

Abstract
We built a whole‐body computational model to study the role of the poorly understood vascular endothelial growth factor (VEGF)165b splice isoform in peripheral artery disease (PAD). This model was built and validated using published and new experimental data from cells, mice, and humans, and explicitly accounts for known properties of VEGF165b: lack of extracellular matrix (ECM)‐binding and weak phosphorylation of vascular endothelial growth factor receptor‐2 (VEGFR2) in vitro. The resulting model captures all known information about VEGF165b distribution and signaling in human PAD, and provides novel, nonintuitive insight into VEGF165b mechanism of action in vivo. Although VEGF165a and VEGF165b compete for VEGFR2 in vitro, simulations show that these isoforms do not compete for VEGFR2 at much lower physiological concentrations. Instead, reduced VEGF165a may drive impaired VEGFR2 signaling. The model predicts that VEGF165b does compete for binding to VEGFR1, supporting a VEGFR1‐mediated response to anti‐VEGF165b. The model predicts a key role for VEGF165b in PAD, but in a different way than previously hypothesized.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Despite multiple clinical trials, there are no approved pro‐angiogenic therapies for PAD. A purportedly “anti‐angiogenic” VEGF isoform, VEGF165b, is elevated in PAD. However, understanding of how this isoform contributes to PAD pathology remains lacking.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ We use a multiscale computational model to resolve seeming contradictions in published data, translate from in vitro signaling measurements to predicted signaling in diseased human skeletal muscle, and test prevailing hypotheses about the mechanism of action of VEGF165b.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ Ours is the first model to capture all known information about VEGF165b properties and distribution in the human body. Although competition is observed between VEGF165a and VEGF165b in vitro, our model shows that VEGF165a and VEGF165b do not compete for VEGFR2 at much lower physiological concentrations. Interestingly, VEGF165b does compete with other VEGF and PlGF isoforms for binding to VEGFR1.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This molecular mechanistic insight is key to development of effective pro‐angiogenic strategies for PAD treatment.
Peripheral artery disease (PAD) is a manifestation of chronic atherosclerotic disease in which occlusion of arteries in the legs results in skeletal muscle ischemia, pain, and limited mobility.1 PAD leads to muscle atrophy, capillary rarefaction,2, 3 and other anatomic changes,4 and eventual below‐knee or higher amputation (25–40% 6‐month risk with critical limb ischemia5). Despite this ischemia, sufficient angiogenesis (growth of new capillaries from the existing vascular network) to restore normal perfusion does not seem to occur in PAD. Interestingly, levels of vascular endothelial growth factor (VEGF), considered central to promoting angiogenesis in response to ischemia, are elevated threefold in blood6, 7 and are unchanged at rest in muscle biopsies8 and interstitial fluid of PAD‐afflicted muscle.9 The primary treatments for PAD are: exercise, which can promote VEGF secretion10 but is often painful for patients; and surgical revascularization, for which many patients are not suited and which is not always successful.1 After arterial occlusion, remaining blood flow to the foot occurs via new or remodeled collateral vessels2; angiogenesis is known to precede increases in muscle oxygen uptake in patients with PAD.11 As such, promoting angiogenesis to improve muscle perfusion is considered a promising therapeutic avenue. Despite many clinical trials, there are no approved growth factor‐based therapies (protein or gene‐based delivery of VEGF or fibroblast growth factor‐2, or upregulation of these through transcription factors), due to lack of efficacy and side effects, including edema.1, 12 Although this failure can be partially attributed to poor, spatially inhomogeneous delivery of short duration,13, 14 it is also clear that a lack of understanding of the mechanism behind the signaling impairment in PAD limits selection of appropriate therapeutic strategies.15 Computational models provide a unique potential to examine this signaling complexity, bridging observations in cell culture experiments, animal models that recapitulate human disease only to a limited extent, and human patients with PAD.16
The VEGF family is complex, consisting of five ligand genes, including VEGFA and placental growth factor (PlGF), three receptors (vascular endothelial growth factor receptors (VEGFR)1–3), and multiple co‐receptors, including neuropilin‐1 (NRP1).17 The VEGFRs can be alternatively spliced, producing soluble isoforms (e.g., soluble VEGFR1 (sVEGFR1)) that bind to VEGF, PlGF, and heparin sulfate proteoglycans (HSPGs). The sVEGFR1 levels are increased in mice following hindlimb ischemia (HLI), but not in human PAD.18, 19 VEGFA (hereafter referred to as VEGF), considered the primary proangiogenic protein, can be spliced into numerous isoforms, each with different ability to bind to NRP1 and to HSPGs. The most prevalent in the human body are: VEGF121, which binds to neither the extracellular matrix (ECM) nor NRP1; VEGF165, which binds to both ECM and NRP1; and VEGF189, which binds to ECM more strongly than VEGF165 and also binds to NRP1.20, 21 These isoform‐specific properties have physiological relevance; in murine systems and in human tumors implanted in mice, expression of VEGF121 alone leads to formation of vascular networks consisting of small numbers of wide‐diameter vessels; expression of VEGF165 alone produces a relatively normal phenotype; and expression of VEGF189 alone results in a highly branched network of thin vessels.21
Recently, altered expression of additional splice isoforms—the “VEGFxxxb” isoforms, with different C‐terminal six amino acids (in exon 8)22, 23—has been measured in several disease conditions, including PAD,24, 25 cancer,22, 26 systemic sclerosis,27 and pre‐eclampsia.28 Changes in VEGF splicing can be induced by specific growth factors,29, 30 by exercise,31, 32 and by ECM stiffness,33 although the mechanisms involved in disease‐induced splicing, and even tissue‐specific splicing,34 are not well‐established. Despite only a small change in sequence, VEGF165b, the counterpart of VEGF165a, does not bind to NRP1, and did not bind to heparin or HSPGs in three independent in vitro studies.35, 36, 37 Additionally, despite binding to VEGFR2 with the same affinity as VEGF165a 36, 37 (Figure 1 b), VEGF165b phosphorylates VEGFR2 only weakly, a property hypothesized to result from its lack of NRP1‐binding.36 This poor activation of VEGFR2 suggested that VEGF165b may be anti‐angiogenic, acting as a “brake” to prevent binding of “a” isoforms to VEGFR2 and reduce signaling,23, 25 although other studies have suggested that “b” isoforms are indeed weakly pro‐angiogenic in vitro and in tumors.35, 38 Study of the “a” and “b” isoforms in vivo has been complicated by difficulties in achieving consistent measurements, detection of both classes of isoforms by commonly used antibodies and/or lack of clarify about which isoforms are measured in a specific study, splicing differences between mice and humans,39 and the difficulty of detecting low‐abundance VEGFxxxb mRNA in mice.40, 41 As such, although VEGF165b has been detected in healthy humans,42 it is secreted at much higher levels in the blood of patients with PAD than in healthy controls,25 and is increased in the adipose tissue of obese patients,43 reliable quantification of the levels of total and free VEGF165a and VEGF165b protein in healthy and diseased human tissues (e.g., via biopsy vs. microdialysis) and blood remains elusive. Nonetheless, promising improvements in blood flow observed in diabetic mice subjected to HLI following treatment with an anti‐VEGF165b antibody25 suggest that VEGF165b may be an important, albeit poorly understood, missing piece in the PAD puzzle. Using a computational model, we can screen potential ranges of relative secretion of these isoforms, to understand the implications of splicing changes on VEGF distribution and endothelial receptor signaling. This will deepen our understanding of how signaling is perturbed in disease, a critical step in the design of the next generation of pro‐angiogenic therapies.
Figure 1.
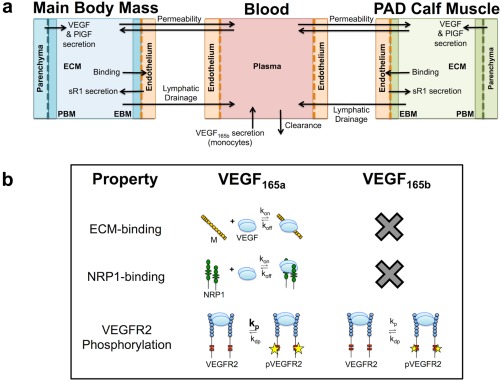
Overview of model structure and vascular endothelial growth factor (VEGF)165b properties. (a) Structure of the multiscale whole‐body compartment model, incorporating peripheral artery disease (PAD)‐specific changes in geometry and molecular expression of the calf muscle, and secretion of VEGF165b into the blood. (b) VEGF165b lacks the ability to bind to heparin sulfate proteoglycans (HSPGs) and neuropilin‐1 (NRP1), and phosphorylates vascular endothelial growth factor receptor (VEGFR)2 weakly. EBM, endothelial basement membrane; ECM, extracellular matrix; PBM, parenchymal basement membrane; PlGF, placental growth factor; pVEGFR2, phosphorylated VEGFR2.
OBJECTIVES
Our objective was to develop a systems pharmacology model of endogenous VEGF165b and other VEGF isoforms in PAD in order to better understand: (1) the distribution of VEGF165b in the body, as compared to that of VEGF165a; (2) the effects of VEGF165b on VEGFR1 and VEGFR2 activation; and (3) resulting signaling changes in PAD (due to altered VEGF165b expression) that may be responsible for the observed impaired angiogenic response to ischemia. We aim to develop a platform that can be used to screen potential pro‐angiogenic therapies for PAD. In achieving these objectives, we improve greatly on a previous pharmacokinetic/pharmacodynamic model of PAD,44 which was unable to capture the signaling impairment observed in PAD, by incorporating separate simulation of VEGFR2 ligand‐binding and site‐specific phosphorylation,45, 46 and by incorporating recent discoveries about VEGF165b (Figure 1 b) and its relevance to PAD.8, 25, 43 By iteratively building upon and validating our models using both previously unpublished and published data, in vitro and in vivo, we improve the predictive capabilities without adding many parameters at a time. These improvements allow us to predict clinically relevant quantities that are difficult or impossible to measure in vivo, such as VEGF distribution and VEGFR1 and VEGFR2 signaling in muscle tissue, explicitly accounting for physiological processes (Figure 1 a), and maintaining physiological levels of ligands and receptors.
METHODS
To study the distribution of VEGF165b within the human body, we modified our previously published three‐compartment model,46 which includes the blood, the main bulk of body tissue (main body mass), and a calf muscle (gastrocnemius + soleus) with PAD‐specific changes in geometry and molecular expression44 (Figure 1). Within the tissue compartments, the relative fractions of interstitial space, ECM and basement membrane, and endothelial and other cells (including myocytes) are estimated based on histology and other measurements44 (Supplementary Figure S1). Detailed model parameterization and experimental protocols can be found in the Supplemental Methods and Tables S1–S14.
RESULTS
Modeling the role of VEGF165b in PAD
To capture the role of VEGF165b in PAD, we incorporated: (1) its measured binding properties (Figure 1 b); (2) changes in expression of VEGF165a and VEGF165b in skeletal muscle; and (3) secretion of VEGF165b into blood (e.g., by monocytes). In the tissue compartments, we screened the possible range of relative VEGF165a and VEGF165b expression, maintaining constant free VEGF levels in plasma to mimic the roughly unchanged total VEGF protein and free VEGF in interstitial fluid in human PAD8, 9 (Supplementary Figure S2e, Supplementary Model Fitting). In the bloodstream, we then increased secretion of VEGF165b to capture the roughly threefold higher observed serum VEGF in patients with PAD than healthy humans.25 Inclusion of VEGF165b secretion into the bloodstream was necessary to achieve target blood VEGF levels without the unrealistic tissue VEGF concentrations observed in previous models.44 The resulting model matches all known information about VEGF distribution in PAD (Figure 2 a).
Figure 2.
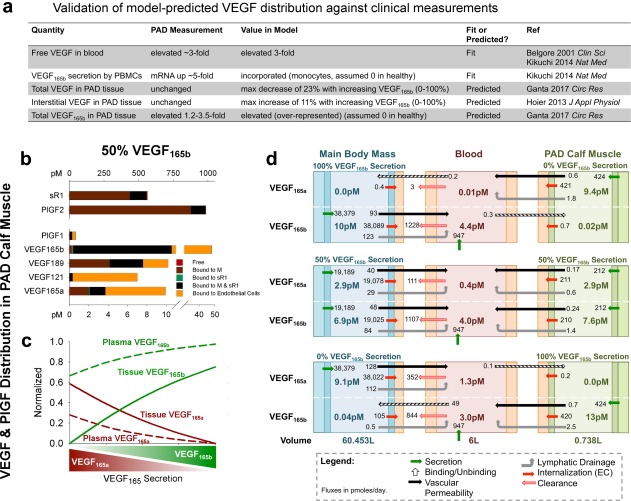
Vascular endothelial growth factor (VEGF)165b is predicted to be over‐represented in tissue and blood compared to VEGF165a. (a) Comparison of model‐predicted VEGF distribution to clinical measurements in patients with peripheral artery disease (PAD) vs. healthy control subjects. The VEGF splicing switch was modeled as a modification of the relative secretion rates of VEGF165a and VEGF165b, in agreement with measured changes in VEGF mRNA in PAD.25 (b) Predicted distribution of VEGF and placental growth factor (PlGF) isoforms and soluble vascular endothelial growth factor receptor (sVEGFR)1 in the PAD calf muscle, with equal secretion of VEGF165a and VEGF165b by parenchymal cells. Bound to M: ligand bound to extracellular matrix or basement membrane; bound to sVEGFR1: ligand bound to soluble VEGFR1 alone; bound to M and sVEGFR1: ligand bound to both matrix protein and soluble VEGFR1. (c) Fraction of total VEGF in plasma and PAD calf muscle (tissue) that is VEGF165a and VEGF165b, as a function of varying VEGF165 splicing (in both tissue compartments). (d) Steady‐state net flow profiles for VEGF165a and VEGF165b between the PAD calf muscle, blood, and main body mass, with different relative secretion of VEGF165a and VEGF165b in the PAD calf muscle and main body mass. PBMC, peripheral blood mononuclear cell.
Pharmacokinetics of VEGF165b: Predicted over‐representation in tissue and blood
To understand the pharmacokinetics of VEGF165b, as compared to VEGF165a, we examined the predicted distributions of these isoforms in the PAD calf muscle and plasma at steady‐state. When VEGF165a and VEGF165b were secreted at equal rates in tissue (fractional VEGF165b secretion = 50%), the model predicts that VEGF165b protein is over‐represented compared to VEGF165a in tissue (Figure 2 b), both as extracellular ligand (Supplementary Figure S2c) and endothelial cell‐bound ligand (Figure 2 b, orange). This over‐representation (relative to fractional secretion) results from: (a) lack of ECM‐binding, leading to 2.4‐fold more free VEGF165b than VEGF165a in the PAD calf muscle; combined with (b) lack of NRP1‐binding slowing binding to VEGFR2 and subsequent recycling, and thus slowing turnover of VEGF165b‐VEGFR2 complexes.45, 46, 47 The model predicts that this over‐representation of VEGF165b in total tissue VEGF and free VEGF in blood (Figure 2 c) is predicted to occur at all VEGF165b levels, with a larger difference in blood than tissue due to secretion of VEGF165b (e.g., by monocytes) into the bloodstream.
To further probe the differential distribution of VEGF165a and VEGF165b, we calculated the net steady‐state secretion, transport, consumption, and clearance of each isoform in each compartment, at different fractional VEGF165b secretion rates in the PAD calf muscle and main body mass (Figure 2 d). The first thing to note here is that, consistent with our previous models,44, 46 most tissue‐produced VEGF is consumed by local endothelial cells. As such, VEGF isoform secretion in one tissue compartment has minimal effect on VEGF isoform concentrations in the other compartment, suggesting that local VEGF isoform secretion is the key driver of local tissue signaling. A small amount of intravasation of VEGF165b and VEGF165a from blood into tissues is predicted only when the two tissue compartments exclusively produce different VEGF isoforms. Over‐representation of VEGF165b in free tissue VEGF is evident with equal secretion of the two isoforms (Figure 2 d, middle).
We next examined the potential of plasma VEGF165b as a biomarker of VEGFR signaling in the PAD calf muscle. We found that, due to its larger size, the main body mass is predicted to contribute the bulk of tissue‐derived VEGF in the bloodstream (Supplementary Figure S2b); thus, blood VEGF isoform levels are likely a poor biomarker of VEGF isoform levels in the PAD calf muscle. This prediction is consistent with the lack of correlation between serum VEGF165b and ankle‐brachial index in patients with PAD, as measured by Kikuchi et al.,25 and highlights the need for tissue biopsy measurements of signaling or microdialysis measurements of local VEGF isoform concentrations to accurately predict patient‐specific signaling state.
VEGF165b is over‐represented in binding to endothelial VEGFR1 and VEGFR2
We next “zoomed in” on the endothelial‐bound fraction of tissue VEGF and PlGF to examine growth factor binding to endothelial VEGFR1 and VEGFR2. With equal secretion of VEGF165b and VEGF165a in the PAD calf muscle, VEGF165b is predicted to dominate binding to both VEGFR1 and VEGFR2 (Figure 3 a), with higher (but still low) receptor occupancy (Figure 3 c; 14% and 10% surface occupancy, respectively) than previously predicted in healthy tissue46 (Supplementary Figure S3a). This is a direct result of lack of binding to NRP1 and ECM by VEGF165b (see Pharmacokinetics section, Supplementary Results). As fractional secretion of VEGF165b increases, the model predicts increasing dominance in receptor binding by VEGF165b, with equivalent binding of VEGF165a and VEGF165b to VEGFR2 when only 25% of secreted VEGF165 is VEGF165b, and even more dramatic increases in VEGF165b‐VEGFR1 binding (Figure 3 b). As VEGF165b increases, surface endothelial VEGFR1 occupancy is predicted to increase, whereas surface VEGFR2 occupancy is predicted to decrease, and total VEGFR2 occupancy remains constant (Figure 3 c), suggesting a shift in relative signaling by VEGFR2 vs. VEGFR1.
Figure 3.
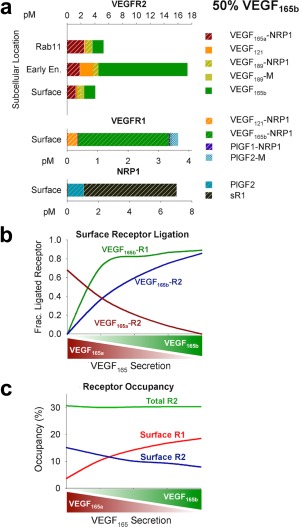
Vascular endothelial growth factor (VEGF)165b is predicted to dominate endothelial receptor binding. (a) Ligands bound to endothelial vascular endothelial growth factor receptor (VEGFR)2 on the cell surface, in early (Rab4/5) endosomes, and recycling (Rab11) endosomes, and cell surface VEGFR1 and neuropilin‐1 (NRP1). Unoccupied receptor levels not shown. Units: pM of total tissue in the peripheral artery disease (PAD) calf muscle. Complexes not listed are present at levels too low to be seen in the figure. (b) Fraction of ligand‐bound endothelial cell surface VEGFR1 and VEGFR2 bound to VEGF165a and VEGF165b, as a function of fractional VEGF165b secretion. Note that VEGF165a‐VEGFR1 binding is too low to be visualized here (see a). (c) Percentage of endothelial cell surface VEGFR1 and VEGFR2 bound to any ligand, as a function of fractional VEGF165b secretion. PlGF, placental growth factor.
Novel insight gained by testing mechanistic hypotheses
What seems to be conflicting information in the literature can sometimes be resolved by using a computational model to directly compare experiments performed under different conditions. Here, we use our model to resolve confusion over two key VEGF165b hypotheses.
Hypothesis 1: VEGF165b is a weak agonist of VEGFR2
By explicitly simulating VEGFR2 ligand‐binding and phosphorylation as separate processes, we can now for the first time account for weak phosphorylation of VEGFR2 by VEGF165b, and explore the in vivo endothelial signaling implications of increased VEGF165b expression in PAD. We modified the phosphorylation rates as opposed to dephosphorylation rates because dephosphorylation is dependent on tyrosine site and subcellular location45 as opposed to VEGF isoform.
We fit the phosphorylation rate constant (kp) for VEGFR2 upon binding of VEGF165b (as compared to VEGF165a) to in vitro data from porcine aortic endothelial cells transfected with VEGFR2 and NRP1 by Kawamura et al.35 using our previously published cell‐level model45 (Figure 4a 35, 43, 45 and Supplementary Figure S4a). The required reduction in kp to fit experimental data (from 1 s−1 for VEGF165a to 8x10−4 s−1 for VEGF165b) demonstrates that lack of binding to NRP1 by VEGF165b, which is accounted for in our simulations, is not sufficient to explain the weak phosphorylation of VEGFR2 observed following stimulation with VEGF165b. Together, the experimental data and our model show that, although phosphorylation of VEGF165a‐VEGFR2 is fast, activation of VEGF165b‐VEGFR2 is slow compared to VEGF‐VEGFR2 binding. We validated this prediction against independent data from ex vivo fat pads (Figure 4 b)43; the optimized kp from above (red line) captured a reduction in VEGFR2 phosphorylation as VEGF165b increased, which is not captured with strong (as VEGF165a) phosphorylation of VEGFR2 (green line). This result demonstrates the need to explicitly account for weak activation of VEGFR2 by VEGF165b to accurately predict signaling in tissues.
Figure 4.
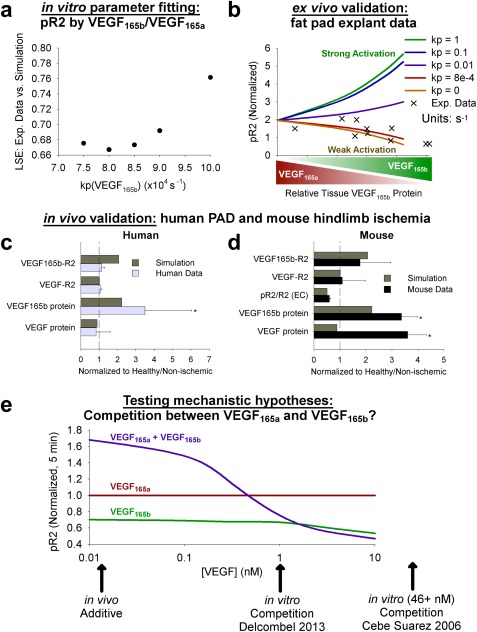
Implications of weak vascular endothelial growth factor receptor (VEGFR)2 phosphorylation by vascular endothelial growth factor (VEGF)165b in vitro and in vivo. (a) The phosphorylation rate for VEGFR2 bound to VEGF165b was fit to in vitro data from Kawamura et al.,35 on pR2 following stimulation with VEGF165b or VEGF165a at different concentrations. Axes units: 10−4 s−1. The optimized value for use in the model for VEGF165b was chosen as the value that minimized the least squared error (LSE; y‐axis) between simulations and experimental data, as elaborated upon in the Supplementary Methods. (b) Validation of optimized kp value for VEGF165b (8*10−4 s−1) against measurements of pR2 as a function of relative VEGF165b in ex vivo human fat pads as measured by Ngo et al.43 (c,d) Validation of in vivo compartment model against human peripheral artery disease (PAD) and mouse hindlimb ischemia measurements. Simulations use 75% fractional VEGF165b secretion in the PAD calf muscle, normalized to the 25% VEGF165b secretion case. (c) Human data are total tissue measurements from PAD muscle biopsies, normalized by healthy patient values. Asterisks denote significance using an unpaired, two‐tailed t test with P ≤ 0.05 (n = 10 PAD subjects; 5 normal subjects for VEGF protein; and 6 normal subjects for VEGF‐VEGFR binding). (d) Mouse measurements are from gastrocnemius muscle 3 days after femoral artery ligation, and represent total tissue measurements (receptor‐bound ligand and VEGF protein) or CD31+ cells (pR2/R2), normalized by equivalent quantities in the contralateral gastrocnemius muscle. Asterisks denote significance using an unpaired, two‐tailed t test with P ≤ 0.05 (n = 4). (e) Dose‐dependent competition between VEGF165a and VEGF165b. The pR2 at 5 minutes after VEGF addition, normalized by VEGF165a at each concentration. Simulations performed using endothelial cell culture model, including VEGFR2 and NRP1, but not VEGFR1.45
To validate this mechanistic insight in vivo, we compared model predictions of VEGF and VEGF165b protein levels and VEGFR2 ligation and phosphorylation to measurements in human PAD and murine HLI from an extended analysis of the data presented in Ganta et al.8 To match the roughly threefold increase in VEGF165b protein observed in human PAD and murine HLI, we compared simulation results for 75% fractional VEGF165b secretion to those for 25% fractional VEGF165b secretion. These simulations predict the effect of changing VEGF165b secretion only; we made no other changes in tissue anatomy or molecular expression. The model accurately captures the increase in VEGF165b without substantial increases in total VEGF or VEGF‐R2 observed in muscle biopsies of patients with PAD, validating the model's predictive power for human PAD (Figure 4 c), and suggesting altered VEGF splicing is a key driver of the observed signaling changes. We also compared these model predictions to murine HLI; whereas tissue VEGF levels increase substantially in HLI,19 the model accurately captures trends in VEGF binding to VEGFR2 and the experimentally observed lack of increase (nonsignificant decrease) in VEGFR2 phosphorylation (Figure 4 d). This suggests that, although there are many differences between human PAD and murine HLI (e.g., geometric scaling,14 sVEGFR1 expression,19 and time‐scale), receptor‐level signaling seem to be similar in this case (as supported by the recent work of Ganta et al.8), giving us confidence in the relevance of comparisons between model predictions and experimental data in mice.
Hypothesis 2: VEGF165b does not compete with VEGF165a for binding to VEGFR2 at physiological concentrations
We leveraged the newly fit and validated model to test the prevailing hypothesis that VEGF165a and VEGF165b compete for binding to VEGFR2, leading to observed reductions in VEGFR2 phosphorylation in some experiments. To do this, we simulated VEGFR2 phosphorylation in cultured endothelial cells following stimulation with VEGF165a, VEGF165b, or both (Figure 4 e). The model captured experimentally observed competition at in vitro concentrations of 1 nM or higher.36, 37 However, competition is concentration‐dependent, and the model predicts that, due to low receptor occupancy (Figure S4b, dotted lines) at physiological concentrations (1–15 pM),46 VEGF165a and VEGF165b do not compete for VEGFR2 phosphorylation in vivo. We then further examined signaling in vivo using the compartment model, with PAD‐specific molecular expression and physiology in the calf muscle. This model predicted that the impaired VEGF receptor signaling with increasing VEGF165b expression observed in PAD results from reduced expression of other VEGF isoforms (as total VEGF levels are unchanged), rather than from competition between VEGF isoforms for receptor binding, as observed in vitro (Figure 4 e and Figure 5 d). This conclusion, which could not have been reached with experiments alone, has important implications for therapy; it suggests strategies designed to increase local VEGFxxxa secretion or delivery will have a larger impact on VEGFR2 phosphorylation than antibody‐based therapies designed to remove VEGF165b.
Figure 5.
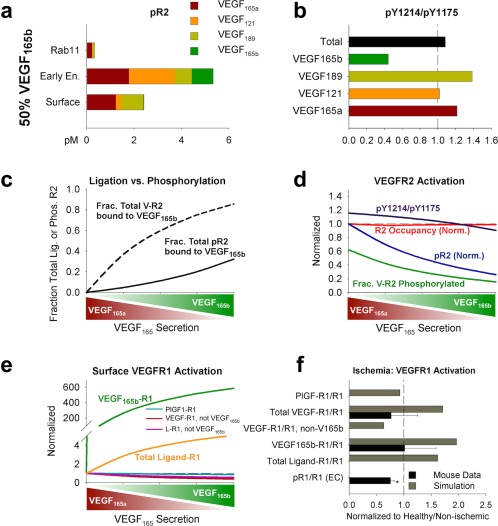
In vivo vascular endothelial growth factor receptor (VEGFR) activation varies with vascular endothelial growth factor (VEGF)165b levels in simulated human peripheral artery disease (PAD). (a) VEGFR2 phosphorylation (on ≥1 tyrosine) as a function of VEGF isoform and subcellular location in the PAD calf muscle, with 50% fractional VEGF165b secretion. (b) VEGF isoform‐specific, neuropilin‐1 (NRP1)‐dependent trafficking and subcellular location‐specific dephosphorylation rates lead to isoform‐specific predictions of relative phosphorylation on Y1175 and Y1214. Fifty percent fractional VEGF165b secretion. (c) VEGFR2 ligation and phosphorylation by VEGF165b, as a function of local fractional VEGF165b secretion. (d) Total (cell surface + endosomes) VEGFR2 phosphorylation as a function of local fractional VEGF165b secretion. (e) Endothelial surface VEGFR1 ligation as a function of local fractional VEGF165b secretion. Note that expression of placental growth factor (PlGF) remains constant across conditions. L: ligand (VEGF or PlGF). (f) Comparison of experimental VEGF‐VEGFR1 and pR1 in murine hindlimb ischemia and model predictions of VEGFR1 ligation. Simulations use the PAD calf muscle, with 75% secretion of VEGF165b, normalized to the 25% VEGF165b secretion case. Mouse measurements are from gastrocnemius muscle 3 days after femoral artery ligation, and represent CD31+ cells, normalized to equivalent quantities in the contralateral muscle. Asterisks denote significance using an unpaired, two‐tailed t test with P ≤ 0.05 (n = 10 for each group).
Putting the above together, we can conclude that VEGF165b is a weak agonist of VEGFR2, but does not compete with VEGF165a for binding to VEGFR2 at physiological concentrations.
VEGF165b regulates signaling of both VEGFR2 and endothelial VEGFR1 in vivo
We next explored model predictions of VEGFR2 phosphorylation and VEGFR1 ligand binding in vivo. Despite being the dominant ligand bound to VEGFR2 in our simulations, VEGF165b is predicted to contribute only modestly to pR2, even without competing with other isoforms for VEGFR2, due to its weak ability to phosphorylate VEGFR2 (Figure 5 , Supplemental Results). The fraction of ligand‐bound VEGFR2 phosphorylated at steady‐state decreases from 62% with no VEGF165b secretion to 16% with 100% relative VEGF165b secretion (Figure 5 a,d, and Supplementary Figure S5a).
A lack of detailed understanding of VEGFR1 phosphorylation by different ligands makes explicit prediction of VEGFR1 signaling difficult, although VEGF and PlGF activate different tyrosine sites,48 and VEGF165b seems not to phosphorylate Y1333 on VEGFR1, upstream of signal transducer and activator of transcription 3 (STAT3) in PAD.8 As a step toward this end, we examined the profile of ligands predicted to bind endothelial cell surface VEGFR1 at steady‐state, with varying relative VEGF165b secretion (Figure 5 e and Supplementary Figure S5b). With increasing VEGF165b, ligation of VEGFR1 by other VEGF isoforms and by PlGF is predicted to decrease. This reduction in PlGF‐VEGFR1 suggests that, unlike VEGFR2, and consistent with recent data from Ganta et al.,8 competition between VEGF165b and other ligands does occur on VEGFR1. Comparing these model predictions of VEGFR1 binding to reduced VEGFR1 Y1333 phosphorylation in murine HLI8 suggests that both PlGF and non‐VEGF165b VEGF isoforms may contribute to VEGFR1 Y1333 phosphorylation (Figure 5 f). This result emphasizes the need for careful quantitative studies to discriminate between physiological and molecular conditions under which competition does or does not play a role, and to further elucidate the role of VEGFR1 on endothelial and other cells in PAD.
VEGF165b overexpression experiments confirm competition for VEGFR1 but not VEGFR2
To this point, we have focused on a switch in expression of VEGF165a and VEGF165b, with total VEGF remaining constant. However, this is not an accurate reflection of murine HLI, in which total VEGF increases,19 or time‐varying changes in VEGF secretion in exercising humans or during intermittent claudication. As such, we studied the impact of changes in VEGF165b expression, independent of VEGF165a secretion. We first examined experimental overexpression of VEGF165b. Using an extended analysis of the data presented in Ganta et al.,8 and assuming changes in expression of VEGF165b only, measurements of total VEGFA and VEGF165b suggest that in nonischemic Balb/c gastrocnemius muscle, VEGF165b represents ∼24% of total VEGF, increasing to 64% in the VEGF165b overexpression experiment. As such, we used 25% fractional VEGF165b secretion as our model baseline, to match the experimentally observed protein levels, and increasing VEGF165b secretion 3.5‐fold in the overexpression case to match the observed relative increases in protein. The model mirrors the small (∼10%), nonsignificant increase in VEGFR2 phosphorylation observed experimentally (Figure 6 a), and predicts decreased PlGF‐VEGFR1 binding (Figure 6 b), potentially consistent with the nonsignificant decrease in VEGFR1 phosphorylation observed, and again supporting the hypothesis that ligands compete for VEGFR1 but not VEGFR2. We further investigated the sensitivity of VEGFR1 and VEGFR2 signaling to small changes in VEGF165b expression (Figure 6 c and Supplementary Figure S6a). The model predicts that VEGFR1 is consistently more sensitive to changes in VEGF165b expression than VEGFR2, and that signaling changes less in response to varying VEGF165b than varying VEGF165a (Figure 6 c and Supplementary Figure S6b).
Figure 6.
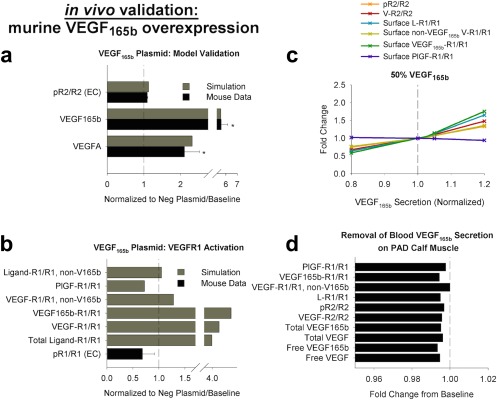
Increased expression of vascular endothelial growth factor (VEGF)165b with constant VEGF165a alters vascular endothelial growth factor receptor (VEGFR)1 activation more than VEGFR2 activation. (a,b) Validation of in vivo compartment model against mouse VEGF165b overexpression. Simulations in the peripheral artery disease (PAD) calf muscle use 25% VEGF165b secretion as the nonischemic baseline. Mouse measurements are from nonischemic gastrocnemius muscle 7 days after transfection with VEGF165b plasmid or a control plasmid, and represent total tissue measurements (receptor‐bound ligand and VEGF protein) or CD31+ cells (pR1/R1), normalized by equivalent quantities in the control group. (a) VEGF protein and endothelial VEGFR2 phosphorylation. Asterisks denote significance using an unpaired, two‐tailed t test with P ≤ 0.05 (n = 4 per group). (b) Experimental endothelial VEGFR1 phosphorylation, compared to simulated VEGFR1 ligand‐binding (n = 4 per group). (c) Simulation of direct increases or decreases in local VEGF165b secretion in the PAD calf muscle, at 50% fractional VEGF165b secretion in both tissue compartments, normalized to baseline quantities. (d) Minimal impact of removing monocyte secretion of VEGF165b into the bloodstream is predicted on quantities in the PAD calf muscle. PlGF, placental growth factor.
DISCUSSION
Our objective in building this model was to investigate in detail the implications of the experimentally measured properties of VEGF165b—lack of ECM binding, lack of NRP1 binding, and weak phosphorylation of VEGFR2—on the role of this isoform in PAD. We leveraged a previously built and validated computational model that accounts explicitly for differences in ECM and NRP1 binding by VEGF isoforms, as well as simulating binding, trafficking, and tyrosine site‐specific phosphorylation of VEGFR2 as distinct, although related, processes.46 This framework enabled us to directly implement the unique properties of VEGF165b, making predictions of disease‐specific in vivo concentrations and signaling that are difficult, if not impossible, to quantify experimentally. In doing so, we built a model that is qualitatively consistent with all observed in vitro behaviors of VEGF165b and all available knowledge of VEGF distribution in human PAD (Figure 2 a). This process sheds light onto the mechanism of action of VEGF165b in PAD (Table 1) more accurately and more completely than previous models have done44, 49 (Supplementary Table S15), providing insight that is critical for design of future pro‐angiogenic therapies.
Table 1.
Key model predictions
| Prediction | Experimental basis or validation | Therapeutic implications |
|---|---|---|
| VEGF165b is over‐represented in tissue | Elevated muscle VEGF165b in PAD and murine hindlimb ischemia ( 8, 49) (Figure 2) | Understand pharmacokinetics of VEGF165b to better predict its role in disease and therapy response |
| VEGF165b secretion into the blood has minimal effect on baseline VEGFR signaling | Unchanged total muscle VEGFA in PAD ( 8, 9) (Figures 2 and 6) | Blood VEGF165b is neither a good biomarker nor a therapeutic target for pro‐angiogenic therapy |
| VEGF165b is a weak agonist of VEGFR2 phosphorylation in vivo | Consistent with in vitro observations ( 35, 36, 37), ex vivo measurements (fat pads) ( 43), and in vivo data ( 8) (Figures 4 and 6) | Translate in vitro observations into an in vivo, physiological context to predict changes in signaling in disease |
| Reduced VEGF165a in PAD contributes to reduced VEGFR2 phosphorylation | Prediction is result of properties measured in vitro ( 35, 36, 37) placed in a physiological context (Figure 4) | VEGF165b‐VEGFR2 binding alone is not responsible for reduced angiogenic signaling in PAD. Affects therapy design. |
| VEGF165b does not compete for binding to VEGFR2, but does compete for binding to VEGFR1 | VEGFR1 phosphorylation is increased by delivery of anti‐VEGF165b and decreased by overexpression of VEGF165b, but VEGFR2 phosphorylation is not substantially affected ( 8) (Figures 4, 5, 6) | Understand mechanism of action of VEGF165b, and how anti‐VEGF165b induces improved perfusion recovery in mice ( 25). Leverage for design of pro‐angiogenic therapies. |
PAD, peripheral artery disease; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
Interpretation of model predictions
The results presented in this paper demonstrate that VEGF165b does indeed play a role in the pathology of PAD, but in a different way than previously hypothesized.25 For example, the model predicts that, contrary to in vitro observations, VEGF165b does not compete with other VEGF isoforms for binding to VEGFR2 in vivo, due to the low VEGF concentrations and VEGFR2 occupancy predicted in physiological conditions (in both healthy and ischemic tissue compartments). Instead, as total VEGF levels are roughly constant in PAD‐afflicted tissue, the model suggests that reduced VEGF165a, concomitant with increasing VEGF165b, is the source of reduced VEGFR2 phosphorylation observed in some studies.25, 43 As another example, consistent with experimental data,8 the model predicts that modest increases in VEGF165b will indeed slightly increase pR2, not reduce it, and decreased VEGF165b will decrease pR2 slightly. Interestingly, and again consistent with Ganta et al.8 and the previously unpublished experimental data presented here, the model does predict competition between VEGF165b and other ligands for binding to VEGFR1, which seems to be poorly or not at all phosphorylated by VEGF165b on tyrosine‐1333. This supports a VEGFR1‐mediated pro‐angiogenic response to anti‐VEGF165b treatment, as opposed to a VEGFR2‐mediated response. The model also predicts that increased secretion of VEGF165b into the bloodstream does not play a major role in VEGFR signaling in tissue, with locally produced VEGF dominating the local signaling environment. The model does predict over‐representation of VEGF165b protein and VEGF165b receptor binding in tissue, suggesting that microdialysis or muscle biopsy measurements of VEGF165b may be a good predictor of local angiogenic impairment.
Open questions
There are still many open questions about the role and properties of VEGF165b, which limit our ability to fully interpret our model predictions, but which, with new experimental data, this model can be leveraged to answer. These measurements would increase our ability to confidently predict the effectiveness of potential pro‐angiogenic therapies. For example, understanding at the molecular level exactly how VEGF165b binds with normal affinity to VEGFR2 but induces only weak phosphorylation would be instructive. One hypothesis involves changes in homodimerization and heterodimerization of ligands or receptors. This study and the work of Ganta et al.8 also motivate a better understanding of VEGFR1 binding, trafficking, and differences in activation by VEGF165b, other VEGF isoforms, and PlGF, in order to better target this pathway in PAD and other diseases (see Supplementary Results for details).
Finally, and perhaps most critically, we are limited by available quantitative measurements of absolute and relative levels of VEGF165a and VEGF165b in blood, healthy tissue, and diseased tissue. Particularly, there is a need for careful attention to which isoforms are detected by a given antibody, to ensure that VEGFxxxb isoforms are accounted for. Although measurements of difference in total protein between healthy and diseased tissue are available in the form of Western blot data, which was used here to construct and validate this model, accurate quantitative measurements (e.g., enzyme‐linked immunosorbent assay) are key to pin down the distribution of these isoforms, as well as other VEGF and PlGF isoforms.46 For example, how much VEGF165b is present in healthy tissue remains an open question, although we know it decreases in several types of cancer,22, 23 and increases in PAD and white adipose tissue.8, 25, 43 We used our model to explore the dynamic range of relative VEGF165a and VEGF165b secretion and the implications of this splicing switch for signaling in a way that has not been possible experimentally. However, to fully understand signaling in disease, we need to know where patients reside on this spectrum. In this model, we assumed high VEGF165b only in the blood and in a relatively small PAD calf muscle, whereas in real patients with extensive PAD or systemic cardiovascular disease and/or large quantities of adipose tissue, the relative amounts of “healthy” and “diseased” tissue with high VEGF165b expression may be very different, potentially altering the VEGF165b pharmacokinetic predictions presented here. Although such reliable and quantitative measurements remain challenging,40 there is hope that the future will bring the required tools (e.g., consistent quantitative VEGF isoform‐specific enzyme‐linked immunosorbent assay and/or tissue microdialysis), which, with the help of quantitative frameworks to integrate the data, will continue to improve our understanding of PAD and VEGF165b, leading to more successful therapy design and clinical outcomes.
CONCLUSIONS
This model, which we believe is the first to translate in vitro observations of VEGF165b properties into the context of human PAD, provides novel insight into questions that have remained challenging to answer due to a lack of reliable, quantitative, and feasible measurement techniques. We integrated existing knowledge and previously unpublished data to test prevailing hypotheses about VEGF165b mechanism of action in PAD, and highlight important future questions and measurements on the path toward more effective treatments for PAD. The model's ability to capture key aspects of VEGF signaling in human PAD and murine HLI, as well as predict response to perturbation (VEGF165b overexpression) gives us confidence that the insight elucidated here is meaningful and relevant. In the future, this work can be extended to examine VEGF165b in other diseases (e.g., cancer, obesity, and pre‐eclampsia), and to study promising pro‐angiogenic therapies.
Conflict of Interest
The authors declared no conflict of interest.
Source of Funding
This work was funded by National Defense Science and Engineering Graduate (NDSEG) Fellowship (to L.E.C.), R01HL101200 (to F.M.G. and B.H.A.), 1R01 HL116455 (to B.H.A.), 1R01 HL121635 (to B.H.A.), R00HL093219 (to F.M.G.), a Sloan Research Fellowship (to F.M.G.), and the American Heart Associate Scientist Development Grant 16SDG30340002 (to V.C.G.).
Author Contributions
L.E.C. and F.M.G. wrote the manuscript. L.E.C. and F.M.G. designed the research. L.E.C. and V.C.G. performed the research. L.E.C., V.C.G., B.H.A., and F.M.G. analyzed the data.
Supporting information
Supporting Information 1
Supporting Information 2
Supporting Information 3
Supporting Information 4
Supporting Information 5
Supporting Information 6
Supporting Information 7
Supporting Information 8
Supporting Information 9
Supporting Information 10
Supporting Information 11
Supporting Information 12
Supporting Information 13
Supporting Information 14
Supporting Information 15
Supporting Information 16
Supporting Information 17
Supporting Information 18
Supporting Information 19
Supporting Information 20
Supporting Information 21
Supporting Information 22
Supporting Information 23
Supporting Information 24
Supporting Information 25
Supporting Information 26
Supporting Information 27
Supporting Information 28
Supporting Information 29
Supporting Information 30
References
- 1. Annex, B.H. Therapeutic angiogenesis for critical limb ischaemia. Nat. Rev. Cardiol. 10, 387–396 (2013). [DOI] [PubMed] [Google Scholar]
- 2. Robbins, J.L. et al Relationship between leg muscle capillary density and peak hyperemic blood flow with endurance capacity in peripheral artery disease. J. Appl. Physiol. (1985) 111, 81–86 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Askew, C.D. et al Skeletal muscle phenotype is associated with exercise tolerance in patients with peripheral arterial disease. J. Vasc. Surg. 41, 802–807 (2005). [DOI] [PubMed] [Google Scholar]
- 4. Baum, O. , Djonov, V. , Ganster, M. , Widmer, M. & Baumgartner, I. Arteriolization of capillaries and FGF‐2 upregulation in skeletal muscles of patients with chronic peripheral arterial disease. Microcirculation 12, 527–537 (2005). [DOI] [PubMed] [Google Scholar]
- 5. Norgren, L. et al Inter‐Society Consensus for the Management of Peripheral Arterial Disease (TASC II). J. Vasc. Surg. 45 (suppl. S), S5–S67 (2007). [DOI] [PubMed] [Google Scholar]
- 6. Belgore, F.M. , Blann, A.D. & Lip, G.Y. Measurement of free and complexed soluble vascular endothelial growth factor receptor, Flt‐I, in fluid samples: development and application of two new immunoassays. Clin. Sci. (Lond). 100, 567–575 (2001). [PubMed] [Google Scholar]
- 7. Blann, A.D. , Belgore, F.M. , McCollum, C.N. , Silverman, S. , Lip, P.L. & Lip, G.Y.H. Vascular endothelial growth factor and its receptor, Flt‐1, in the plasma of patients with coronary or peripheral atherosclerosis, or type II diabetes. Clin. Sci. (Lond). 102, 187–194 (2002). [PubMed] [Google Scholar]
- 8. Ganta, V.C. , Choi, M. , Kutateladze, A. & Annex, B.H. VEGF165b modulates endothelial VEGFR1‐STAT3 signaling pathway and angiogenesis in human and experimental peripheral arterial disease. Circ. Res. 120, 282–295 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hoier, B. et al Angiogenic response to passive movement and active exercise in individuals with peripheral arterial disease. J. Appl. Physiol. (1985) 115, 1777–1787 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hoier, B. & Hellsten, Y. Exercise‐induced capillary growth in human skeletal muscle and the dynamics of VEGF. Microcirculation 21, 301–314 (2014). [DOI] [PubMed] [Google Scholar]
- 11. Duscha, B.D. et al Angiogenesis in skeletal muscle precede improvements in peak oxygen uptake in peripheral artery disease patients. Arterioscler. Thromb. Vasc. Biol. 31, 2742–2748 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grochot‐Przeczek, A. , Dulak, J. & Jozkowicz, A. Therapeutic angiogenesis for revascularization in peripheral artery disease. Gene 525, 220–228 (2013). [DOI] [PubMed] [Google Scholar]
- 13. Ozawa, C.R. et al Microenvironmental VEGF concentration, not total dose, determines a threshold between normal and aberrant angiogenesis. J. Clin. Invest. 113, 516–527 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ylä‐Herttuala, S. , Markkanen, J.E. & Rissanen, T.T. Gene therapy for ischemic cardiovascular disease: some lessons learned from the first clinical trials. Trends Cardiovasc. Med. 14, 295–300 (2004). [DOI] [PubMed] [Google Scholar]
- 15. Briquez, P.S. , Clegg, L.E. , Martino, M.M. , Gabhann, F.M. & Hubbell, J.A. Design principles for therapeutic angiogenic materials. Nat. Rev. Mater. 1, 15006 (2016). [Google Scholar]
- 16. Clegg, L.E. & Mac Gabhann, F. Molecular mechanism matters: benefits of mechanistic computational models for drug development. Pharmacol. Res. 99, 149–154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mac Gabhann, F. & Popel, A.S. Systems biology of vascular endothelial growth factors. Microcirculation 15, 715–738 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu, F.T. Stefanini, M.O. , Mac Gabhann, F. , Kontos, C.D. , Annex, B.H. & Popel, A.S. A systems biology perspective on sVEGFR1: its biological function, pathogenic role and therapeutic use. J. Cell. Mol. Med. 14, 528–552 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hazarika, S. , Dokun, A.O. , Li, Y. , Popel, A.S. , Kontos, C.D. & Annex, B.H. Impaired angiogenesis after hindlimb ischemia in type 2 diabetes mellitus: differential regulation of vascular endothelial growth factor receptor 1 and soluble vascular endothelial growth factor receptor 1. Circ. Res. 101, 948–956 (2007). [DOI] [PubMed] [Google Scholar]
- 20. Koch, S. & Claesson‐Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect. Med. 2, a006502 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vempati, P. , Popel, A.S. & Mac Gabhann, F. Extracellular regulation of VEGF: isoforms, proteolysis, and vascular patterning. Cytokine Growth Factor Rev. 25, 1–19 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bates, D.O. et al VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down‐regulated in renal cell carcinoma. Cancer Res. 62, 4123–4131 (2002). [PubMed] [Google Scholar]
- 23. Harper, S.J. & Bates, D.O. VEGF‐A splicing: the key to anti‐angiogenic therapeutics? Nat. Rev. Cancer 8, 880–887 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jones, W.S. et al Alteration in angiogenic and anti‐angiogenic forms of vascular endothelial growth factor‐A in skeletal muscle of patients with intermittent claudication following exercise training. Vasc. Med. 17, 94–100 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kikuchi, R. et al An antiangiogenic isoform of VEGF‐A contributes to impaired vascularization in peripheral artery disease. Nat. Med. 20, 1464–1471 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Varey, A.H. et al VEGF 165 b, an antiangiogenic VEGF‐A isoform, binds and inhibits bevacizumab treatment in experimental colorectal carcinoma: balance of pro‐ and antiangiogenic VEGF‐A isoforms has implications for therapy. Br. J. Cancer 98, 1366–1379 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Manetti, M. et al Overexpression of VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, leads to insufficient angiogenesis in patients with systemic sclerosis. Circ. Res. 109, e14–e26 (2011). [DOI] [PubMed] [Google Scholar]
- 28. Bills, V.L. , Varet, J. , Millar, A. , Harper, S.J. , Soothill, P.W. & Bates, D.O. Failure to up‐regulate VEGF165b in maternal plasma is a first trimester predictive marker for pre‐eclampsia. Clin. Sci. (Long). 116, 265–272 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nowak, D.G. et al Expression of pro‐ and anti‐angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J. Cell Sci. 121 (Pt 20), 3487–3495 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nowak, D.G. et al Regulation of vascular endothelial growth factor (VEGF) splicing from pro‐angiogenic to anti‐angiogenic isoforms: a novel therapeutic strategy for angiogenesis. J. Biol. Chem. 285, 5532–5540 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gustafsson, T. , Ameln, H. , Fischer, H. , Sundberg, C.J. , Timmons, J.A. & Jansson, E. VEGF‐A splice variants and related receptor expression in human skeletal muscle following submaximal exercise. J. Appl. Physiol. (1985) 98, 2137–2146 (2005). [DOI] [PubMed] [Google Scholar]
- 32. Jensen, L. , Pilegaard, H. , Neufer, P.D. & Hellsten, Y. Effect of acute exercise and exercise training on VEGF splice variants in human skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R397–R402 (2004). [DOI] [PubMed] [Google Scholar]
- 33. Bordeleau, F. et al Tissue stiffness regulates serine/arginine‐rich protein‐mediated splicing of the extra domain B‐fibronectin isoform in tumors. Proc. Natl. Acad. Sci. USA 112, 8314–8319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ng, Y.S. , Rohan, R. , Sunday, M.E. , Demello, D.E. & D'Amore, P.A. Differential expression of VEGF isoforms in mouse during development and in the adult. Dev. Dyn. 220, 112–121 (2001). [DOI] [PubMed] [Google Scholar]
- 35. Kawamura, H. , Li, X. , Harper, S.J. , Bates, D.O. & Claesson‐Welsh, L. Vascular endothelial growth factor (VEGF)‐A165b is a weak in vitro agonist for VEGF receptor‐2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res. 68, 4683–4692 (2008). [DOI] [PubMed] [Google Scholar]
- 36. Cébe Suarez, S. et al A VEGF‐A splice variant defective for heparan sulfate and neuropilin‐1 binding shows attenuated signaling through VEGFR‐2. Cell. Mol. Life Sci. 63, 2067–2077 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Delcombel, R. et al New prospects in the roles of the C‐terminal domains of VEGF‐A and their cooperation for ligand binding, cellular signaling and vessels formation. Angiogenesis 16, 353–371 (2013). [DOI] [PubMed] [Google Scholar]
- 38. Catena, R. et al VEGF121b and VEGF165b are weakly angiogenic isoforms of VEGF‐A. Mol. Cancer 9, 320 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu, J. et al The evolution of alternative splicing exons in vascular endothelial growth factor A. Gene 487, 143–150 (2011). [DOI] [PubMed] [Google Scholar]
- 40. Bates, D.O. et al Detection of VEGF‐A(xxx)b isoforms in human tissues. PLoS One 8, e68399 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harris, S. et al Do anti‐angiogenic VEGF (VEGFxxxb) isoforms exist? A cautionary tale. PLoS One 7, e35231 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Woolard, J. et al VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 64, 7822–7835 (2004). [DOI] [PubMed] [Google Scholar]
- 43. Ngo, D.T.M. et al Antiangiogenic actions of vascular endothelial growth factor‐A165b, an inhibitory isoform of vascular endothelial growth factor‐A, in human obesity. Circulation 130, 1072–1080 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu, F.T. , Stefanini, M.O. , Mac Gabhann, F. , Kontos, C.D. , Annex, B.H. & Popel, A.S. VEGF and soluble VEGF receptor‐1 (sFlt‐1) distributions in peripheral arterial disease: an in silico model. Am. J. Physiol. Heart Circ. Physiol. 298, H2174–H2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Clegg, L.W. & Mac Gabhann, F. Site‐specific phosphorylation of VEGFR2 is mediated by receptor trafficking: insights from a computational model. PLoS Comput. Biol. 11, e1004158 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Clegg, L.E. & Mac Gabhann, F. A computational analysis of in vivo VEGFR activation by multiple co‐expressed ligands. PLoS Comput. Biol. 13, e1005445 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ballmer‐Hofer, K. , Andersson, A.E. , Ratcliffe, L.E. & Berger, P. Neuropilin‐1 promotes VEGFR‐2 trafficking through Rab11 vesicles thereby specifying signal output. Blood 118, 816–826 (2011). [DOI] [PubMed] [Google Scholar]
- 48. Autiero, M. et al Role of PIGF in the intra‐ and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat. Med. 9, 936–943 (2003). [DOI] [PubMed] [Google Scholar]
- 49. Chu, L.H. et al A multiscale computational model predicts distribution of anti‐angiogenic isoform VEGF165b in peripheral arterial disease in human and mouse. Sci. Rep. 6, 37030 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information 1
Supporting Information 2
Supporting Information 3
Supporting Information 4
Supporting Information 5
Supporting Information 6
Supporting Information 7
Supporting Information 8
Supporting Information 9
Supporting Information 10
Supporting Information 11
Supporting Information 12
Supporting Information 13
Supporting Information 14
Supporting Information 15
Supporting Information 16
Supporting Information 17
Supporting Information 18
Supporting Information 19
Supporting Information 20
Supporting Information 21
Supporting Information 22
Supporting Information 23
Supporting Information 24
Supporting Information 25
Supporting Information 26
Supporting Information 27
Supporting Information 28
Supporting Information 29
Supporting Information 30