

Abstract
We aimed to reanalyze the differences in the pharmacokinetics (PKs) of meloxicam in East Asian populations based on a population approach using previously published data and to investigate the factors found in population PK analysis that affect the pharmacodynamics (PDs) of meloxicam. Population PK analysis was performed in 119 healthy male subjects (30 Japanese, 30 Chinese, 29 Korean, and 30 white) under strictly controlled trial conditions with regulated meals and a single lot of the drug. We found that CYP2C9 genotype and lean body mass were statistically significant predictors of clearance and volume of distribution, respectively. A statistical significant difference in the PK parameters between ethnic groups could not be identified. Simulations using PK/PD models showed that CYP2C9 genotype is the factor that affects the PDs of meloxicam. The genetic polymorphisms highlighted in this study would be beneficial for conducting clinical trials in East Asians with similar genetic backgrounds.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Clinical trial data have not been fully utilized among East Asian countries, such as Japan, China, and South Korea, because ethnic differences in PK and PD variables remain unclear.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ We re‐analyzed the PK properties meloxicam in three East Asian populations and one white population using a population modeling approach with published data. Moreover, PK covariates that affect the PD properties of meloxicam were investigated in PK/PD modeling and simulation analyses.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ We showed that CYP2C9 genotypes and LBM are statistically significant predictors of the CL and Vc of meloxicam, respectively, but that, of these predictors, only CYP2C9 genotypes affect the PDs of meloxicam. In addition, no statistically significant differences in PK parameters were identified between the present East Asian populations.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ The roles of CYP2C9 polymorphisms demonstrated herein may be applicable to future studies of ethnic differences, and the ensuing insights may facilitate clinical trials in East Asian populations with similar genetic backgrounds.
During recent decades, numbers of global clinical trials have increased because of the need for timely drug approval in multiple countries.1 In these clinical trials, interethnic differences in pharmacokinetic (PK) and pharmacodynamic (PD) properties should be considered. Moreover, because interethnic differences in PK properties have been reported between East Asian and white populations, the clinical trial results of one group cannot be readily extrapolated to the other.2, 3, 4 It also remains unclear whether interethnic differences in PK and PD properties of medications are present among East Asian populations, although East Asian clinical trials have been effective for some high prevalence diseases, such as gastric cancer.5
Thus, it may be necessary to clarify ethnic differences in PKs and PDs to facilitate the utilization of clinical trial data across East Asian countries, such as Japan, China, and South Korea. Accumulation of this information may also improve the planning of East Asian clinical trials by decreasing costs and shortening study durations. The factors associated with interethnic differences in the PKs of medications include intrinsic factors, such as genotype, and extrinsic factors, such as dietary patterns and coadministered medications. Therefore, clinical trials that are strictly controlled for these extrinsic factors could be used to evaluate interethnic differences in the PKs of drugs in terms of the associated intrinsic factors. To clarify and explain ethnic differences in PK parameters, Hasunuma et al.6 investigated differences in exposure levels of moxifloxacin, simvastatin, and meloxicam among Japanese, Chinese, and South Korean healthy male subjects. In that research, data from a single lot of the drug was used; food calories, nutritional content, and hardness of the drinking water were matched; and plasma meloxicam concentrations were measured from a single site. However, no detailed ethnic evaluations of the PKs of meloxicam have been performed among East Asian populations using a population approach.
Meloxicam is a cyclooxygenase inhibitor commonly used to treat patients with pain due to rheumatoid arthritis7 and surgery.8 The dose proportionality between orally administered doses of 7.5–30 mg has been confirmed,9 and the total clearance, volume of distribution, and absolute bioavailability have been reported to be 7.2 mL/min, 10.7 L, and 89% in healthy subjects, respectively. In addition, the terminal elimination half‐life of meloxicam was 20 hours, and the plasma protein binding consistently reached 99.5% over the concentration range used in clinical practice.10 Approximately 0.6% of the intravenously administered drug was excreted in the urine,11 and meloxicam is primarily metabolized to its 5′‐hydroxymethyl metabolite by cytochrome P450 2C9 (CYP2C9). Meloxicam enters enterohepatic and/or enteroenteric circulations, as indicated by increased meloxicam clearance after intravenous administration with cholestyramine coadministration.10, 12 Lehr et al.13 also reported a population PK model of meloxicam that incorporates the enterohepatic circulation. The thromboxane B2 (TXB2) generation is commonly used as a biomarker of cyclooxygenase activity,14, 15, 16 and it is a stable product of cyclooxygenase‐mediated thromboxane A2 catabolism. A sequential population PK/PD model of meloxicam was previously reported by Tegeder et al.,17 who monitored cyclooxygenase inhibition according to decreased TXB2 generation. Rohatagi et al.18 also reported a relationship between serum TXB2 contents and the incidence of gastroduodenal ulcers and erosions.
The study objective was to reanalyze differences in PKs of meloxicam among East Asian (Japanese, Chinese, and South Koreans) and white populations using population PK analyses of published clinical trial data. Accordingly, we investigated how factors found in population PK modeling affect PDs of meloxicam using PK/PD models and simulations. Understanding of the factors that affect TXB2 generation after meloxicam administration may reveal useful biomarkers of meloxicam efficacy and safety in East Asian populations.
METHODS
Data source
Population pharmacokinetic modeling
Plasma meloxicam concentrations and clinical data from Hasunuma et al.6 were used as the source of population PK modeling data. Subject characteristics are shown in Table 1. One hundred nineteen healthy male subjects (CYP2C9 genotypes: Japanese, 26 subjects with *1/*1 and 4 subjects with *1/*3; Chinese, 26 subjects with *1/*1, 1 subject with *1/*2, and 3 subjects with *1/*3; Korean, 25 subjects with *1/*1 and 4 subjects with *1/*3; and white, 20 subjects with *1/*1, 5 subjects with *1/*2, 1 subject with *2/*2, and 4 subjects with *1/*3) were administered single oral doses of meloxicam (7.5 mg).6 Blood samples were collected before and at 1, 2, 3, 4, 5, 6, 8, 12, 24, 36, 48, 60, and 72 hours after drug administration, and plasma meloxicam concentrations were determined using liquid chromatography‐tandem mass spectrometry.6 The trial was registered in the UMIN Clinical Trials Registry system (UMIN000004173), and the population PK study was approved by the Ethics Committee of the School of Pharmacy, Nihon University.
Table 1.
Demographic data
| Characteristics | Ethnicity | |||
|---|---|---|---|---|
| Japanese | Chinese | Korean | White | |
| No. of subjects | 30 | 30 | 29 | 30 |
| Smoking history | 9 | 1 | 2 | 6 |
| Age, years | 24 (21–30) | 31 (23–34) | 24 (21–29) | 26 (21–35) |
| Weight, kg | 63.5 (52.1–84.5) | 67.0 (51.0–91.0) | 69.1 (56.3–84.4) | 74.5 (55.9–100) |
| BMI, kg/m2 | 21.7 (18.6–29.1) | 23.5 (19.2–29.0) | 22.6 (19.2–26.3) | 24.6 (19.9–29.8) |
| BSA, m3 | 1.75 (1.56–2.00) | 1.78 (1.53–2.08) | 1.86 (1.64–2.06) | 1.93 (1.62–2.30) |
| LBM, kg | 52.5 (44.5–62.9) | 54.0 (43.6–66.3) | 57.3 (47.6–65.2) | 59.8 (47.1–75.2) |
| Height, cm | 172 (161–180) | 168 (160–180) | 177 (168–186) | 177 (162–195) |
| eGFR, mL/min | 97.4 (74.1–138) | 120 (92.8–152) | 116 (98.9–151) | 121 (85.4–158) |
| ALB, g/dL | 4.5 (4.0–4.9) | 4.4 (4.0–5.0) | 4.4 (4.0–5.2) | 4.5 (3.9–4.9) |
| ALT, IU/L | 15 (8–38) | 16 (6–29) | 13 (3–38) | 19 (9–63) |
| AST, IU/L | 16 (12–25) | 20 (14–35) | 16 (11–31) | 20 (12–82) |
| DBIL, mg/dL | 0.2 (0.1–0.6) | 0.2 (0.1–0.5) | 0.3 (0.1–0.7) | 0.1 (0.0–0.27) |
| HDL, mg/dL | 52 (37–100) | 49 (29–69) | 51 (29–97) | 50 (30–78) |
| LDH, IU/L | 135 (106–189) | 120 (83–152) | 159 (130–469) | 133 (98–259) |
| LDL, mg/dL | 86 (53–177) | 105 (41–159) | 104 (59–130) | 88 (45–141) |
| TBIL, mg/dL | 1.0 (0.4–2.2) | 0.9 (0.5–1.6) | 1.2 (0.6–2.0) | 0.6 (0.3–2.1) |
| TG, mg/dL | 84 (46–384) | 108 (65–203) | 84 (46–172) | 105 (39–304) |
| TP, g/dL | 7.1 (6.2–7.7) | 7.8 (6.9–8.3) | 6.9 (6.3–8.6) | 6.9 (6.1–8.2) |
| GGT, IU/L | 20 (10–50) | 16 (11–34) | 15 (9–34) | 16 (6–58) |
Data are expressed as median (range).
ALB, serum albumin; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; BSA, body surface area; DBIL, direct bilirubin; eGFR, estimated glomerular filtration rate; GGT, gamma‐glutamyl transpeptidase; HDL, high‐density lipoprotein cholesterol; LBM, lean body mass; LDH, lactate dehydrogenase; LDL, low‐density lipoprotein cholesterol; TBIL, total bilirubin; TG, triglyceride; TP, total protein.
Pharmacokinetic/pharmacodynamic modeling
Plasma meloxicam concentrations and the inhibition of TXB2 generation were retrieved from the data of Bae et al.14 and were used as source data for PK/PD modeling. Graphs of plasma meloxicam concentration‐time data and percentage inhibition of TXB2 generation relative to baseline levels were scanned, and all graphed data points were digitized using UN‐SCAN‐IT software (Silk Scientific, Orem, UT). These graphs represent the averaged data and are only separated by CYP2C9*1/*1 or CYP2C9*1/*13 genotypes. Demographic data (numbers or means ± SDs) for subjects of the CYP2C9*1/*1 group includes 12 healthy Korean volunteers of age 23.2 ± 2.6 years and body weights of 71.8 ± 8.1 kg. The data for subjects in the CYP2C9*1/*13 group includes 9 healthy Korean volunteers of age 24.4 ± 2.5 years and body weights of 70.7 ± 7.3 kg. Percent serum TXB2 generation relative to basal values was calculated as the difference between 100% and the scanned percent inhibition of TXB2 generation. Twenty‐one healthy male volunteers carrying either CYP2C9*1/*1 or CYP2C9*1/*13 genotypes were administered single oral doses of meloxicam (15 mg).14 In that report, blood samples for determinations of plasma meloxicam concentrations and TXB2 generation were collected before and at 1, 2, 3, 4, 5, 6, 8, 10, 12, 24, 48, and 72 hours after the administration of meloxicam.14 The SDs of plasma concentrations and percent inhibition of TXB2 generation at 1–8 hours after administration were not digitized because the error bars were not recognized for all groups.
Population pharmacokinetic modeling
Population analyses were performed using NONMEM version 7.2,19 PsN,20 Wings for NONMEM (http://wfn.sourceforge.net/), and Xpose21 software packages. Population PK parameters were estimated using first‐order conditional estimations with interactions. Model selection was based on the objective function value (OFV) calculated by NONMEM, the relative SE of the parameter estimates, and model diagnosis plots.22
To describe plasma meloxicam concentration profiles during the absorption phase, several structural models were estimated using one‐compartment or two‐compartment models with first‐order absorption, transit absorption, and zero‐order and first‐order parallel absorption with or without lag time. Additionally, to describe fluctuating plasma concentration profiles during the elimination phase, the enterohepatic circulation model13 was tested. In these analyses, interindividual variability in PK parameters was assumed to follow log‐normal distributions and was modeled using exponential functions, whereas residual variability was evaluated using a proportional error model and a combined additive plus proportional error model. Parameters with individual values between 0 and 1 were modeled as follows:
| (1) |
| (2) |
where A is a temporary variable, θF corresponds with a typical value of a fraction in the jth individual, Fj corresponds with the estimated fraction of the jth individual, and represents normally distributed interindividual variability with a mean of zero and a variance of ωF 2.23
Potential covariates were investigated using stepwise forward addition and backward elimination with P values of 0.01 and 0.001, respectively. An ethnic difference was defined as the difference that is not explained by demographic data, including age, body size, renal function, smoking history, other clinical laboratory data, and CYP2C9 genotypes. Potential covariates in the forward step were investigated step‐by‐step in the following order (i.e., at each step, the model with the lowest OFV was forwarded to the next step if it was statistically significant): age, body size index (body weight, body mass index, lean body mass (LBM) using the James formula,24 and body surface area), renal function data (estimated glomerular filtration rate25, 26, 27), smoking history, other clinical laboratory data, CYP2C9 genotypes, and ethnic differences. Relationships between parameters and tested covariates are shown in Supplementary Table S1.
Covariates were introduced into the model as either continuous or categorical functions. Continuous covariates were centered on the median value of the population and were modeled using the following power equation:
| (3) |
where θ is a covariate scale factor that describes the magnitude of the covariate‐parameter relationships. Categorical covariates were modeled as follows:
| (4) |
where n is an indicator variable (0/1), and CYP2C9 polymorphisms were modeled as follows:
| (5) |
where CLpop is the apparent clearance of meloxicam, CL*1/*1 is the total apparent clearance for subjects with the CYP2C9*1/*1 genotype, and n*x and θ*x are numbers of alleles and the fractional changes of CL*1/*1 attributable to respective genes.28, 29, 30 Ethnic differences were modeled using the following expression:
| (6) |
where Ppop is the PK parameter, Pja is the PK parameter for Japanese patients, n and θ are indicator variables (0/1) representing fractional changes in PK parameters of Chinese (ch), Korean (ko), and white (ca) subjects compared with those of Japanese subjects.
Model qualification
Uncertainty in parameter estimates was assessed using bootstrapping, and resampling was performed 1,000 times and was stratified by the CYP2C9 genotypes. Median values and 2.5th and 97.5th percentiles of parameter estimates from these analyses were compared with those of the final model. The performance of the final population PK model was evaluated using a prediction‐corrected visual predictive check (pc‐VPC),22, 31 which was generated by simulating 1,000 replicates of the study from the final models. For visual inspection, 95% confidence intervals for the 5th, 50th, and 95th percentiles of simulated concentrations at each time point are shown as areas, and 5th, 50th, and 95th percentiles of the observed data are represented as lines.
To compare predictions from the final population PK model with those from previously reported data, we simulated 1,000 individuals who were administrated meloxicam.32 To compare the profiles reported by Bae et al.,14 plasma concentration profiles were simulated after single‐dose administration. In these computations, the CYP2C9 genotype was set to *1/*1 and the LBMs of 1,000 individuals were generated using a parametric approach based on original datasets. Plasma concentration‐time profiles after multiple administrations were also simulated and compared with those reported by the “interview form” of Morbic tablets (drug information media used in Japan).33 The CYP2C9 genotype distribution was generated at the same ratio as for the original dataset, and the LBMs of 1,000 individuals were parametrically derived from the original dataset.
Pharmacokinetic/pharmacodynamic modeling
The PK parameters were estimated using the structure model that was selected by population PK analyses. Specifically, PD modeling was performed using a sequential approach using PD observations and PK predictions based on the PK model. The PK/PD model comprised the PK model and the indirect response model with input inhibition. On the basis of decreased TXB2 generation because of the inhibition of cyclooxygenase by meloxicam, rates of change in percent serum TXB2 generation relative to basal values were described by the following physiological indirect response model34, 35, 36:
| (7) |
where R is percent serum TXB2 generation relative to the basal value, kin is the zero‐order rate constant for the increase in the percent serum TXB2 generation of the basal value, Cp is the plasma meloxicam concentration, IC50 is the plasma meloxicam concentration that decreases kin by 50%, kout is the first‐order rate constant for the decrease in percent serum TXB2 generation of the basal value, and γ is a sigmoidicity parameter. Under basal conditions, percent serum TXB2 generation value was 100%, with kin set as 100*kout.
Simulation
To quantify the impact of covariates on clearance (CL) and volume of distribution in the central compartment (Vc) to PDs, the population mean of the final population PK model was used to simulate meloxicam steady‐state concentrations at a dosing schedule of 7.5 mg once daily. The CL was calculated for CYP2C9*1/*1, *1/*2, *1/*3, *2/*2, *2/*3, and *3/*3 genotypes, and Vc was calculated at LBMs of 75.2, 55.0, and 43.6 kg, reflecting maximum, median, and minimum LBMs of the populations in the original dataset, respectively. Simulated meloxicam concentrations and PD parameter estimates from the PK/PD model were used to simulate steady‐state effects of meloxicam. These simulations are deterministic and the time‐weighted average percent serum TXB2 generation over 24 hours relative to the basal value was used as a summary parameter to characterize time courses of each endpoint after meloxicam administration.
RESULTS
Population pharmacokinetic modeling
Concentration‐time profiles of ethnic groups are shown in Figure 1 a, and double logarithmic concentration‐time profiles in the absorption phase of ethnic groups are shown in Figure 1 b. The profiles of the subjects had convex downward curves and straight lines during the absorption phase, as observed in Figure 1 b. Therefore, a one‐compartment model with a transit absorption model,37, 38 and zero‐order and first‐order parallel absorption with lag time model39 were tested. In the latter model, the estimated time (1.42 hours) for meloxicam to enter the central compartment according to the zero‐order process was close to the estimated absorption lag time of 1.9 hours with the first‐order rate process. However, because discontinuation of absorption between 1.42 and 1.9 hours is physiologically unlikely, the estimated time for the zero‐order process was assumed equal to the absorption lag time even though the OFV was increased (ΔOFV = 49.928; df = 1). Moreover, the OFV for the parallel absorption model was lower than that of the transit absorption model (ΔOFV = 249.780; df = 2) and the first‐order absorption with the lag time model (ΔOFV = 388.343; df = 2). The OFV for the two‐compartment model with zero‐order and first‐order parallel absorption with the lag time model was lower than that of the one‐compartment model (ΔOFV = 129.673; df = 2). Tests of the enterohepatic circulation model13 were hampered by convergence. The OFV for combined additive and proportional model was the same as that for the proportional error model, and the SD of additive error in the residual variability was 0.00546 ng/mL. Thus, residual variability was modeled using a proportional error model. We concluded that the two‐compartment model with the parallel absorption model described the data adequately, and we used this for the final structure model (Figure 2). Interindividual variability was estimated reliably for apparent CL, apparent Vc, the first‐order absorption rate constant (ka), and the dose fraction absorbed by zero‐order absorption (F), which showed a correlation between ka and F.
Figure 1.
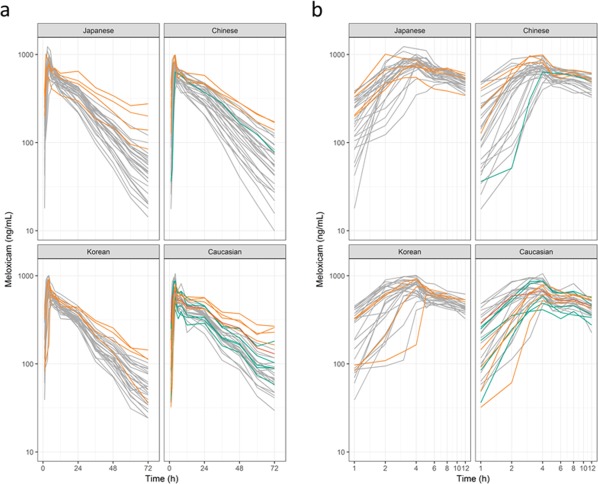
Plasma concentration‐time profiles of meloxicam for (a) single and (b) double logarithmic plots. Individuals are represented by lines. Colors represent CYP2C9 genotypes; gray line, *1/*1; green line, *1/*2; orange line, *1/*3; and red line, *2/*2.
Figure 2.
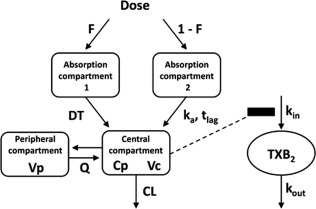
Pharmacokinetic/pharmacodynamic model for meloxicam. Absorption phase profiles are described by zero‐order absorption and first‐order absorption with the lag time model. CL, apparent clearance; Cp, plasma meloxicam concentrations; DT, duration of meloxicam entry into the central compartment from the absorption compartment 1 by zero‐order rate; F, fraction of the dose absorbed through the zero‐order absorption process; ka and tlag, first‐order rate constant and lag time, respectively; kin, zero‐order rate constant for increases in percent serum thromboxane B2 (TXB2) generation relative to basal values; kout, first‐order rate constant for decreases in percent serum TXB2 generation; Q, apparent intercompartmental clearance; Vc, apparent volume of distribution in the central compartment; Vp, apparent volume of distribution in the peripheral compartment.
In the covariate model summarized in Supplementary Table S2, the full model included LBM and serum albumin as covariates for Vc, and CYP2C9 genotypes as covariates for CL after the forward step. In the backward step, exclusion of CYP2C9 genotypes from CL (ΔOFV = 57.854; df = 2; P < 0.001) and LBM from Vc (ΔOFV = 41.335; df = 1; P < 0.001) caused statistically significant increases in OFV. No statistically significant ethnic differences were identified in PK parameters. The bootstrapping results and final population PK parameter estimates of meloxicam are shown for the final model in Table 2. The model predicted CL for meloxicam was decreased by 15%, 29%, 40%, 55%, and 80% in subjects with CYP2C9*1/*2, *2/*2, *1/*3, *2/*3, and *3/*3 genotypes, respectively, compared with that in subjects with the CYP2C9*1/*1 genotype. We also stratified concentration‐time profiles by LBM (Figure 3) and present model diagnosis plots in Supplementary Figure S1.
Table 2.
Parameter estimates and bootstrap confidence intervals for the population pharmacokinetic model
| Original dataset | Bootstrap result | |||
|---|---|---|---|---|
| Parameter | Estimate (±1.96 × SE) | Median (95% CI) | ||
| CL (L/h) | 0.391 | (0.375 to 0.407) | 0.390 | (0.375 to 0.407) |
| CYP2C9 *2 on CL | −0.147 | (−0.234 to −0.0604) | −0.147 | (−0.215 to −0.0410) |
| CYP2C9 *3 on CL | −0.400 | (−0.488 to −0.312) | −0.400 | (−0.483 to −0.301) |
| Vc (L) | 7.79 | (7.24 to 8.34) | 7.80 | (7.01 to 8.35) |
| LBM on Vd | 1.05 | (0.695 to 1.40) | 1.06 | (0.746 to 1.41) |
| Q (L/h) | 1.24 | (0.948 to 1.53) | 1.24 | (1.00 to 1.68) |
| Vp (L) | 2.73 | (2.20 to 3.26) | 2.72 | (2.22 to 3.49) |
| Ka (/h) | 2.00 | (1.38 to 2.62) | 2.05 | (1.44 to 2.84) |
| DT (h) | 1.91 | (1.86 to 1.96) | 1.91 | (1.83 to 1.94) |
| F | 0.425 | (0.367 to 0.483) | 0.423 | (0.364 to 0.481) |
| ωCL (CV%) | 21.3 | (18.4 to 23.9) | 21.0 | (18.2 to 23.5) |
| ωVd (CV%) | 17.2 | (13.4 to 20.3) | 17.1 | (14.0 to 20.0) |
| ωKa (CV%) | 131 | (68.1 to 201) | 130 | (81.5 to 193) |
| ω2 F | 2.03 | (1.46 to 2.60) | 2.00 | (1.45 to 2.62) |
| ωKa‐ωF | 0.243 | (0.0269 to 0.459) | 0.262 | (0.0421 to 0.482) |
| σ (CV%) | 12.3 | (11.3 to 13.2) | 12.3 | (11.4 to 13.2) |
CI, confidence interval; CL, apparent clearance; CLpop, population mean of apparent clearance; DT, duration of meloxicam entry into the central compartment from the absorption compartment by zero‐order rate; F, fraction of the dose absorbed through the zero‐order absorption process; Ka, first‐order absorption rate constant; LBM, lean body mass; n*2, number of CYP2C9*2 alleles; n*3, number of CYP2C9*3 alleles; Q, apparent intercompartmental clearance; tlag, absorption lag time for meloxicam entry into the central compartment from the absorption compartment with the first‐order rate; Vc, apparent volume of distribution in the central compartment; Vcpop, population mean of apparent volume of distribution in the central compartment; Vp, apparent volume of distribution in the peripheral compartment; ωCL, interindividual variability of CL; ω2 F, interindividual variability of F; ωKa, interindividual variability of Ka; ωKa‐ωF, correlation coefficient between interindividual variability of Ka and that of F; ωVd, interindividual variability of Vd; σ, residual variability.
DT and tlag were set to the same value. Eta‐shrinkage: CL, 1.89%; Vc, 8.81%; Ka, 12.0%; F, 6.30%. Epsilon‐shrinkage: 13.7%. Calculations: tlag = DT (h). The final population pharmacokinetic equation was , where .
Figure 3.
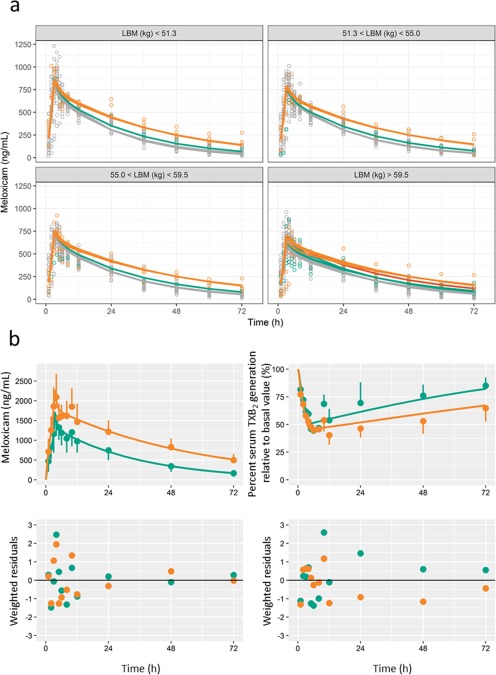
Graphical fit of the population pharmacokinetic (PK) model and the PK/pharmacodynamic (PD) model. (a) Plasma concentration‐time profiles of meloxicam stratified by lean body mass; circles represent observed data from Hasunuma et al.6; lines represent population predictions; colors represent CYP2C9 genotypes; gray, *1/*1; green, *1/*2; orange, *1/*3; and red, *2/*2. (b) Plasma meloxicam concentration and percent serum thromboxane B2 (TXB2) generation relative to basal value‐time profiles; left upper panel, plasma concentration profile after administration of 15 mg of meloxicam; right upper panel, profiles for percent serum TXB2 generation relative to the basal value; left and right lower panel, PK and PD residuals plots. Plasma meloxicam concentration plots represent data from Bae et al.,14 and plots of percent serum TXB2 generation were calculated as the difference between 100% and the scanned percent inhibition of TXB2 generation. Circles represent observed average data. Lines represent predictions. Colors represent CYP2C9 genotypes; green, *1/*1; and orange, *1/*13. Error bars represent SDs. The SDs at 1–8 hours after administration were not digitized because the error bars were not recognized for all groups.
The present pc‐VPC results (Supplementary Figure S2a) were stratified by ethnicity and CYP2C9 genotypes and are presented in Supplementary Figures S3 and S4, respectively. In the pc‐VPC, the percentile intervals describing prediction‐corrected observations were mostly within the 95% confidence intervals of percentile intervals for the simulated concentrations. Supplementary Figures S2b and S2c show the reported concentrations for single‐dosing and multiple‐dosing strategies, respectively, and the results of the simulation are compared with those of previous studies,14, 33 which provide external validation of our model. The pc‐VPC of the final model indicates the absence of model misspecification and good simulation properties. The external validation indicates that the model seems to systemically underpredict the external data.
Pharmacokinetic/pharmacodynamic modeling
The PK/PD model (Figure 2) and the PK/PD parameter estimates and their SEs (Table 3) were estimated using the same structure model as selected for the population PK analysis. Time courses of observed and predicted meloxicam concentrations and percent serum TXB2 generation relative to basal values are shown with residual plots in Figure 3. Reasonable agreement was demonstrated between observed and predicted effects of meloxicam, as there is a tendency for bias at later time points for each group.
Table 3.
Parameter estimates for the pharmacokinetic/pharmacodynamic model
| CYP2C9*1/*1 | CYP2C9*1/*13 | |
|---|---|---|
| Parameter | Estimate (%RSE) | Estimate (%RSE) |
| CL (L/h) | 0.320 (1.35) | 0.147 (2.12) |
| Vc (L) | 5.24 (6.35) | 3.38 (22.9) |
| Q (L/h) | 3.41 (18.5) | 3.38 (22.4) |
| Vp (L) | 5.18 (6.89) | 4.55 (20.4) |
| ka (/h) | 5.01 (9.18) | 5.27 (8.48) |
| DT (h) | 2.99 (0.140) | 2.96 (0.242) |
| F | 0.653 (2.25) | 0.713 (8.79) |
| kout (/h) | 0.912 (17.2) | |
| γ | 0.739 (9.62) | |
| IC50 (ng/mL) | 1390 (10.2) | |
| tlag = DT, kin = 100*kout | ||
CL, apparent clearance; DT, duration for meloxicam to enter the central compartment from the absorption compartment by zero‐order rate; F, fraction of the dose absorbed by zero‐order absorption; IC50, plasma meloxicam concentrations that decrease kin by 50%; ka, first‐order absorption rate constant; kout, first‐order rate constant for the decrease in percent serum thromboxane B2 relative to the basal value; Q, apparent intercompartmental clearance; Vc, apparent volume of distribution in the central compartment; Vp, apparent volume of distribution in the peripheral compartment; γ, sigmoidicity parameter.
Simulation
Figure 4 shows the relationships between changes in covariates and the effects of meloxicam. The time‐weighted average percentage serum TXB2 generation values for 24 hours at steady state in typical subjects with CYP2C9*1/*1, *1/*2, *1/*3, *2/*2, *2/*3, or *3/*3 genotypes were 60.5%, 57.6%, 51.0%, 54.0%, 45.7%, or 31.8%, respectively, and corresponding LBMs were 75.2, 55.0, or 43.6 kg and 60.3%, 60.5%, or 60.7%, respectively. The simulated time‐weighted average percentage serum TXB2 generation value of a typical subject with the CYP2C9*3/*3 genotype was 0.53‐fold that of a typical subject with the CYP2C9*1/*1 genotype.
Figure 4.
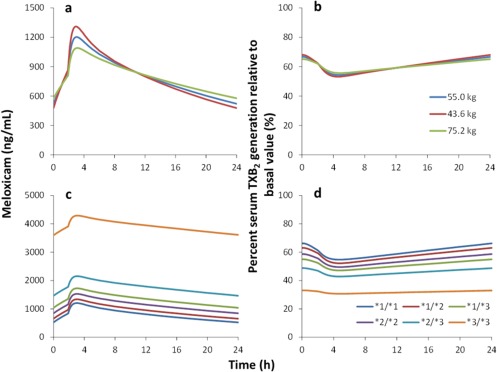
Relationships between changes in covariates and the effects of meloxicam. The graphs show the impacts of lean body mass (a, b) and CYP2C9 genotypes (c, d) on plasma meloxicam concentrations and percent serum thromboxane B2 (TXB2) generation relative to basal value‐time profiles. The simulation was performed by varying CYP2C9 genotypes or lean body mass for 7.5‐mg daily meloxicam administration under steady‐state conditions. a and b, CYP2C9 *1/*1 genotype; c and d, the lean body mass is 55.0 kg.
DISCUSSION
Quantitative investigations of differences in PK and PD properties of medications among Japanese, Chinese, and South Korean populations may provide fundamental information for improving the efficacy and safety of medications and the efficiency of drug development. Therefore, we investigated the ethnic differences in the PKs of meloxicam using a population modeling approach based on data obtained from the trial under strict conditions.
In this study, the structural model of meloxicam was characterized by a two‐compartment model with zero‐order and first‐order parallel absorption with lag time. Lehr et al.13 previously developed an enterohepatic circulation model controlled by a sine function for meloxicam PKs following intravenous administration. In addition, Hasunuma et al.6 showed that the second peak or delayed absorption in the PK profile of meloxicam is observed at around 8 hours after administration. Herein, we tested the enterohepatic circulation model13 to describe this phenomenon, but were hampered by convergence. Specifically, we observed fluctuating plasma concentration profiles during the elimination phase in white subjects (Figure 1 a) but observed little or no fluctuations among East Asian subjects. In the enterohepatic circulation model, plasma concentration data for the second and third peaks of the elimination phase and reabsorption processes are required. However, because blood sampling was performed every 12 hours during the elimination phase, reabsorption processes in enterohepatic circulation were not detected. The fluctuation of conditional weighted residuals after 12 hours may be improved by incorporating the enterohepatic circulation model (Supplementary Figure S1c). Hence, further studies are required to collect data after intravenous administration and at multiple time points during the elimination phase to describe enterohepatic circulation. Moreover, to describe the second peak or delayed absorption in terms of reabsorption after first‐pass effect, we tested a zero‐order and first‐order parallel absorption with lag time model39 (Figure 1 b). In these computations, subjects had convex downward curves or straight lines during the absorption phase, and these interindividual differences were described by the interindividual variability of F.
CYP2C9 genotypes and LBMs were statistically significant predictors of CL and Vc, respectively, and similarly decreased clearance in carriers of CYP2C9*2 and/or *3 mutations has been demonstrated for many other drugs. In particular, previous population PK analyses indicated that phenobarbital clearance was decreased by 48% in a Japanese cohort of CYP2C9*1/*3 carriers,40 whereas glimepiride clearance was reportedly decreased by 38% in a Korean cohort of subjects with the CYP2C9*1/*3 genotype.41 In consistent comparisons with CL in patients with the CYP2C9*1/*1 genotype, Lane et al.42 showed that (S)‐warfarin clearance was decreased by 15%, 33%, 55%, 50%, and 71% in subjects with CYP2C9*1/*2, *2/*2, *1/*3, *2/*3, and *3/*3 genotypes, respectively. Hence, decreased clearance ratios in carriers of CYP2C9*2 and/or *3 mutations are similar for both drugs. These data can also be extrapolated to other drugs that are mainly metabolized by hepatic CYP2C9 and have low hepatic extraction ratios. In addition, Meineke & Türck7 reported that body weights and age influence the CL of meloxicam, and that body weights affected the volume of distribution of meloxicam in patients with rheumatoid arthritis. However, neither body weights nor ages were statistically significant covariates for CL in the present study, whereas CYP2C9 genotype was a strong predictor of the CL of meloxicam. Potentially, the influences of body weight and age on CL were obscured by the narrow ranges of these variables herein. However, LBM was a significant covariate for Vc in this study, as shown in patients with rheumatoid arthritis. Moreover, because body size indexes may reflect distributions of meloxicam in extracellular fluid, they were considered a contributor to interindividual variability of Vc, which was reportedly 10.7 L in a noncompartmental analysis.9
To simulate the influence of covariates on drug effects, data were retrieved from Bae et al.14 and used to build a PK/PD model for meloxicam. The estimated meloxicam IC50 value (1,390 ng/mL shown in Table 3) is similar to that (1,100 ng/mL) in a previous study of cyclooxygenase‐1 inhibition in whole human blood.43 Thus, using these data, we applied the PK/PD model (the PD factors) and the final population PK model to evaluate the impacts by CYP2C9 genotypes and LBM on PD value‐time profiles (percent serum TXB2 generation relative to the basal value) for 7.5 mg/day meloxicam administration. Rinder et al.44 showed that the relative serum TXB2 generation was decreased by 37% from baseline at 6 hours after 7.5‐mg/day meloxicam administrations under steady‐state conditions, but did not evaluate CYP2C9 genotypes. Thus, we compared this value in simulations of CYP2C9*1/*1 and *1/*2, which are common in white populations. In agreement with Rinder et al.,44 our model of subjects with a mean LBM of 55 kg indicated that relative serum TXB2 generation was decreased by 45% and 47% because of CYP2C9*1/*1 and CYP2C9*1/*2 genotypes, respectively (Figure 4 d). Overall, the impact of LBM on the changes in TXB2 generation (Figure 4 b) were small because changes in meloxicam concentrations varied little with LBM (Figure 4 a) and the estimated sigmoidicity parameter was 0.739 in the PD model. Therefore, no dosage adjustment was needed on the basis of LBM, which was considered a statistically significant predictor of Vc. We also predicted that the impact of the CYP2C9*3/*3 genotype was greater than that of the other genotypes, and the time‐weighted average relative serum TXB2 generation in carriers of CYP2C9*3/*3 was 0.53‐fold that of CYP2C9*1/*1 carriers. Hence, analgesic effects of meloxicam are likely stronger among patients with CYP2C9*3/*3 genotypes, warranting consideration of the risk of gastroduodenal injury in CYP2C9*3/*3 patients.
The present study is limited primarily by the absence of covariates in the PD models, reflecting the computational availability of average profile data only in published PK/PD studies.14 However, in a study by Rohatagi et al.,18 no statistically significant covariates for PD parameters of the cyclooxygenase inhibitor CS‐706 were detected in population PK/PD analyses. Because meloxicam and CS‐706 have similar modes of action, covariates of the PD of meloxicam may also be undetectable.
Finally, only one of the present subjects carried the rare allele combination CYP2C9*2/*2 genotype. Thus, we modeled CYP2C9 polymorphisms with assumptions of proportional effects for the subjects with rare allele combinations, as shown in Eq. 5. This proportional effect model has been used in several population PK analyses, including those for dextromethorphan,28 tolbutamide,29 and warfarin.30 However, the present calculated CL value of 0.0782 L/h in subjects with CYP2C9*3/*3 is greater than the meloxicam oral clearance of 0.043 ± 0.004 L/h, which was calculated using a noncompartmental analysis of Korean subjects (mean ± SD; n = 3) with the CYP2C9*3/*3 genotype,45 further indicating a comparatively large impact of the CYP2C9*3/*3 genotype. Thus, further studies are necessary to investigate the PK of rare allele combinations.
In conclusion, we constructed a population PK model using data from our previously published clinical trial data with a single lot of drug, with matched food calories and nutritional contents, and plasma meloxicam concentration measurements that were performed at the same site. No statistically significant ethnic differences in these PK parameters were identified using population analysis, thus expanding the data reported by Hasunuma et al.6 Although the present population PK model showed that CYP2C9 genotypes and LBM are statistically significant predictors of CL and Vc, respectively, among the factors found in population PK analysis, only CYP2C9 genotypes affected the PDs of meloxicam during our simulations. Moreover, the effects of the CYP2C9*3/*3 genotype were larger than those of the other genotypes, warranting careful consideration of the potential for adverse effects in CYP2C9*3/*3 patients. Finally, the present simulations provided important information for future clinical trials in East Asians with similar genetic backgrounds.
Conflict of Interest
The authors have no conflicts of interest that are directly relevant to the contents of this research article.
Supporting information
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
Supplementary Files
Acknowledgments
This study was supported by the Research on Regulatory Science of Pharmaceuticals and Medical Devices from the Japan Agency for Medical Research and Development (AMED). The authors thank Daisuke Suzuki and Tomomi Ogino for their assistance.
Author Contributions
T.A., A.M., and Y.M. wrote the manuscript. M.T. and S.K. designed research. M.T. and Y.S. performed the research. T.A., Y.I., M.K., and Y.M. analyzed the data.
References
- 1. Mori, K. & Toyoshima, S. Recent approaches by the PMDA to promoting new drug development: change in the status of the PMDA in relation to new drug development over the last five years. Drug Inf. J. 43, 47–55 (2009). [Google Scholar]
- 2. Fukunaga, S. , Kusama, M. , Arnold, F.L. & Ono, S. Ethnic differences in pharmacokinetics in new drug applications and approved doses in Japan. J . Clin. Pharmacol. 51, 1237–1240 (2011). [DOI] [PubMed] [Google Scholar]
- 3. Nakajima, M. , Kuroiwa, Y. & Yokoi, T. Interindividual differences in nicotine metabolism and genetic polymorphisms of human CYP2A6. Drug Metab. Rev. 34, 865–877 (2002). [DOI] [PubMed] [Google Scholar]
- 4. Birmingham, B.K. et al Rosuvastatin pharmacokinetics and pharmacogenetics in Caucasian and Asian subjects residing in the United States. Eur. J. Clin. Pharmacol. 71, 329–340 (2015). [DOI] [PubMed] [Google Scholar]
- 5.International Agency for Research on Cancer, GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence worldwide in 2012. <http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx#> (2012). Accessed 20 July 2017.
- 6. Hasunuma, T. et al Absence of ethnic differences in the pharmacokinetics of moxifloxacin, simvastatin, and meloxicam among three East Asian populations and Caucasians. Br. J. Clin. Pharmacol. 81, 1078–1090 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meineke, I. & Türck, D. Population pharmacokinetic analysis of meloxicam in rheumatoid arthritis patients. Br. J. Clin. Pharmacol. 55, 32–38 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aoki, T. et al Premedication with cyclooxygenase‐2 inhibitor meloxicam reduced postoperative pain in patients after oral surgery. Int. J. Oral Maxillofac. Surg. 35, 613–617 (2006). [DOI] [PubMed] [Google Scholar]
- 9. Türck, D. , Busch, U. , Heinzel, G. & Narjes, H. Clinical pharmacokinetics of meloxicam. Arzneimittelforschung 47, 253–258 (1997). [PubMed] [Google Scholar]
- 10. Türck, D. , Roth, W. & Busch, U. A review of the clinical pharmacokinetics of meloxicam. Br. J. Rheumatol. 35 Suppl 1, 13–16 (1996). [DOI] [PubMed] [Google Scholar]
- 11. Schmid, J. , Busch, U. , Heinzel, G. , Bozler, G. , Kaschke, S. & Kummer, M. Pharmacokinetics and metabolic pattern after intravenous infusion and oral administration to healthy subjects. Drug Metab. Dispos. 23, 1206–1213 (1995). [PubMed] [Google Scholar]
- 12. Busch, U. , Heinzel, G. & Narjes, H. The effect of cholestyramine on the pharmacokinetics of meloxicam, a new non‐steroidal anti‐inflammatory drug (NSAID), in man. Eur. J. Clin. Pharmacol. 48, 269–272 (1995). [DOI] [PubMed] [Google Scholar]
- 13. Lehr, T. , Staab, A. , Tillmann, C. , Trommeshauser, D. , Schaefer, H.G. & Kloft, C. A quantitative enterohepatic circulation model: development and evaluation with tesofensine and meloxicam. Clin. Pharmacokinet. 48, 529–542 (2009). [DOI] [PubMed] [Google Scholar]
- 14. Bae, J.W. , Choi, C.I. , Jang, C.G. & Lee, S.Y. Effects of CYP2C9*1/*13 on the pharmacokinetics and pharmacodynamics of meloxicam. Br. J. Clin. Pharmacol. 71, 550–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Perini, J.A. , Vianna‐Jorge, R. , Brogliato, A.R. & Suarez‐Kurtz, G. Influence of CYP2C9 genotypes on the pharmacokinetics and pharmacodynamics of piroxicam. Clin. Pharmacol. Ther. 78, 362–369 (2005). [DOI] [PubMed] [Google Scholar]
- 16. Riendeau, D. et al Biochemical and pharmacological profile of a tetrasubstituted furanone as a highly selective COX‐2 inhibitor. Br. J. Pharmacol. 121, 105–117 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tegeder, I. , Lötsch, J. , Krebs, S. , Muth‐Selbach, U. , Brune, K. & Geisslinger, G. Comparison of inhibitory effects of meloxicam and diclofenac on human thromboxane biosynthesis after single doses and at steady state. Clin. Pharmacol. Ther. 65, 533–544 (1999). [DOI] [PubMed] [Google Scholar]
- 18. Rohatagi, S. et al Predictive population pharmacokinetic/pharmacodynamic model for a novel COX‐2 inhibitor. J. Clin. Pharmacol. 47, 358–370 (2007). [DOI] [PubMed] [Google Scholar]
- 19. Beal S.L., Sheiner L.B. & Boeckmann A.J. eds. NONMEM Users Guide (1989–2011). (Icon Development Solutions, Ellicott City, MD, 2011). [Google Scholar]
- 20. Lindbom, L. , Pihlgren, P. & Jonsson, E.N. PsN‐Toolkit–a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput. Methods Programs Biomed. 79, 241–257 (2005). [DOI] [PubMed] [Google Scholar]
- 21. Jonsson, E.N. & Karlsson, M.O. Xpose–an S‐PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed. 58, 51–64 (1999). [DOI] [PubMed] [Google Scholar]
- 22. Nguyen, T.H. et al Model evaluation of continuous data pharmacometric models: metrics and graphics. CPT Pharmacometrics Syst. Pharmacol. 6, 87–109 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bouzom, F. , Laveille, C. , Merdjan, H. & Jochemsen, R. Use of nonlinear mixed effect modeling for the meta‐analysis of preclinical pharmacokinetic data: application to S 20342 in the rat. J. Pharm . Sci. 89, 603–613 (2000). [DOI] [PubMed] [Google Scholar]
- 24. Green, B. & Duffull, S.B. What is the best size descriptor to use for pharmacokinetic studies in the obese? Br. J. Clin. Pharmacol. 58, 119–133 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Imai, E. , Yasuda, Y. & Makino, H. Japan Association of Chronic Kidney Disease Initiatives (J‐CKDI). JMAJ 54, 403–405 (2011). [Google Scholar]
- 26. Ma, Y.C. et al Modified glomerular filtration rate estimating equation for Chinese patients with chronic kidney disease. J. Am. Soc. Nephrol. 17, 2937–2944 (2006). [DOI] [PubMed] [Google Scholar]
- 27. Lee C.S. et al Ethnic coefficients for glomerular filtration rate estimation by the Modification of Diet in Renal Disease study equations in the Korean population. J. Korean Med. Sci. 25, 1616–1625 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abduljalil, K. et al Assessment of activity levels for CYP2D6*1, CYP2D6*2, and CYP2D6*41 genes by population pharmacokinetics of dextromethorphan. Clin. Pharmacol. Ther. 88, 643–651 (2010). [DOI] [PubMed] [Google Scholar]
- 29. Kirchheiner, J. et al Impact of CYP2C9 and CYP2C19 polymorphisms on tolbutamide kinetics and the insulin and glucose response in healthy volunteers. Pharmacogenetics 12, 101–109 (2002). [DOI] [PubMed] [Google Scholar]
- 30. Hamberg, A.K. et al A pharmacometric model describing the relationship between warfarin dose and INR response with respect to variations in CYP2C9, VKORC1, and age. Clin. Pharmacol. Ther. 87, 727–734 (2010). [DOI] [PubMed] [Google Scholar]
- 31. Bergstrand, M. , Hooker, A.C. , Wallin, J.E. & Karlsson, M.O. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 13, 143–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lindauer, A. et al Pharmacokinetic/pharmacodynamic modeling of biomarker response to sunitinib in healthy volunteers. Clin. Pharmacol. Ther. 87, 601–608 (2010). [DOI] [PubMed] [Google Scholar]
- 33.“Interview form” of Morbic® Tablets. <http://www.info.pmda.go.jp/go/interview/1/650168_1149035F1020_1_118_1F.pdf> (2011). Accessed 31 October 2013.
- 34. Aoyama, T. et al Pharmacokinetic/pharmacodynamic modeling and simulation of rosuvastatin using an extension of the indirect response model by incorporating a circadian rhythm. Biol. Pharm. Bull. 33, 1082–1087 (2010). [DOI] [PubMed] [Google Scholar]
- 35. Matsumoto, Y. et al Population pharmacokinetic‐pharmacodynamic modeling of TF‐505 using extension of indirect response model by incorporating a circadian rhythm in healthy volunteers. Biol. Pharm. Bull. 28, 1455–1461 (2005). [DOI] [PubMed] [Google Scholar]
- 36. Rose, R.H. , Neuhoff, S. , Abduljalil, K. , Chetty, M. , Rostami‐Hodjegan, A. & Jamei, M. Application of a physiologically based pharmacokinetic model to predict OATP1B1‐related variability in pharmacodynamics of rosuvastatin. CPT Pharmacometrics Syst. Pharmacol. 3, e124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ide, T. , Sasaki, T. , Maeda, K. , Higuchi, S. , Sugiyama, Y. & Ieiri, I. Quantitative population pharmacokinetic analysis of pravastatin using an enterohepatic circulation model combined with pharmacogenomic information on SLCO1B1 and ABCC2 polymorphisms. J. Clin. Pharmacol. 49, 1309–1317 (2009). [DOI] [PubMed] [Google Scholar]
- 38. Savic, R.M. , Jonker, D.M. , Kerbusch, T. & Karlsson, M.O. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J. Pharmacokinet. Pharmacodyn. 34, 711–726 (2007). [DOI] [PubMed] [Google Scholar]
- 39. Abe, S. , Chiba, K. & Suwa, T. Low‐molecular‐weight heparin pharmacokinetics: a dual absorption model approach. Int. J. Clin. Pharmacol. Ther. 51, 482–489 (2013). [DOI] [PubMed] [Google Scholar]
- 40. Goto, S. et al Population estimation of the effects of cytochrome P450 2C9 and 2C19 polymorphisms on phenobarbital clearance in Japanese. Ther. Drug Monit. 29, 118–121 (2007). [DOI] [PubMed] [Google Scholar]
- 41. Yoo, H.D. , Kim, M.S. , Cho, H.Y. & Lee, Y.B. Population pharmacokinetic analysis of glimepiride with CYP2C9 genetic polymorphism in healthy Korean subjects. Eur. J. Clin. Pharmacol. 67, 889–898 (2011). [DOI] [PubMed] [Google Scholar]
- 42. Lane, S. et al The population pharmacokinetics of R‐ and S‐warfarin: effect of genetic and clinical factors. Br. J. Clin. Pharmacol. 73, 66–76 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Haeringen, N.J. , van Sorge, A.A. , Van Delft, J.L. & Carballosa Coré‐Bodelier, V.M. Flurbiprofen and enantiomers in ophthalmic solution tested as inhibitors of prostanoid synthesis in human blood. J. Ocul. Pharmacol. Ther. 16, 345–352 (2000). [DOI] [PubMed] [Google Scholar]
- 44. Rinder, H.M. , Tracey, J.B. , Souhrada, M. , Wang, C. , Gagnier, R.P. & Wood, C.C. Effects of meloxicam on platelet function in healthy adults: a randomized, double‐blind, placebo‐controlled trial. J. Clin. Pharmacol. 42, 881–886 (2002). [DOI] [PubMed] [Google Scholar]
- 45. Lee, H.I. et al Strongly increased exposure of meloxicam in CYP2C9*3/*3 individuals. Pharmacogenet. Genomics 24, 113–117 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
Supplementary Files