

Abstract
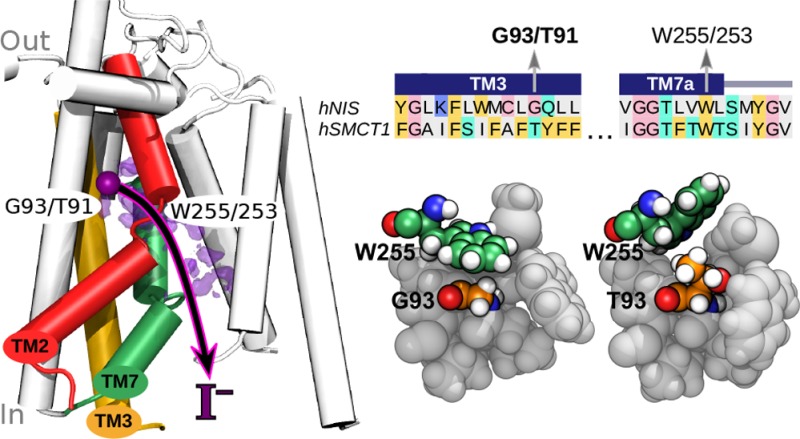
Several apical iodide translocation pathways have been proposed for iodide efflux out of thyroid follicular cells, including a pathway mediated by the sodium-coupled monocarboxylate transporter 1 (SMCT1), which remains controversial. Herein, we evaluate structural and functional similarities between SMCT1 and the well-studied sodium-iodide symporter (NIS) that mediates the first step of iodide entry into the thyroid. Free-energy calculations using a force field with electronic polarizability verify the presence of a conserved iodide-binding pocket between the TM2, TM3, and TM7 segments in hNIS, where iodide is coordinated by Phe67, Gln72, Cys91, and Gln94. We demonstrate the mutation of residue Gly93 of hNIS to a larger amino acid expels the side chain of a critical tryptophan residue (Trp255) into the interior of the binding pocket, partially occluding the iodide binding site and reducing iodide affinity, which is consistent with previous reports associating mutation of this residue with iodide uptake deficiency and hypothyroidism. Furthermore, we find that the position of Trp255 in this hNIS mutant mirrors that of Trp253 in wild-type hSMCT1, where a threonine (Thr91) occupies the position homologous to that occupied by glycine in wild-type hNIS (Gly93). Correspondingly, mutation of Thr91 to glycine in hSMCT1 makes the pocket structure more like that of wild-type hNIS, increasing its iodide affinity. These results suggest that wild-type hSMCT1 in the inward-facing conformation may bind iodide only very weakly, which may have implications for its ability to transport iodide.
Introduction
Iodide transport into the thyroid gland is a crucial step in thyroid hormone biosynthesis. Thus, defective iodide accumulation perturbs thyroid hormone status, thereby seriously affecting metabolism, growth, and maturation of a variety of organ systems.1 Uptake of iodide from the blood plasma to the thyroid follicular cells is mediated by a glycoprotein expressed at the basolateral membrane—the sodium-iodide symporter (NIS; SLC5A5 gene), which actively cotransports two sodium cations per iodide anion.2,3 NIS couples an inward “uphill” translocation of iodide against its electrochemical gradient to the inward sodium gradient maintained by the Na+/K+-ATPase.4 Significant efforts have been directed to study NIS-associated thyroid pathologies and have provided a detailed understanding of NIS function. Notably, NIS not only mediates active iodide transport in the thyroid but also in other tissues, including salivary glands, gastric mucosa, and lactating mammary gland.5
Following NIS-mediated entry, iodide must move from the intracellular space and into the follicular lumen. This process has been attributed to different proteins that function as channels and/or transporters located on the apical side of the thyroid follicular cells.6 Initially, because mutations in SLC26A4 lead to Pendred syndrome, an autosomal recessive disorder characterized by sensorineural deafness, goiter, and impaired iodide organification, Pendrin (PDS), an anion transporter capable of exchanging chloride by iodide, was implicated in the apical exit of iodide.7,8 Subsequently, the physiological role of Pendrin as the singular or predominant apical iodide transporter has been questioned. In vivo biochemical assays showed that Pendrin may participate in the iodide efflux into thyroid lumen but not uniquely.9,10 Moreover, Pendrin knockout mice showed no disruption of thyroid function.11 Thus, other proteins have been suggested as mediators of apical iodide efflux in thyroid. These include, for example: ClC-5, a voltage-gated chloride channel;12 CFTR, the cystic fibrosis transmembrane conductance regulator;13 TMEM16A (also known as anoctamin-1), a Ca2+-activated chloride channel;14 and SMCT1, a sodium-coupled monocarboxylate transporter.15,16 In particular, SMCT1, a sodium-dependent cotransporter of monocarboxylates and short-chain fatty acids encoded by the SLC5A8 gene, warrants further scrutiny. SMCT1 is localized on the apical membrane of diverse epithelia such as colon, kidney, brain, and thyroid.17 Both SMCT1 and NIS share significant homology on both nucleotide and amino acid levels. Because NIS and SMCT1 localize to opposite surfaces of thyroid epithelial cells—NIS basolaterally and SMCT1 apically, a role for SMCT1 in passive iodide efflux was proposed.16 Intriguingly, Xenopus oocyte expression studies demonstrated that SMCT1 transports a wide variety of monocarboxylates, but transport of iodide or other inorganic anions was not observed under the conditions of the study.17 Moreover, findings of normal thyroid function in Slc5a8 knockout mice called into question the role for SMCT1 in thyroid iodide transport.18 Subsequently, functional expression studies indicated that, at low external sodium concentrations, SMCT1 can permit an anionic leak current bearing NO3– > I– > Br– > Cl– selectivity. These findings, therefore, argue that SMCT1 plays a role in iodide accumulation within the thyroid follicular lumen.15
Previous work on hNIS identified key residues for basolateral sodium and iodide transport in thyroid. Based on the significant sequence similarity between hNIS and hSMCT1, we therefore hypothesize that homologous residues in hSMCT1 may confer similar function. For example, sodium ion binding essential for the transport cycle of hNIS has been associated with residue T354;19 this is homologous to T352 in hSMCT1. With respect to iodide transport, a compound heterozygous G93R/T354P hNIS mutation was found in patients with iodide transport defects. Expression of either T354P or G93R mutants in COS-7 cells demonstrated minimal iodide uptake activity, confirming that these hNIS mutations directly cause the iodide transport deficiency observed in these patients.20 The implications of these observations prompted Paroder-Belenitsky et al.21 to evaluate iodide transport for various amino acid substitutions at position 93 in hNIS. Each mutant displayed a different level of iodide transport activity with a clear pattern: the longer the side chain, the lower the iodide uptake. The G93T hNIS mutant is particularly relevant for comparing hNIS and hSMCT1. Compared to wild-type hNIS, G93T hNIS has an approximately 18-fold higher Km (lower apparent affinity) for iodide and 5-fold higher Km for sodium.21 Notably, hSMCT1 contains a threonine residue at position 91, corresponding to position 93 in hNIS. These observations suggest the hypothesis that hSMCT1 can transport iodide under particular conditions, albeit with a lower iodide affinity and at a lower transport rate than hNIS. Thus, the specific residue at this key position might determine the distinct iodide transport behavior of the two proteins.
Taken together, these lines of evidence prompted us to perform a computational study of hSMCT1, with special emphasis on the T91 and T352 residues. Given that the interaction of iodide in hNIS has been much more comprehensively studied, we applied the same computational techniques to hNIS as validation, and, furthermore, compared the roles of the T91 and T352 residues of hSMCT1 to the homologous residues of hNIS (G93 and T354). To address the controversy regarding iodide transport in hSMCT1, here we examine molecular models for both proteins and utilize the precision offered by these models to characterize putative sodium and iodide binding pockets. Although it is currently computationally infeasible to perform molecular dynamics (MD) simulations of the complete transport cycle, here we make use of MD and free-energy calculation techniques22,23 to determine if regions of high iodide affinity appear in the proteins. The absence of a region of high iodide affinity strongly suggests a lack of significant iodide transport, while the presence of such a region makes iodide transport plausible. It should be noted, however, that the simulations cannot provide unequivocal evidence of iodide transport. Our simulations employ the CHARMM Drude polarizable force field,24 which provides a physically sound representation of the interaction between the highly polarizable iodide ion and the protein. Indeed, the iodide ion has the greatest atomic polarizability of any atom type currently represented in this force field.
Methods
Molecular Modeling
To compare hSMCT1 and hNIS (UniProtKB codes Q8N695 and Q92911), we built molecular models of both proteins, using the identical modeling protocol. We searched for a template structure using Psi-Blast v2.2.3025 on the nonredundant Protein Data Bank (PDB) database, as well as HHPred from HHsuite v3.026 on a nonredundant PDB sequence database clustered at 70% sequence identity. The structure of the sodium/galactose transporter from Vibrio parahemolyticus (vSGLT), in an inward-facing conformation (PDB code 3DH4),27 was selected as the most suitable template for the two target proteins, exhibiting in both cases ≈72% coverage. In preparation for modeling, preliminary pairwise sequence alignments—i.e., hSMCT1/vSGLT and hNIS/vSGLT—were constructed using the AlignMe server v1.1 in PS mode.28 Adjustments to the initial alignments were made to optimize the agreement with PSIPRED v3.329 secondary structure and TOPCONS v2.030 transmembrane predictions and to reduce the number of residues with low stereochemical quality as evaluated by PROCHECK v3.5.31 After each refinement of the alignments, 100 iterations of model building were performed and the effect on the residues was evaluated using the PROCHECK and ProQM32 scores. In the final alignments, 20.1% of the residues were identical for hNIS and vSGLT as estimated over the whole sequence, excluding residues 1–52 in the N terminus, residues 466–517 between TM12 and TM13, and residues 550–643 in the C terminus, for which there is not a structural template. In the case of hSMCT1 and vSGLT, the identity was 22.1% excluding residues 1–50 in the N terminus, residues 463–518 between TM12 and TM13, and residues 545–610 in the C terminus. The refined alignments were then used to build a total of 2000 homology models of hSMCT1 and hNIS on the basis of vSGLT using MODELLER v9.13.33 The residues excluded in the final alignments were not modeled. The best hSMCT1 and hNIS models were selected as those with the lowest Molpdf energy value of MODELLER and the highest PROCHECK and global ProQM scores. The final models were of excellent quality according to PROCHECK, with 99.7% of the residues in the favored and additional allowed regions of the Ramachandran plot for both proteins. The global ProQM score was 0.721 for hNIS and hSMCT1 reaches a value of 0.734, which compares well with that of the template structure (0.731). Values of 0.7 are typical of membrane protein structures solved by X-ray crystallography. To identify residues involved in sodium and iodide binding in hSMCT1 and hNIS based on previous reported biochemical data, the final models in an inward-facing conformation were structurally compared using PyMOL v1.7.7 (Schrödinger, LLC). The position of both protein models in the membrane was defined by their superposition onto that of vSGLT, which was determined with the Orientation of Proteins in Membranes (OPM) server.34 The final representative hNIS and hSMCT1 models are available from the Protein Model DataBase (https://bioinformatics.cineca.it/PMDB/) with accession number PM0080745 and PM0080746, respectively.
Sequence Analysis
Protein sequences of all known members of the solute:sodium symporter family (SSS, transporter classification database [TCDB] 2.A.21) were searched against the UniprotKB database.35 A total of 1055 sequences were found, which were sorted by length and clustered at 90% identity using the UCLUST v1.2 algorithm.36 Short sequences (<400 amino acids long) were excluded, leaving 405 homologues of vSGLT, hNIS, and hSMCT1. A multiple-sequence alignment was then built with the MAFFT server37 and analyzed with Jalview v2.8.2.38 The conservation of residues was estimated as residue probabilities for the whole multiple-sequence alignment using the Weblogo v3.4 server.39
Computational Docking
For the purpose of inserting iodide into putative binding pockets predicted for hSMCT1 and hNIS molecular models, docking simulations using AutoDock v4.2.640 were performed. Both the iodide ion and the proteins were prepared for docking by AutoDockTools v1.5.6,40 assigning Gasteiger partial charges. A grid box of size 40 Å × 40 Å × 40 Å and spacing 0.375 Å was built around the putative iodide binding pocket for each target protein, with the grid center defined as the mass center of the residues F67, F87, M90, G93, Q94, W255, and Y257 for hNIS and the residues F65, F85, F88, T91, Y92, W253, and Y257 for hSMCT1. Those binding pockets include residues within 10 Å of G93 and T91 for hNIS and hSMCT1, respectively. The grid parameters were generated using AutoGrid v4.2.6 and the Lamarckian genetic algorithm41 was used to perform a search of the configurational space of the iodide–protein system. In order to select the best scoring docked conformation, analysis of the results was performed using AutoDockTools v1.5.6. It should be noted that AutoDock was used only to choose the initial position of the iodide ion for the equilbration calculations and to obtain a very rough idea of the affinity. Free energy calculations, sampling a large region of the interior of the proteins, were subsequently carried out to estimate the binding position of iodide using the more accurate Drude force field.
MD Simulation
The lowest energy conformation of each ion-bound complex—namely, hNIS:iodide and hSMCT1:iodide—were used as starting point to evaluate the specific interactions between iodide and the two target wild-type (WT) proteins through MD simulations. In addition, hNIS and hSMCT1 ion-bound mutants were simulated, which were built by replacing G93 by Thr in the WT hNIS:iodide complex, and T91 by Gly in hSMCT1:iodide. Each WT and mutated complex was embedded in a pre-equilibrated 2,3 dipalmitoyl-d-glycero-1-phosphatidylcholine (DPPC) bilayer solvated with explicit water molecules. The position of the complexes in the membrane was defined after their superposition onto the vSGLT structure, for which the membrane orientation was determined with the OPM server.34 Sodium and chloride ions (0.15 M NaCl) were added to the aqueous phase to ensure charge neutrality. The systems were initially prepared in the context of the CHARMM36 nonpolarizable additive force field;42 then, systems compatible with the Drude polarizable force field43 were generated using the “Drude Prepper” module from the CHARMM-GUI server.44 TIP3P water molecules were automatically turned into the simple water model, 4-point with negative Drude particle (SWM4-NDP).45 The initial configurations of the systems were optimized by means of an energy minimization algorithm, initially on only the Drude particles and then all atoms. This was followed by an equilibration and relaxation in a ≈22 ns MD simulation at 323 K in the isobaric–isothermal ensemble. This temperature was required to maintain the fluidity of the DPPC membrane (currently DPPC is the only lipid available for the Drude force field).46 Soft harmonic restraints were applied to the protein backbone (N, C, Cα, and O atoms) during the first 2.5 ns of simulation, which were gradually decreased from 60 to 0.5 kcal mol–1 Å–2. The 0.5 kcal mol–1 Å–2 levels were maintained throughout all simulations. Constant temperature (323 K) was enforced using a Langevin thermostat with a damping coefficient of 5 ps–1. Constant pressure (101.325 kPa) was enforced using the Langevin piston method.47 Long-range electrostatic interactions were computed using the particle-mesh Ewald summation method,48 with a smooth real-space cutoff applied between 10 and 12 Å. All covalent bonds involving hydrogen as well as the intramolecular geometries of water were constrained.49 The Verlet-I/r-RESPA multiple time-step integrator50 was used with a time step of 1 fs. The temperature of the Drude oscillator was fixed at 1 K, and a reflective hard-wall constraint was applied to impose a maximum distance of 0.25 Å between the Drude particle and its parent atom’s nucleus.24 All MD simulations were performed using the program NAMD v2.1051 and analyzed with VMD v1.9.2.52 The root mean-square deviation (RMSD) for the position of backbone and side chain atoms in the simulated systems was utilized to appraise protein stability and thermodynamic equilibrium. The bulk of the calculations were performed using resources provided by the Extreme Science and Engineering Discovery Environment.53
Free Energy Calculation
To determine the relative iodide affinity in the putative binding pockets predicted for hNIS and hSMCT1, the adaptive biasing force (ABF) method22,23 was applied. The ion-bound WT (hNIS and hSMCT1) and mutant (G93T hNIS and T91G hSMCT1) structures of both proteins were analyzed. After minimization and equilibration of the WT and mutated systems using the MD protocol described above, a transition coordinate emulating the iodide transport pathway was defined with the Caver v3.0.1 program.54 The starting point for the calculation of the transport pathway was the center of mass of the Cα atoms of residues F67, F87, M90, G93, Q94, W255, and Y257 for WT and G93T hNIS, and the residues F65, F85, F88, T91, Y92, W253, and Y257 for WT and T91G hSMCT1. In all cases, the transport pathways were not linear, so the path was divided into seven points p1 to p7 situated about 3.5 Å from each other. For each protein system, six ABF calculations were performed, each associated with a line segment between consecutive points on the path. The potential of mean force (PMF) for each window was calculated as a function of the distance along the vector from point pi to point pi+1, i.e. the transition coordinate for the segment beginning at point i was si = (R – pi)·(pi+1 – pi)/|pi+1 – pi|, where R is the position of the iodide ion. The ABF calculations were performed using the Colvars module55 of NAMD v2.10.51 The domain of each calculation was 0 ≤ si ≤ 3.5 Å, and force samples were collected in bins having widths of 0.1 Å to capture small-scale variations in the PMF. All calculations were performed using the simulation conditions described in the preceding section (MD Simulation) for 24–68 ns of simulated time for windows 1 and 2, and 14–22 ns for the remaining windows, totaling 202 ns for WT hNIS, 214 ns for G93T hNIS, 141 ns for WT hSMCT1, and 115 ns for T91G hSMCT1. The convergence of the calculations was evaluated by comparing the gradient of the free energy of the first and second half of the length of the simulations (see the Supporting Information (SI)).
The PMF for each path segment were computed along different directions, and therefore could not simply be concatenated. Instead, the resulting PMFs wi(si) were combined with the statistics of the iodide ion position to construct full three-dimensional PMFs W(x, y, z). For each segment i, we consider an orthonormal coordinate frame (si, ti, ui) with the first axis parallel to the line segment pi+1 – pi with an origin at pi. In each segment, we calculated Pi(ti, ui|si), the probability distribution of the coordinates orthogonal to the line segment for a given value of si during the ABF simulations. From this, we generated a three-dimensional PMF by
| 1 |
where x, y, and z were computed from (si, ti, ui) by a transformation to global coordinate frame. In practice, the values of wi(si) and Pi(ti, ui|si) were defined on evenly spaced grids in the (si, ti, ui) coordinate frame, and cubic interpolation56 was used to calculate Wi(x, y, z) on a grid aligned with the global (x, y, z) coordinate frame. This grid had a resolution of 0.3 Å in all three directions. As the 3D PMFs, Wi(x, y, z), had arbitrary reference potentials, the potential values were shifted so that overlapping regions among different Wi(x, y, z) possessed the same average value. The shifted PMFs were then combined, with the values in overlapping regions calculated as averages weighted by the number of samples contributing to Pi(ti, ui|si). The result was a single 3D PMF, W(x, y, z), for each protein system, which described the free-energy at accessible positions along the pathway. The PMFs were anchored so that the average in a small region about 10 Å along the pathway from the center of the pocket to the cytoplasmic exit is zero. The free energies should be comparable between the WT and mutant of the same cotransporter (hNIS or hSMCT1) because the distance of this anchor point from the mutated residue is about 17 Å; however, direct comparison between the free energies of hNIS and hSMCT1 is questionable. Uncertainties in the free energies were estimated by partitioning the ABF samples into two halves, repeating all calculations on each half separately, and taking the uncertainty to be the difference in the two resulting values. Further details on the convergence of the ABF calculations are provided in Figure S2–5 of the SI.
Simulation Analysis
The residues within 6–8 Å of the iodide ion were analyzed for both WT and mutated hNIS and hSMCT1 proteins over the trajectories of MD simulation. Particularly, the distance between the iodide and indole NH group of W255 for hNIS and W253 for hSMCT1, as well as the distance between this NH group and the Cα atom of residue 93 (hNIS) or residue 91 (hSMCT), was determined. The initial nonequilibrium portion of each trajectory was discarded for the analyses. All figures were generated using PyMOL v1.7.7 (Schrödinger, LLC) and VMD v1.9.2.52
Calculations with Protein Fragments
To further corroborate the estimations of iodide affinity, we performed free energy calculations using the ABF method22,23 on fragments of our hNIS and hSMCT1 models and mutants. The fragments consisted of portions of TM2, TM3, TM7, and TM11, specifically residues 53 to 100, 242 to 298, and 412 to 437, inclusive, for hNIS, and residues 51 to 98, 240 to 296, and 410 to 435 for hSMCT1. The proteins were simulated in periodic boxes of aqueous NaCl solution (0.15 M), with equilibrium sizes of about 46 Å × 50 Å × 57 Å. The simulation conditions were similar to those described in the section MD Simulation, except that a 0.5 fs time step was used. The transition coordinate (u) in the ABF simulations was the position of the iodide ion projected along a line passing through the position of lowest iodide free-energy for WT hNIS predicted in the section Free Energy Calculation and another point in the solution far from the protein. ABF was applied along the transition coordinate on the interval −0.9 ≤ u ≤ 13.0 Å using a bin size of 0.1 Å. A harmonic restraint was applied when the distance between the iodide ion and the line defining the transition coordinate surpassed 4.7 Å, keeping the ion within a cylinder of approximately this radius. The standard binding free energy was calculated by57
| 2 |
where β = 1/(kBT) is the inverse thermal energy, R = 4.7 Å is the radius of the restraining cylinder, and C0 is the standard concentration (1/1660.5389 Å3). The integration limits, a and b, where chosen to be a = −0.9 Å and b = 4.0 Å. The choice of a and b mattered little for the hNIS models, as long as the free energy minima near u = 0 were included. However, the weak affinity seen for the hSMCT1 models made it difficult to define the bound region, and the choice of b could have a large effect on ΔG°. The ABF simulations comprised 290–320 ns of simulated time for each of the four systems. Uncertainties in the resulting free energies were estimated by comparing the force samples from the two halves of each simulation.58
Results
Models of hNIS and hSMCT1 in an Inward-Facing Conformation
Paroder-Belenitsky and colleagues previously reported a hNIS homology model.21 However, in order to ensure consistency in the present comparisons of sequence and structural similarity between hSMCT1 and hNIS, we built models of both proteins using an identical, standard modeling protocol. Considering that both the sodium-iodide symporter (NIS, TCDB 2.A.21.5.1) and the sodium-coupled monocarboxylate transporter (SMCT1, TCDB 2.A.21.5.3) belong to the SSS protein family, the crystal structure of vSGLT, a member of this family, was selected as the most suitable template for modeling. The vSGLT structure was captured in an inward-facing conformation, with a galactose-binding site occluded from the outside solution by hydrophobic residues and a predicted sodium-binding site based on a comparison with the LeuT transporter.27 Thus, as in the template, the hNIS and hSMCT1 models also represent an inward-facing state of the protein. Notably, vSGLT has 14 TM segments with both the N- and C-termini exposed to the extracellular milieu; however, a core of 13 TM segments is a shared feature of the SSS family. In fact, NIS and SMCT1 from Homo sapiens were predicted to have 13 α-helical TM segments, with the N-terminus facing the extracellular medium and the C-terminus facing the cytoplasm (Figure 1 A). Because the conformation of TM1 was not physically well represented in the vSGLT crystal structure used for modeling, resultant hNIS and hSMCT1 models contained 12 of 13 membrane-spanning helices.
Figure 1.

Structural modeling of hSMCT1 and hNIS using the vSGLT transporter as a template. (A) Schematic representation of hSMCT1/hNIS topology with the N- and C-terminus exposed to the extracellular medium and cytoplasm, respectively. The helices on the red triangular background comprise repeat 1, while repeat 2 is composed of the helices on the blue triangular background. (B) Helix representation of the hSMCT1 model, rendered with Bendix,89 in an inward-facing conformation viewed from the plane of the membrane, with the extracellular side at the top. The helices are colored according to the topology with triangular backgrounds indicating the orientation of repeats. The position of the protein in the membrane was defined after its superposition onto that of vSGLT, which was determined with the OPM server.34 The hNIS model shows a similar fold. (C) Ensemble of the refined sequence alignments (vSGLT/hNIS and vSGLT/hSMCT1) used for modeling. The alignment is colored according to the chemical properties of the residues: gray, aliphatic (A, I, L, M, and V); cyan, polar uncharged (N, Q, S, and T); yellow, aromatic (F, W, and Y); red, acidic (D and E); purple, basic (K, R, and H); pink, exceptional (C, G and P). The secondary structure (helix) assignment for the vSGLT crystal structure was obtained with DSSP90 and is indicated by dark blue rectangles.
As shown in Figure 1B, for the hSMCT1 model (and hNIS model as well) the transmembrane helices are arranged in two structural repeats of five TMs each (TM2–6 and TM7–11), which are related by 2-fold pseudosymmetry with an axis that runs normal to the membrane and between the two halves. The inverted pseudosymmetric fold has been identified as a common pattern of secondary active transporters59,60 and appears to facilitate the alternating-access mechanism of substrate transport across the membrane. Although not related by function, the vSGLT template and both hNIS and hSMCT1 proteins modeled here share an alternate-access mechanism with tight coupling between sodium and substrate transport. Moreover, for hNIS and hSMCT1, the sequence identity with vSGLT is 20.1% and 22.1%, respectively, while the similarity reaches 44.3% for hNIS and 42.8% for hSMCT1 (Figure 1C). Members of the same protein family often share the same tertiary fold, despite having relatively low sequence identity.61 Because vSGLT and hNIS/hSMCT1 are members of the SSS family, our models can be expected to have correct large-scale structure. Further facilitating our comparative study is the considerable conservation between hNIS and hSMCT1 themselves; overall, they show 51.1% sequence identity and 71.4% sequence similarity.
Conserved Sodium Binding Site in hNIS and hSMCT1
Previous biochemical analysis of iodide uptake in hNIS revealed that five polar residues, including four with hydroxy moieties (T351, S353, T354, S356) and one amide (N360), located in the TM9 segment play a key role in Na+/iodide cotransport.19 Superimposing the hNIS model onto the vSGLT crystal structure showed that these residues were found in the same region as the sodium binding site indicated by the crystal structure of vSGLT.27 We sought, therefore, to identify all residues near this region in the hNIS and hSMCT1 models possibly contributing to sodium binding. In particular, we analyzed polar and acidic side chains within 8 Å of the superimposed sodium ion of vSGLT. In the hNIS model, we found 12 residues putatively coordinating a sodium ion in this position, including: S66 and S69 in TM2; T190, D191 and Q194 in TM6; S349, T351, S353, T354, S356 and T357 in TM9; and, Q263 in TM7 (Figure 2A). Superimposing the hSMCT1 model onto vSGLT reveals a sodium-binding pocket nearly identical to that predicted for hNIS. The sodium binding pocket comprises the homologous residues S64 and S67 in TM2; T188, D189 and Q192 in TM6; S347, T349, S351, T352, S354 and S355 in TM9; and, Q261 in TM7 (Figure 2B). Notably, while this work was being developed, Ferrandino et al. reported computational and biochemical data supporting the role of residues S66, D191, Q194, and Q263 in coordinating sodium in hNIS at the Na2 site,62 providing independent confirmation of our findings. We propose, therefore, that at least one sodium ion involved in hNIS and hSMCT1 substrate transport is coordinated by a binding pocket conserved between both proteins.
Figure 2.
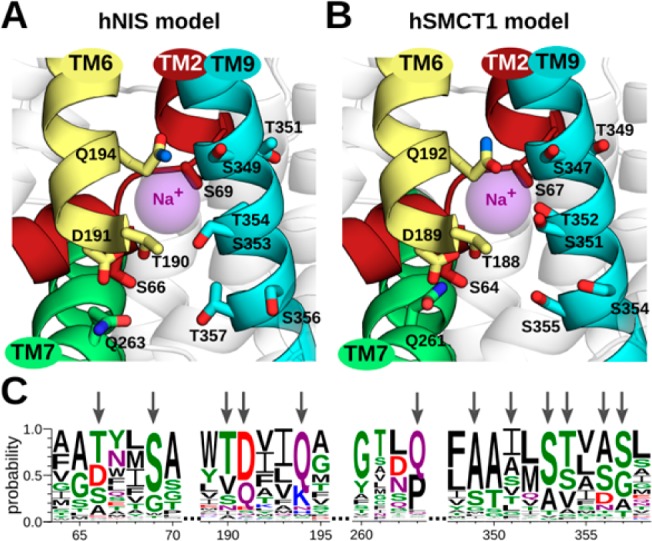
Predicted sodium-binding sites in hNIS and hSMCT1 and the conservation of associated residues. After superimposing the models on the vSGLT crystal structure, a putative sodium-binding site was predicted for (A) hNIS and (B) hSMCT1. The sodium ion reported in vSGLT, used as a reference point for our analysis, is shown as a purple sphere. The predicted sodium-binding site conserved in both models involves residues in TM2, TM6, TM7, and TM9 segments, which are shown in a cartoon representation and colored as in Figure 1. The putative sodium-coordinating residues are displayed as sticks colored by atom type (oxygen in red and nitrogen in blue). The participation of residues S66, D191, Q194, and Q263 in hNIS-mediated iodide uptake has been recently demonstrated by experiments.62 The hNIS and hSMCT1 models are oriented with the extracellular side toward the top of the page. (C) Sequence logo illustrating conservation of the residues (indicated with arrows) predicted to coordinate the Na+ ion. The conservation was evaluated among 405 members of the SSS family. The residue colors are as follows: polar (G, S, T, Y, and C) in green, amide-terminated (N and Q) in purple, basic (K, R, and H) in blue, acidic (D and E) in red, and hydrophobic (A, V, L, I, P, W, F, and M) in black. Numbering follows the sequence of hNIS.
Notwithstanding the fact that hNIS and hSMCT1 have been described as possessing different functions, the two transporters seem to share important residues coordinating sodium transport. Here, we evaluate the conservation of these residues in all members reported of the SSS family (Figure 2C). At position 66/64 of hNIS/hSMCT1, this analysis showed a preponderance of polar or acidic residues, namely T, D, or S; while at position 69/67, serine was highly conserved. The TM6 segment (residues 190–194 in hNIS and 188–192 in hSMCT1) exhibited a T[DQ]xx[QK] motif, and at position 263/261 in the TM7 segment a glutamine residue was mostly conserved. Between residues 349–357/347–355 in TM9, a S[TS]xϕ[SG] motif was observed, where ϕ is usually A, S, D, or N. Curiously, a motif similar to that found in TM9, including polar and acidic residues, has been described previously to coordinate sodium ions in transporters of the divalent anion/Na+ symporter (DASS, TCDB 2.A.47) family;63 and recently, the same pattern was found in the p-aminobenzoyl-glutamate transporter (AbgT, TCDB 2.A.68) family.64 It should be noted, that in DASS and AbgT these motifs were identified in helical hairpins, whereas in SSS the S[TS]xϕ[SG] motif is part of a canonical helix.
Putative Residues Involved in Iodide Binding in hNIS and hSMCT1
It is well-known that active iodide uptake across the basolateral membrane in the thyroid is mediated by NIS. However, the precise molecular mechanisms by which this protein binds and transports iodide from the blood plasma to the thyroid follicular cells has not yet been completely elucidated. Informed by a previous site-directed mutagenesis study of the G93, W255 and Y259 hNIS residues,21 our hNIS model representing an inward-facing conformation of the protein enabled us to identify a putative iodide-binding pocket. The binding site comprises residues situated within the membrane-spanning helices TM2, TM3, and TM7, including: F67, Q72, F87, M90, C91, G93, Q94, W255, and Y259 (Figure 3A). All these residues line an aqueous cavity of the protein, except G93, which is in close contact with W255, the role of which is further described in following sections. Interestingly, after superimposing the hNIS model onto the vSGLT crystal structure, we observed that the predicted iodide-binding pocket is equivalent in position to the galactose-binding site in vSGLT, suggesting an underlying mechanistic unity for substrate transport across the SSS family members.
Figure 3.
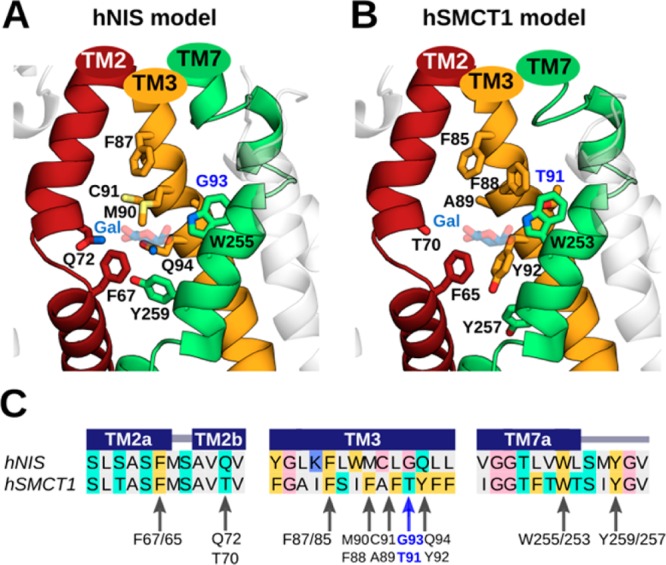
Predicted iodide-binding pockets in hNIS and hSMCT1 and the conservation of associated residues. For the (A) hNIS and (B) hSMCT1 models, these sites involve the transmembrane TM2, TM3, and TM7 segments, which are shown in a cartoon representation and colored as in Figure 1. The nine putative residues coordinating iodide in both models are displayed as sticks colored by atom type (oxygen in red, nitrogen in blue, and sulfur in light yellow). Residues G93/T91 are highlighted in blue. G93 in hNIS was found to be mutated in patients with goitrous hypothyroidism. The galactose (Gal) molecule reported in vSGLT is located near the predicted iodide position in the models and is shown here for reference as transparent blue and red sticks. The predicted iodide binding cavities are displayed with the extracellular side toward the top of the page. (C) Sequence alignment between hNIS and hSMCT1 showing the conservation of residues proposed to coordinate iodide. The alignment is colored according to the chemical properties of the residues as in Figure 1. The secondary structure assignments obtained with PSIPRED (helix) are indicated by dark blue rectangles. Arrows indicate the residues involved in iodide binding according to our prediction.
We compared our hSMCT1 model with hNIS to determine whether a similar substrate-binding cavity appeared in the former. Coady et al. recently reported that SMCT1 possesses an anionic leak current (with selectivity NO3– > I– > Br– > Cl–) that becomes significant at low external sodium concentration (under conditions of their experiment), which may represent a second function for SMCT1 in addition to coupled short chain fatty acid transport.15 Through a structural comparison between hNIS and hSMCT1, we observed that both proteins possess a pocket with many similarities. In hSMCT1, that cavity is formed by residues F65, T70, F85, F88, A89, T91, Y92, W253, and Y257 (Figure 3B). Interestingly, four of these residues are conserved between hNIS and hSMCT1 (F67/F65, F87/F85, W255/W253, and Y259/Y257). However, the remaining residues critical to iodide binding in hNIS—Q72, M90, C91, G93, and Q94—are replaced, respectively, by T70, F88, A89, T91, and Y92 in hSMCT1 (Figure 3C). We hypothesize that the difference between G93 in hNIS and the homologous residue T91 in hSMCT1 may play a crucial role in the iodide transport mediated by both proteins, considering the pathological effect observed in patients expressing a compound heterozygous G93R/T354P in hNIS.20 Indeed, Paroder-Belenitsky et al.21 performed several substitutions at position 93 in hNIS—including G93T/S/N/Q/E/D/A, with a clear pattern: the longer the side chain at this position, the higher the Km for the anion substrates and the lower the iodide affinity.
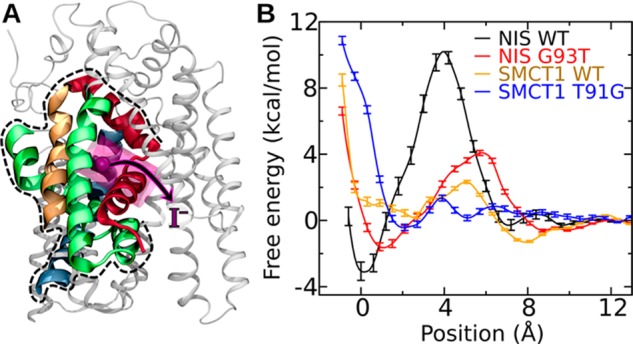
Iodide Free Energy in hNIS and hSMCT1
The potential role of hSMCT1 in iodide transport has been widely discussed. The first overexpression studies in two mammalian cell lines, African green monkey kidney (COS-7) and Chinese hamster ovary (CHO), suggested this capability,16 whereas SMCT1 expression in MDCK (Madin-Darby Canine Kidney) cells exhibited no iodide transport and the protein was implicated in Na+-dependent monocarboxylate transport in thyroid cells.17 In a later study, however, hSMCT1 showed a sodium-sensitive anionic leak current that could potentially explain the electrogenic transport of iodide in the thyroid.15 Thus, to determine the potential for iodide transport by hSMCT1, we evaluated the binding free energy of iodide in the binding pocket predicted for wild-type hNIS and hSMCT1. The critical role of residues G93 in hNIS and T91 in hSMCT1 in determining this parameter also was evaluated by assessing the effects of the reciprocal mutations, G93T and T91G, in hNIS and hSMCT1, respectively. Figure 4A shows that, in their inward-facing conformations, the hNIS and hSMCT1 models exhibit an access tunnel from which the presumed iodide-binding pocket connects with the cytoplasm. We defined a transition coordinate along this tunnel to calculate the free-energy landscape for iodide binding in the inward-facing conformation. Particularly, we analyzed the statistics of the iodide ion position along this coordinate to construct fully three-dimensional potentials of mean force of iodide in the access tunnels of hNIS WT, hNIS G93T, hSMCT1 WT, and hSMCT1 T91G, represented in Figure 4B–E.
Figure 4.

Free-energy calculation of iodide binding to hNIS and hSMCT1. (A) Image showing the putative binding site and proposed iodide exit pathway through hNIS, indicating their position relative to the complete protein. (B–E) Magnification of the binding site and iodide entry/exit pathway in wild-type and mutant hNIS and hSMCT1. The 3D free-energy map is represented by purple surfaces, where regions with free energies < −1 and < −3 kcal/mol relative to the value at a reference position are enclosed by transparent and solid surfaces, respectively. The C atoms of the W255/253 are green, and those of residue 93/91 are orange. The residues with C atoms in cyan are those within 4.5 Å of the minimum free energy.
For WT hNIS (Figure 4B), a relatively large region of low free energy was observed, having a deep minimum (Table 1) located within 3.5 Å of the side chains of F67, Q72, C91, and Q94. As shown in Figure 4C, the position of this free energy minimum in the WT is occupied by W255 in the hNIS G93T mutant, displacing the free energy minimum toward Q72 and leading to a smaller region of low free energy and a weaker overall affinity. The iodide at the minimum free energy position is coordinated (within 3.5 Å) by the same residues as in the WT, with the addition of I292. Additional regions of low free-energy were also seen along the transition coordinate used for the ABF calculations, suggesting an iodide transport pathway through hNIS to reach the interior of the thyroid follicular cells.
Table 1. Minimum Free Energy Values in the 3D Free Energy Map of Iodide in Each Complete Cotransporter Model and the Estimated Standard Binding Free Energy Calculated for Fragments of Each Model.
| system | min free energy for full system (kcal/mol)a | standard ΔG for protein fragment (kcal/mol) |
|---|---|---|
| hNIS WT | –7.6 ± 0.5 | –0.9 ± 0.4 |
| hNIS G93T | –6.3 ± 0.6 | 0.3 ± 0.2 |
| hSMCT1 WT | –2.8 ± 0.6 | 1.9 ± 0.2 |
| hSMCT1 T91G | –6.1 ± 0.9 | 1.4 ± 0.2 |
Values may not be directly comparable between hNIS and hSMCT1 (see text).
Figure 4B and C (hNIS WT and hNIS G93T) can be directly compared because, as described in Methods, the free energy maps are anchored at point far from the mutation. Likewise, Figure 4D and E (hSMCT1 WT and hSMCT1 T91G) should also be directly comparable. However, comparisons between hNIS and hSMCT1 could be called into question due to a slightly different environment of iodide at the point to which the potential of mean force was anchored. Despite this caveat, there are clear similarities in the location of low free energy regions along the tunnel between the 3D free-energy maps for hNIS and hSMCT1. For instance, a conserved phenylalanine (F67 in hNIS and F65 hSMCT1) lies close to the position of minimum free energy for all four systems. On the other hand, both hSMCT1 WT and hSMCT1 T91G appear to show weaker iodide affinity than either hNIS systems. Indeed, for hSMCT1 WT, the region of low free energy is poorly defined and hardly exists at all (Figure 4D). The glutamine residues that make contact with iodide in hNIS (Q72 and Q94) are replaced with threonine and tyrosine (T70 and Y92) in hSMCT1. T70 remains within 3.5 Å of the minimum free energy position of iodide in hSMCT1 WT, whereas Y92 appears key for binding iodide in hSMCT1 T91G. Like the hNIS G93T mutant, a portion of hSMCT1 WT’s pocket is occupied by a tryptophan (W253), apparently discouraging iodide binding. In the hSMCT1 T91G mutant, the side chain of W253 is packed into the wall of the pocket, similar to its position in hNIS WT. Overall, our analysis suggests that iodide binding is facilitated by interactions with aromatic and primary amide side chains, as well as sulfur-containing groups.
It should be noted that the goal of our simulations was simply to identify a putative iodide-binding pocket in hNIS to be compared with the homologous structure in hSMCT1. Our simulations cannot directly reveal iodide transport. Observing the effects of major conformational changes in the protein is precluded by the fact that the homology models were restrained to the inward-facing conformation during the simulations. However, the free energy results are consistent with a kinetic analysis of the G93T hNIS mutant performed by Paroder-Belenitsky et al.,21 showing an important loss of iodide affinity (an approximately 18-fold higher Km) for the mutant as compared with the WT protein, which would correspond to an apparent free-energy difference of ≈2 kcal/mol.
Role of Residue 93/91
Residue 93 in hNIS was originally found to be mutated in patients affected with congenital hypothyroidism and shown by Paroder-Belenitsky et al.21 to be important for iodide transport. However, neither it nor its homologue in hSMCT1 makes direct contact with the most favorable site for iodide in our simulations, lying at a distance >8 Å. Thus, the effect of mutations at residue 93/91 appears to be indirect. Figure 4B–D suggests that the two systems with glycine at residue 93/91 have W255/253 retracted from the pocket, while those with threonine at this position have a pocket partially occupied by the bulky tryptophan side chain. Therefore, we propose that a small glycine residue allows the tryptophan side chain to insert into the wall of the iodide-binding pocket (Figure 5A), while a larger residue at position 93/91 expels the tryptophan into the interior space of this pocket (Figure 5B). Figure 5C demonstrates that the positions of W255/253 in this figure are typical: W255/253 spends a majority of the time within 4.8 Å of G93/91 in hNIS WT and hSMCT1 T91G, while it is more than 7 Å from T93/91 in hNIS G93T and hSMCT1 WT.
Figure 5.
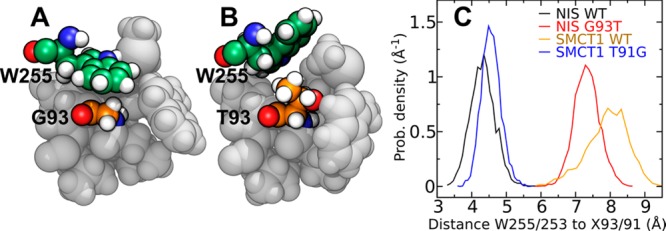
Role of residue 93 of hNIS and the homologous residue 91 of hSMCT1 in iodide binding. (A) In this simulation snapshot of hNIS WT, the tryptophan (W255) forms the wall of the putative iodide-binding pocket, making contact with G93. (B) In the hNIS G93T mutant, the steric bulk of the T93 side chain expels W255 into the pocket. (C) Histogram of the distance between the ring nitrogen of W255 (W253 in hSMCT1) and the Cα atom of the residue at position 93 (91 in hSMCT1).
Standard Free Energies in Transporter Fragments
The free energy calculations described in the previous section do not yield absolute binding free energies because this requires the iodide to reach an appropriate reference state. To overcome this limitation, we performed free energy calculations using fragments of the proteins, illustrated in Figure 6A, in which the iodide could be brought from the putative binding site to a position in the solvent sufficiently far from the protein to serve as a reference state. Although the correspondence between the fragment systems and the full systems is only approximate, particularly with respect to the long-range effects of the electrostatic environment, the calculations with these fragments allow local interactions with the iodide binding site to be rigorously compared among the four systems. The same backbone restraints used for the full systems were used for the fragments, ensuring that the fragments did not unravel. The potentials of mean force as a function of distance from the putative binding site are shown in Figure 6B. In qualitative agreement with the calculations for the complete systems, hNIS WT exhibits the deepest free-energy minimum, while the minimum for hSMCT1 WT is the weakest. Consistent with the 3D free-energy map in Figure 4, the free energy minima for hNIS G93T, hSMCT1 WT, and hSMCT1 T91G are displaced a few angstroms along the transition coordinate from the minimum in hNIS WT. It is notable that the free-energy barrier to exit the binding site for hNIS WT is much larger than for any of the other systems (13.3 ± 0.7 kcal/mol). Using the derivation of Limongelli et al.,57 we estimated the standard binding free energies of iodide in the fragment systems, which are shown in Table 1. In the case of the hNIS systems, the results of this estimation are robust to the choice of integral limits in eq 2, as long as the global minima are included. However, for the hSMCT1 systems, the minima near the putative binding site are weak and the result is sensitive to the limits, particularly for hSMCT1 WT, where the global minimum occurs outside of the putative binding site at a distance of 7.9 Å along the transition coordinate. Limits of a = −0.9 Å and b = 4.0 Å were used in Table 1. The calculations with the fragments seem to show that the iodide affinity for the hSMCT1 T91G mutant, while greater than that of the hSMCT1 WT, is significantly lower than that for hNIS G93T. On the other hand, the calculations with the full systems suggested similar affinities for the two mutants; however, as mentioned previously, direct comparison of the full-system results for hSMCT1 T91G and hNIS G93T may not be valid.
Figure 6.
Free energy calculations on fragments of wild-type hNIS, hSMCT1, and their mutants. (A) Structure of the fragment. The portion of the hNIS WT included in the fragment is shown in color (TM2, red; TM3, orange; TM7 green; TM11, blue) by broad ribbons, while the part not included is shown by thin gray ribbons. The position of lowest free energy for iodide is represented by a purple sphere, while the region to which the iodide was restrained during the calculation is shown as a transparent purple cylinder. (B) Free energy as a function of the position of iodide along the axis of the cylinder shown in panel A.
Discussion
To initiate comparisons between the hNIS and hSMCT1 transporters and pinpointing functionally conserved residues, we generated homology models of both proteins using exactly the same modeling protocol. Analysis of the available crystal structures of SSS family revealed that only the sodium/galactose transporter from Vibrio parahemolyticus (vSGLT)27,65 has been elucidated to date. Therefore, we used vSGLT as the template for molecular modeling of hNIS and hSMCT1 structures. According to the alternating-access mechanisms, a transporter must be able to reach at least three distinctive states: an inward-facing, occluded and outward-facing conformation.66 The available vSGLT structure adopts an inward-facing conformation; thus, the models built here represent such conformations of hNIS and hSMCT1, including 12 of the 13 transmembrane segments in which helices 2–6 and 7–11 are related by inherent pseudosymmetry. Interestingly, although the overall sequence identity between the vSGLT template and modeled proteins is only ∼20%, we demonstrate that all three proteins share common structural features. Here we further show in detail the underlying structural determinants of hNIS and hSMCT1 function.
Members of the SSS family use an inwardly directed sodium electrochemical gradient, generated and maintained by the Na+/K+ ATPase, to transport a diverse set of solutes such as sugars, amino acids, nucleosides, vitamins, or inorganic ions from the extracellular fluid into cells.67 NIS and SMCT1 couple movement of sodium to the transport of iodide and monocarboxylates, respectively. However, their precise substrate binding and transport mechanisms await full elucidation. A variety of residues have been shown to be crucial for efficient coupling of sodium in protein members of the SSS family;68 for example, conserved residues in TM1, TM2, and TM9 have been associated with the sodium binding in the PutP transporter69,70 and residues in TM2 and TM9 are positioned appropriately to coordinate sodium in vSGLT.27 For hNIS, the residues S66, D191, Q194, and Q263 in TM2, TM6, and TM7 have recently been associated with the sodium coordination at the Na2 site,62 whereas Ser and Thr residues in TM9 were shown previously to be required to sodium binding and its translocation during substrate transport.19 Our results, obtained using the hNIS and hSMCT1 models, reveal that equivalent OH-containing amino acids and glutamine residues in the segments TM2, TM6, TM7, and TM9 constitute a putative sodium-binding site and may participate in ion translocation in both proteins. The predicted sodium binding sites are highly conserved in hNIS and hSMCT1, including a threonine residue at position 354 and 352, respectively. In hNIS, T354 plays a key role in sodium-dependent transport of iodide.19,20,71 Moreover, a T354P mutation of hNIS in the TM9 segment was identified in a patient who developed goitrous hypothyroidism.20 Subsequent site-directed mutagenesis studies demonstrated the requirement for a hydroxy group at position 354 for hNIS function.71 Thus, in hSMCT1, T352 may prove similarly critical for sodium-dependent transport. In PutP, a hydroxy moiety at position T341—equivalent to T354 in hNIS—was shown to be essential for sodium binding;72 and an alanine mutation in S365 for vSGLT—also homologous to T354—completely abolished the sodium-dependent substrate transport.27 Collectively, these observations support our predicted sodium binding pockets. A second sodium-binding site is expected to exist for both hNIS and hSMCT1, given that iodide transport in hNIS involves cotransport of two sodium ions73,74 and that hSMCT1 has exhibited a 2:1 stoichiometry for sodium-substrate cotransport.15 This second site remains unidentified. It should be noted that the conservation of residues involving the sodium binding site in hNIS and hSMCT1 was evaluated in all members of the SSS family, revealing important sodium-associated sequence motifs that are even conserved in other sodium-dependent protein families such as DASS and AbgT.63,64 The fact that a similar pattern is found in three different families of proteins having highly divergent sequences overall, underscores the importance of these regions for function and the likelihood of their involvement in sodium binding.
With regard to iodide transport, experiments yielding a set of residues for which mutation leads to severe defects in NIS-mediated iodide uptake—namely, mutations situated in the transmembrane or intracellular segments—3,75include the following: (1) mutants of the phosphorylated amino acid residue S43, T49, S227, T577, and S581,76 (2) kinetic analysis of the extracellular charged H226 residue,77 (3) deletions in the region spanning residues 234 to 280 of the TM7 segment,78 (4) mutations of conserved charged amino acids in the extracellular region,79 and (5) site-directed mutagenesis of the G93, W255, and Y259 residues21 suggested by a G93R/T354P spontaneous mutation expressed in a patient with a diffuse goiter.20 Based on the studies of Paroder-Belenitsky et al.,21 we identified a putative iodide-binding pocket comprising transmembrane segments TM2, TM3, and TM7 in hNIS. Four aromatic residues F67, F87, W255, and Y259 within this binding region are conserved in hSMCT1 (F65, F85, W253, and Y257), allowing us to assume certain similarities in the specificity and the substrate transport mechanism between the two proteins. Aromatic residues play specialized roles for substrate transport in members of the LeuT family of sodium-solute symporters. For example, vSGLT and hSGLT1 exhibit a triad of aromatic residues that form part of an interactive network comprising their substrate and sodium binding sites.80 Precisely, F67, F87, W255, and Y259 in hNIS and F65, F85, W253, and Y257 in hSMCT1 are homologous residues, suggesting conserved substrate binding pockets. In addition to the aromatic residues, our hNIS and hSMCT1 models also predict important roles for polar amino acids, including glutamine and threonine, in coordination of iodide. Our simulations seem to reveal a mechanism by which mutation of G93 in hNIS causes reduced iodide transport in experiment21 and pathological effects observed in patients with congenital hypothyroidism.20 Namely, mutation of G93 expels W255 into the iodide-binding site. Notably, our calculations suggest that the presence of a threonine residue at the corresponding position of hSMCT1 (T91) contributes a significant factor to its reduced ability to transport iodide relative to hNIS.
Computationally demanding MD simulations using the polarizable CHARMM Drude force field and the ABF free-energy calculation technique were performed to investigate iodide affinity in the predicted binding pockets of the hNIS and hSMCT1 models and to gain insight into hSMCT1’s potential for apical iodide transport in thyroid. Recently, Cappet et al. demonstrated a high level of predictability to calculate binding free energy using homology models with very low identity (∼22%)81 similar to levels found between hNIS, hSMCT1 and the vSGLT template. Moreover, it should be noted that the ABF method has been applied successfully in evaluations of substrate transport in transmembrane proteins,82,83 whereas the Drude polarizable force field has shown superior accuracy in describing ion–protein interactions.84 Iodide has the highest polarizability among common monatomic ions85 and polarizability is particularly important in describing interactions between ions and tryptophan,43 a residue located in the predicted iodide binding pockets. Using the CHARMM3686 force field, which does not represent atomic polarizability, we support this assertion by demonstrating that the potential of mean force for iodide in the fragment of hNIS WT (analogous to those shown in Figure 6B) does not possess a global minimum near the putative iodide binding site (see Figure S1, in the SI).
Wild-type proteins and mutants with an interchange of G93 by threonine in hNIS and T91 by glycine in hSMCT1 were analyzed. For iodide in complex with WT hNIS, the PMF values reached an overall minimum in the region surrounding G93, whereas the G93T mutant showed a displacement of the predicted iodide binding pocket and a decrease in affinity. Table 1 suggests a decrease in affinity by a factor of 8 or 9 for this mutation, which roughly agrees with the 18-fold decrease in the apparent iodide affinity (Km) observed by Paroder-Belenitsky et al.21 for mutations at position 93 of hNIS. With this validation of our methodology, we further evaluated the iodide interaction with WT hSMCT1, which exhibits a binding pocket comparable to hNIS, albeit with significantly lower iodide affinity. Moreover, an increase in iodide affinity was observed when the hSMCT1 iodide-binding pocket becomes more like that of hNIS through the T91G mutation. Nevertheless, additional structural elements likely are required for hSMCT1 to attain the high iodide affinity calculated for hNIS. Particularly, our structural analyses revealed that the orientation of homologous conserved aromatic residues W255/253 (in hNIS/hSMCT1) is a key factor in iodide affinity. Previous experimental studies have suggested important roles for aromatic residues in iodide binding, e.g. in an inhibitory anion-selective Cys-loop receptor, the glutamate-gated chloride channel α.87 In addition, given that sodium binding is a required step in the transport cycle, it would be interesting to consider the effect of the presence of sodium in its binding site on the affinity of iodide for its putative site. Likely the addition of a positive charge to the protein would cause the iodide affinity to slightly increase. Taking the dielectric constant of the interior of the protein to be >488 and the distance between the binding sites to be ≈15 Å, the change in the free energy should be in the range −5.5 < ΔΔG < 0 kcal/mol.
The present work reveals much lower iodide affinity in the pocket of hSMCT1 analogous to the putative iodide-binding pocket of hNIS. While the calculations presented here have a number of limitations—the use of approximate homology models, representation of only the inward facing conformation, and the inability to exhibit conformational changes associated with the transport cycle, they suggest that hSMCT1 is unlikely to function as an iodide transporter comparable to hNIS. Indeed, one could hypothesize that any transport of iodide by hSMCT1 occurs by a qualitatively different path than the dominant iodide-transport mechanism of hNIS. On the other hand, the low affinity in the binding site that we have calculated for hSMCT1 may still be adequate for an apical iodide leak into the follicular lumen, in contrast to robust and highly specific hNIS transport activity required for intracellular iodide accumulation. Our study is envisioned to prove useful for better understanding iodide binding and transport by sodium-coupled cotransporters and providing a molecular basis to treat iodide-associated pathologies.
Acknowledgments
This work was supported by Kansas Bioscience Authority funds to the Institute of Computational Comparative Medicine (ICCM) at Kansas State University and to the Nanotechnology Innovation Center of Kansas State University (NICKS), as well as National Institutes of Health/National Institute of General Medical Sciences Award 1R15GM101674-01A1. A.V.-J. thanks the academic program “Desarrollo de Núcleo Científico Multidisciplinario”, L’Oreal-UNESCO Rising Talent Award, and FONDECYT research initiation grant no. 11170223. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575. This work was supported in part by a grant from the National Science Foundation (CHE-1726332).
Glossary
Abbreviations
- SMCT1
Na+/monocarboxylate transporter 1
- NIS
Na+/I– symporter
- SGLT
Na+/galactose transporter
- SSS
sodium:solute symporter
- ABF
adaptive biasing force
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.7b00521.
Figure comparing results of the Drude force field and a nonpolarizable force field. Figure indicating convergence of the ABF calculations for wild type and mutated hNIS and hSMCT1 proteins interacting with iodide (PDF)
Author Contributions
A.V.-J., P.F. and J.C. conceived the project. A.V.-J. and J.C. carried out computational modeling and simulations and analyzed data. A.V.-J., P.F., and J.C. wrote the manuscript. All authors contributed to the editing of the paper and to scientific discussions.
The authors declare no competing financial interest.
Supplementary Material
References
- Werner S. C.; Ingbar S. H.; Braverman L. E.; Utiger R. D.. Werner & Ingbar’s the Thyroid: A Fundamental and Clinical Text; Lippincott Williams & Wilkins, 2005. [Google Scholar]
- Dai G.; Levy O.; Carrasco N. Cloning and Characterization of the Thyroid Iodide Transporter. Nature 1996, 379 (6564), 458–460. 10.1038/379458a0. [DOI] [PubMed] [Google Scholar]
- Dohán O.; De la Vieja A.; Paroder V.; Riedel C.; Artani M.; Reed M.; Ginter C. S.; Carrasco N. The Sodium/Iodide Symporter (NIS): Characterization, Regulation, and Medical Significance. Endocr. Rev. 2003, 24 (1), 48–77. 10.1210/er.2001-0029. [DOI] [PubMed] [Google Scholar]
- Bagchi N.; Fawcett D. M. Role of Sodium Ion in Active Transport of Iodide by Cultured Thyroid Cells. Biochim. Biophys. Biochim. Biophys. Acta, Biomembr. 1973, 318 (2), 235–251. 10.1016/0005-2736(73)90117-X. [DOI] [PubMed] [Google Scholar]
- Dohán O.; Carrasco N. Advances in Na+/I– Symporter (NIS) Research in the Thyroid and Beyond. Mol. Cell. Endocrinol. 2003, 213 (1), 59–70. 10.1016/j.mce.2003.10.059. [DOI] [PubMed] [Google Scholar]
- Fong P. Thyroid Iodide Efflux: A Team Effort?. J. Physiol. 2011, 589 (24), 5929–5939. 10.1113/jphysiol.2011.218594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett L. A.; Glaser B.; Beck J. C.; Idol J. R.; Buchs A.; Heyman M.; Adawi F.; Hazani E.; Nassir E.; Baxevanis A. D.; Sheffield V. C.; Green E. D. Pendred Syndrome Is Caused by Mutations in a Putative Sulphate Transporter Gene (PDS). Nat. Genet. 1997, 17 (4), 411–422. 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]
- Karniski L. P.; Wang R.; Kreman T. M.; Sheffield V. C.; Scott D. A. The Pendred Syndrome Gene Encodes a Chloride-Iodide Transport Protein. Nat. Genet. 1999, 21 (4), 440–443. 10.1038/7783. [DOI] [PubMed] [Google Scholar]
- Gillam M. P.; Sidhaye A. R.; Lee E. J.; Rutishauser J.; Stephan C. W.; Kopp P. Functional Characterization of Pendrin in a Polarized Cell System. Evidence for Pendrin-Mediated Apical Iodide Efflux. J. Biol. Chem. 2004, 279 (13), 13004–13010. 10.1074/jbc.M313648200. [DOI] [PubMed] [Google Scholar]
- Bizhanova A.; Kopp P. Controversies Concerning the Role of Pendrin as an Apical Iodide Transporter in Thyroid Follicular Cells. Cell. Physiol. Biochem. 2011, 28 (3), 485–490. 10.1159/000335103. [DOI] [PubMed] [Google Scholar]
- Wangemann P.; Kim H.-M.; Billings S.; Nakaya K.; Li X.; Singh R.; Sharlin D. S.; Forrest D.; Marcus D. C.; Fong P. Developmental Delays Consistent with Cochlear Hypothyroidism Contribute to Failure to Develop Hearing in Mice Lacking Slc26a4/Pendrin Expression. Am. J. Physiol. - Ren. Physiol. 2009, 297 (5), F1435–F1447. 10.1152/ajprenal.00011.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hove M.-F.; Croizet-Berger K.; Jouret F.; Guggino S. E.; Guggino W. B.; Devuyst O.; Courtoy P. J. The Loss of the Chloride Channel, ClC-5, Delays Apical Iodide Efflux and Induces a Euthyroid Goiter in the Mouse Thyroid Gland. Endocrinology 2006, 147 (3), 1287–1296. 10.1210/en.2005-1149. [DOI] [PubMed] [Google Scholar]
- Li H.; Ganta S.; Fong P. Altered Ion Transport by Thyroid Epithelia from CFTR(−/−) Pigs Suggests Mechanisms for Hypothyroidism in Cystic Fibrosis. Exp. Physiol. 2010, 95 (12), 1132–1144. 10.1113/expphysiol.2010.054700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyffels L.; Strickaert A.; Virreira M.; Massart C.; Van Sande J.; Wauquier C.; Beauwens R.; Dumont J. E.; Galietta L. J.; Boom A.; Kruys V. Anoctamin-1/TMEM16A Is the Major Apical Iodide Channel of the Thyrocyte. Am. J. Physiol. Cell Physiol. 2014, 307 (12), C1102–1112. 10.1152/ajpcell.00126.2014. [DOI] [PubMed] [Google Scholar]
- Coady M. J.; Wallendorff B.; Bourgeois F.; Lapointe J.-Y. Anionic Leak Currents through the Na+/Monocarboxylate Cotransporter SMCT1. Am. J. Physiol. Cell Physiol. 2010, 298 (1), C124–131. 10.1152/ajpcell.00220.2009. [DOI] [PubMed] [Google Scholar]
- Rodriguez A.-M.; Perron B.; Lacroix L.; Caillou B.; Leblanc G.; Schlumberger M.; Bidart J.-M.; Pourcher T. Identification and Characterization of a Putative Human Iodide Transporter Located at the Apical Membrane of Thyrocytes. J. Clin. Endocrinol. Metab. 2002, 87 (7), 3500–3503. 10.1210/jcem.87.7.8797. [DOI] [PubMed] [Google Scholar]
- Paroder V.; Spencer S. R.; Paroder M.; Arango D.; Schwartz S.; Mariadason J. M.; Augenlicht L. H.; Eskandari S.; Carrasco N. Na(+)/Monocarboxylate Transport (SMCT) Protein Expression Correlates with Survival in Colon Cancer: Molecular Characterization of SMCT. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (19), 7270–7275. 10.1073/pnas.0602365103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank H.; Gröger N.; Diener M.; Becker C.; Braun T.; Boettger T. Lactaturia and Loss of Sodium-Dependent Lactate Uptake in the Colon of SLC5A8-Deficient Mice. J. Biol. Chem. 2008, 283 (36), 24729–24737. 10.1074/jbc.M802681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Vieja A.; Reed M. D.; Ginter C. S.; Carrasco N. Amino Acid Residues in Transmembrane Segment IX of the Na+/I- Symporter Play a Role in Its Na+ Dependence and Are Critical for Transport Activity. J. Biol. Chem. 2007, 282 (35), 25290–25298. 10.1074/jbc.M700147200. [DOI] [PubMed] [Google Scholar]
- Kosugi S.; Inoue S.; Matsuda A.; Jhiang S. M. Novel, Missense and Loss-of-Function Mutations in the Sodium/Iodide Symporter Gene Causing Iodide Transport Defect in Three Japanese Patients. J. Clin. Endocrinol. Metab. 1998, 83 (9), 3365. 10.1210/jcem.83.9.5243. [DOI] [PubMed] [Google Scholar]
- Paroder-Belenitsky M.; Maestas M. J.; Dohán O.; Nicola J. P.; Reyna-Neyra A.; Follenzi A.; Dadachova E.; Eskandari S.; Amzel L. M.; Carrasco N. Mechanism of Anion Selectivity and Stoichiometry of the Na+/I- Symporter (NIS). Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (44), 17933–17938. 10.1073/pnas.1108278108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darve E.; Pohorille A. Calculating Free Energies Using Average Force. J. Chem. Phys. 2001, 115 (20), 9169–9183. 10.1063/1.1410978. [DOI] [Google Scholar]
- Comer J.; Gumbart J. C.; Hénin J.; Lelièvre T.; Pohorille A.; Chipot C. The Adaptive Biasing Force Method: Everything You Always Wanted to Know but Were Afraid to Ask. J. Phys. Chem. B 2015, 119 (3), 1129–1151. 10.1021/jp506633n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemkul J. A.; Huang J.; Roux B.; MacKerell A. D. An Empirical Polarizable Force Field Based on the Classical Drude Oscillator Model: Development History and Recent Applications. Chem. Rev. 2016, 116 (9), 4983–5013. 10.1021/acs.chemrev.5b00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F.; Madden T. L.; Schäffer A. A.; Zhang J.; Zhang Z.; Miller W.; Lipman D. J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25 (17), 3389–3402. 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söding J.; Biegert A.; Lupas A. N. The HHpred Interactive Server for Protein Homology Detection and Structure Prediction. Nucleic Acids Res. 2005, 33 (Web Server), W244–W248. 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faham S.; Watanabe A.; Besserer G. M.; Cascio D.; Specht A.; Hirayama B. A.; Wright E. M.; Abramson J. The Crystal Structure of a Sodium Galactose Transporter Reveals Mechanistic Insights into Na+/Sugar Symport. Science 2008, 321 (5890), 810–814. 10.1126/science.1160406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm M.; Staritzbichler R.; Khafizov K.; Forrest L. R. AlignMe—a Membrane Protein Sequence Alignment Web Server. Nucleic Acids Res. 2014, 42 (W1), W246–W251. 10.1093/nar/gku291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D. T. Protein Secondary Structure Prediction Based on Position-Specific Scoring Matrices. J. Mol. Biol. 1999, 292 (2), 195–202. 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Tsirigos K. D.; Peters C.; Shu N.; Käll L.; Elofsson A. The TOPCONS Web Server for Consensus Prediction of Membrane Protein Topology and Signal Peptides. Nucleic Acids Res. 2015, 43, W401. 10.1093/nar/gkv485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283. 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Ray A.; Lindahl E.; Wallner B. Model Quality Assessment for Membrane Proteins. Bioinformatics 2010, 26 (24), 3067–3074. 10.1093/bioinformatics/btq581. [DOI] [PubMed] [Google Scholar]
- Eswar N.; Webb B.; Marti-Renom M. A.; Madhusudhan M. S.; Eramian D.; Shen M.-Y.; Pieper U.; Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Protein Sci. 2007, 2.9.1. 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- Lomize M. A.; Lomize A. L.; Pogozheva I. D.; Mosberg H. I. OPM: Orientations of Proteins in Membranes Database. Bioinformatics 2006, 22 (5), 623–625. 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- Boutet E.; Lieberherr D.; Tognolli M.; Schneider M.; Bansal P.; Bridge A. J.; Poux S.; Bougueleret L.; Xenarios I. UniProtKB/Swiss-Prot, the Manually Annotated Section of the UniProt KnowledgeBase: How to Use the Entry View. Methods Mol. Biol. 2016, 1374, 23–54. 10.1007/978-1-4939-3167-5_2. [DOI] [PubMed] [Google Scholar]
- Edgar R. C. Search and Clustering Orders of Magnitude Faster than BLAST. Bioinformatics 2010, 26 (19), 2460–2461. 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Katoh K.; Standley D. M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30 (4), 772–780. 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A. M.; Procter J. B.; Martin D. M. A.; Clamp M.; Barton G. J. Jalview Version 2—a Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25 (9), 1189–1191. 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks G. E.; Hon G.; Chandonia J.-M.; Brenner S. E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14 (6), 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30 (16), 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Goodsell D. S.; Halliday R. S.; Huey R.; Hart W. E.; Belew R. K.; Olson A. J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19 (14), 1639–1662. . [DOI] [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiórkiewicz-Kuczera J.; Yin D.; Karplus M. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102 (18), 3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Lopes P. E. M.; Huang J.; Shim J.; Luo Y.; Li H.; Roux B.; MacKerell A. D. Polarizable Force Field for Peptides and Proteins Based on the Classical Drude Oscillator. J. Chem. Theory Comput. 2013, 9 (12), 5430–5449. 10.1021/ct400781b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29 (11), 1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Lamoureux G.; Harder E.; Vorobyov I. V.; Roux B.; MacKerell A. D. Jr. A Polarizable Model of Water for Molecular Dynamics Simulations of Biomolecules. Chem. Phys. Lett. 2006, 418 (1–3), 245–249. 10.1016/j.cplett.2005.10.135. [DOI] [Google Scholar]
- Chowdhary J.; Harder E.; Lopes P. E. M.; Huang L.; MacKerell A. D.; Roux B. A Polarizable Force Field of Dipalmitoylphosphatidylcholine Based on the Classical Drude Model for Molecular Dynamics Simulations of Lipids. J. Phys. Chem. B 2013, 117 (31), 9142–9160. 10.1021/jp402860e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller S. E.; Zhang Y.; Pastor R. W.; Brooks B. R. Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J. Chem. Phys. 1995, 103 (11), 4613–4621. 10.1063/1.470648. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103 (19), 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Miyamoto S.; Kollman P. A. Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13 (8), 952–962. 10.1002/jcc.540130805. [DOI] [Google Scholar]
- Tuckerman M.; Berne B. J.; Martyna G. J. Reversible Multiple Time Scale Molecular Dynamics. J. Chem. Phys. 1992, 97 (3), 1990–2001. 10.1063/1.463137. [DOI] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26 (16), 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14 (1), 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Towns J.; Cockerill T.; Dahan M.; Foster I.; Gaither K.; Grimshaw A.; Hazlewood V.; Lathrop S.; Lifka D.; Peterson G. D.; Roskies R.; Scott J. R.; Wilkens-Diehr N. XSEDE: Accelerating Scientific Discovery. Comput. Sci. Eng. 2014, 16 (5), 62–74. 10.1109/MCSE.2014.80. [DOI] [Google Scholar]
- Chovancova E.; Pavelka A.; Benes P.; Strnad O.; Brezovsky J.; Kozlikova B.; Gora A.; Sustr V.; Klvana M.; Medek P.; Biedermannova L.; Sochor J.; Damborsky J. CAVER 3.0: A Tool for the Analysis of Transport Pathways in Dynamic Protein Structures. PLoS Comput. Biol. 2012, 8 (10), e1002708. 10.1371/journal.pcbi.1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorin G.; Klein M.; Hénin J. Using Collective Variables to Drive Molecular Dynamics Simulations. Mol. Phys. 2013, 111, 3345–3362. 10.1080/00268976.2013.813594. [DOI] [Google Scholar]
- Wells D. B.; Abramkina V.; Aksimentiev A. Exploring Transmembrane Transport through Alpha-Hemolysin with Grid-Steered Molecular Dynamics. J. Chem. Phys. 2007, 127 (12), 125101. 10.1063/1.2770738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limongelli V.; Bonomi M.; Parrinello M. Funnel Metadynamics as Accurate Binding Free-Energy Method. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (16), 6358–6363. 10.1073/pnas.1303186110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poblete H.; Miranda-Carvajal I.; Comer J. Determinants of Alanine Dipeptide Conformational Equilibria on Graphene and Hydroxylated Derivatives. J. Phys. Chem. B 2017, 121 (15), 3895–3907. 10.1021/acs.jpcb.7b01130. [DOI] [PubMed] [Google Scholar]
- Myers-Turnbull D.; Bliven S. E.; Rose P. W.; Aziz Z. K.; Youkharibache P.; Bourne P. E.; Prlić A. Systematic Detection of Internal Symmetry in Proteins Using CE-Symm. J. Mol. Biol. 2014, 426 (11), 2255–2268. 10.1016/j.jmb.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest L. R. Structural Symmetry in Membrane Proteins. Annu. Rev. Biophys. 2015, 44, 311–337. 10.1146/annurev-biophys-051013-023008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockner T.; Jurik A.; Weissensteiner R.; Freissmuth M.; Ecker G. F.; Sitte H. H.. Development of Refined Homology Models: Adding the Missing Information to the Medically Relevant Neurotransmitter Transporters. In Membrane Transport Mechanism; Krämer R., Ziegler C., Eds.; Springer Series in Biophysics; Springer: Berlin Heidelberg, 2014; pp 99–120. [Google Scholar]
- Ferrandino G.; Nicola J. P.; Sánchez Y. E.; Echeverria I.; Liu Y.; Amzel L. M.; Carrasco N. Na+ Coordination at the Na2 Site of the Na+/I– Symporter. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (37), E5379–E5388. 10.1073/pnas.1607231113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancusso R.; Gregorio G. G.; Liu Q.; Wang D.-N. Structure and Mechanism of a Bacterial Sodium-Dependent Dicarboxylate Transporter. Nature 2012, 491 (7425), 622–626. 10.1038/nature11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara-Jaque A.; Fenollar-Ferrer C.; Mulligan C.; Mindell J. A.; Forrest L. R. Family Resemblances: A Common Fold for Some Dimeric Ion-Coupled Secondary Transporters. J. Gen. Physiol. 2015, 146 (5), 423–434. 10.1085/jgp.201511481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe A.; Choe S.; Chaptal V.; Rosenberg J. M.; Wright E. M.; Grabe M.; Abramson J. The Mechanism of Sodium and Substrate Release from the Binding Pocket of VSGLT. Nature 2010, 468 (7326), 988–991. 10.1038/nature09580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardetzky O. Simple Allosteric Model for Membrane Pumps. Nature 1966, 211 (5052), 969–970. 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- Wright E. M.; Loo D. D. F.; Hirayama B. A.; Turk E. Surprising Versatility of Na+-Glucose Cotransporters: SLC5. Physiology 2004, 19, 370–376. 10.1152/physiol.00026.2004. [DOI] [PubMed] [Google Scholar]
- Perez C.; Ziegler C. Mechanistic Aspects of Sodium-Binding Sites in LeuT-like Fold Symporters. Biol. Chem. 2013, 394 (5), 641–648. 10.1515/hsz-2012-0336. [DOI] [PubMed] [Google Scholar]
- Jung H. The Sodium/Substrate Symporter Family: Structural and Functional Features. FEBS Lett. 2002, 529 (1), 73–77. 10.1016/S0014-5793(02)03184-8. [DOI] [PubMed] [Google Scholar]
- Mazier S.; Quick M.; Shi L. Conserved Tyrosine in the First Transmembrane Segment of Solute:Sodium Symporters Is Involved in Na+-Coupled Substrate Co-Transport. J. Biol. Chem. 2011, 286 (33), 29347–29355. 10.1074/jbc.M111.263327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy O.; Ginter C. S.; De la Vieja A.; Levy D.; Carrasco N. Identification of a Structural Requirement for Thyroid Na+/I- Symporter (NIS) Function from Analysis of a Mutation That Causes Human Congenital Hypothyroidism. FEBS Lett. 1998, 429 (1), 36–40. 10.1016/S0014-5793(98)00522-5. [DOI] [PubMed] [Google Scholar]
- Hilger D.; Böhm M.; Hackmann A.; Jung H. Role of Ser-340 and Thr-341 in Transmembrane Domain IX of the Na+/Proline Transporter PutP of Escherichia Coli in Ligand Binding and Transport. J. Biol. Chem. 2008, 283 (8), 4921–4929. 10.1074/jbc.M706741200. [DOI] [PubMed] [Google Scholar]
- Eskandari S.; Loo D. D. F.; Dai G.; Levy O.; Wright E. M.; Carrasco N. Thyroid Na+/I– Symporter Mechanism, Stoichiometry, and Specificity. J. Biol. Chem. 1997, 272 (43), 27230–27238. 10.1074/jbc.272.43.27230. [DOI] [PubMed] [Google Scholar]
- Nicola J. P.; Carrasco N.; Amzel L. M. Physiological Sodium Concentrations Enhance the Iodide Affinity of the Na+/I– Symporter. Nat. Commun. 2014, 5, 3948. 10.1038/ncomms4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Vieja A.; Dohan O.; Levy O.; Carrasco N. Molecular Analysis of the Sodium/Iodide Symporter: Impact on Thyroid and Extrathyroid Pathophysiology. Physiol. Rev. 2000, 80 (3), 1083–1105. [DOI] [PubMed] [Google Scholar]
- Vadysirisack D. D.; Chen E. S.-W.; Zhang Z.; Tsai M.-D.; Chang G.-D.; Jhiang S. M. Identification of in Vivo Phosphorylation Sites and Their Functional Significance in the Sodium Iodide Symporter. J. Biol. Chem. 2007, 282 (51), 36820–36828. 10.1074/jbc.M706817200. [DOI] [PubMed] [Google Scholar]
- Wu S.-L.; Ho T.-Y.; Liang J.-A.; Hsiang C.-Y. Histidine Residue at Position 226 Is Critical for Iodide Uptake Activity of Human Sodium/Iodide Symporter. J. Endocrinol. 2008, 199 (2), 213–219. 10.1677/JOE-08-0249. [DOI] [PubMed] [Google Scholar]
- Liang J.-A.; Chen C.-P.; Huang S.-J.; Ho T.-Y.; Hsiang C.-Y.; Ding H.-J.; Wu S.-L. A Novel Loss-of-Function Deletion in Sodium/Iodide Symporter Gene in Follicular Thyroid Adenoma. Cancer Lett. 2005, 230 (1), 65–71. 10.1016/j.canlet.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Li C.-C.; Ho T.-Y.; Kao C.-H.; Wu S.-L.; Liang J.-A.; Hsiang C.-Y. Conserved Charged Amino Acid Residues in the Extracellular Region of Sodium/Iodide Symporter Are Critical for Iodide Transport Activity. J. Biomed. Sci. 2010, 17, 89. 10.1186/1423-0127-17-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Loo D. D. F.; Hirayama B. A.; Wright E. M. The Importance of Being Aromatic: Pi Interactions in Sodium Symporters. Biochemistry 2012, 51 (47), 9480–9487. 10.1021/bi301329w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappel D.; Hall M. L.; Lenselink E. B.; Beuming T.; Qi J.; Bradner J.; Sherman W. Relative Binding Free Energy Calculations Applied to Protein Homology Models. J. Chem. Inf. Model. 2016, 56 (12), 2388–2400. 10.1021/acs.jcim.6b00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov I.; Cheng X.; Sine S. M.; McCammon J. A. Barriers to Ion Translocation in Cationic and Anionic Receptors from the Cys-Loop Family. J. Am. Chem. Soc. 2007, 129 (26), 8217–8224. 10.1021/ja070778l. [DOI] [PubMed] [Google Scholar]
- Zhang J.-L.; Zheng Q.-C.; Yu L.-Y.; Li Z.-Q.; Zhang H.-X. Effect of External Electric Field on Substrate Transport of a Secondary Active Transporter. J. Chem. Inf. Model. 2016, 56 (8), 1539–1546. 10.1021/acs.jcim.6b00212. [DOI] [PubMed] [Google Scholar]
- Li H.; Ngo V.; Da Silva M. C.; Salahub D. R.; Callahan K.; Roux B.; Noskov S. Y. Representation of Ion–Protein Interactions Using the Drude Polarizable Force-Field. J. Phys. Chem. B 2015, 119 (29), 9401–9416. 10.1021/jp510560k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.; Whitfield T. W.; Harder E.; Lamoureux G.; Vorobyov I.; Anisimov V. M.; MacKerell A. D.; Roux B. Simulating Monovalent and Divalent Ions in Aqueous Solution Using a Drude Polarizable Force Field. J. Chem. Theory Comput. 2010, 6 (3), 774–786. 10.1021/ct900576a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B.; Zhu X.; Shim J.; Lopes P. E. M.; Mittal J.; Feig M.; Mackerell A. D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone φ, ψ and Side-Chain χ(1) and χ(2) Dihedral Angles. J. Chem. Theory Comput. 2012, 8 (9), 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs R. E.; Gouaux E. Principles of Activation and Permeation in an Anion-Selective Cys-Loop Receptor. Nature 2011, 474 (7349), 54–60. 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Li C.; Zhang Z.; Alexov E. On the Dielectric “Constant” of Proteins: Smooth Dielectric Function for Macromolecular Modeling and Its Implementation in DelPhi. J. Chem. Theory Comput. 2013, 9 (4), 2126–2136. 10.1021/ct400065j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl A. C. E.; Chavent M.; Sansom M. S. P. Bendix: intuitive helix geometry analysis and abstraction. Bioinformatics 2012, 28, 2193. 10.1093/bioinformatics/bts357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W.; Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577. 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.