

Key Points
Biallelic RTEL1 mutations generate a large clinical spectrum ranging from classical Hoyeraal-Hreidarsson syndrome to isolated aplastic anemia.
Abstract
Telomeres are repetitive hexameric sequences located at the end of linear chromosomes. They adopt a lariat-like structure, the T-loop, to prevent them from being recognized as DNA breaks by the DNA repair machinery. RTEL1 is a DNA helicase required for proper telomere replication and stability. In particular, it has been postulated that RTEL1 is involved in the opening of the T-loop during telomere replication to avoid sudden telomere deletion and telomere circle (T-circle) formation. In humans, biallelic RTEL1 mutations cause Hoyeraal-Hreidarsson syndrome (HH), a rare and severe telomere biology disorder characterized by intrauterine growth retardation, bone marrow failure, microcephaly and/or cerebellar hypoplasia, and immunodeficiency. To date, 18 different RTEL1 mutations have been described in 19 cases of HH with short telomeres. The impaired T-loop resolution has been proposed to be a major cause of telomere shortening in RTEL1 deficiency. However, the biological and clinical consequences of this disorder remain incompletely documented. Here, we describe 4 new patients harboring biallelic RTEL1 mutations, including 2 novel missense mutations located in the C-terminal end of RTEL1 (p.Cys1268Arg and p.Val1294Phe). Clinical characteristics from these 4 patients were collected as those from 4 other RTEL1-deficient patients previously reported. In addition, we assessed whether T-circles, the product of improper T-loop resolution, were detected in our RTEL1-deficient patients. Overall, our study broadens and refines the clinical and biological spectrum of human RTEL1 deficiency.
Visual Abstract
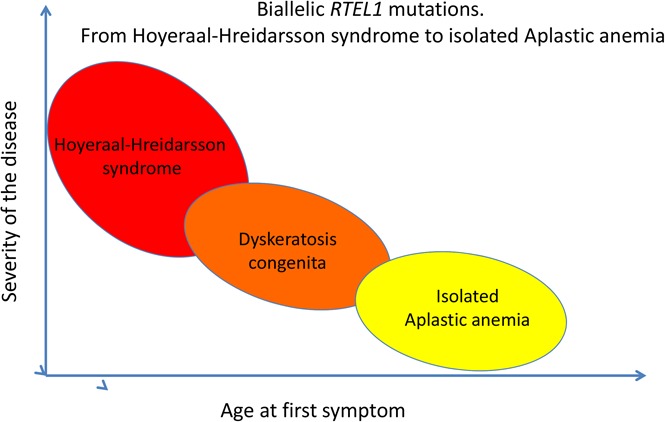
Introduction
Hoyeraal-Hreidarsson syndrome (HH) is a rare systemic disorder typically caused by germline mutations in genes involved in telomere maintenance.1 It is considered to be an extreme phenotype of the telomere biology disorder (TBD) dyskeratosis congenita. First described by Hoyeraal et al in 1970 and Hreidarsson et al in 1988,2,3 HH syndrome associates intrauterine growth retardation, cerebellar hypoplasia, immunodeficiency, and progressive bone marrow failure. HH is recognized as a telomere biology disorder because most patients with HH display very short leukocyte telomere lengths. Moreover, all the HH-causing genes identified so far are participating in the telomere length regulation and/or protection.1 To date, Dyskerin encoded by the DKC1 gene; TERC, TERT, and TPP1 encoded by the ACD gene; TIN2 encoded by the TINF2 gene; RTEL1; and PARN represent the 7 factors found deficient in HH.1,4-6 Other researchers and we recently reported patients with HH carrying mutations in RTEL1, a gene encoding a DNA helicase.7-12 RTEL1 belongs to a family of iron–sulfur-cluster-containing DNA helicases; other members include XPD, FANCJ, and DDX11/ChlR1.13 The helicase domain of RTEL1 exerts an in vitro antirecombinogenic activity by displacing DNA recombination intermediates, the so-called D-loop.14 Via this antirecombinogenic activity, RTEL1 has been proposed to disassemble telomere loops (T-loops), thus facilitating telomere replication and maintaining the integrity of chromosome ends.15,16 In addition to its helicase domain, RTEL1 possesses a tandem of Harmonin-like domains likely involved in interaction with factors that remain to be characterized,17 and a PIP domain allowing interaction with PCNA, a protein participating in DNA replication and repair.18 Likewise, RTEL1 is important to cope with replicative stress and to repair DNA interstrand crosslinks.7,18,19 In humans, splice variants generate 2 main RTEL1 isoforms of 1219 amino acids and 1300 amino acids, respectively.11 The longer RTEL1 isoform possesses an additional RING C4C4 domain at its C-terminal part.11 Most of the studies focusing on RTEL1 functions originate from analyses performed in animal models (mostly mice and Caenorhabditis elegans). Because telomere lengths are significantly longer in laboratory mice compared with humans (50-150 and 5-15 kb, respectively),20 distinct requirement and/or function of factors involved in telomere stability and replication might exist across species. Thus, the analysis of TBDs in humans constitutes a unique opportunity to decipher the function of distinct telomere factors. Here, we describe 4 new RTEL1-deficient patients and 2 novel RTEL1 mutations. We further report the detailed clinical and biological features of these patients, as well as of 4 previously characterized RTEL1-deficient patients.10,11
Materials and methods
Patients
The 8 patients were diagnosed in the Genome Dynamics in the Immune System Laboratory (Institut Imagine, Paris France), and the Genetic Department of Bichat Hospital (GHU Paris Nord-Val de Seine, Paris, France). Informed consent for our study was obtained from the families in accordance with the Declaration of Helsinki. This study was approved by the Institutional Review Board of INSERM and Assistance Publique des Hôpitaux de Paris. Clinical and biological data were retrieved directly from the clinicians in charge of the patients.
Identification of RTEL1 mutations
Whole-exome sequencing was performed on affected individuals from our cohort of genetically uncharacterized Hoyeraal-Hreidarsson and bone marrow failure families, as described previously.11 Whole-exome sequencing was performed with DNA extracted from blood samples. Four patients from this cohort (P1, P2, P3, P4) have been included in previous reports.10,11 The DNA mutation numbering system used is based on cDNA sequence from the long RTEL1 isoform of 1300 amino acids (Uniprot ID, Q9NZ71; NCBI ID, NM_001283009.1). Nucleotide numbering reflects cDNA numbering, with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence. The initiation codon is codon 1.
Telomere length assessment
Telomere length was determined by using the telomere restriction fragment (TRF) assay. DNA (800 ng) extracted from whole blood cells was digested with HinfI and RsaI enzymes, resolved by a 0.7% agarose gel, and transferred to a nylon membrane. Hybridization was performed overnight at 42°C, using EasyHyb solution (Roche) with γ-32P-labeled telomeric (TTAGGG)4 probe. After washes, membranes were exposed over a PhosphorImager (Agfa). PhosphorImager exposures of telomere-probed Southern blots were analyzed with the ImageJ program. The digitalized signal data were then transferred to Microsoft Excel and served as the basis for calculating mean TRF length, using the formula L = (ODi)/(ODi/Li), where ODi is the integrated signal intensity at position i and Li is the length of DNA fragment in position i.
Telomere circle (T-circle) detection
2-dimensional-neutral neutral gel.
Digestion of genomic DNA (20-30 µg) was performed as described earlier in TRF protocol. Digested genomic DNA was separated on a 0.4% agarose gel in 1× TAE (Tris, acetate, EDTA) at 1.5 V/cm over the course of 14 hours (first dimension), and then on a 0.9% agarose gel containing ethidium bromide in 1× TAE at 6 V/cm over the course of 6 hours. The DNA was then transferred onto a nylon membrane and hybridized overnight at 42°C, using EasyHyb solution (Roche) with γ-32P-labeled telomeric (TTAGGG)4 probe. After washes (5 min in 2× saline-sodium citrate [SSC] and 10 min in 0.2× SSC 0.1% sodium dodecyl sulfate) membranes were exposed over a PhosphorImager (Agfa).
T-circle amplification.
T-circle amplification assay used to detect T-circles was adapted from Zellinger et al.21 Briefly, 0.5 to 2 µg of genomic DNA was digested with HinfI and RsaI enzymes overnight at 37°C. DNA was then precipitated and resuspended in an annealing buffer (0.2 M Tris at pH 7.5, 0.2 M KCl, and 1 mM EDTA) with 1 µM (CCCTAA)4 primer containing a thiophosphate at the 3′ end. The mix was heated at 99° C for 5 minutes and let cool down at room temperature. DNA was then precipitated and resuspended in 20 µL of the T-circle amplification reaction buffer (33 mM Tris-acetate at pH 7.9, 10 mM magnesium acetate, 66 mM potassium acetate, 0.1% Tween 20, 1 mM DTT, and 0.37 mM dNTPs). The rolling circle amplification was carried out with 7.5 U Phi29 DNA polymerase (Fermentas) at 30°C for 16 hours. The reaction was then stopped by a 65°C incubation for 15 minutes. The product of T-circle amplification was resolved on an 0.7% agarose gel and transferred to a nylon membrane. Hybridization was performed overnight at 42°C, using EasyHyb solution (Roche) with γ-32P-labeled telomeric (TTAGGG)4 probe. After washes (5 min in 2× SSC and 10 min in 0.2× SSC 0.1% sodium dodecyl sulfate), membranes were exposed over a PhosphorImager (Agfa).
Results
Identification of biallelic RTEL1 mutations in 4 patients with short telomeres
We previously reported 4 patients with HH (P1, P2, P3, P4) carrying biallelic RTEL1 mutations.10,11 We performed whole-exome sequencing of other affected individuals from our cohort with Hoyeraal-Hreidarsson phenotype or bone marrow failure to identify the molecular origin of these conditions. We filtered the results with the hypothesis of autosomal-recessive inheritance of the disease and retained only genes harboring a single homozygous variant or compound heterozygous variants. In addition, given the rarity and the severity of the disease, we suspected that the pathogenic gene variants would be rarely present (<1%) in the public SNP databases (dbSNPs, EVS, 1000 Genome, and ExAC) or in our in-house database (8319 individuals). This analysis led us to identify 4 new patients (P5, P6, P7, P8) from 3 unrelated families carrying biallelic RTEL1 mutations (Figure 1A). Patients P5 and P6, 2 siblings from a consanguineous family, carry a homozygous c.3880G>T mutation leading to the substitution of a valine to a phenylalanine at position 1294 (p.Val1294Phe; Figure 1A). P7, a unique boy from a consanguineous family, carries a homozygous variant leading to a missense mutation c.2869C>T; p.Arg957Trp in the first harmonin-like domain. To our knowledge, this mutation, previously reported in a compound heterozygous context (Table 1),12 has never been found as homozygous. P8 carries compound heterozygous RTEL1 missense mutations: the already reported c.2892T>G; p.Phe964Leu, and a new mutation, c.3802T>C; p.Cys1268Arg. Sanger sequencing confirmed the presence of biallelic RTEL1 mutations in all patients and further indicated that the parents were heterozygous for the mutated RTEL1 allele, whereas the healthy siblings were either heterozygous or homozygous for the wild-type allele (Figure 1B). All RTEL1-deficient patients reported so far exhibit abnormally short telomeres, indicating the crucial role of RTEL1 in telomere maintenance.7-12 Accordingly, the telomere length determined by TRF was reduced in whole blood cells from patients P5, P6, P7, and P8 in comparison with the telomere length of their parents and siblings (Figure 1C).
Figure 1.

RTEL1 mutations identified in 4 novel patients. (A) Pedigrees of the patients’ families. (B) Direct Sanger sequencing of the RTEL1 gene in the patients, the healthy siblings, and their parents. Sequencing was performed on DNA extracted from blood samples. Arrows indicate the position of the mutations. (C) Mean telomere lengths of whole blood cells from patients, their parents, and healthy siblings were estimated by the TRF method. The estimated telomere lengths are indicated.
Table 1.
Characteristics of the RTEL1 mutations affecting its coding region
| Patient | Chromosome | Pos, hg19 | Ref | Alt | cDNA | Amino acid | Genetic status | Affected domain | CADD | ExAC freq. | Mutation described in references |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P4 | 20 | 62309527 | A | T | c.949A>T | Lys317* | Heterozygous | Helicase, HD1 | 40 | 0 | Jullien et al, 201610 |
| P1, P2 | 20 | 62321174 | C | G | c.2097C>G | Ile699Met | Heterozygous | Helicase, HD2 | 25.9 | 0 | Le Guen et al, 201311 |
| P3 | 20 | 62321531 | G | A | c.2233G>A | Val745Met | Heterozygous | Helicase, HD2 | 24.4 | 0 | Le Guen et al, 201311 |
| P7 | 20 | 62324513 | C | T | c.2869C>T | Arg957Trp | Homozygous | Harmonin, HNL1 | 33 | 6/119930 | This study; Walne et al, 201312 |
| P8 | 20 | 62324536 | T | A | c.2892T>G | Phe964Leu | Heterozygous | Harmonin, HNL1 | 23.9 | 0 | This study; Walne et al, 201312 |
| P1, P2 | 20 | 62326911 | T | C | c.3730T>C | Cys1244Arg | Heterozygous | RING C4C4 | 23.2 | 2/117900 | Le Guen et al, 201311 |
| P8 | 20 | 62326983 | T | C | c.3802T>C | Cys1268Arg | Heterozygous | RING C4C4 | 26.0 | 0 | This study |
| P5, P6 | 20 | 62327188 | G | T | c.3880G>T | Val1294Phe | Homozygous | RING C4C4 | 25.4 | 0 | This study |
Alt, alternative; CADD, Combined Annotation-Dependent Depletion; ExAC freq, ExAC frequency; hg19, human genome 19; Pos, position; Ref, reference.
To gain a broader view of the clinical phenotype of individuals with biallelic RTEL1 mutations, we collected data from patients P5, P6, P7, and P8, as well as 4 previously reported RTEL1-deficient patients (P1, P2, P3, P4).10,11 In total, the 8 patients carry 10 different RTEL1 mutations. Two mutations are located in intronic regions leading to splice anomalies,10,11 and 8 are located in coding regions (Figure 2; Table 1). Only 1 mutation creates a premature stop codon (K317* in P4;10 Figure 2). The 7 missense mutations are located in different domains of the protein (Table 1; Figure 2): 2 are in the helicase domain (p.Val745Met for P3, and p.Ile699Met for P1 and P2),11 2 are in the first harmonin N-like (HNL1) domain (p.Arg957Trp for P7, and p.Phe964Leu for P8), and 3 are in the C-terminal C4C4 RING finger domain (p.Cys1244Arg for P1 and P2,11 p.Cys1268Arg for P8, and p.Val1294Phe for P5 and P6) that is only present in the long RTEL1 isoform containing 1300 amino acids (Figure 1; Table 1). To our knowledge, the p.Cys1268Arg and p.Val1294Phe RTEL1 mutations have not been reported previously. Predicted structure data indicated that p.Ile699Met, p.Val745Met, and p.Arg957Trp may affect the functions of the concerned domains, whereas p.Phe964Leu should interfere with their folding or the stability of the harmonin-like domains. The 2 cysteines (p.Cys1244Arg and p.Cys1268Arg) are directly involved in zinc binding.11,17,22 The 8 mutations affecting the RTEL1 coding regions are predicted to be highly deleterious, as estimated by the Combined Annotation-Dependent Depletion method,23 and are absent or found at a very low frequency in ExAC Browser (Table 1).
Figure 2.

Domain architecture of the human RTEL1 with the localization of the different experimentally identified mutations affecting the coding region in the 8 patients. Mutations in boxes are homozygous. HD1 and HD2, helicase domain. The RTEL1 mutations in P1,P2, P3, and P4 have been reported previously.10,11
Absence of T-circle detection in RTEL1-deficient patients
The antirecombinogenic activity of RTEL1 was recently proposed to be important to open the T-loop during telomere replication and to inhibit sudden telomere loss and subsequent T-circle formation.15,16 We thus analyzed the presence of T-circles by using the neutral-neutral 2-dimensional gel technique in cells from P1 and P3. As shown in Figure 3A, the presence of T-circles leads to an additional signal observed in SV40-transformed fibroblasts overexpressing the catalytic telomerase subunit hTERT (positive control). No such signal was detected in SV40-transformed fibroblasts from P1 or activated T cells (T-blasts) from P3 (Figure 3A). To further analyze T-circle formation, we used the T-circle amplification assay, a technique that is presumably more sensitive, and therefore requiring less DNA, than 2-dimensional gel analysis. This assay is based on the detection by Southern blot of T-circles after their isothermic linear amplification by the highly processive Phi29 DNA-polymerase.16,21 Alternative lengthening of telomere is a telomerase-independent mechanism that maintains telomere length and that spontaneously generates T-circles.24 Alternative lengthening of telomere–positive cells (SUSM1, VA13, and U2OS) gave rise to a significant augmented signal in the presence of Phi29, as expected (Figure 3B). In contrast, we did not detect any significant increase in T-circle signal in the patients’ cells, whatever the origin of cells analyzed (SV40-transformed fibroblasts, peripheral blood cells, B lymphoblastoid cell line, and activated T-cells; Figure 3B). From these 2 sets of experiments, we conclude that RTEL1 deficiency in the patients tested did not result in aberrant formation of T-circles.
Figure 3.

T-circle detection by 2-dimensional neutral-neutral gels and T-circle amplification assays. (A) Two-dimensional gel analysis of telomeric DNA from a T-circle positive control (SV40 transformed fibroblasts overexpressing the telomerase TERT-encoding gene) reveals an additional signal corresponding to T-circles. Two-dimensional gel analysis of telomeric DNA from SV40-transformed fibroblasts from P1 and activated T cells (T-blasts) from P3 did not reveal T-circle signal. (B) Detection of T-circles by T-circle amplification in different positive controls (ie, alternative lengthening of telomere–positive cells SUSM1, VA13, and U2OS), but not in SV40-fibroblats, T-blasts, B lymphoblastoid cell line, and blood cells from healthy controls and RTEL1-deficient patients P1-P8.
Patients’ clinical features
Among the 8 patients with biallelic RTEL1 mutations included in our study, 3 were born from consanguineous parents (Figure 1; Table 2). Sex ratio (M/F) was 3/5 (Table 2). Clinical presentations leading to the diagnosis of telomere biology disorder were progressive pancytopenia (n = 2), opportunistic infections (n = 1), and protracted diarrhea (n = 3). Two patients (P2 and P6) were diagnosed because of a similar phenotype in siblings (ie, P1 and P5, respectively). Median age at diagnosis was 23 months (range, 15-183 months) for patients with no prior family cases. Although 7 of 8 patients presented with classical features of HH (ie, intrauterine growth retardation, microcephaly, developmental delay), it is noteworthy that 1 patient (P8) was diagnosed through a systematic genetic screening for isolated aplastic anemia and was free of nonhematologic symptoms (Table 2).
Table 2.
Clinical features of RTEL1-deficient patients
| Characteristic | P1* | P2† | P3 | P4 | P5† | P6† | P7 | P8 |
|---|---|---|---|---|---|---|---|---|
| Sex | Male | Female | Female | Female | Female | Male | Male | Female |
| Consanguinity | No | No | No | No | Yes | Yes | Yes | No |
| Developmental features | ||||||||
| IGR | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No |
| Prematurity | Yes (29 WG) | Yes (30 WG) | Yes (36 WG) | No (38 WG) | No (37 WG) | Yes (32 WG) | Yes (33 WG) | No (38 WG) |
| Hypotrophy | <3rd percentile | <3rd percentile | <3rd percentile | <3rd percentile | <3rd percentile | <3rd percentile | <3rd percentile | No |
| Dysmorphia | No | Yes | Yes | Yes | No | Yes | No | No |
| Aplastic anemia | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Immunodeficiency | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Neurological features | ||||||||
| Microcephaly | <1st percentile | <1st percentile | <1st percentile | <1st percentile | <1st percentile | <1st percentile | <1st percentile | No |
| Developmental delay | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No |
| Spastic diplegia | Yes | Yes | No | No | No | No | Yes | No |
| Cerebellar dysfunction | No | No | Mild | Yes | No | No | Yes | No |
| Cerebellar atrophy | Yes | Yes | Yes | Yes | Yes | No | Yes | No |
| Gastrointestinal features | ||||||||
| Colitis | Yes | Yes | Yes | No | Yes (NI) | Yes (CMV) | Yes (CMV) | No |
| Esophageal stenosis | No | No | Yes | No | No | No | No | No |
| Mucocutaneous features | ||||||||
| Oral leukoplakia | Yes | Yes | Yes | No | Yes | No | No | No |
| Skin hyperpigmentation | No | Yes | Yes | No | Yes | Yes | No | No |
| Nail dystrophy | No | No | Yes | No | No | No | No | No |
| Outcome | Death (4 y), Aspergillosis | Death (18 mo), Aspergillosis | Alive (9 y old) | Alive (8 y old) | Death (26 mo) | Death (16 mo), sepsis | Alive (5 y old) | Alive (19 y old) |
NI, noninfectious; WG, week of gestation.
P1 and P2 are siblings.
P5 and P6 are siblings.
Neurological features.
Most of the children suffered from delayed speech motor and development. Three patients (P3, P4, and P7) had clinical cerebellar dysfunction with dysmetria and nystagmus. Spastic diplegia was present in 3 patients. Seven of the 8 patients presented with microcephaly (less than the first percentile) (Table 2). Magnetic resonance imaging revealed cerebellar hypoplasia (6 of the 7 patients with microcephaly), dysmorphia of corpus callosum (n = 3), delayed myelination (n = 1), and periventricular leukomalacia (n = 1).
Infections.
Severe infections were a common complication of patients in our cohort. Bacterial sepsis caused by infection with Stenotrophomonas maltophilia, Pseudomonas aeruginosa, Pantoea agglomerans, and Enterococcus faecium occurred in 3 patients (P1, P5, and P7). Three patients presented with fungal infection: a sepsis with Candida non-albicans (P5), and invasive pulmonary aspergillosis (P1 and P2) with brain abscess (P1). These severe bacterial and fungal infections occurred in the absence of severe neutropenia: all patients had more than 500 neutrophils per microliter at the time of the infections. Moreover, 4 patients developed opportunistic infections reminiscent of patients with primary combined immunodeficiency. Patients P5, P6, and P7 suffered cytomegalovirus (CMV) viremia associated with colitis in 2 of them (P6 and P7) (Table 2). One patient (P1) developed severe primary varicella zoster virus infection that required treatment with intravenous acyclovir. The infection was resolutive with no relapse for P1. Of note, P1’s first clinical presentation was respiratory distress caused by a Pneumocystis jirovecii infection that was successfully treated with cotrimoxazole. The importance of the infectious symptomatology, even in the absence of severe cytopenia, emphasizes the importance of the immune deficiency in this TBD.
Immunodeficiency.
Lymphopenia was present in all patients (Table 3). Although all lymphocyte subsets were involved, B and NK cells were particularly affected (Table 3). The CD4/CD8 ratio was normal in all patients. Proportion of naive CD4+ T cells (evaluated by the CD45RA marker; n = 6) and proportion of recent thymus emigrant (characterized by the CD45RA+CD31+ markers) were mostly conserved with a median of 76.5% (range, 58%-83%) and 55% (range, 51%-67%), respectively. CD8+ T-cell subsets were evaluated in 2 patients (P3 and P7), showing reduced CCR7+CD45RA+ naive and increased effector subsets (CCR7−CD45RA− effector for P3 and CCR7−CD45RA+ terminal effector for P7). T-cell proliferation in response to phytohemagglutinin and/or antigens was reduced in 5 of the 7 patients tested. Hypogammaglobulinemia affected 5 of 7 evaluated patients, with impairment of some antibody response toward specific antigens. Virtual absence of B lymphocytes was observed in P5, whereas P2 presented with agammaglobulinemia, despite the persistence of circulating B cells. NK cell deficiency was present in all but 2 (P4 and P8) evaluated patients with a median NK count of 24/μL of blood (range, 7-78/μL). Intravenous immunoglobulin replacement was initiated in 6 patients, and cotrimoxazole prophylaxis in 7 patients.
Table 3.
Immunological features of the RTEL1-deficient patients
| Characteristic | P1* | P2* | P3 | P4 | P5† | P6† | P7 | P8 | Normal Values | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at evaluation | 20 mo | 6 mo | 21 mo | 44 mo | 25 mo | 15 mo | 32 mo | 15 y | 6-24 mo* | 24 mo-6 y | >12 y |
| Lymphocyte counts | |||||||||||
| Total count, /μL | 800 | 1300 | 1200 | 1300 | 700 | 840 | 1000 | 600 | 3400-8900 | 2300-5400 | 1400-3300 |
| CD3+ count, /μL | 640 | 1170 | 1080 | 1040 | 679 | 790 | 910 | 548 | 1900-5900 | 1400-3700 | 1000-2200 |
| CD3+CD4+, /μL | 424 | 780 | 720 | 592 | 532 | 585 | 420 | 275 | 1400-4300 | 700-2200 | 530-1300 |
| CD45RA+/CD4+, % | 81 | 95 | 72 | NA | 65 | 59 | 83 | NA | 88-95 | 58-70 | 58-70 |
| CD45RA+CD31+/CD4+, % | 57 | 67 | 51 | NA | NA | NA | 53 | NA | 60-72 | 43-55 | 43-55 |
| CD3+CD8+, /μL | 184 | 247 | 216 | 337 | 154 | 172 | 400 | 254 | 500-1700 | 490-1300 | 330-920 |
| CCR7+CD45RA+/CD8+, % | NA | NA | 10 | NA | NA | NA | 45 | NA | 52-68 | 52-68 | 52-68 |
| CCR7+CD45RA−/CD8+, % | NA | NA | 1 | NA | NA | NA | 2 | NA | April 3 | April 3 | April 3 |
| CCR7−CD45RA−/CD8+, % | NA | NA | 76 | NA | NA | NA | 20 | NA | November 20 | November 20 | November 20 |
| CCR7−CD45RA+/CD8+, % | NA | NA | 13 | NA | NA | NA | 32 | NA | 16-28 | 16-28 | 16-28 |
| CD19+, /μL | 16 | 0 | 72 | 65 | 0 | 18 | 40 | 74 | 610-3500 | 390-1400 | 110-570 |
| CD3−CD56+CD16+, /μL | 24 | 78 | 24 | 149 | 7 | 8 | 50 | 179 | 160-950 | 130-720 | 70-480 |
| Ig levels at first evaluation | |||||||||||
| IgG, g/L | 2.2 | 0.4 | 1.92 | 8.46 | 2.92 | 6.98 | 4 | NA | 3.5-8.96 | 5.82-11.54 | 6.55-12.29 |
| IgA, g/L | 0.13 | <0.06 | 0.15 | 0.62 | 0.9 | 1.12 | 0.75 | NA | 0.33-1.22 | 0.46-1.57 | 0.5-2.03 |
| IgM, g/L | 0.04 | <0.06 | 0.2 | 0.56 | 0.54 | 0.56 | 0.3 | NA | 0.5-1.52 | 0.54-1.55 | 0.53-1.62 |
| Serologic testing | NA | NA | Tetanos+; diphteriae+/− | NA | NA | NA | Tetanos+/−; anti-HBs+;S Pneumo- | NA | |||
| T-cell proliferation | |||||||||||
| PHA | Diminished | Normal | Normal | NA | Diminished | Normal | Diminished | NA | |||
| OKT3 | NA | Normal | Normal | NA | NA | NA | NA | NA | |||
| Tetanus toxoid | Diminished | Diminished | Normal | NA | NA | NA | NA | NA | |||
| Candidin | Diminished | Diminished | Diminished | NA | NA | NA | NA | NA | |||
P1 and P2 are siblings.
P5 and P6 are siblings.
Bone marrow failure.
Early onset of bone marrow failure occurred in all patients (Table 4). It represented the first indication of telomere biology disorder for 2 patients. All patients had 1 or more cytopenia at diagnosis: 4 had thrombocytopenia, 6 had normocytic or macrocytic anemia, and 2 had neutropenia. Seven of 8 patients with bone marrow failure required regular transfusion support. The median age for starting transfusion was 32.5 months (range, 11-52 months). P7 and P8 still remain independent of any transfusion 2 and 4 years after diagnosis, respectively, with moderate cytopenia (supplemental Table 1).
Table 4.
Hematological features of the RTEL1-deficient patients
| Normal values | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Characteristic | P1* | P2* | P3 | P4 | P5† | P6† | P7 | P8 | 6 mo-6 y | After 6 y |
| Age at first evaluation | 20 mo | 6 mo | 21 mo | 44 mo | 25 mo | 15 mo | 32 mo | 15 y | ||
| Blood count at first evaluation | ||||||||||
| Total WBC | 3.4 | 7.8 | 8.4 | 2.1 | 2.5 | 1.94 | 4 | 4.1 | 6-17.5 | 04-10 |
| Neutrophils | 2.2 | 2.7 | 5.4 | 0.5 | 1.7 | 0.8 | 2.4 | 3.3 | 1.5-8.5 | 1.5-7 |
| Lymphocytes | 0.8 | 1.2 | 1.7 | 1.1 | 0.7 | 0.84 | 1 | 0.6 | 3-9.5 | 1.5-4 |
| Hemoglobin (g/dL) | 8.7 | 11.7 | 10.5 | 5.8 | 9.5 | 7.5 | 10.4 | 13.2 | 10.5-12 | 12-17 |
| Platelets (G/L) | 178 | 199 | 403 | 8 | 60 | 14 | 324 | 37 | 150-450 | 150-450 |
| Bonne marrow cellularity | — | — | Moderately hypocellular | Hypocellular | Hypocellular | — | — | — | ||
| Age at start of transfusion support | 41 mo | 11 mo | 52 mo | 40 mo | 25 mo | 15 mo | — | — | ||
Bone marrow cellularity on bone marrow smear.
Patients 1 and 2 are siblings.
†Patients 5 and 6 are siblings.
Gastrointestinal features.
Protracted diarrhea was observed in 6 of 8 patients and was the inaugural symptom of the TBD in 2 cases. All these patients presented with either an infectious colitis with CMV inclusions (P6 and P7) or an inflammatory colitis. Histological features consisted of nonspecific lymphoplasmacytic infiltrates with chorionic edema in P6 and granulomatous lesions associated with mononuclear and eosinophilic infiltrates in P3. P3 also presented with esophageal strictures that required endoscopic dilatation.
Other features.
Mucocutaneous features consisted of pigmentation abnormalities (n = 3) and oral leukoplakia (n = 4). P3 progressively developed nail dystrophy classical of dyskeratosis congenita. Facial dysmorphia was reported in 4 patients. No skeletal abnormalities were noted.
Outcomes.
Four patients died of infection: bacterial sepsis (n = 2), invasive aspergillosis (n = 2). Death occurred at a median age of 26 months (range, 18-48 months). Four patients remain alive. Two of them are dependent on transfusion support. The last whole blood counts of these 4 patients are provided in supplemental Table 1.
Discussion
We here report a series of 8 patients with biallelic RTEL1 mutations. Our study shows that biallelic RTEL1 mutations are associated with not only a classical HH phenotype but also with isolated aplastic anemia (P8). The heterogeneity observed in clinical presentation is reminiscent of what has been described for other genetic defects associated with TBDs (eg, Dyskerin-deficient or TERC-deficient patients) underlying the need of a systematic screening for TBD also in patients with “isolated” bone marrow failure.25 Importantly, we did not find any correlation between telomere length and the severity of phenotype (evaluated through age at first symptom or age at first transfusion). However, future genotype–phenotype correlation studies should provide clues about the clinical heterogeneity observed in patients carrying biallelic RTEL1 mutations.
Immunodeficiency is the most underrecognized feature of HH, even though it was specified by Berthet et al in the third case of HH published in 1994.26-28 It is noteworthy that all RTEL1-deficient patients in our cohort presented with combined immunodeficiency revealed in 1 by opportunistic infections (eg, P. jirovecii in P1). Thus, the diagnosis of HH should be considered in any child presenting with combined immunodeficiency associated with other developmental features (such as microcephaly and/or cerebellar hypoplasia). The prognosis of TBD associated with biallelic RTEL1 mutation remains poor. Death occurred in 4 patients of the 8 reported, mostly because of infection favored by immunodeficiency. Systematic prophylaxis with immunoglobulin replacement cotrimoxazole and antifungal prophylaxis should be proposed to all patients. However, the management of aplastic anemia remains a dilemma. Until now, the only curative treatment to bone marrow failure is allogeneic stem cell transplantation (HSCT), a procedure harboring a significantly increased risk for transplantation-related morbidity and mortality for patients with telomere disorders when myeloablative conditioning with alkylating agents is used. If some teams are investigating the use of a reduced-intensity conditioning regimen for HSCT for TBD, the outcome of this procedure remains largely dependent on active posttransplant infections, donor cell engraftment, and the occurrence of graft-versus-host disease. Indeed, inflammation associated with graft-versus-host disease might theoretically worsen the disease-related organ senescence. In line with this assumption, it is noteworthy that some transplanted patients with HH have presented with lethal post-HSCT pulmonary or liver fibrosis.1,9,29 Nevertheless, some experts still recommend transplanting these patients when required.30 A point to be investigated in the next studies will be the toxicity of transplant according to the involved gene. Patients ineligible for HSCT may benefit from androgen therapy, as some recent studies have shown improvement of the hematopoiesis in patients with TBD treated with androgen.31,32 Interestingly, this improvement is associated with telomere elongation in peripheral blood mononuclear cells.32 However, androgen therapy remains associated with frequent adverse effects, and its chronic administration could eventually result in stem cell exhaustion in the treated patients.33 In our cohort, androgen therapy was introduced in P4, but was stopped because of the lack of biological improvement.
Heterozygous RTEL1 mutations leading to haploinsufficiency have been recently associated with primary pulmonary fibrosis and short telomeres.22,34,35 However, none of the patients in our series presented with such features, probably because of their young age. Although the patients’ parents have not developed pulmonary fibrosis so far, they should probably avoid exposure to environmental stress (eg, smoking, fibrogenic dust inhalation) and be followed-up by a pulmonologist. Moreover, one can imagine that in some cases, RTEL1 heterozygous mutations might lead to late-onset bone marrow failure.
Most of the RTEL1-deficient patients described to date carry compound heterozygous mutations, whereas homozygous RTEL1 mutations have only been reported for p.Arg1264His and p.Phe964Leu.7,12 We here describe 4 novel patients (P5, P6, P7, and P8), 3 of whom carry homozygous RTEL1 mutations (p.Val1294Phe and p.Arg957Trp). To our knowledge, the p.Val1294Phe has never been reported, whereas the p.Arg957Trp mutation has only been described as heterozygous. Given that the complete loss of RTEL1 function is lethal in mice,36 one can hypothesize that the p.Arg957Trp and p.Val1294Phe mutations are hypomorphic, although severe, and retain some residual activity. Interestingly, p.Val1294Phe is located in the C-terminal part of RTEL1 specific to the longer isoform. This indicates that the expression of the short wtRTEL11219 isoform is not sufficient to protect patients P5 and P6 from telomere dysfunction. This observation also suggests that the RTEL11300 isoform plays a critical role in the regulation of telomere length. The RTEL11300 isoform specifically harbors a RING domain that has been shown to interact with the telomeric factor TRF2 and be crucial for the T-loop opening.15 Nevertheless, we did not detect T-circles in patients’ cells, using either 2-dimensional neutral-neutral gels or T-circle amplification assays. This suggests that RTEL1 deficiency causing telomere shortening, terminal deletion, and sister telomere loss is not inevitably associated with T-circle accumulation.10,11 One cannot exclude, however, that T-loop excision and T-circle formation in RTEL1-defective cells only occur in a context of long telomeres, such as in mice, as previously reported,16 or in human cell lines with long telomeres or in which RTEL1 expression is suddenly downregulated (eg, via knockdown).
The precise function of the C-terminal part of the long RTEL1 isoform containing the RING C4C4 domain remains elusive, but its in vivo importance has been clearly shown by the human phenotype as a result of the p.Val1294Phe mutation in P5 and P6 in a homozygous state. Further investigations are warranted to delineate the structural and functional effects of the identified mutations, especially the p.Val1294Phe variant, and to elucidate how human RTEL1 participates in telomere stability and genome integrity.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This work is dedicated to the patients and their families. The authors are indebted to Christine Bole-Feysat and Patrick Nitschké for their technical assistance. This work has been supported by institutional grants from INSERM, Ligue Nationale contre le Cancer (Equipe Labellisée La Ligue), Groupement d'intérêt scientifique–Institut des maladies rares, Agence pour la recherche sur le cancer, Institut National du Cancer/Canceropôle Ile de France, Institut Imagine. This program has received a state subsidy managed by the National Research Agency (ANR) under the “Investments for the Future” program bearing the reference ANR-10-IAHU-01. P.R., I.C. and A.L-V. are recipients of a collaborative ANR grant ANR-14-CE10-0006. P.R. is a scientist from Centre National de la Recherche Scientifique.
Authorship
Contribution: F.T wrote the manuscript, designed the study, collected the data, and participated in the clinical care of the patients; L.K., C.M., and L.J., performed the genetic testing; D.M., A.I., F.D., S.S., V.G.-M., A.E., A.F., and T.L. participated in the clinical care of the patients and critically read the manuscript; J.S. performed biological analysis and critically read the manuscript; A.L.-V. provided scientific advice and critically read the manuscript; I.C. performed structural analysis of RTEL1 and critically read the manuscript; J.-P.d.V. provided scientific advice and critically read the manuscript; C.K. performed the genetic testing and critically read the manuscript; and P.R. designed the study, performed experiments, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Fabien Touzot, Département de Biothérapie, Hôpital Necker–Enfants Malades, 75015 Paris, France; e-mail: fabien.touzot@aphp.fr; and Patrick Revy, Laboratory of Genome Dynamics in the Immune System, Institut Imagine, 24 Boulevard du Montparnasse, 75015 Paris, France; e-mail: patrick.revy@inserm.fr.
References
- 1.Glousker G, Touzot F, Revy P, Tzfati Y, Savage SA. Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Br J Haematol. 2015;170(4):457-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoyeraal HM, Lamvik J, Moe PJ. Congenital hypoplastic thrombocytopenia and cerebral malformations in two brothers. Acta Paediatr Scand. 1970;59(2):185-191. [DOI] [PubMed] [Google Scholar]
- 3.Hreidarsson S, Kristjansson K, Johannesson G, Johannsson JH. A syndrome of progressive pancytopenia with microcephaly, cerebellar hypoplasia and growth failure. Acta Paediatr Scand. 1988;77(5):773-775. [DOI] [PubMed] [Google Scholar]
- 4.Dhanraj S, Gunja SM, Deveau AP, et al. . Bone marrow failure and developmental delay caused by mutations in poly(A)-specific ribonuclease (PARN). J Med Genet. 2015;52(11):738-748. [DOI] [PubMed] [Google Scholar]
- 5.Moon DH, Segal M, Boyraz B, et al. . Poly(A)-specific ribonuclease (PARN) mediates 3′-end maturation of the telomerase RNA component. Nat Genet. 2015;47(12):1482-1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tummala H, Walne A, Collopy L, et al. . Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Invest. 2015;125(5):2151-2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ballew BJ, Joseph V, De S, et al. . A recessive founder mutation in regulator of telomere elongation helicase 1, RTEL1, underlies severe immunodeficiency and features of Hoyeraal Hreidarsson syndrome. PLoS Genet. 2013;9(8):e1003695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ballew BJ, Yeager M, Jacobs K, et al. . Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum Genet. 2013;132(4):473-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng Z, Glousker G, Molczan A, et al. . Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal-Hreidarsson syndrome. Proc Natl Acad Sci USA. 2013;110(36):E3408-E3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jullien L, Kannengiesser C, Kermasson L, et al. . Mutations of the RTEL1 Helicase in a Hoyeraal-Hreidarsson Syndrome Patient Highlight the Importance of the ARCH Domain. Hum Mutat. 2016;37(5):469-472. [DOI] [PubMed] [Google Scholar]
- 11.Le Guen T, Jullien L, Touzot F, et al. . Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet. 2013;22(16):3239-3249. [DOI] [PubMed] [Google Scholar]
- 12.Walne AJ, Vulliamy T, Kirwan M, Plagnol V, Dokal I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet. 2013;92(3):448-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uringa EJ, Youds JL, Lisaingo K, Lansdorp PM, Boulton SJ. RTEL1: an essential helicase for telomere maintenance and the regulation of homologous recombination. Nucleic Acids Res. 2011;39(5):1647-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Youds JL, Mets DG, McIlwraith MJ, et al. . RTEL-1 enforces meiotic crossover interference and homeostasis. Science. 2010;327(5970):1254-1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarek G, Vannier JB, Panier S, Petrini JH, Boulton SJ. TRF2 recruits RTEL1 to telomeres in S phase to promote t-loop unwinding. Mol Cell. 2015;57(4):622-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell. 2012;149(4):795-806. [DOI] [PubMed] [Google Scholar]
- 17.Faure G, Revy P, Schertzer M, Londono-Vallejo A, Callebaut I. The C-terminal extension of human RTEL1, mutated in Hoyeraal-Hreidarsson syndrome, contains harmonin-N-like domains. Proteins. 2014;82(6):897-903 [DOI] [PubMed] [Google Scholar]
- 18.Vannier JB, Sandhu S, Petalcorin MI, et al. . RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science. 2013;342(6155):239-242. [DOI] [PubMed] [Google Scholar]
- 19.Barber LJ, Youds JL, Ward JD, et al. . RTEL1 maintains genomic stability by suppressing homologous recombination. Cell. 2008;135(2):261-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calado RT, Dumitriu B. Telomere dynamics in mice and humans. Semin Hematol. 2013;50(2):165-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zellinger B, Akimcheva S, Puizina J, Schirato M, Riha K. Ku suppresses formation of telomeric circles and alternative telomere lengthening in Arabidopsis. Mol Cell. 2007;27(1):163-169. [DOI] [PubMed] [Google Scholar]
- 22.Kannengiesser C, Borie R, Ménard C, et al. . Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur Respir J. 2015;46(2):474-485. [DOI] [PubMed] [Google Scholar]
- 23.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cesare AJ, Griffith JD. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol Cell Biol. 2004;24(22):9948-9957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirwan M, Dokal I. Dyskeratosis congenita: a genetic disorder of many faces. Clin Genet. 2008;73(2):103-112. [DOI] [PubMed] [Google Scholar]
- 26.Berthet F, Caduff R, Schaad UB, et al. . A syndrome of primary combined immunodeficiency with microcephaly, cerebellar hypoplasia, growth failure and progressive pancytopenia. Eur J Pediatr. 1994;153(5):333-338. [DOI] [PubMed] [Google Scholar]
- 27.Berthet F, Tuchschmid P, Boltshauser E, Seger RA. The Hoyeraal-Hreidarsson syndrome: don’t forget the associated immunodeficiency. Eur J Pediatr. 1995;154(12):998. [DOI] [PubMed] [Google Scholar]
- 28.Cossu F, Vulliamy TJ, Marrone A, Badiali M, Cao A, Dokal I. A novel DKC1 mutation, severe combined immunodeficiency (T+B-NK- SCID) and bone marrow transplantation in an infant with Hoyeraal-Hreidarsson syndrome. Br J Haematol. 2002;119(3):765-768. [DOI] [PubMed] [Google Scholar]
- 29.Touzot F, Gaillard L, Vasquez N, et al. . Heterogeneous telomere defects in patients with severe forms of dyskeratosis congenita. J Allergy Clin Immunol 2012;129(2):473-482. [DOI] [PubMed]
- 30.Peffault de Latour R, Peters C, Gibson B, et al. ; Pediatric Working Party of the European Group for Blood and Marrow Transplantation; Severe Aplastic Anemia Working Party of the European Group for Blood and Marrow Transplantation. Recommendations on hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Bone Marrow Transplant. 2015;50(9):1168-1172. [DOI] [PubMed] [Google Scholar]
- 31.Khincha PP, Wentzensen IM, Giri N, Alter BP, Savage SA. Response to androgen therapy in patients with dyskeratosis congenita. Br J Haematol. 2014;165(3):349-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Townsley DM, Dumitriu B, Liu D, et al. . Danazol Treatment for Telomere Diseases. N Engl J Med. 2016;374(20):1922-1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang QS, Benedetti E, Deater M, et al. . Oxymetholone therapy of fanconi anemia suppresses osteopontin transcription and induces hematopoietic stem cell cycling. Stem Cell Rep. 2015;4(1):90-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cogan JD, Kropski JA, Zhao M, et al. . Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am J Respir Crit Care Med. 2015;191(6):646-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stuart BD, Choi J, Zaidi S, et al. . Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. 2015;47(5):512-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ding H, Schertzer M, Wu X, et al. . Regulation of murine telomere length by Rtel: an essential gene encoding a helicase-like protein. Cell. 2004;117(7):873-886. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.