

Key Points
Fanconi anemia patients have exacerbated cytogenetic clonal mosaicism as detected by molecular karyotyping of blood DNA with SNP assays.
Bone marrow clonal abnormalities can be detected in blood DNA and used as biomarkers of cancer risk and poor prognosis.
Abstract
Detectable clonal mosaicism for large chromosomal events has been associated with aging and an increased risk of hematological and some solid cancers. We hypothesized that genetic cancer predisposition disorders, such as Fanconi anemia (FA), could manifest a high rate of chromosomal mosaic events (CMEs) in peripheral blood, which could be used as early biomarkers of cancer risk. We studied the prevalence of CMEs by single-nucleotide polymorphism (SNP) array in 130 FA patients’ blood DNA and their impact on cancer risk. We detected 51 CMEs (4.4-159 Mb in size) in 16 out of 130 patients (12.3%), of which 9 had multiple CMEs. The most frequent events were gains at 3q (n = 6) and 1q (n = 5), both previously associated with leukemia, as well as rearrangements with breakpoint clustering within the major histocompatibility complex locus (P = 7.3 × 10−9). Compared with 15 743 age-matched population controls, FA patients had a 126 to 140 times higher risk of detectable CMEs in blood (P < 2.2 × 10−16). Prevalent and incident hematologic and solid cancers were more common in CME carriers (odds ratio [OR] = 11.6, 95% confidence interval [CI] = 3.4-39.3, P = 2.8 × 10−5), leading to poorer prognosis. The age-adjusted hazard risk (HR) of having cancer was almost 5 times higher in FA individuals with CMEs than in those without CMEs. Regarding survival, the HR of dying was 4 times higher in FA individuals having CMEs (HR = 4.0, 95% CI = 2.0-7.9, P = 5.7 × 10−5). Therefore, our data suggest that molecular karyotyping with SNP arrays in easy-to-obtain blood samples could be used for better monitoring of bone marrow clonal events, cancer risk, and overall survival of FA patients.
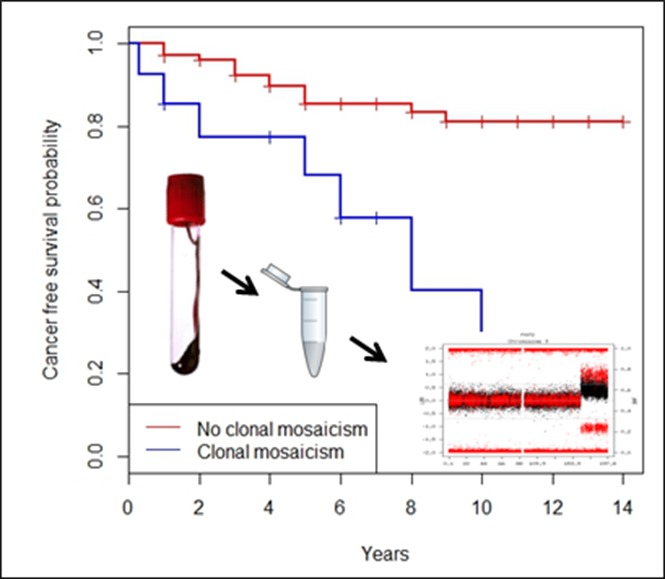
Visual Abstract
Introduction
Mosaicism is the coexistence of cells with different genetic composition within an individual, caused by postzygotic mutations during development that are propagated to a subset of adult cells.1 The frequency of mosaicism is likely underestimated because somatic events may not be associated with phenotypic effects or may be negatively selected.2-4 However, mosaicism can contribute to tumor development resulting from the accumulation of events, which can serve as tumor markers during clonal evolution or arise as a consequence of the malignancy itself.2,3,5 Although the identification of mosaic events will greatly benefit from single-cell analyses, chromosomal rearrangements >1 to 2Mb can also be inferred from studies of DNA from cell populations by genome-wide single-nucleotide polymorphism (SNP) genotypes when the proportion of clonally affected cells is larger than a certain threshold, that is, 7% to 18% depending on the type of rearrangement and the quality of the array.6 In this context, the term “detectable clonal mosaicism” refers to chromosomal mosaic events (CMEs) that occur in a proportion of cells sufficient for their detection. This operational term underestimates the true rate of CMEs. The frequency of detectable CMEs in autosomes is low in individuals <50 years (<0.5%), but it has been shown to increase with age and to be strongly associated with a higher risk of hematological cancer (OR = 22-30) and slightly related to some solid tumors (OR = 4).2,3,7 The frequency of CMEs is also higher in conditions of accelerated aging, such as type 2 diabetes, with a higher prevalence of cardiovascular complications among individuals with CMEs.8
We hypothesized that genome instability disorders with impaired DNA repair, such as FA, could show a higher rate of CMEs at an early age and that CME detection could herald the high risk of hematological and mucosal cancers in these patients. FA is a rare, genetic tumor predisposition disease characterized by impaired DNA interstrand crosslink repair and secondary genomic and chromosomal instability.9 The diagnostic hallmark of FA is increased chromosomal breakage when patients’ cells are cultured with diepoxybuthane or mitomycin C. There are at least 21 FA and FA-like genetic subtypes, each one resulting from mutations in distinct FA genes.9,10 FA patients clinically present with some congenital malformations, progressive bone marrow failure (BMF), and cancer susceptibility,11 particularly hematologic malignancies and typical solid tumors, including head, neck, esophageal, and gynecological squamous cell carcinomas (SCC) and treatment-related liver tumors. Given the extraordinary risk of hematological and mucosal cancers at early ages, stringent follow-up protocols are recommended for FA patients, including periodic bone marrow biopsies, starting in the first decade of life.12 Because of the invasiveness of these control tests, clinical follow-up of some patients is difficult. Thus, less invasive but equally effective screening approaches would be useful to improve the quality of life of FA patients during their medical follow-up. In this study, we investigated the detection of cytogenetic events in blood DNA by molecular karyotyping and whether the presence of such abnormal events is a prognostic biomarker for leukemia, solid cancer, and survival in FA.
Methods
Patient cohort and samples
Total genomic DNA was extracted from peripheral blood samples from 130 FA patients by using a standard phenol–chloroform extraction method or the Puregene DNA Isolation Kit (Qiagen, Inc). There was no cell fractionation other than red cell lysis before DNA extraction. All patients (mean age: 14.4 years old, range: 0-50 years old) had been diagnosed with FA based on clinical criteria and a positive chromosome fragility test followed by genetic complementation and mutational analysis (67.4% with FANCA, 24.1% from other complementation groups, and 8.5% not yet classified). DNA of a paraffin-embedded sample of FA013 (anal SCC) was extracted by using the QIAamp DNA FFPE Tissue Kit (Qiagen, Inc). Reverse mosaicism was studied by the quantification of chromosome fragility in FA cells following diepoxybuthane or mitomycin C assays. Reverse mosaicism was considered when chromosomal aberrations were observed in <50% of the cells after diepoxybuthane treatment.13 Studies were all performed under institutional review board–approved protocols by the involved research centers.
SNP array analyses
DNA samples were analyzed with the SNP arrays Illumina Human1M-Duo v3 (∼1 000 000 SNP probes, 17 samples), HumanOmniExpressExome-8 v1.2 (∼960 000 SNP probes, 1 sample), HumanOmniExpress-12 v1-BeadChip (∼730 000 SNP probes, 96 samples), or InfiniumHumanCore-12 v1 (∼250 000 SNP probes, 16 samples), at the Spanish National Center for Genotyping. A total of 9 samples from FA patients with CMEs were also analyzed with Illumina InfiniumHumanCore-12 v1 (∼250 000 SNP probes). The detection of mosaic rearrangements was based on the assessment of allelic imbalance and copy number changes using the Mosaic Alteration Detection (MAD) algorithm implemented in R Genomic Alteration Detection Analysis software.6
We defined the breakpoint interval for each CME as the region located between 2 informative SNP probes (the first probe within and outside the event). We restricted the search to breakpoints mapped to intervals <200 Kb in order to compare our results with reported data generated with SNP arrays of lower probe density. Then, we analyzed putative enrichment on genomic features at these regions by calculating the percentage of total breakpoint interval length overlapping with each genomic feature (segmental duplications [SD], copy number variants [CNV], genes, and meiotic recombination hotspots [MRH]), and comparing these values with the established distribution of each genomic feature in the human genome (University of California, Santa Cruz genome browser [http://genome.ucsc.edu/cgi-bin/hgGateway] and HapMap database [http://hapmap.ncbi.nlm.nih.gov/]).
The cellular proportion with a specific rearrangement was calculated by using the b allele frequency (BAF) split in the altered region. The formulae used to obtain the cellular proportion for each event type (gain, loss, uniparental disomy [UPD]) was reported elsewhere.4 To calculate the proportion of cells for mosaic multicopy gains with allelic imbalance (genotypes AAA/B and A/B), we adjusted the previous formulae (G2 [proportion of cells with a 2-copy gain] = Bdev/[0.5 – Bdev]), where Bdev is the deviation from the expected BAF value of 0.5 for heterozygous SNPs, and also included the dosage values estimated by the average log R ratio (LRR) signal in the region. In some tetrasomies without allelic imbalance (genotypes AA/BB and A/B), there is not a BAF split, and the proportion of cells cannot be calculated by using BAF values. In these cases, we only used the median-adjusted LRR signal of the region to obtain the cellular proportion. The LRR signal in detected events was adjusted by the autosomal average LRR in samples with only 1 CME. In samples with multiple events, the LRR signal was adjusted by the average LRR in a region of the same chromosome without structural variants. Considering that nonmosaic simple gain (AA/B) has the same genetic dosage as a mosaic tetrasomy in a 0.5 proportion of cells (genotypes AA/BB or AAA/B and A/B), we can define the function between the LRR and cellular proportion as half the function in simple gains.
Control datasets
In order to estimate the frequency of CMEs in the population of patients <18 years old and between 19 and 50 years old, we analyzed SNP array data obtained from blood DNA of unaffected individuals (siblings and parents) from several datasets: (1) the Simons Simplex Collection (https://sfari.org/),14 a characterized sample consisting of 2644 families of autistic probands that includes nonaffected parents and siblings; we analyzed SNP array data (Illumina Human 1MDuo and Omni2.5) of 2209 children (siblings) and 4354 parents; (2) the Autism Genome Project (https://www.autismspeaks.org/science/initiatives/autism-genome-project),15 which consists of 2611 families, each one with a child affected by an autism spectrum disorder and nonaffected parents; we analyzed SNP array data (Illumina Human 1M) of 4086 parents (between 19 and 50 years old); and (3) we added the published data also generated with the MAD algorithm as described above from the Avon Longitudinal Study of Parents and Children (http://www.bristol.ac.uk/alspac/) (N = 3290 children) and the Twins Early Development Studies (http://www.teds.ac.uk/) (N = 1804 children).16
Microsatellite analyses
Microsatellites were amplified from blood and tumor DNA by standard polymerase chain reaction protocols, and the products were analyzed on an ABI PRISM 3100 genetic analyzer according to the manufacturer’s instructions. For relative allelic peak quantitative analysis, trace data were retrieved by using the accompanying software (GeneScan, Applied Biosystems).
Multiplex ligation-dependent probe amplification
Approximately 100 ng of genomic DNA from each sample was subjected to multiplex ligation-depended probe amplification (MLPA) using 2 commercial panels with multiple subtelomeric probes (P036-E1 and P070-B2) according to the manufacturer’s instructions (MRC-Holland, Amsterdam, The Netherlands). Each MLPA signal was normalized and compared with the corresponding mean peak height obtained from 2 control DNA samples.
Statistical analyses
Statistical tests for 2 × 2 tables comparing CME prevalence for FA individuals and controls were based on Fisher’s exact test due to the small numbers. P values, odds ratios (ORs) and 95% confidence intervals at were computed using the Fisher’s test R function.17 Binomial or Bernoulli distribution was used to compute the probability of observing a number of chromosomal breakpoints in a defined genomic region, for example, the MHC region, by chance. The formula was ; , where n was the number of experiments (breakpoint intervals), k was the number of succeeds (breakpoint intervals overlapping with the MHC region), p was the probability of succeeding (size of the MHC region versus the whole genome), and q was the probability of failure (1 – p).
To estimate the survival function from lifetime data, we used the Kaplan-Meier estimator.18 Here, cancer-free time was calculated as the difference between the year of diagnosis of the first cancer after blood sample collection and the year of blood sample collection in individuals with cancer development; in those cancer-free FA patients, cancer-free time was calculated as the difference between the year of the last clinical examination or the year of exitus (when it occurred) and the year when the blood sample was obtained. A total of 6 out of 130 FA patients were excluded from the cancer-free time analysis due to incomplete information. In addition, in order to evaluate the real incidence of cancer after sample collection, 15 out of 124 FA patients were excluded from the cancer-free time reanalysis because of having cancer (n = 14) or dying (n = 1) at the time of sample collection. Age-adjusted cancer risk was computed by using a Cox proportional hazard model, including CMEs and age as covariates. Overall survival was also analyzed using Cox proportional hazard models.
Results
Exacerbated clonal mosaicism in FA blood samples
We evaluated the presence of CMEs in peripheral blood DNA of 130 FA patients (mean age: 14.5 years old, range: 0-50 years old), previously collected for the purpose of genetic diagnosis. SNP arrays were used to build molecular karyotypes that were analyzed with the MAD software.6 In total, 16 out of 130 FA patients (12.3%) had detectable CMEs that were 4.4 to 159 Mb in size (Figure 1, Table 1; supplemental Table 1). Five of the CME carriers (5/91, 5.5%) were children or adolescents <18 years old, whereas 11 (11/39, 28.2%) were in the 19- to 50-years-old range. The mean age at the time of sample collection was 23.2 years for the group with CMEs and 13.3 years for the group without CMEs. Although there is information from multiple case-control studies of cancer,7 we analyzed an age-matched, healthy population of 15 743 controls with SNP arrays of similar density and used the same methods to establish unbiased risks in FA. The frequency of detectable CMEs in the general population of individuals <18 years old was 0.04% (3/7,303) and was 0.3% (26/8,440) in individuals between 19 and 50 years old (supplemental Table 2). Thus, the risk of having detectable mosaicism in FA patients <18 years was almost 140 times higher than in control individuals (OR = 139.9, 95% CI = 26.8-897.9, P = 1.37 × 10−8), whereas the risk was slightly lower in FA patients between 19 and 50 years of age (OR = 126.3, 95% CI = 51.1-297.9, P < 2.2 × 10−16). Out of the 16 FA patients with CMEs, 9 had >1 event with a total of 51 rearrangements, with 1 patient harboring 16 CMEs (FA351). The chromosomal distribution of the 51 CMEs detected in FA is shown in Figure 1A. We detected all types of CMEs, including segmental copy-neutral losses of heterozygosity due to uniparental isodisomies (n = 6), segmental losses (n = 18), segmental single (n = 17) or multicopy gains (n = 6), entire chromosome monosomies (n = 2), and trisomies (n = 2) (Figure 1B). Most CMEs included the telomeres (80.4%), 7.8% spanned centromeres, and only 19.6% were interstitial. CMEs were observed in different FA genetic subtypes (11 FA-A, 1 FA-C, 3 FA-D2, and 1 FA-J), suggesting that CMEs do not preferentially affect a specific complementation group. We validated 41 out of 42 CMEs (97.6%) by independent experimental methods using the same DNA source. Dosage at the subtelomeric regions was analyzed by MLPA, whereas different microsatellites were used to quantify allelic balance at heterozygous sites (supplemental Figure 1; supplemental Table 1). Some rearrangements could not be studied due to sample unavailability. There was a single event discrepant on reanalysis likely due to a failure of the validation method.
Figure 1.

Genomic distribution of CMEs in FA patients. (A) Genomic distribution of the 51 CMEs detected in blood DNA of 16 of 130 FA patients. The circular plot shows the chromosomal location of each of the 51 mosaic events detected (red bars: losses; blue bars: gains; orange bars: multicopy gains; green bars: copy-neutral events or UPDs). (B) Ratios of the dosage values (LRR) as a function of the type of rearrangement and the estimated fraction of cells. LRR signals follow a linear correlation with the percentage of cells with mosaicism for each type of rearrangement (losses, copy-neutral events or UPDs, gains, and multicopy gains).
Table 1.
Features of the 16 FA individuals with CMEs
| Sample | Genetic group | CMEs (size in Mb) | Start (Mb) | End (Mb) | Cell fraction (%) | Age at DNA sampling (y) | Clinical features (age at BMF in y) | Years from DNA sampling to cancer | Cancer diagnosis (age in y) | Evolution (age in y) | Cause of death |
|---|---|---|---|---|---|---|---|---|---|---|---|
| FA466 | A | UPD 4p (6.6) | pter | 6.6 | 9 | 0.7 | BRH, CaLS, BMF (0.7) | — | None | Alive (8) | — |
| FA106 | A | UPD 6p (26.1) | pter | 26.1 | 13 | 15 | BMF (8) | — | None | Exitus (22) | H1N1 viral infection |
| FA117 | A | UPD 10q (70.4) | 65.1 | qter | 17 | 30 | BMF (8) | 6 | AML (36) | Exitus (36) | BM transplant complications |
| FA648 | A | Gain 1q (104.3)* | cen | qter | 29 | 32 | BRH, ID, KM, BMF (32) | 0 | Lymphoma (32) | Exitus (32) | Lymphoma |
| 041607 | A | Monosomy 7 (159.1) | pter | qter | 59 | 50 | GR, BMF (23) | 0 | Breast (37,45), AML (50) | Exitus (50) | AML |
| FA647 | A | Gain 3q (36.3)† | 161.5 | qter | 30 | 4 | GIM, BMF (4) | 1 | MDS (5) | Exitus (5) | BMF |
| Loss 5q (49.3)† | 131.4 | qter | 32 | ||||||||
| EGF058 | A | Gain 1q (76.7) | cen | 222.8 | 59 | 23 | GR, BMF (23) | 8 | HNSCC (31) | Exitus (31) | Cancer treatment toxicity |
| Gain 3q (64.7) | 133.1 | qter | 27 | ||||||||
| FA013 | A | UPD 6p (32.7) | pter | 32.7 | 28 | 24 | BRH, CaLS, BMF (7) | 10 | Anal SCC (34) | Exitus (35) | Metastatic Anal SCC |
| Gain 6p (22.8) | 32.7 | 55.5 | 27 | ||||||||
| FA360 | A | Gain 1q (102.7)† | cen | qter | 76 | 30 | BMF (13) | 0 | AML (30) | Exitus (30) | Multiorganic failure after HSCT |
| Loss 11q (30.4)† | 104.5 | qter | 87 | ||||||||
| Trisomy 8 (146.1) | pter | qter | 26 | ||||||||
| Trisomy 9 (141.0) | pter | qter | 27 | ||||||||
| 041869 | A | Gain 1p (33.5) | 18.5 | 52.0 | 45 | 39 | GR, BRH, CaLS, KM, BMF (8) | 5 | Vulvar SCC (27), esophagus SCC (44) | Exitus (44) | Esophagus SCC |
| Gain 1q (105.9)* | cen | qter | 45 | ||||||||
| Loss 7q (11.1) | 148.0 | qter | 48 | ||||||||
| Loss 8p (4.4) | pter | 4.4 | 32 | ||||||||
| FA178 | A | Loss 1p (29.6) | pter | 29.6 | 41 | 19 | CaLS, UHM, BMF (9) | 0 | AML (19) | Exitus (19) | AML therapy complications (sepsis) |
| Gain 1p (25.8) | 29.6 | 55.4 | 39 | ||||||||
| Gain 3q (63.1)* | 134.7 | qter | 51 | ||||||||
| Loss 5q (98.7) | 82.0 | qter | 38 | ||||||||
| Loss 18q (8.8) | 69.2 | qter | 42 | ||||||||
| FA351 | C | Loss 3p (4.1) | pter | 4.1 | 29 | 2.7 | BMF (2.7) | 0.3 | AML (3) | Exitus (3) | AML therapy complications (multiorganic failure) |
| Gain 3q (18.5) | 159.7 | 178.7 | 69 | ||||||||
| Gain 3q (18.9)* | 178.8 | qter | 62 | ||||||||
| Loss 4p (12.3) | pter | 12.3 | 50 | ||||||||
| Loss 6p (28.6) | pter | 28.6 | 16 | ||||||||
| Loss 6q (63.8) | 107.1 | qter | 80 | ||||||||
| Loss 7p (57.5) | pter | 57.5 | 42 | ||||||||
| Loss 7q (15.3) | 143.8 | qter | 75 | ||||||||
| Gain 8q (20.6) | 56.1 | 76.7 | 26 | ||||||||
| Gain 8q (16.9) | 76.7 | 94.1 | 56 | ||||||||
| Gain 8q (52.0) | 94.2 | qter | 32 | ||||||||
| Gain 13q (93.4) | 21.7 | qter | 19 | ||||||||
| UPD 16p (29.7) | pter | 29.7 | 73 | ||||||||
| Loss 18q (20.7) | 57.4 | qter | 36 | ||||||||
| Gain 21q (12.0) | 36.1 | qter | 29 | ||||||||
| Gain Xp (42.7) | pter | 42.7 | 40 | ||||||||
| 110243 | D2 | UPD 3p (53.6) | pter | 53.6 | 27 | 22 | GR, BRH, HL, ID, KM, BMF (9) | — | None | Alive (27) | — |
| FA531 | D2 | Loss 1p (21.2) | pter | 21.2 | 14 | 26 | GR, BRH | — | None | Alive (28) | — |
| 000644 (+) | D2 | Monosomy 7 (159.1) | pter | qter | 27 | 27 | GR, BRH | 0 | Vulvar SCC (23), oral premalignant findings (24), AML (27) | Alive (37) | — |
| Loss 12p (27.1)‡ | pter | 27.1 | 27 | ||||||||
| Gain 20p (30.8)‡ | pter | 30.8 | 27 | ||||||||
| Loss 20q (20.6)‡ | 30.8 | 51.4 | 27 | ||||||||
| Gain 20q (11.5)‡ | 51.4 | qter | 27 | ||||||||
| FA072 | J | Gain 1q (105.8)* | cen | qter | 66 | 28 | CaLS, CM, BMF (20) | 2 | AML (30) | Exitus (31) | AML therapy complications (pneumonia) |
| Gain 3q (33.6)* | 164.2 | qter | 98 | ||||||||
| Loss 6p (29.9)† | pter | 29.9 | 51 | ||||||||
| Gain 16p (22.6)† | pter | 22.6 | 44 |
Columns contain information about patient, genetic group, detected CMEs including genomic location and type, start and end of each rearrangement and the estimated proportion of affected cells, the age at DNA sampling and BMF, the main clinical features, the years from sampling to the first cancer diagnosis, the type of cancer and the evolution including cause of death. (+) A single case with CMEs, 000644, presented reverse mosaicism of the FA mutation in blood. The rearrangements detected by SNP array in blood also found by cytogenetic analysis of BM aspirates are shown in bold and italic letters.
AML, acute myeloid leukemia; BM, bone marrow; BMF, bone marrow failure; BRH, bilateral radial hypoaplasia; CaLS: café-au-lait spot; cen, centromere; CM, cardiac malformation; GIM, gastrointestinal malformation; GR, growth restriction; HL, hearing loss; HNSCC, head and neck squamous cell carcinoma; HSCT, hematopoietic stem cells transplantation; ID, intellectual disability; KM, kidney malformation; MDS, myeloid dysplastic syndrome; NoM: no relevant malformation; pter, p terminal; qter: q terminal; UHM, unilateral hand malformation.
Gains of >1 extra copy (triplication-tetrasomy).
Possible unbalanced translocation.
Complex rearrangements.
In addition, we also used lower resolution (∼250 000) SNP arrays in 9 samples to further validate the CMEs and check for the clinical validity of this type of low-cost molecular karyotype. All CMEs detected with higher resolution arrays were also detectable with lower resolution (Figure 2; supplemental Figure 2).
Figure 2.

SNP array plots detecting CMEs in a FA patient. Illustrative example of the chromosome 6 plot (black dots represent total intensity values, LRR; red dots represent BAF values) from 1 individual (FA013) whose CMEs (terminal 6p UPD and proximal 6p gain) were both detected by high-density (730 000) (A) and lower-density (250 000) SNP arrays (B). Additional examples are shown in the supplemental annex (supplemental Figures 1 and 2).
Mapping of chromosomal breakpoints and recurrent rearrangements
To further investigate the mutational mechanisms generating CMEs in FA somatic cells, we determined the breakpoint interval for each CME. We detected significant gene enrichment in the FA breakpoint intervals because the odds of having genes in that region were 4.5 times higher than in the whole genome (OR = 4.5, 95% CI = 2.4-8.7, P = 5.2 × 10−7). However, there was no difference in the content of SD, polymorphic CNV, or previously defined MRH (Table 2).19,20 Interestingly, the breakpoints of 5 CMEs (2 terminal losses, 1 interstitial gain, and 2 terminal UPDs) occurred at or near the MHC region (chr6:29570005-33377657, hg19) (Figure 3A). Given the very low probability for the occurrence of 5 of 51 breakpoints in this genomic region by chance (P = 7.3 × 10−9), the MHC locus seems to be a hotspot for somatic rearrangements in FA. Other recurrent CMEs in FA were gains at 3q (n = 6) and 1q (n = 5), with both rearrangements strongly associated with hematologic cancer development and poor prognosis21-23 (Figure 3B,C). On this direction, we were able to detect CMEs that are typically associated with hematologic malignancy (1q/3q gains and chromosome 7 monosomy) in 10 out of 15 FA patients with both CMEs and hematologic problems (bone marrow failure, hematologic premalignancy or malignancy) (Table 1).
Table 2.
Genomic features at the chromosomal breakpoint regions in FA somatic rearrangements
| GF | % of BPi overlapping with the GF | % of the genome overlapping with the GF | Ratio BPi/genome | P |
|---|---|---|---|---|
| SDs | 8.2 | 519 | 1.6 | .56 |
| CNVs | 65.1 | 77.9 (DGV) | 0.8 | .059 |
| Genes | 64.3 | 27.520 | 2.3 | 5.2 × 10−7 |
| MRH | 7.5 | 6.3 (HapMap database) | 1.1 | .78 |
From left to right, this table shows genomic features whose overlap was studied in FA BPis. The total length of the defined BPis was obtained in order to calculate the global percentage overlapping with each feature. The proportion (%) of BPis overlapping with each genomic feature (column 2) was compared with the genomic proportion of the feature reported in the literature (column 3) to detect an increased or decreased overlapping with respect to the expected rate (column 4). We detected a statistically significant enrichment of genes in FA BPis.
BPi, breakpoint interval; GF, genomic feature; DGV, Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home).
Figure 3.

Clustering of somatic chromosomal rearrangements in FA patients at the MHC locus (6p21) and chromosome arms 1q and 3q. (A) Out of 51 CMEs in FA patients, 5 (9.8%) had breakpoints within or near the MHC–HLA locus at 6p21 (P = 7.3 × 10−9 [2 terminal 6p losses in FA351 and FA072, 2 terminal 6p UPD in FA013 and FA106, and an interstitial gain in FA013]), suggesting that this region is a hotspot for somatic chromosomal rearrangements. Dashed lines delimitate the CIs for the specific breakpoints, spanning as much as 11 Mb in 6p UPD in FA013. (B) A total of 6 CMEs (11.7%) involved 3q in 5 FA individuals (EGF058, FA072, FA178, FA351, and FA647), all of them genomic gains. (C) Five additional gain-type CMEs (9.8%) in 5 individuals (FA072, 041869_b; FA648; EGF058; FA360) were located in 1q as shown. The plots, generated with the University of California, Santa Cruz genomic browser (http://genome.ucsc.edu/), show the distribution of rearrangements in FA patients along with those previously reported in the population, based on multiple case-control studies of cancer.7 Purple: gains; red: losses; green: neutral; gray: complex rearrangements.
Increased cancer risk and poor prognosis associated with clonal mosaicism in FA
Among the FA individuals with CMEs, 5 were diagnosed with hematologic malignancies at the time of sampling, whereas 4 more patients were diagnosed 3 months to 6 years later (Table 1). Common chromosomal rearrangements associated with leukemia (1q gain, 3q gain, and/or chromosome 7 monosomy) were detected by SNP array in blood DNA of 8 of the FA cases with hematologic malignancies.21-24 We compared findings by conventional cytogenetic testing in bone marrow aspirates in 5 patients with our findings detected by SNP arrays in peripheral blood DNA, and, generally, we observed that the SNP array of blood DNA detected all CMEs present in the bone marrow along with extra findings (Table 1). Other types of cancer were also diagnosed in FA patients with CMEs, either before sampling (mouth or vulvar SCC and breast cancer) or during follow-up (mouth, anal, or esophagus SCC). Of note, the 6p gain and 6p UPD detected in the peripheral blood of FA013 were also found by microsatellite assays in the anal SCC developed 10 years later (supplemental Figure 3), indicating that these CMEs, which were present in 2 tissues of different ontogenetic origin, probably arose very early during embryonic development. Globally, we observed that 12 of 16 (75%) FA individuals with CMEs versus 23 of 112 (20.5%, 2 cases departed from medical follow-up) without CMEs developed a malignancy 0 to 10 years after DNA sampling, revealing a significantly increased risk of cancer in FA patients with CMEs (OR = 11.6, 95% CI = 3.4-39.3, P = 2.8 × 10−5). Additionally, the diagnosis of prevalent or incident cancer was clearly increased in FA patients with >1 CME (9/9 individuals) compared with those with a single CME (3/7 individuals) (Fisher’s exact test, P = .019) (Table 1). Using the Kaplan-Meier algorithm,18 we observed that the FA patients with CMEs in blood had a shortened cancer-free time after sampling compared with the FA patients without CMEs (hazard risk [HR] = 5.1, 95% CI =2.5-10.3, P = 6.78 × 10−6) (Figure 4A). Given that the mean age was different between both groups of FA patients (with and without CMEs), we obtained an age-adjusted HR of having cancer, which was 4.2 times higher in individuals with CMEs than in individuals without CMEs (age-adjusted HR = 4.2, 95% CI = 2.0-8.7, P = 1.2 × 10−4). Importantly, we also observed shortened cancer-free time in FA patients with CMEs when repeating the analysis by excluding FA patients with cancer (n = 14) or exitus (n = 1) at the time of sample collection (HR = 5.8, 95% CI = 2.3-14.5, P = 1.8 × 10−4). In this case, the age-adjusted HR of having cancer was almost 5 times higher in CME individuals than in individuals without CMEs (age-adjusted HR = 4.9, 95% CI = 1.9-12.7, P = 1.1 × 10−3) (Figure 4B). We detected 11 individuals with CMEs who did not have any type of cancer at the time of sample collection. Seven of these cases developed cancer (4 hematologic tumors and 3 SCC) during an average follow-up of 4.61 years (0.3-10 years) (Table 1). On the other side, among the 98 FA patients without CMEs who did not have cancer at the time of sample collection, 14 developed a malignancy during an average follow-up of 3.85 years (1-9 years) (7 individuals with hematologic malignancies, 3 cases of SCC, 1 patient with both myelodysplastic syndrome and SCC, and 3 patients with other tumors) (supplemental Table 3). Accordingly, there is an increased risk of developing cancer after mosaicism detection that is statistically significant (OR = 10.5, 95% CI = 2.71-40.6, P = 7.6 × 10−4).
Figure 4.

Reduced cancer-free time in FA patients with CMEs compared with patients without CMEs. (A) We calculated cancer-free time (years) for 124 FA patients (incomplete information for the remaining 6 cases), 16 with CMEs and 108 without CMEs, from the time of blood collection until the first cancer diagnosis or the last follow-up (if cancer-free); correction by exitus year was done when required. A significantly abridged cancer-free time was observed in FA patients with CMEs using Kaplan-Meier statistical analysis (HR = 5.1, 95% CI = 2.5-10.3, P = 6.78 × 10−6). The age-adjusted HR of having cancer was 4.2 times higher in CME carriers than noncarriers (age-adjusted HR = 4.2, 95% CI = 2.0-8.7, P = 1.2 × 10−4). (B) In order to consider the incident cancer after blood sample collection, we performed a Kaplan-Meier reanalysis discarding 14 cases with cancer diagnosis at the same time of sample collection and 1 case of exitus at the same year of sampling out of the 124 FA patients included in the initial analysis. A significantly shortened cancer-free time was detected again in FA patients with CMEs compared with those without CMEs (HR = 5.8, 95% CI = 2.3-14.5, P = 1.8 × 10−4). As shown in the figure, we obtained the same results when adjusting by age (age-adjusted HR = 4.9, 95% CI = 1.9-12.7; P = 1.1 × 10−3).
Regarding survival, the HR of dying was 4 times higher in individuals having CMEs than those without CMEs (HR = 4.0, 95% CI = 2.0-7.9, P = 5.7 × 10−5). Although the main causes of death were cancer or related complications in both groups, the proportion of cancer-related deaths was much higher in individuals with CMEs (11/12) than in those without CMEs (12/29, 1 case with no information about the exitus’ cause) (OR = 15.5, 95% CI = 1.7-137.7, P = 4.7 × 10−3).
Clonal mosaicism is related to FA functional deficiency
It is widely known that, in about 20% to 25% of FA patients, reverse mosaicism can arise in vivo from site-specific reversion of a germ line mutation to wild-type sequence either in stem cells or in more committed lineage-specific hematopoietic progenitor cells, usually resulting in a less severe hematologic phenotype. However, the reverted clone rarely repopulates the entire bone marrow, and a proportion of mutated cells remains.25,26 Reverse mosaicism, revealed by a diepoxybutane test resulting in <50% of the cells with chromosomal aberrations,13 was detected a few years after initial sample collection in 27 of the 130 studied FA patients. Only 1 case with reverse mosaicism presented with CMEs in blood along with a diagnosis of vulvar SCC, and none developed hematopoietic cancer. Thus, clonal mosaicism appears clearly related to the functional impairment of DNA interstrand crosslink repair and secondary chromosomal instability that occurs in cells with biallelic mutations in FA genes.
Discussion
CMEs are rare in young, healthy individuals, but have been strongly associated with aging and an increased risk of hematological cancer and also slightly related to some solid cancers in the general population. Importantly, these studies estimate that the risk of hematological cancer is 10-fold higher for mosaic than for nonmosaic individuals.2,3,5 Thus, in this article, we hypothesized that cancer predisposition disorders, such as FA,9,10 could manifest a high rate of CMEs, even at an early age, and that CMEs could be indicative of a higher risk of cancer in the coming years. Our study, based on high-resolution SNP array analysis of 130 patients and 15 743 age-matched controls, revealed an exacerbated risk of detectable mosaicism in FA individuals, indicating that proper DNA repair suppresses genomic heterogeneity of the soma in vivo in healthy people. We also observed an increased risk of cancer and a shortened survival due to cancer after DNA sampling in FA patients with CMEs when compared with FA patients without CMEs. Interestingly, considering only the incident cases of cancer after blood sampling, we detected a reduced cancer-free time in FA patients with CMEs after blood sample collection, showing a tendency of more diagnosis of cancer after mosaicism detection. As somehow expected, a significant proportion of the CMEs detected in blood DNA has been clearly associated with hematologic malignancies (1q gains, 3q gains, and chromosome 7 monosomy), representing two-thirds of FA patients with both CMEs and hematologic complications. These data, together with the fact that 2 mosaic rearrangements (6p gain and 6p UPD) were found both in blood and solid tumor samples in a FA patient, reinforce the idea that CMEs can lead to tumor development or arise as a consequence of tumor clonal expansion.
Cancer development in FA patients has been traditionally associated with chromosome copy gains and losses, as previously mentioned. With this work, we have discovered that copy-neutral changes could play an important role in FA malignancies, because 11.8% of CMEs detected in FA patients were UPDs. We have also contributed to increasing the knowledge regarding hotspots for CMEs in FA, because we discovered that the MHC locus is a hotspot for somatic rearrangements in FA, in addition to the already known 1q and 3q regions. Because structural variation at the MHC locus is a likely substrate for somatic rearrangements in cells of the immune system, derogated repair of MHC rearrangements may facilitate their clonal proliferation in FA cells. Interestingly, copy-neutral loss of heterozygosity in 6p by UDP involving the MHC locus has been reported in acquired aplastic anemia27 and primary nervous system lymphoma.28 Loss of the mismatched HLA haplotype by UPD has also been found after haploidentical hematopoietic stem cell transplantation and infusion of donor T cells in leukemic patients, leading to relapse through the escape of leukemic cells from the donor's antileukemic T cells.29-31
Elucidating the genomic features of breakpoint intervals of CMEs can provide information about the mechanisms underlying these rearrangements as well as the phenotypic consequences for the patient. The enrichment of coding genes in FA breakpoint intervals reinforces the growing evidence coupling transcriptional activity with DNA repair,32,33 which is perturbed in FA patients. Importantly, we detected 2 CMEs in both blood and tumor (anal SCC) samples in 1 FA patient. Although we cannot completely rule out the possibility of some blood or cell free DNA contamination or the presence of lymphocytes in the tumor sample, the different allelic imbalance, indicative of a different proportion of cells with each of the aberrant rearrangements in the tumor with respect to the blood, would suggest a common embryonic origin with different progression in different cells. Although the evidence establishing a mechanistic link between CME detection and solid tumor development is still weak, it is also supported by previous data showing a similar proportion of cells carrying CMEs in the blood and bladder mucosa of 4 individuals with bladder cancer.4
Regarding reverse mosaicism, which is described in up to a quarter of FA patients, we detected CMEs in the blood of a single individual who was also diagnosed with vulvar SCC. Importantly, none of the 27 patients with reverse mosaicism was diagnosed with hematologic malignancy, reinforcing the idea that reversion of a germ line mutation in hematopoietic cells protects against genomic instability, leading to CMEs and hematopoietic cancer. In fact, the reversion of FA mutation could become visible following the same clonal process as any CME. A preexisting reverted wild-type clone, which initially contributes to the somatic genomic heterogeneity, could undergo progressive monoclonal expansion until repopulating an important fraction of the bone marrow in a similar way in which a clone with a chromosomal event is positively selected and leads to a mosaicism.
Given the high prevalence of CMEs in blood samples of FA patients and the documented association of detectable CMEs with hematologic and, to a lesser extent, solid cancer, it is logical to propose that early detection of CMEs could help in clinical decision-making. It is well known that strict follow-up according to the clinical guidelines recommending annual bone marrow sampling for FA patients is very difficult for patients and families, especially when the patient’s hematology remains stable. Although prospective data on sequential sampling of a large cohort are required to better define the sensitivity, specificity, and most appropriate sampling intervals, we propose that SNP arrays of DNA from easy-to-obtain samples, such as blood cells, should be included in the regular follow-up of patients with FA, with the goal of identifying individuals at higher risk of cancer in order to better direct additional exams and treatments. The possibility of studying additional samples that might better detect nonhematological tumors, such as plasma-free DNA or saliva, should also be evaluated in future studies, as well as the utility of these tools in other chromosome instability and bone marrow failure syndromes.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the FA families and FA parent organizations, namely Asociación Española de Anemia de Fanconi and Ralf Dietrich, for their collaboration and continuous support, as well as the Spanish FA Clinical Network for their contributions in taking care of these patients (supplemental Table 4). The authors would also like to thank Francisco X. Real for his critical review of the manuscript and Tania Lobato for technical support in DNA extraction of tumor samples.
J.R.-C. was supported by a fellowship from the Spanish Ministry of Education (FPU13/00782) and M.A.-C. was supported by a Juan de la Cierva postdoctoral fellowship (JCI-2011-10660). J. Surrallés' team is funded by the Generalitat de Catalunya (SGR0489-2009, SGR317-2014), the ICREA-Academia program, Marató de TV3 (project 464/C/2012), the Spanish Ministry of Health (projects FANCOSTEM and FANCOLEN), the Spanish Ministry of Science and Innovation (projects CB06/07/0023, SAF2012-31881, and SAF2015-64152-R), the European Commission (EUROFANCOLEN project HEALTH-F5-2012-305421 and FANCODRUG project H2020-703521), the Fanconi Anemia Research Fund, Inc, and the European Regional Development (FEDER). L.A.P.-J.’s team is funded by the Spanish Ministry of Health (FIS-PI1302481 cofunded by FEDER), the Generalitat de Catalunya (SRG1468-2014), and the ICREA-Academia program, and also acknowledges support from the Spanish Ministry of Economy and Competiveness Programa de Excelencia Maria de Maeztu (MDM-2014-0370). CIBERER is an initiative of the Instituto de Salud Carlos III (Madrid, Spain). D.S. was supported by the Schroeder-Kurth Fund.
Footnotes
The SNP array analyses reported in this article have been deposited in the Gene Expression Omnibus data repository (https://www.ncbi.nlm.nih.gov/geo/, accession numbers GSE93692 and GPL22924).
Authorship
Contribution: L.A.P.-J. and J. Surrallés designed the study, supervised the data, and wrote the manuscript; J.R.-C. performed the validation studies and contributed to writing the manuscript; J.R.-C., M.L.-S., and B.R.-S. performed the CME detection and associated analyses; M.L.-S. and J.R.G. contributed to the population control data and statistical analyses; R.P. and M.A.-C. contributed to the patient characterization, classification, and clinical data; the rest of the coauthors (J.A.C., J.A.B., J.S., I.B., A.C., C.B., M.A.D., C.D.d.H., J. Soulier, and D.S.) provided blood and tumor samples and clinical and genetic data; and all authors approved and commented on the manuscript.
Conflict-of-interest disclosure: B.R.-S. is an employee of, and L.A.P.-J. is a scientific advisor for, qGenomics, SL. The remaining authors declare no competing financial interests.
Correspondence: Luis Alberto Pérez-Jurado, Genetics Unit, Department of Experimental and Health Sciences, Universitat Pompeu Fabra, 88 Dr. Aiguader St, PRBB Building, 08003 Barcelona, Spain; and Jordi Surrallés, Genetics Department, Hospital de Sant Pau, Laboratories floor-2, 89 Sant Quintí St, 08041 Barcelona, Spain; e-mail: jsurralles@santpau.cat.
References
- 1.Fernández LC, Torres M, Real FX. Somatic mosaicism: on the road to cancer. Nat Rev Cancer. 2016;16(1):43-55. [DOI] [PubMed] [Google Scholar]
- 2.Jacobs KB, Yeager M, Zhou W, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012;44(6):651-658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laurie CC, Laurie CA, Rice K, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012;44(6):642-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodríguez-Santiago B, Malats N, Rothman N, et al. Mosaic uniparental disomies and aneuploidies as large structural variants of the human genome. Am J Hum Genet. 2010;87(1):129-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schick UM, McDavid A, Crane PK, et al. Confirmation of the reported association of clonal chromosomal mosaicism with an increased risk of incident hematologic cancer. PLoS One. 2013;8(3):e59823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.González JR, Rodríguez-Santiago B, Cáceres A, et al. A fast and accurate method to detect allelic genomic imbalances underlying mosaic rearrangements using SNP array data. BMC Bioinformatics. 2011;12(1):166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Machiela MJ, Zhou W, Sampson JN, et al. Characterization of large structural genetic mosaicism in human autosomes. Am J Hum Genet. 2015;96(3):487-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonnefond A, Skrobek B, Lobbens S, et al. Association between large detectable clonal mosaicism and type 2 diabetes with vascular complications. Nat Genet. 2013;45(9):1040-1043. [DOI] [PubMed] [Google Scholar]
- 9.Bogliolo M, Surralles J.. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev. 2015;33:32-40 [DOI] [PubMed] [Google Scholar]
- 10.Wang AT, Smogorzewska A. SnapShot: Fanconi anemia and associated proteins. Cell. 2015;160(1-2):354-354.e1. [DOI] [PubMed] [Google Scholar]
- 11.Kutler DI, Wreesmann VB, Goberdhan A, et al. Human papillomavirus DNA and p53 polymorphisms in squamous cell carcinomas from Fanconi anemia patients. J Natl Cancer Inst. 2003;95(22):1718-1721. [DOI] [PubMed] [Google Scholar]
- 12.Hays L, ed. Fanconi Anemia: Guidelines for Diagnosis and Management. 4th ed Eugene, OR: Fanconi Anemia Research Fund, Inc; 2014:421. [Google Scholar]
- 13.Castella M, Pujol R, Callén E, et al. Chromosome fragility in patients with Fanconi anaemia: diagnostic implications and clinical impact. J Med Genet. 2011;48(4):242-250. [DOI] [PubMed] [Google Scholar]
- 14.Fischbach GD, Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010;68(2):192-195. [DOI] [PubMed] [Google Scholar]
- 15.Pinto D, Delaby E, Merico D, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94(5):677-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King DA, Jones WD, Crow YJ, et al. ; Deciphering Developmental Disorders Study. Mosaic structural variation in children with developmental disorders. Hum Mol Genet. 2015;24(10):2733-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 18.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53(282):457-481. [Google Scholar]
- 19.Sharp AJ, Locke DP, McGrath SD, et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77(1):78-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gregory TR. Synergy between sequence and size in large-scale genomics. Nat Rev Genet. 2005;6(9):699-708. [DOI] [PubMed] [Google Scholar]
- 21.Tönnies H, Huber S, Kühl JS, Gerlach A, Ebell W, Neitzel H. Clonal chromosomal aberrations in bone marrow cells of Fanconi anemia patients: gains of the chromosomal segment 3q26q29 as an adverse risk factor. Blood. 2003;101(10):3872-3874. [DOI] [PubMed] [Google Scholar]
- 22.Quentin S, Cuccuini W, Ceccaldi R, et al. Myelodysplasia and leukemia of Fanconi anemia are associated with a specific pattern of genomic abnormalities that includes cryptic RUNX1/AML1 lesions. Blood. 2011;117(15):e161-e170. [DOI] [PubMed] [Google Scholar]
- 23.Meyer S, Bristow C, Wappett M, et al. Fanconi anemia (FA)-associated 3q gains in leukemic transformation consistently target EVI1, but do not affect low TERC expression in FA. Blood. 2011;117(22):6047-6050. [DOI] [PubMed] [Google Scholar]
- 24.Mehta PA, Harris RE, Davies SM, et al. Numerical chromosomal changes and risk of development of myelodysplastic syndrome--acute myeloid leukemia in patients with Fanconi anemia. Cancer Genet Cytogenet. 2010;203(2):180-186. [DOI] [PubMed] [Google Scholar]
- 25.Lo Ten Foe JR, Kwee ML, Rooimans MA, et al. Somatic mosaicism in Fanconi anemia: molecular basis and clinical significance. Eur J Hum Genet. 1997;5(3):137-148. [PubMed] [Google Scholar]
- 26.Waisfisz Q, Morgan NV, Savino M, et al. Spontaneous functional correction of homozygous fanconi anaemia alleles reveals novel mechanistic basis for reverse mosaicism. Nat Genet. 1999;22(4):379-383. [DOI] [PubMed] [Google Scholar]
- 27.Ogawa S. Clonal hematopoiesis in acquired aplastic anemia. Blood. 2016;128(3):337-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwindt H, Vater I, Kreuz M, et al. Chromosomal imbalances and partial uniparental disomies in primary central nervous system lymphoma. Leukemia. 2009;23(10):1875-1884. [DOI] [PubMed] [Google Scholar]
- 29.Vago L, Perna SK, Zanussi M, et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N Engl J Med. 2009;361(5):478-488. [DOI] [PubMed] [Google Scholar]
- 30.Villalobos IB, Takahashi Y, Akatsuka Y, et al. Relapse of leukemia with loss of mismatched HLA resulting from uniparental disomy after haploidentical hematopoietic stem cell transplantation. Blood. 2010;115(15):3158-3161. [DOI] [PubMed] [Google Scholar]
- 31.Dubois V, Sloan-Béna F, Cesbron A, et al. Pretransplant HLA mistyping in diagnostic samples of acute myeloid leukemia patients due to acquired uniparental disomy. Leukemia. 2012;26(9):2079-2085. [DOI] [PubMed] [Google Scholar]
- 32.García-Rubio ML, Pérez-Calero C, Barroso SI, et al. The Fanconi anemia pathway protects genome integrity from R-loops. PLoS Genet. 2015;11(11):e1005674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwab RA, Nieminuszczy J, Shah F, et al. The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol Cell. 2015;60(3):351-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.