Real-time qPCR analysis
Total RNA was isolated from cells using RNAiso Plus according to the manufacturer’s instructions (Takara). Reverse transcription of total RNA was performed using the Primescript Reverse Transcriptase kit with oligo-dT primers used for gene expression analysis and random hexamers used for HIV long terminal repeat (LTR) according to the manufacturer’s instructions (Takara). DNA was extracted from cells using the DNeasy kits (QIAGEN). RNA and DNA were eluted using Ultrapure DNase/RNase-Free Distilled Water (Invitrogen) and concentrations determined using Nanodrop 8000 (Thermo Scientific). For complementary DNA (cDNA) analysis by real-time PCR on the ViiA7 instrument (Life Technologies) using the SYBR Premix Ex Taq II reagent (Takara), the following primer pairs were used: SAMHD1, 5′- GGATTACTAAAAACCAGGTTTCACAACT-3′, 5′-GCTCTGCAAATTTCTCTGGCAG-3′; APOBEC3G, 5′- GGTCAGAGGACGGCATGAGA-3′, 5′- GCAGGACCCAGGTGTCATTG-3′; tetherin, 5′-CTGCAACCACACTGTGATG-3′, 5′-ACGCGTCCTGAAGCTTATG-3′; Mx2, 5′-CAGCCACCACCAGGAAACA-3′, 5′-TTCTGCTCGTACTGGCTGTACAG-3′; LTR, 5′-GCCTCAATAAAGCTTGCCTTGA-3′, 5′-TCCACACTgACTAAAAgggTCTgA-3′; UL138, 5′-GGTTCATCGTCTTCGTCGTC-3′, 5′-CACGGGTTTCAACAGATCG-3′; UL111.5A, 5′-CCCGACACGCGGAAAA-3′, 5′-TTCATCGAGTAAAACCTACGTTGGT-3′; LUNA, 5′-GAGCCTTGACCACTTGGTAC-3′, 5′-GGAAAACACGCGGGGGA-3′, interferon-α4 (IFN-α4), 5′-GAAGAGACTCCCCTGATGAATGT-3′, 5′-GCACAGGTATACACCAAGCTTCTTC-3′; IFN-β, 5′-AGCTGAAGCAGTTCCAGAAG-3′, 5′-AGTCTCATTCCAGCCAGTGC-3′; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-ACAGTCCATGCCATCACTGCC-3′, 5′-GCCTGCTTCACCACCTTCTTG-3′; UL54, 5′-GCACCGAGACGCGCACCGAA-3′, 5′-CAGCCTCTACCCTTCCATCA-3′; UL55, 5′-GTAGCTGGCATTGCGATTGGT-3′, 5′-TCCAACACCCACAGTACCCGT-3′; UL86, 5′-CACGGTCCCGGTTTAGCA-3′, 5′-CGTAACGTGGACCTGACGTTT-3′; UL99, 5′-TTCACAACGTCCACCCACC-3′, 5′-GTGTCCCATTCCCGACTCG-3′; US3, 5′-ACCGTGGATATGGTGGACAT-3′, 5′-AACAGCAGACCCCAATTGTC-3′. Relative expression was calculated by normalization to GAPDH and by ΔΔCT method. For HIV-1 DNA analysis, real-time PCR using TaqMan Universal PCR Master Mix (Life Technologies) with specific probes and primers was performed for genes HIV-1 p17 and CCR5 as reported.33 Cell numbers were calculated by determining CCR5 copies using a CCR5-encoding plasmid standard, then divided by 2 (2 DNA copies of CCR5 per cell). Late reverse transcription (RT) and 2-LTR circle copies determination was performed by TaqMan qPCR as described.34
Digital PCR
DNA copy numbers for HCMV were performed for UL55 and UL123exon4 genes according to a previous study.35 The instrument QuantStudio 3D Digital PCR system (Life Technologies) was used. Briefly, PCR reactions with templates were partitioned into chips with 20 000 wells and positive signals were assessed by a scanner. Analysis was performed using the QuantStudio 3D AnalysisSuite Cloud Software (Life Technologies) to calculate the number of copies from known cell numbers based on CCR5 copy number determination.
RT-PCR
Total RNA was extracted from mock or HCMV-infected PB-CD34 cells at day 5 PI; 0.5 μg of total RNA was reverse transcribed using oligo-dT primers and Primescript Reverse Transcriptase kit in a 20 μL reaction (Takara Inc). A total of 2 μL of cDNA was used as template for PCR thermal cycling in a 50-μL reaction with CD4, US28, and GAPDH primers as described elsewhere.36,37
Flow cytometry
The following antibodies were used for immunostaining for phenotypic analysis using the FACSCalibur or Aria III instruments (BD Biosciences): anti-CD34- phycoerythrin (PE; Miltenyi Biotec); anti-CD38-PE (Biolegend), anti-CD45RA-PE/Cy7 (Biolegend), anti-CD123-PerCP/Cy5.5 (Biolegend), anti-CD4-PerCP/Cy5.5 (eBioscience), anti-HLA-DR-PE/Cy7 (eBioscience), anti-HLA-DR-FITC (Biolegend), anti-HLA-A,B,C-Pacific Blue (Biolegend), anti-CD80-APC (Biolegend), anti-CD86-PerCP/Cy5.5 (Biolegend), anti-CD11c-fluorescein isothiocyanate (FITC; Biolegend), anti-p24-FITC (Beckman Coulter), anti-CXCR4-PE/Cy7 (Biolegend), anti-CCR5-PE (BD Biosciences), and anti-gB (Novus Biologicals) plus anti-mouse IgG2a Alexa Fluor 647 (Invitrogen). Analysis was performed using FlowJo software (Tree Star). To assess gB and p24 intercellular staining in PB-CD34 cells, cells were fixed by fixation/permeabilization buffer (BD Biosciences), and stained with anti-gB and anti-p24 FITC antibodies overnight at 4°C before secondary antibody anti-mouse IgG2a Alexa Fluor 647 was used for staining for 1 hour at 4°C, before being analyzed.
Western blotting
PB-CD34 cells were lysed for total protein with denaturing lysis buffer consisting of 10 mM Tris-HCl (pH 7.5), 200 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol, 0.5% NP-40, 1 mM phenylmethylsulfonyl fluoride, 10 μg/mL aprotinin, 10 μg/mL leupeptin, and 1.25 μg/mL pepstatin A. Cells were lysed on ice for 30 minutes before being centrifuged for 13 000g at 4°C. Supernatant collected was assessed for total protein concentration using the BCA kit (Pierce). Equal amounts of protein were loaded onto sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, electrophoresed and transferred to polyvinylidene fluoride membrane (Millipore). Blocking occurred with PBS/0.05% Tween-20 with 5% blotting grade skim milk (Bio-Rad) and 0.5% bovine serum albumin. Primary antibodies against SAMHD1 (Novus Biological), APOBEC3G (Sigma-Aldrich), Mx2 (Novus Biologicals), tetherin (Thermo Scientific), and β-actin (Cell Signaling Technologies) were used for immunostaining followed by horseradish peroxidase–conjugated antibodies (Amersham). Protein signals were assessed by WesternBright ECL horseradish peroxidase substrate (Advansta) and signals captured by Amersham Hyperfilm ECL (GE Healthcare) after exposure.
Dual-reporter RGH pseudovirus
To construct pseudoviruses, red-green-HIV (RGH) plasmid,38 and plasmids encoding envelope proteins from X4-tropic HxB2 (ie, X4-RGH) and R5-tropic JR-FL (ie, R5-RGH) were used to cotransfect 293T cells cultured in Dulbecco’s modified Eagle medium supplemented with 20% fetal bovine serum (GIBCO). After 2 days, supernatants were collected and centrifuged at 4000g for 10 minutes before ultracentrifuged at 20 000g for 1.5 hours at 4°C. Pellets were resuspended before another ultracentrifugation through underlaid 20% sucrose gradient at 20 000g for 1.5 hours. Virus stock p24 concentration was determined using the RETRO-TEK HIV-1 p24 Antigen enzyme-linked immunosorbent assay kit (ZeptoMetrix). Testing of pseudovirus was performed on Jurkat cells and fluorescence signals were acquired using the Carl Zeiss LSM 700 confocal microscope and images analyzed by ImageJ.
Statistics
All statistical analyses were performed using a paired 2-tailed Student t test to calculate P values. P < .05 was considered statistically significant. Data are presented as mean ± standard error of the mean (SEM) of at least 4 independent experiments unless indicated in the figure legends.
Results
Peripheral CD34+ cells can be expanded in vitro to enrich progenitor cells
To determine if PB CD34+ progenitor can support HCMV latency, we first isolated CD34+ cells from healthy human PBMCs for characterization. Typically, 0.5 ± 0.14 × 106 CD34+ cells could be isolated from 2 × 108 PBMCs with a purity >70% (supplemental Figure 1A-B). To further determine CD34+ subsets in PB, we adapted the hematopoiesis model by Dick’s group to examine cell surface markers by flow cytometry.39 Heterogeneous CD34+ progenitor subpopulations are found in PBMCs, where a majority is common myeloid progenitors (26.9%; CD34+CD38+CD45RA−CD123+), followed by hematopoietic stem cells (HSCs 20.5%; CD34+CD38−CD45RA−) and granulocyte-monocyte progenitors (GMPs 13.9%; CD34+CD38+CD45RA+CD123+), with lower frequency of multilymphoid progenitors (28.7%; CD34+CD38−CD45RA+) and megakaryocyte-erythrocyte progenitors (6.6%; CD34+CD38+CD45RA−CD123−) (supplemental Figure 1B). Moreover, in vitro culture of CD34+ cells using cytokine-conditioned media (GM-CSF, stem cell factor, and IL-3) can improve cell numbers, colony formation, purity, and proportion of HSCs (supplemental Figure 1A,C).40 After 30 days of culture, the purity of CD34+ cells increased to 93.5% and cell numbers expanded ∼sixfold (supplemental Figure 1A), with the phenotype of the cells reverted to mainly HSCs (average 66.4% of CD34+ cells, n = 6), whereas other subsets diminished over time (Figure 1A; supplemental Figure 1B).
Figure 1.

PB-CD34 cell culture model for the establishment of HCMV latent infection. (A) CD34+ cells isolated from healthy PBMCs were cultured for up to 30 days and the phenotype of cell subsets were assessed by flow cytometry over time (n = 7). (B) Digital PCR analysis of HCMV UL55 and UL123ex4 viral genes for DNA copy number from known cell inputs determined by CCR5 qPCR from mock or HCMV strain Towne- or Merlin-infected PB-CD34 cells in a time course experiment (n ≥ 3). (C) Real-time qPCR analysis (log10) of latency-associated transcripts UL111.5A, LUNA, and UL138 at day 5 PI from mock- or HCMV-infected PB-CD34 cells (n = 6) or productive infection genes UL54, UL55, UL86, UL99, and US3 (n = 4) (D). MRC5 infection served as positive control (n = 3). Data show mean ± SEM. *P < .05, **P < .01, ***P < .001. (E) Plaque assay of mock or HCMV-infected PB-CD34 cells, or supernatant from infected cultures, was applied to fibroblast monolayers and observed for CPE over time. Representative bright field microscope images are shown. Original magnification ×40, except rightmost panels (expanded insets) ×100. CMP, common myeloid progenitor; MEP, megakaryocyte-erythrocyte progenitor; MLP, multilymphoid progenitor.
Establishment of HCMV latent infection in PB-derived CD34+ cells
To determine if latent HCMV infection can be established in PB-CD34 cells, infection at multiplicity of infection = 3 using HCMV strain Towne was performed as previously described.1 By 5 days PI, total DNA was harvested to quantify the number of viral DNA copies found in these cells using digital PCR with 2 sets of HCMV-specific primers for UL55 and UL123exon4.37 To accurately determine the number of cells input, genomic DNA copy was quantified using CCR5 gene (because 2 copies exist in 1 cell).35 Combining the HCMV gene and CCR5 data, in a time course experiment, ∼15 copies of HCMV DNA could be detected per infected PB-CD34 cell at days 1, 3, 5, 7, and 14 PI (Figure 1B; supplemental Figure 2). This level of infection is consistent to other in vitro latent HCMV CD34+ cell infection models.1,41 Note that HCMV DNA was not detected in mock infection. To determine the state of HCMV infection in PB-CD34 cells, we examined if HCMV latency-associated genes are expressed, including UL111.5A, LUNA, and UL138.3,5,6 qPCR was performed on PB-CD34 cells infected with mock, HCMV strain Towne, or the clinical isolate Merlin for 5 days (Figure 1C). Consistent with previous findings, we found that both HCMV strains express UL111.5A and LUNA significantly in PB-CD34 cells. Because UL138 gene is encoded by clinical isolates, only Merlin infection resulted in its expression (Figure 1C). In contrast, none of the lytic genes (UL54, UL55, UL86, UL99, US3) showed significant expression compared with productively infected MRC5 cells (Figure 1D). To verify if any HCMV productive infection occurred in these cells, PB-CD34 culture supernatants collected at day 5 PI were added to permissive human fibroblasts. However, no plaque formation was observed after >10 days (Figure 1E). On the other hand, infected PB-CD34 cells cocultured with fibroblast monolayers resulted in plaque formation by day 7, indicative of reactivation (Figure 1E, arrows). In summary, HCMV strains Towne and Merlin can establish latency with similar characteristics in PB-CD34 cells.
HCMV latent infection could modulate human fetal liver-derived CD34+ cell immune molecules4,42; therefore, we examined cell surface expression of HLA-DR, MHC class I, CD80, CD83, and CD86 by flow cytometry following mock- or Merlin-infected PB-CD34 cells. Consistent to previous reports,4,43 HLA-DR expression was downmodulated to the level of uninfected cells (supplemental Figure 3), whereas MHC class I molecules and the costimulatory molecules CD80 and CD83 (but not CD86) were significantly upregulated (supplemental Figure 3). Thus, HCMV can establish latent infection in PB-CD34 cells and appears to modulate immune molecules.42,44
HCMV latently infected PB-CD34 cells are more susceptible to HIV-1 infection
Because AIDS progresses faster in HCMV-seropositive individuals, we next examined if latently infected PB-CD34 cells can favor HIV-1 infection. HCMV or mock infection of PB-CD34 cells at day 5 PI were infected by HIV-1NL4-3 (X4-tropic; X4-HIV). Three days after HIV-1 infection, DNA was extracted for digital PCR (for HIV-1 p17) and qPCR (for CCR5) analysis. As shown in Figure 2A, HIV-1 DNA was not detected in mock- and HCMV-infected cells, but direct HIV-1 infection resulted in approximately 50 copies being detected in 500 cells (ie, 10% infection). In contrast, HIV-1 infection of PB-CD34 cells harboring latent HCMV resulted in ∼threefold increase in proviral DNA copies (ie, 26% infection; Figure 2A). In addition, there was a higher level of intracellular p24 staining among CD34+ cells in HIV-1 infection with latent HCMV compared with mock infection (Figure 2B). Similarly, a higher level of LTR expression was observed with HIV-1 infection of HCMV latently infected PB-CD34 cells than infection of mock CD34+ cells that increased over time in a time course experiment (Figure 2C). To determine if there is a discrepancy between X4- or R5-tropic HIV-1, we next infected mock or latent HCMV PB-CD34 cells at day 5 PI with HIV-1NL4-3 and HIV-1JR-FL (R5-tropic; R5-HIV) for 2 days to assess integrated DNA and found increased copy numbers in the latter (Figure 2D).
Figure 2.

HCMV latent infection of PB-CD4 cells results in increased HIV-1 infection. PB-CD34 cells mock- or HCMV strain Towne-infected at day 5 PI were infected by HIV-1NL4-3 for 3 days. (A) Proviral DNA copy number determined by digital PCR with known input cell numbers calculated from CCR5 qPCR is shown (n = 12). (B) Representative flow cytometry plots and column graph for intracellular staining for CD34+p24 on PB-CD34 cells with or without HCMV latent infection at day 5 PI followed by HIV-1 infection for 48 hours with X4-tropic HIV-1NL4-3 (n = 4). Numbers represent percentages on plots. (C) LTR activity from cDNA determined by qPCR in a time course experiment (n = 4). PB-CD34 cells infected with mock or HCMV strain Merlin day 5 PI were infected with HIV-1NL4-3 or R5-tropic HIV-1JR-FL for 12 hours and qPCR was performed (n = 4) to assess (D) proviral DNA, (E) late RT products, (F) 2-LTR circles, and (G) HCMV DNA copies. Data represent mean ± SEM. *P < .05.
Success of HIV-1 infection and integration is accompanied by a low level of late RT products and 2-LTR circles, which are otherwise characteristic of abortive infection.36,45 Using qPCR, analysis of late RT products found a significant decrease with X4-HIV infection but not R5-HIV, despite the presence of HCMV (Figure 2E). On the other hand, 2-LTR circles appeared to be higher for R5-HIV compared with X4-HIV infection of PB-CD34 cells and were not affected by latent HCMV (Figure 2F). Together, these results indicate that both X4-HIV and R5-HIV could successfully infect PB-CD34 cells that and latent HCMV could support an increased level of HIV-1 infection. Finally, to determine if HIV-1 infection could reactivate latent HCMV, we examined supernatants from HCMV/HIV-1–infected PB-CD34 cells up to 20 days of culture and no CPE was observed in the plaque assay (data not shown). In addition, HCMV DNA copy number following X4- or R5-HIV infection had no significant increase, verifying that HCMV remains latent in PB-CD34 cells despite HIV-1 (Figure 2G).
To further illustrate that PB-CD34 cells can support HIV-1 infection, we made use of the dual-reporter construct RGH.34 RGH differentiates between silent latent infection and active infection based on Gag-enhanced green fluorescence protein (eGFP; green) and Nef-mCherry (red) fluorescence: early eGFP indicates active infection, mCherry indicates latent infection, and dual positive of both signals in 1 cell depicts de novo Gag expression and integrated latent virus (Figure 3A). Using this tool, pseudoviruses with X4-tropic HxB2 (X4-RGH) and R5-tropic JR-FL (R5-RGH) envelopes were generated. The infectious ability of the RGH pseudovirus (inoculum 20 ng p24) was first tested in Jurkat cells. As shown in Figure 3B, eGFP and mCherry signals can be detected by day 2 PI using X4-RGH, and some cells have dual signals. Similar results were obtained for R5-RGH (data not shown). Next, mock or HCMV-infected PB-CD34 cells at day 5 PI were infected with X4- or R5-RGH pseudoviruses. After 2 days, PB-CD34 cells infected with RGH viruses had mainly red signals in cells without HCMV, suggesting that latent infections were established by the pseudovirus, albeit a low level of subsequent replication. Consistently, latent HCMV in these cells resulted in significantly higher active infection (yellow) for both X4- and R5-RGH, whereas some cells still remained latent (Figure 3B-C). However, for R5-RGH, a slight but significant increase in active infection was observed and most cells still retained HIV-1 latency (Figure 3B-C). Together, these results further demonstrate that HCMV latent infection in CD34+ cells led to an increased permissiveness for HIV-1 infection.
Figure 3.

HIV-1 integration and infection of PB-CD34 cells using a dual-reporter pseudovirus. Pseudoviruses constructed with RGH backbone and envelope from HxB2 (X4-tropic) and JR-FL (R5-tropic). (A) Schematic representation for active infection indicated by eGFP fluorescence (green), integrated viral DNA (latency) by mCherry fluorescence (red), and active replication from integrated genome (yellow; arrows in panels B-C). Confocal microscopy was used to examine eGFP and mCherry fluorescence signals 2 days after infection. Representative images of cells showing single or dual mCherry/eGFP signals in Jurkat cells (B) or mock or HCMV-infected PB-CD34 cells (C) at day 2 after RGH/HxB2 pseudovirus infection, with cell count data shown from 4 independent PB-CD34 experiments. Columns represent data as mean ± SEM. Average number of cells with integrated genome is shown below. Original magnification ×400 for panels B-C. *P < .05, **P < .01.
HCMV/HIV-1 coinfection and differentiation of PB-CD34 cells
Because these data show that latent HCMV exists in a majority of PB-CD34 cells and that ∼10% can harbor HIV-1 proviral DNA, we next sought to demonstrate that coinfection of the same cell exists. By immunofluorescence and confocal microscopy, HCMV gB and HIV-1 p24 proteins can be detected in the same cell following HCMV/HIV-1 infection of PB-CD34 cells and reactivation by GM-CSF/TNF-α (Figure 4A). Furthermore, intracellular flow cytometry staining revealed that approximately 15.6 ± 4.4% of PB-CD34 cells are coinfected by the 2 viruses and that HIV-1 preferably infected those cells with preexisting latent HCMV (Figure 4B; supplemental Figure 4). In addition, HCMV appears to influence the differentiation state of the cells. Compared with mock, the GMP subset was higher in latent HCMV-infected PB-CD34 cells (supplemental Figure 5). Furthermore, HCMV/HIV-1 coinfected cultures retained a high proportion of HSC similar to mock or HIV-1 alone, whereas HCMV cultures had lower HSC and higher GMP subsets (supplemental Figure 6). Also, by lineage staining over the course of experiment, HCMV latent infection alone does indeed induce differentiation; there was an increase in lin+ cells that was otherwise reverted after HIV-1 infection (supplemental Figure 7).
Figure 4.

HCMV/HIV-1 coinfected PB-CD34+ cells and enhanced transfer of HIV-1 to autologous CD4+ T cells by latent HCMV. PB-CD34 cells were infected with mock or HCMV strain Merlin for 5 days before HIV-1NL4-3 infection for 3 days. (A) GM-CSF/TNF-α reactivation treatment for 14 hours to detect HIV-1 Gag p24 (red) and HCMV gB (green) by immunofluorescence and counterstained for nucleus (Hoechst, blue). Representative images with bright field and a scale bar of 10 μm are shown. (B) Flow cytometry detection of p24 signals analyzed on gB+- (shaded) and gB−-gated (dotted) for HCMV-infected samples. Representative histograms of 2 independent experiments are shown. (C) After 7 days post-HIV infection of mock or HCMV PB-CD34 cells, coculture with autologous naïve CD4+ T cells (1:5) was performed for 48 hours before being immunostained for flow cytometry. Representative plots with arrows indicating gating for analyzing intracellular p24 signals in CD4+ CD25+, or CD25− subpopulations in mock or HCMV- and/or HIV-1–infected PB-CD34 cells are shown. (C) Column graph for data from 4 independent experiments as a column graph showing mean ± SEM. Numbers represent percentages. *P < .05, **P < .01.
Latent HCMV enhances HIV-1 transmission between infected PB-CD34 cells and autologous CD4+ T cells
To determine if HIV-1–infected PB-CD34 cells can transmit virus to permissive CD4+ T cells, infected PB-CD34 cells were cocultured with negatively isolated autologous naïve CD4+ T cells in a 1:5 ratio as previously described.4 Forty-eight hours after coculture, cells were analyzed for intracellular p24 signals on CD34−CD4+ cells comparing between CD25+ and CD25− subpopulations (Figure 4C). As shown in Figure 4C, p24 signals were only found in those CD4+ T cells that express the activation marker CD25. Compared with mock, HCMV-infected or HCMV/HIV-1–infected PB-CD34 cell cocultures, HIV-1–infected PB-CD34 cells induced higher naïve CD4+ T-cell activation indicated by upregulation of CD25 expression (Figure 4C). However, HCMV/HIV-1 coculture resulted in an significantly increased level of p24 among the CD4+CD25+ T cells compared with HIV-1 (Figure 4C). Transwell showed that cell–cell interaction is required for viral transfer for coinfected cells to CD4+ T cells (Figure 4C).
Latent HCMV upregulates HIV coreceptors expressed on PB-CD34 cells
Previous studies have indicated that CD34+ progenitor cells express both CXCR4 and CCR59,46,47; hence, we next examined if latent HCMV infection of PB-CD34 cells alters HIV receptors expression, which may allow increased infection by HIV-1. By flow cytometry, there was an increase in expression of both CXCR4 and CCR5 on the cell surface in 2 independent experiments at day 5 PI when compared with HCMV-infected and mock PB-CD34 cells (Figure 5A). No significant surface expression of CD4 was found by flow cytometry (Figure 5B); however, RT-PCR using 2 sets of primers detected CD4 gene expression in PB-CD34 cells that was not affected by latent HCMV, nor was the internal control GAPDH altered (Figure 5C).38 Interestingly, the virally encoded chemokine receptor US28 gene was found to be expressed in HCMV-infected PB-CD34 cells, which is consistent with previous findings using other latent CD34+ models and further validates that HCMV established latency.1,2,48 US28 was reported to be a possible cofactor for HIV-1 entry.49 Blocking of CD4 or coreceptors, however, did not completely abolish HIV-1 infection on PB-CD34 cells with or without latent HCMV (supplemental Figure 8), which probably suggests that entry receptor alone is not solely responsible for HIV-1 infection of these cells.
Figure 5.

Analysis of HIV receptors expressed by PB-CD34 cells and modulated by latent HCMV. Mock or HCMV strain Merlin was used for infection of PB-CD34 cells from 2 independent donors. On day 5 PI, cells were immunostained with isotype or specific antibodies (Abs) for (A) CXCR4 and CCR5, with subgating to illustrate coexpression with frequencies depicted for 2 donors (right) analyzed by flow cytometry. (B) CD4 expression on PB-CD34 cells were analyzed by flow cytometry based on isotype controls. (C) Agarose gel electrophoresis of RT-PCR products performed for CD4, HCMV US28, and GAPDH. Arrows point to the expected product size. Ladder, 100-bp DNA ladder; Neg, no template control.
HIV-1 restriction factors are downregulated by HCMV latent infection in PB-CD34 cells
Myeloid-lineage cells are less permissive to HIV-1 infection and replication because of their high constitutive expression of HIV-1 restriction factors, including SAMHD1, Mx2, tetherin, APOBEC3G, and APOBEC3A.50 Interestingly, the precursors of monocytes or macrophages, which are CD34+ myeloid progenitor cells, restrict HIV-1 replication and could in turn induce the virus into latency.9,10 It remains unclear whether HCMV latent infection would affect the expression of HIV-1 restriction factors in CD34+ cells. Therefore, to examine this, SAMHD1, APOBEC3G, Mx2, and tetherin gene expression was determined by qPCR in a time course experiment in PB-CD34 cells. Compared with mock-infected cells, HCMV infection led to the significant decrease of these restriction factors from day 1 PI and remained low through day 14 PI (Figure 6A). The downregulation of these proteins was confirmed by western blotting, in which the proteins were reduced upon HCMV latent infection in PB-CD34 cells at day 5 PI (Figure 6B).
Figure 6.

HCMV latent infection downregulates HIV-1 restriction factors in PB-CD34 cells. (A) qPCR analysis of SAMHD1, APOBEC3G, Mx2, and tetherin gene expression following mock and HCMV strain Towne infection of PB-CD34 cells in a time course experiment (n = 8). GAPDH was used for normalization. (B) Western blotting analysis of protein expression of SAMHD1, APOBEC3G, Mx2, tetherin, and β-actin between mock and HCMV strain Towne-infected PB-CD34 cells at day 5 PI. (C) PB-CD34 cells were infected with mock, Merlin, or UV-inactivated Merlin for 5 days PI before being analyzed for expression of restriction factors and β-actin by qPCR normalized to GAPDH (n = 4). (D) Supernatants from mock- or HCMV-infected PB-CD34 cells at day 5 PI were added to other independent PB-CD34 cells (n = 4) and expression of genes measured by qPCR. Column graphs show data as mean ± SEM. *P < .05, **P < .01, ***P < .001.
Because a unique subset of viral genes facilitates HCMV latency,1,2 we next investigated if the downregulation of HIV-1 restriction factors is dependent on de novo HCMV protein expression. HCMV strain Merlin was inactivated by UV exposure before infecting PB-CD34 cells for 5 days (Figure 6C)1 with live virus and mock infection conducted in parallel. UV-inactivated Merlin did not downregulate HIV-1 restriction factors, suggesting that de novo synthesis of HCMV latent gene products is likely responsible for this phenomenon. As a control, β-actin was not affected by live or UV-inactivated virus infections. Further, to illustrate if soluble factors can lead to the downregulation of HIV-1 restriction factors, supernatants from mock or HCMV-infected PB-CD34 cultures were applied to fresh PB-CD34 cells for 5 days, but no difference was found (Figure 6D), suggesting that direct infection may be required for such effect.
Discussion
HIV/AIDS remains a devastating human disease without effective cure and poses a challenge for eradication despite the success of antiretroviral therapy. One of the largest hurdles to cure HIV-1 is the clearance of latent reservoirs that exist in different cell types and in different tissue compartments. Apart from memory CD4+ T cells,51 several studies have proposed that CD34 hematopoietic progenitor cells could be another HIV-1 latent reservoir.9,10,52 Although still controversial, the presence of HIV-1 restriction factors and innate immune response could affect the efficiency of HIV-1 infection into these cells. Naturally, it becomes complicated when the ubiquitous HCMV establishes latency in CD34+ progenitor cells.1,2,4,41,53 Although HCMV/HIV-1 coinfected individuals had faster progression toward AIDS,28,29 in this study, we delineated the interplay of these viruses in an in vitro model of CD34+ progenitor cells.
Our model used a cytokine-conditioned long-term culture to enhance the early HSC subset and total cell numbers (Figure 1A; supplemental Figure 1). Comparatively, other models with CD34+ cells derived from human bone marrow, cord blood, or fetal liver often have limited cell numbers with heterogeneous subsets. Also, these sources are more difficult to obtain compared with PB. Using this PB-CD34 model, the hallmark characteristics of HCMV latency for 2 HCMV strains (Towne and Merlin) were shown. The copy number of HCMV genomes and infection rate is similar to those found in HCMV latent infection of fetal liver-derived CD34+ cells1 (Figure 1B). The lack of increase in DNA copy number over time and the absence of virus in the supernatant of infected PB-CD34 cells indicate that no productive infection can be detected (Figure 1B,E). Critically, the detection of latency-associated transcripts UL111.5A, LUNA, UL138, and US28 (Figures 1C and 5C); absence of apparent productive genes (Figure 1D); and viral reactivation by coculture with fibroblasts (Figure 1E), shows that HCMV can successfully establish latency in the PB-CD34 cell culture model. The versatility of this model may extend to the study of clinical blood samples from HIV-1 and different diseases.
Establishment of HCMV latency is facilitated by a unique subset of viral genes as others and we have identified previously.1,2 Latent HCMV genes can downregulate MHC-II molecules; regulate TNFR1 and MRP1, silencing of the MIEP viral gene region; and maintain the viral genome.54 Our study identified additional functions encoded by HCMV during latency in CD34+ cells. Given that HIV-1 infects myeloid cells with difficulties because of intrinsic restriction factors including SAMHD1, APOBEC3A, APOBEC3G, and Mx2,18,20,27 HCMV could likely affect these genes to favor HIV-1 infection in CD34+ cells. Our data show that latent HCMV can downregulate HIV restriction factors (Figure 6) and upregulate CXCR4 and CCR5 (Figure 5A,C).47,55 Whether other receptors including DC-SIGN and mannose receptor that were demonstrated for HIV-1 entry into astrocytes, dendritic cells, and monocytes,56-58 play a role in CD34+ cells or are affected by HCMV remains elusive. Recent reports demonstrated that type I IFNs could induce HIV-1 restriction factors in myeloid-lineage cells such as Mx2 and APOBEC3G.27,59-61 HCMV has also been shown to suppress type I IFN responses during productive infection,51 but whether it occurs during latent infection in PB-CD34 cells remains to be investigated. If so, this would also affect interferon-induced innate sensors IFI16, cGAS, and RIG-I as well as restriction factors that would potentially affect HIV-1 infection and replication.59-62 Therefore, latent HCMV can influence HIV-1 infection at multiple levels in CD34+ cells. Following viral entry, our data suggest that HIV-1 can successfully integrate into the CD34+ cell genome. As a consequence, infected CD34+ cells may facilitate cell–cell transfer to CD4+ T cells under certain conditions in vivo; therefore, the role of latent HCMV appears to enhance HIV-1 infection as well as reactivation. One recent study has shown that latent HCMV increases the expression of histone deacetylases and thereby repress lytic replication during latency.63 As with reactivating HIV-1, treatment with histone deacetylase inhibitor leads to a transient induction of HCMV lytic genes, suggesting that similar mechanisms exist for both viruses to remain latent and that cells with latent HCMV have an increased latency potential advantage for HIV-1.
HIV-1 infection is influenced by the differentiation state of the target cell. CD4+ T cells are infected more efficiently following activation; infection of other cell types such as CD14-derived osteoclasts are also affected by their differentiation status.64 However, myeloid cells (such as dendritic cells and monocytes) that HIV-1 infects poorly because of viral restriction factors, are often associated with low activation state and T-cell–stimulating potential.65 Interestingly, our analysis of the differentiation state of CD34+ cells indicates that HIV-1 infection can maintain the cell in an early progenitor state. Although HCMV latently infected PB-CD34 cells are more inclined to differentiate to GMP over time, HIV-1 infection can somehow revert this process (supplemental Figure 6) within a margin consistent with the frequency of HCMV/HIV-1 coinfected cells detected in our experiments (∼16%). Therefore, HIV-1 may be advantageous for HCMV latency and further show their cooperation in the infection of CD34+ cells.
In the natural setting, it is estimated that a very small number of myeloid progenitor cells harbor latent HCMV (0.004 to 0.01%) of total bone marrow or mobilized mononuclear cells66 and is more prominent in bone marrow CD34+ cells, where the frequency ranges from 0.02% to 0.1%.67 Thus, it may be difficult to identify coinfected cells in natural specimens, especially in normal PB. It is postulated that nondetectable HCMV reactivation and replication can lead to new infections in other cell types and in turn repopulate the latent viral genome in CD34+ cells. Therefore, the immune modulation of CD34+ cells by HCMV latency would clearly offer a relatively “safe” environment for HIV-1 survival, forming a latent reservoir, and assist in HIV-1/AIDS progression. One characteristic of chronic HIV-1 infection is persistent immune activation.68 When this occurs, the elevated level of cytokines may reactivate HCMV from CD34+ cells and lead to infections in other cell types or allow HIV-1 to persist or replicate within these cells. Another advantage for HIV-1 in infection of CD34+ HSCs is that these cells can pump out drugs very efficiently,69 so that antiretroviral therapies may not be effective against HIV-1 latently infected HSCs. Others have found that plasma viral sequences in HIV-1 patients appear to be distinct from those in the CD4+ T cells,70 which points to a possibility that HIV-1 replication in CD34+ cell compartment may be influenced by latent HCMV and increase the diversity of the resulting viral progeny.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Johnny He for the pRGH plasmid, Xilin Wu and Yufei Mo for technical assistance, and the US NIH AIDS Reagent Program for the HIV envelope plasmids.
This study was supported by research grants from the Hong Kong Food and Health Bureau, Health and Medical Research Fund (grants 12111162 and 13120612) (A.K.L.C.), the Hong Kong Research Grant Council (grant HKU5/CRF/13G) (Z.C.), the University Development Fund of the University of Hong Kong, the Li Ka Shing Faculty of Medicine Matching Fund to the HKU AIDS Institute, and the San-Ming Project of Medicine in Shenzhen.
Authorship
Contribution: A.K.L.C. and Z.C. designed the research and wrote the paper. A.K.L.C., Y.H., H.Y.K., and M.C. performed experiments. A.K.L.C., Y.H., and Z.C. analyzed data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Zhiwei Chen, AIDS Institute and Department of Microbiology, L5 40-45, Laboratory Block, Li Ka Shing Faculty of Medicine, 21 Sassoon Rd, Pokfulam, Hong Kong; e-mail: zchenai@hku.hk.
References
- 1.Cheung AKL, Abendroth A, Cunningham AL, Slobedman B. Viral gene expression during the establishment of human cytomegalovirus latent infection in myeloid progenitor cells. Blood. 2006;108(12):3691-3699. [DOI] [PubMed] [Google Scholar]
- 2.Goodrum FD, Jordan CT, High K, Shenk T. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc Natl Acad Sci USA. 2002;99(25):16255-16260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jenkins C, Garcia W, Abendroth A, Slobedman B. Expression of a human cytomegalovirus latency-associated homolog of interleukin-10 during the productive phase of infection. Virology. 2008;370(2):285-294. [DOI] [PubMed] [Google Scholar]
- 4.Cheung AKL, Gottlieb DJ, Plachter B, et al. The role of the human cytomegalovirus UL111A gene in down-regulating CD4+ T-cell recognition of latently infected cells: implications for virus elimination during latency. Blood. 2009;114(19):4128-4137. [DOI] [PubMed] [Google Scholar]
- 5.Bego M, Maciejewski J, Khaiboullina S, Pari G, St Jeor S. Characterization of an antisense transcript spanning the UL81-82 locus of human cytomegalovirus. J Virol. 2005;79(17):11022-11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodrum F, Reeves M, Sinclair J, High K, Shenk T. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood. 2007;110(3):937-945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weekes MP, Tan SY, Poole E, et al. Latency-associated degradation of the MRP1 drug transporter during latent human cytomegalovirus infection. Science. 2013;340(6129):199-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poole E, Avdic S, Hodkinson J, et al. Latency-associated viral interleukin-10 (IL-10) encoded by human cytomegalovirus modulates cellular IL-10 and CCL8 Secretion during latent infection through changes in the cellular microRNA hsa-miR-92a. J Virol. 2014;88(24):13947-13955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carter CC, Onafuwa-Nuga A, McNamara LA, et al. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat Med. 2010;16(4):446-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McNamara LA, Ganesh JA, Collins KL. Latent HIV-1 infection occurs in multiple subsets of hematopoietic progenitor cells and is reversed by NF-κB activation. J Virol. 2012;86(17):9337-9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffin DO, Goff SP. HIV-1 is restricted prior to integration of viral DNA in primary cord-derived human CD34+ cells. J Virol. 2015;89(15):8096-8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 2010;38(13):4274-4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jarmuz A, Chester A, Bayliss J, et al. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 2002;79(3):285-296. [DOI] [PubMed] [Google Scholar]
- 14.Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008;4(12):e1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holmes RK, Malim MH, Bishop KN. APOBEC-mediated viral restriction: not simply editing? Trends Biochem Sci. 2007;32(3):118-128. [DOI] [PubMed] [Google Scholar]
- 16.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424(6944):99-103. [DOI] [PubMed] [Google Scholar]
- 17.Yu X, Yu Y, Liu B, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302(5647):1056-1060. [DOI] [PubMed] [Google Scholar]
- 18.Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003;9(11):1404-1407. [DOI] [PubMed] [Google Scholar]
- 19.Lahouassa H, Daddacha W, Hofmann H, et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol. 2012;13(3):223-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laguette N, Sobhian B, Casartelli N, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474(7353):654-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hrecka K, Hao C, Gierszewska M, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474(7353):658-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Behrendt R, Schumann T, Gerbaulet A, et al. Mouse SAMHD1 has antiretroviral activity and suppresses a spontaneous cell-intrinsic antiviral response. Cell Reports. 2013;4(4):689-696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perez-Caballero D, Zang T, Ebrahimi A, et al. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 2009;139(3):499-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galão RP, Le Tortorec A, Pickering S, Kueck T, Neil SJ. Innate sensing of HIV-1 assembly by Tetherin induces NFκB-dependent proinflammatory responses. Cell Host Microbe. 2012;12(5):633-644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tokarev A, Suarez M, Kwan W, Fitzpatrick K, Singh R, Guatelli J. Stimulation of NF-κB activity by the HIV restriction factor BST2. J Virol. 2013;87(4):2046-2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dubé M, Bego MG, Paquay C, Cohen EA. Modulation of HIV-1-host interaction: role of the Vpu accessory protein. Retrovirology. 2010;7:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goujon C, Moncorgé O, Bauby H, et al. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature. 2013;502(7472):559-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doyle M, Atkins JT, Rivera-Matos IR. Congenital cytomegalovirus infection in infants infected with human immunodeficiency virus type 1. Pediatr Infect Dis J. 1996;15(12):1102-1106. [DOI] [PubMed] [Google Scholar]
- 29.Kovacs A, Schluchter M, Easley K, et al. ; Pediatric Pulmonary and Cardiovascular Complications of Vertically Transmitted HIV Infection Study Group. Cytomegalovirus infection and HIV-1 disease progression in infants born to HIV-1-infected women. N Engl J Med. 1999;341(2):77-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cupsa A, Dumitrescu F, Giubelan L, et al. Impact of CMV infection on horizontally transmitted HIV-1 disease progression. J Int AIDS Soc. 2008;11(Suppl 1):P257. [Google Scholar]
- 31.Lathey JL, Spector DH, Spector SA. Human cytomegalovirus-mediated enhancement of human immunodeficiency virus type-1 production in monocyte-derived macrophages. Virology. 1994;199(1):98-104. [DOI] [PubMed] [Google Scholar]
- 32.Fox-Canale AM, Hope TJ, Martinson J, et al. Human cytomegalovirus and human immunodeficiency virus type-1 co-infection in human cervical tissue. Virology. 2007;369(1):55-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu X, Liu L, Zhang X, et al. F18, a novel small-molecule nonnucleoside reverse transcriptase inhibitor, inhibits HIV-1 replication using distinct binding motifs as demonstrated by resistance selection and docking analysis. Antimicrob Agents Chemother. 2012;56(1):341-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dahabieh MS, Ooms M, Simon V, Sadowski I. A doubly fluorescent HIV-1 reporter shows that the majority of integrated HIV-1 is latent shortly after infection. J Virol. 2013;87(8):4716-4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malnati MS, Scarlatti G, Gatto F, et al. A universal real-time PCR assay for the quantification of group-M HIV-1 proviral load. Nat Protoc. 2008;3(7):1240-1248. [DOI] [PubMed] [Google Scholar]
- 36.Butler SL, Hansen MS, Bushman FD. A quantitative assay for HIV DNA integration in vivo. Nat Med. 2001;7(5):631-634. [DOI] [PubMed] [Google Scholar]
- 37.Sedlak RH, Cook L, Cheng A, Magaret A, Jerome KR. Clinical utility of droplet digital PCR for human cytomegalovirus. J Clin Microbiol. 2014;52(8):2844-2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu Y, Rabinowitz R, Steinitz M, Schlesinger M. Correlation between the expression of CD4 and the level of CD4 mRNA in human B-cell lines. Cell Immunol. 2002;215(1):78-86. [DOI] [PubMed] [Google Scholar]
- 39.Doulatov S, Notta F, Eppert K, Nguyen LT, Ohashi PS, Dick JE. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat Immunol. 2010;11(7):585-593. [DOI] [PubMed] [Google Scholar]
- 40.Heike T, Nakahata T. Ex vivo expansion of hematopoietic stem cells by cytokines. Biochim Biophys Acta. 2002;1592(3):313-321. [DOI] [PubMed] [Google Scholar]
- 41.Goodrum F, Jordan CT, Terhune SS, High K, Shenk T. Differential outcomes of human cytomegalovirus infection in primitive hematopoietic cell subpopulations. Blood. 2004;104(3):687-695. [DOI] [PubMed] [Google Scholar]
- 42.Avdic S, Cao JZ, Cheung AK, Abendroth A, Slobedman B. Viral interleukin-10 expressed by human cytomegalovirus during the latent phase of infection modulates latently infected myeloid cell differentiation. J Virol. 2011;85(14):7465-7471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slobedman B, Mocarski ES, Arvin AM, Mellins ED, Abendroth A. Latent cytomegalovirus down-regulates major histocompatibility complex class II expression on myeloid progenitors. Blood. 2002;100(8):2867-2873. [DOI] [PubMed] [Google Scholar]
- 44.Coronel R, Takayama S, Juwono T, Hertel L. Dynamics of human cytomegalovirus infection in CD34+ hematopoietic cells and derived Langerhans-type dendritic cells. J Virol. 2015;89(10):5615-5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wiskerchen M, Muesing MA. Human immunodeficiency virus type 1 integrase: effects of mutations on viral ability to integrate, direct viral gene expression from unintegrated viral DNA templates, and sustain viral propagation in primary cells. J Virol. 1995;69(1):376-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruiz ME, Cicala C, Arthos J, et al. Peripheral blood-derived CD34+ progenitor cells: CXC chemokine receptor 4 and CC chemokine receptor 5 expression and infection by HIV. J Immunol. 1998;161(8):4169-4176. [PubMed] [Google Scholar]
- 47.Aiuti A, Turchetto L, Cota M, et al. Human CD34(+) cells express CXCR4 and its ligand stromal cell-derived factor-1. Implications for infection by T-cell tropic human immunodeficiency virus. Blood. 1999;94(1):62-73. [PubMed] [Google Scholar]
- 48.Beisser PS, Laurent L, Virelizier JL, Michelson S. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J Virol. 2001;75(13):5949-5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pleskoff O, Tréboute C, Brelot A, Heveker N, Seman M, Alizon M. Identification of a chemokine receptor encoded by human cytomegalovirus as a cofactor for HIV-1 entry. Science. 1997;276(5320):1874-1878. [DOI] [PubMed] [Google Scholar]
- 50.Simon V, Bloch N, Landau NR. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol. 2015;16(6):546-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buzon MJ, Sun H, Li C, et al. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat Med. 2014;20(2):139-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nixon CC, Vatakis DN, Reichelderfer SN, et al. HIV-1 infection of hematopoietic progenitor cells in vivo in humanized mice. Blood. 2013;122(13):2195-2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saffert RT, Penkert RR, Kalejta RF. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J Virol. 2010;84(11):5594-5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sinclair JH, Reeves MB. Human cytomegalovirus manipulation of latently infected cells. Viruses. 2013;5(11):2803-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carter CC, McNamara LA, Onafuwa-Nuga A, et al. HIV-1 utilizes the CXCR4 chemokine receptor to infect multipotent hematopoietic stem and progenitor cells. Cell Host Microbe. 2011;9(3):223-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, Liu H, Kim BO, et al. CD4-independent infection of astrocytes by human immunodeficiency virus type 1: requirement for the human mannose receptor. J Virol. 2004;78(8):4120-4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Geijtenbeek TB, Kwon DS, Torensma R, et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100(5):587-597. [DOI] [PubMed] [Google Scholar]
- 58.Yoshii H, Kamiyama H, Goto K, et al. CD4-independent human immunodeficiency virus infection involves participation of endocytosis and cathepsin B. PLoS One. 2011;6(4):e19352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Y, Wang X, Li J, Zhou Y, Ho W. RIG-I activation inhibits HIV replication in macrophages. J Leukoc Biol. 2013;94(2):337-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lahaye X, Satoh T, Gentili M, et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. 2013;39(6):1132-1142. [DOI] [PubMed] [Google Scholar]
- 61.Monroe KM, Yang Z, Johnson JR, et al. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science. 2014;343(6169):428-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Altfeld M, Gale M Jr. Innate immunity against HIV-1 infection. Nat Immunol. 2015;16(6):554-562. [DOI] [PubMed] [Google Scholar]
- 63.Krishna BA, Lau B, Jackson SE, Wills MR, Sinclair JH, Poole E. Transient activation of human cytomegalovirus lytic gene expression during latency allows cytotoxic T cell killing of latently infected cells. Sci Rep. 2016;6:24674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gohda J, Ma Y, Huang Y, et al. HIV-1 replicates in human osteoclasts and enhances their differentiation in vitro. Retrovirology. 2015;12:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu L, KewalRamani VN. Dendritic-cell interactions with HIV: infection and viral dissemination. Nat Rev Immunol. 2006;6(11):859-868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Slobedman B, Mocarski ES. Quantitative analysis of latent human cytomegalovirus. J Virol. 1999;73(6):4806-4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Khaiboullina SF, Maciejewski JP, Crapnell K, et al. Human cytomegalovirus persists in myeloid progenitors and is passed to the myeloid progeny in a latent form. Br J Haematol. 2004;126(3):410-417. [DOI] [PubMed] [Google Scholar]
- 68.Stacey AR, Norris PJ, Qin L, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol. 2009;83(8):3719-3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chaudhary PM, Roninson IB. Expression and activity of P-glycoprotein, a multidrug efflux pump, in human hematopoietic stem cells. Cell. 1991;66(1):85-94. [DOI] [PubMed] [Google Scholar]
- 70.Bailey JR, Sedaghat AR, Kieffer T, et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J Virol. 2006;80(13):6441-6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.