

Abstract
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disease resulting from pathologically low levels of survival motor neuron (SMN) protein. The majority of mRNA from the SMN2 allele undergoes alternative splicing and excludes critical codons, causing an SMN protein deficiency. While there is currently no FDA-approved treatment for SMA, early therapeutic efforts have focused on testing repurposed drugs such as phenylbutyrate (2), valproic acid (3), riluzole (6), hydroxyurea (7), and albuterol (9), none of which has demonstrated clinical effectiveness. More recently, clinical trials have focused on novel small-molecule compounds identified from high-throughput screening and medicinal chemistry optimization such as olesoxime (11), CK-2127107, RG7800, LMI070, and RG3039 (17). In this paper, we review both repurposed drugs and small-molecule compounds discovered following medicinal chemistry optimization for the potential treatment of SMA.
Keywords: Spinal muscular atrophy, survival motor neuron, small molecule, repurposed drugs, clinical trial
Graphical Abstract
1. INTRODUCTION
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disorder characterized by progressive muscle wasting and eventual loss of muscle function due to severe motor neuron dysfunction.1–4 As one of the leading heritable causes of infant mortality worldwide, SMA affects between 1 in 6,000 to 1 in 11,000 live births and has a carrier frequency of approximately 1 in 40 to 50 people.5–8
The first cases of SMA were first published in the 1890s by Guido Werdnig and Johan Hoffman, two physicians whose autopsy studies revealed the key pathological feature of the disease: severe loss of α-motor neurons (MNs) from the anterior horn of the spinal cord.9 A breakthrough study in 1995 mapped the SMA genes to chromosome 5q13, in which two nearly identical genes encode for the same SMN protein.10 Initially named telomeric and centromeric SMN, these genes are now more commonly referred to as SMN1 and SMN2, respectively.11 These two genes are nearly identical and encode the same 294 residue, 38-kDa SMN protein.10,11 Researchers later determined that SMN is ubiquitously expressed at high levels during embryonic and prenatal development before rapidly decreasing after birth.12
SMN1 and SMN2 mRNAs are transcribed at similar levels in all cells, but approximately 85% of SMN2 transcripts result in abnormal SMN protein due to a single nucleotide difference from C to T in the 6th nucleotide of exon 7 (encoding residue 840) of the SMN2 gene.13,14 The resulting Δ7 SMN2 mRNA is truncated, lacking the codons for 16 amino acids within exon 7 as well as the translational termination codon.15,16 The translated Δ7 protein is unstable and quickly degrades in cells.17–19 The majority of SMA patients lack both copies of the SMN1 gene altogether, and instead have two or more copies of the SMN2 gene.20 In such SMN1 null patients, SMN2 expression is the sole source of SMN protein. Accordingly, the SMN2 gene is unable to fully compensate for the loss of SMN1 due to its Δ7 mutation and decreased production of normal, full-length SMN protein.5 About 5% of SMA patients have mutations in the SMN1 gene that render it dysfunctional.
There are four types of SMA, which are classified by age of onset, age of death, achievements in motor development, and number of SMN2 copies.21 Type I (also known as Werdnig-Hoffman disease) patients demonstrate the most severe SMA symptoms and possess only 2 to 3 copies of SMN2. Within 6 months of birth, these patients are unable to sit unsupported due to their lack of muscle function.22,23 Type II SMA (also referred to as Dubowitz syndrome) patients possess 3 to 4 copies of SMN2 and, notably, are able to sit unsupported.24 In general, Types III and IV are milder forms of SMA with much later onsets. Type III SMA, referred to as juvenile SMA or Kugelberg-Welander disease, typically manifests after 18 months in patients with 4 copies of SMN2. Type IV patients possess 4 to 8 copies of SMN2 and often demonstrate muscular dystrophy symptoms after adulthood.21,23,24 Plainly, there is an inverse relationship between the number of SMN2 copies and the phenotypic severity of this illness.25,26 A variety of SMA mouse models containing the transgenic human SMN2 gene were developed to reflect the range of disease severity observed in humans and have been used to evaluate compounds that increase transcription of SMN2, the inclusion of exon 7 in the SMN2 mRNA transcript, and levels of the SMN protein.27 The Δ7 SMA and the “Taiwanese” of “Li” models are the most commonly used severe mouse models of SMA in preclinical studies.28 Outcome measures in the severe models include survival (~10–14 days), motor function, neuromuscular junction (NMJ) number and morphology. Less severe mouse models include the 2B/- and Smn1C/C, which have mutated the single mouse Smn allele so that its exon 7 is skipped, as well as the SMNRT (SMN read through) and “Burgheron” models with longer survival times (~30–35 days).29–31
Currently, there are seven clinical and 11 preclinical agents for SMA treatment, ranging from repurposed drugs, gene therapy, antisense oligonucleotides, and novel small molecules. Gene therapy is an experimental technique that inserts genes into cells via a viral vector. Genes can be delivered systemically by injection of the vectors intravenously or directly into the cerebral spinal fluid (CSF) to infect neurons. AveXis, the first company to test gene therapy in SMA clinical trials, has developed a proprietary intravenous treatment using self-complementary adeno-associated virus (scAAV-9) vectors that carry a codon-optimized SMN1 sequence and a chimeric intron that reintroduces SMN1 into MNs resulting in SMN protein overexpression.32 The effectiveness of the scAAV-9 vector is attributed to its ability to cross the blood-brain barrier (BBB) and to infect neurons by retrograde trafficking from the axon. This modality is in Phase II clinical trials. Another experimental method, antisense oligonucleotide therapy, aims to deliver a synthetic strand of nucleic acids (DNA, RNA, or a chemically modified analogue) that binds a specific regulatory site on pre-mRNA, thereby modifying exon recognition. Ionis (formerly known as Isis) Pharmaceuticals and Biogen have collaborated to develop a chemically modified antisense ribonucleotide, IONIS-SMNRx, that binds to a splice silencer element in intron 7 of SMN2 and results in its inactivation and the inclusion of exon 7.33 Although these two treatment options demonstrate the innovative and new strategies being developed over the past few years, from here on, we will focus only on the biology and medicinal chemistry strategies associated with producing small molecule treatments for SMA. We review repurposed drugs as well as five clinical and six preclinical novel small molecules candidate therapies.
2. REPURPOSED DRUGS
Finding new uses for existing FDA-approved drugs offers several advantages, including shorter development and approval times, reduced costs, and higher success rates relative to the standard practice of developing new compounds. New drug development can take up to 16 years34 and incurs significant costs during the drug discovery and preclinical stages.35 Drug repurposing avoids a number of these expensive processes and takes between three and 12 years for drug approval. Further, the success rates of repurposed drugs are drastically improved; while 10% of new molecular entities from Phase II and 50% from Phase III clinical trials make it to market, the rates for repurposed compounds are 25% and 65%, respectively.36 With no strong clinical prospects in the SMA drug therapy pipeline, the cost-effective and potentially time-saving testing of repurposed drugs was an attractive source of therapeutic drug discovery.
SMA was granted orphan drug designation by the FDA, which assigns orphan status to drugs and biologics intended for the safe and effective treatment, diagnosis, or prevention of (i) rare diseases and/or disorders affecting fewer than 200,000 people in the U.S. or (ii) diseases and/or disorders affecting more than 200,000 persons, but that are not expected to recover the costs of developing and marketing a treatment drug.37
FDA-approved drugs that enhance SMN2 promoter activity, modulate SMN2 splicing, and stabilize SMN2 mRNA or SMN protein, have been explored as potential means of treating SMA (Table 1). Though repurposed drugs examined for the treatment of SMA have resulted in promising early data, including increased levels of SMN protein, none has successfully made it through Phase III clinical trials.
Table 1.
Small molecules that were repurposed for the treatment of SMA.
| Name | Structure | Mode of Action | MWa | cLogPb | PSAc | HBAd | HBDe | Ref |
|---|---|---|---|---|---|---|---|---|
| Sodium butyrate (1) |
|
Weak class I HDAC inhibitor | 110 | 0.6 | 26 | 2 | 1 | 39 |
| Sodium phenylbutyrate (2) |
|
Weak class I HDAC inhibitor | 186 | 2.0 | 26 | 2 | 1 | 40,45–49 |
| Valproic Acid (3) |
|
Weak class I HDAC inhibitor | 144 | 2.8 | 37 | 2 | 1 | 41,50–54 |
| Suberoylanilide hydroxamic acid (4) |
|
Potent HDAC class I and II | 264 | 1.0 | 78 | 5 | 3 | 42,43 |
| Trichostatin A (5) |
|
Potent HDAC class I and IIb | 302 | 1.9 | 70 | 5 | 2 | 44,55,56 |
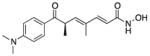
| Riluzole (6) |
|
Neuro-protective | 234 | 3.2 | 48 | 4 | 2 | 57–62 |
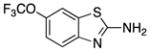
| Hydroxyurea (7) |
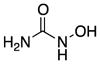
|
Increases Nitric Oxide | 76 | −1.8 | 75 | 4 | 4 | 63–66 |
| Ceftriaxone (8) |
|
Unknown | 554 | 0.02 | 209 | 18 | 5 | 67–69 |
| Albuterol (9) |
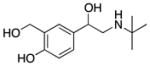
|
β2 Adrenergic Receptor Agonist | 239 | 0.06 | 73 | 4 | 4 | 70–74 |
| Aclarubicin (10) |
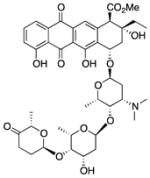
|
Antibiotic | 812 | 3.4 | 217 | 16 | 4 | 75, 76 |
MW, molecular weight;
cLogP, calculated LogP;
PSA, polar surface area;
HBA, hydrogen-bond acceptor;
HBD, hydrogen-bond donor.
2.1 HISTONE DEACETYLASE INHIBITORS
One group of drugs found to increase SMN2 promoter activity is histone deacetylase (HDAC) inhibitors. A basic epigenetic mechanism for regulating gene expression is through acetylation and deacetylation of selected lysines within the histones of chromatin. HDAC inhibitors are enzymes that deacetylate chromatin histones and create a tightly coiled, transcriptionally-repressed region of chromatin, and therefore inhibitors of these enzymes activate gene expression. One potential advantage of HDAC inhibitors is the ability to reverse abnormal cell transcription early on, rather than treating downstream translational endophenotypes. It was proposed that HDAC inhibition could induce transcription of SMN2 and, as a result, could increase the amount of total SMN2 transcripts, which would lead to more functional SMN protein.38 To date, several FDA-approved HDAC inhibitors have been investigated for the treatment of SMA. The first HDAC inhibitor to result in increased SMN2 transcription was sodium butyrate (1).39 Subsequently, phenylbutyrate (2), valproic acid (3), suberoylanilide hydroxamic acid (4), and trichostatin A (5) all were shown to increase SMN2 expression in patient-derived cell lines and animal models.40–44 Importantly, HDAC inhibition is not specific to SMN2, and the expression of multiple genes potentially would be altered, leading to side effects. The results of these approaches are discussed below.
2.1.1 Sodium Butyrate
The HDAC inhibitor 1 is a salt of butyric acid with antineoplastic activity.39 Chang et al. were the first to show that the SMN2 gene could be activated by 1.39 To generate lines of immortalized lymphoid cells, the researchers transformed lymphocytes obtained from SMA patients with Epstein-Barr virus. After exposing the lymphocytes to candidate drugs, they performed quantitative reverse transcriptase PCR (qRT-PCR) to determine which drugs resulted in increased production of full-length SMN2 mRNA. Compound 1 increased both SMN2 transcription and the inclusion of exon 7 in SMN2 transcripts, thereby increasing the production of full-length SMN2 mRNA. A corresponding increase in SMN protein also was seen. Further, there was a 25% extension in the mean survival of Taiwanese SMA mice. The primary disadvantage of 1 is its very short half-life in human serum (T1/2 = 6 min), making it unsuitable for therapy.
2.1.2 Sodium Phenylbutyrate
In a subsequent study, 2, an analog of sodium butyrate, was found to increase full-length SMN2 mRNA and SMN protein in SMA patient-derived fibroblasts.40 Compound 2 is FDA-approved for urea cycle disorder and has been tested for therapeutic benefit in amyotrophic lateral sclerosis (ALS), cystic fibrosis, and several cancers. Compound 2 is more stable, with a 0.8–1 h half-life in human.45 There is evidence suggesting that 2 crosses the BBB; 2 was able to lower long-chain fatty acid levels in the brain of mice with x-linked adrenoleukodystrophy46 and, after IV administration, led to a CSF:plasma ratio of 1:2 in primates.47
An uncontrolled, open-label pilot study investigated the use of 2 in Type II SMA patients and resulted in a 2-fold increase in SMN mRNA and improved Hammersmith Motor Function Scale scores.48 Two additional phase II/III clinical trials were terminated due to slow enrollment and poor compliance. STOPSMA, an open-label trial for 2, was conducted to evaluate the compound in pre-symptomatic infants with Types I and II SMA.49 Although the study was completed in 2013, outcomes have not been reported.
2.1.3 Valproic Acid
Compound 3, an HDAC inhibitor of the aliphatic class, is a carboxylic acid with a pKa of 4.8 that likely is fully ionized at physiological pH.50 However, 3 can cross the BBB via active transport by medium-chain fatty acid transporters. Compound 3 is used clinically for a number of central nervous system (CNS) diseases, including epilepsy and bipolar disorder, and to prevent migraine headaches.50
Compound 3 treatment of SMA patient-derived primary fibroblasts resulted in a 2- to 4-fold increase in SMN protein. This increase coincided with an increase in both total SMN2 transcripts and full-length exon 7-included SMN2 transcripts.41,51 Further analyses revealed increased expression of several splicing factors, each of which had been shown previously to increase SMN2 exon 7 inclusion. A more recent study testing the efficacy of 3 in vitro confirmed an increase in total SMN2 mRNA, but reported no change in exon 7 inclusion levels.52 An in vivo test of 3 efficacy in homozygous 5058 SMA mice resulted in a 100% increase in median survival, a slight increase in SMN protein in the spinal cord, and improvements in muscle and neuron pathology.53 However, the authors noted decreases in growth cone size, axon length, motor neuron survival, and increased excitability in axon terminals, which suggested that 3 might be detrimental to motor neuron survival and function.
Five clinical trials using 3 in SMA have been conducted, but none has reported any clinical benefit. A randomized, prospective, placebo-controlled clinical trial designed to test the efficacy and safety of the combined administration of 3 with L-carnitine in ambulatory adults with Type III SMA was performed, but it demonstrated that 3 was not effective in improving strength or function in this population.54
2.1.4 Suberoylanilide Hydroxamic Acid
Compound 4, known as Vorinostat, is a potent, hydroxamic acid class HDAC inhibitor that inhibits class I and II HDAC inhibitors at submicromolar concentrations.42 Compound 4 is FDA-approved for the treatment of cutaneous T-cell lymphoma. In vitro experiments showed that 4 increased both SMN protein and mRNA in rat hippocampal neurons, motor neuron cultures, and primary SMA patient fibroblasts.42 As with other HDAC inhibitors, these increases coincided with an increase in exon 7 inclusion in SMN2 transcripts. Compound 4 efficacy also was examined in an in vivo study using 5024- and 5058-SMA mouse strains.43 Treatment with 4 increased the lifespan of the SMA mice by 30%, significantly improved motor function abilities, reduced degeneration of motor neurons within the spinal cord, and increased the size of neuromuscular junctions and muscle fibers compared with vehicle-treated SMA mice. SMN RNA and protein levels were elevated significantly in various tissues, including spinal cord and muscle.43 Compound 4 demonstrated positive in vivo effects despite very poor brain access (e.g., brain to plasma ratios between 0.04 and 0.01). Its poor brain penetration likely is due to the fact that 4 is a substrate for the P-glycoprotein (P-gp) and Bcrp1 efflux transporters at the BBB. These transporters are expressed at the blood and spinal cord brain barriers and restrict a number of drug substances access to the CNS. The current clinical status of 4 is not known by the authors.
2.1.5 Trichostatin A
Compound 5 is another HDAC inhibitor of the hydroxamic acid class.55 Compound 5, originally developed as an antifungal drug, is a member of a large class of HDAC inhibitors with a broad spectrum of epigenetic activities. A recent study showed that compound 5-treated SMAΔ7 mice continued to gain weight, maintained stable motor function, and retained intact neuromuscular junctions long after treatment was discontinued.55 Compound 5 treatment resulted in a 19% increase in median survival. When additional nutritional supplements were administered, median survival increased by 170%.55
A 2014 study by Liu et al. found that 5 increased the median lifespan of SMN2B/- mice from 20 days to 8 weeks, significantly attenuated weight loss, and improved motor behavior.56 Interestingly, 5 treatment did not increase the levels of SMN protein in mouse embryonic fibroblasts or myoblasts obtained from SMN2B/- mice. In addition, no change in the levels of SMN transcripts or protein in the brain or spinal cord of 5-treated SMA mice was observed. Furthermore, 5 did not increase SMN protein levels in the hind limb muscle, heart, or liver of SMN2B/- mice, suggesting that 5 acts independently of the SMA gene.
2.1.6 Summary of HDAC Inhibitors
The hydroxamic acid class of HDAC inhibitors was the first to substantially improve the phenotype of SMA mice, with 5 and 4 increasing median survival by 40% and 30%, respectively.43,55 However, because 3 and 2 were already in clinical trials, these compounds were studied in SMA patients relatively quickly despite their low potency as HDAC inhibitors. Results from these studies have shown limited but positive effects in open-label studies in SMA Types II and III. The significantly more potent HDAC inhibitors 4 and 5 demonstrate poor HDAC selectivity and are considered to be too toxic for chronic use to potentially treat SMA. The zinc binding hydroxamic acid group is present in all of the HDAC inhibitors advanced into trials for SMA and is generally associated with poor pharmacokinetics and severe toxicity. To fully evaluate the potential of HDAC inhibitors for the treatment of SMA, more potent, HDAC enzyme-specific, non-hydroxamic acid, CNS penetrant compounds are needed.
2.2 NEUROPROTECTIVES
2.2.1 Riluzole
Riluzole (6), a 2-aminobenzothiazole, is FDA-approved for the treatment of ALS.57 Although 6 was proposed to modulate excitatory neurotransmission mainly through inhibition of glutamate release; the precise neuroprotective mechanisms remain largely speculative. Its pharmacologic properties that may be related to its effect include an inhibitory effect on glutamate release, inactivation of voltage-dependent sodium channels, and the ability to interfere with intracellular events that follow transmitter binding at excitatory amino acid receptors.
Compound 6 exhibits neuroprotective effects in various in vivo experimental models of neuronal injury involving excitotoxic mechanisms. In in vitro models, 6 protected cultured rat motor neurons from the excitotoxic effects of glutamic acid and prevented the death of cortical neurons induced by anoxia.57,58 In ALS patients, 6 resulted in increased life expectancy and reduced rates of neurodegeneration. As a neuroprotective, it did not promote the recovery of lost motor neurons or show an improvement in muscle function. In SMA mouse models, 6 showed stabilization of neuromuscular junctions, but did not prevent the loss of proximal axons.
Compound 6 is able to cross the BBB; however, it is a known substrate for the P-gp efflux transporter. An ALS mouse model showed how the upregulation of P-gp expression correlates with low levels of 6 in the brain. Although it is not known if P-gp is highly expressed in SMA, this concept may explain the inadequate levels of 6 found in in vivo studies. The poor pharmacokinetic properties of 6 also may contribute to its poor long-term efficacy.59,60
A small Phase I clinical trial enrolling seven compound 6-treated and three placebo-treated Type I SMA infants showed 6 to be safe. However, no significant differences in survival or motor abilities were observed.61 Nevertheless, further analysis showed that three patients in the 6 group presented an unusual disease course and were still alive at the age of 30 to 64 months.62 It is unclear whether 6 is beneficial for SMA patients.
2.2.2 Hydroxyurea
Hydroxyurea (7), an FDA-approved compound for the treatment of solid tumors and sickle cell anemia, also has been considered for the treatment of SMA.63 Its ability to increase the expression of fetal hemoglobin suggested it might increase expression of SMN2. Compound 7 treatment of SMA-derived lymphocytes stimulated increases in the levels of total and exon 7-included SMN2 transcripts.63 A recent study suggested that 7 increases nitric oxide, which, in turn, increases SMN2 expression.64 Nevertheless, several clinical trials are underway to evaluate the therapeutic efficacy of 7. Early uncontrolled trials have shown positive trends, although none was statistically significant. A subsequent controlled trial reported no benefit of 7 in SMA treatment.65
Compound 7 has poor brain penetration in rat (e.g., brain to plasma ratio of 0.12–0.25), although it is not considered to be a P-gp substrate.66 However, it has low lipophilicity (e.g., cLogP = −1.8 and hydrogen-bond donors (HBD) = 4). These physicochemical properties likely combine to limit passive transport across the BBB.
2.3 ANTIBIOTICS
2.3.1 Ceftriaxone
Despite having physicochemical properties outside of those normally considered necessary for CNS penetration (e.g., MW = 554 amu, PSA = 208 Å2, hydrogen-bond acceptors (HBA) = 15), the β-lactam antibiotic ceftriaxone (8) crosses the BBB through active transport.67 Though 8 was demonstrated to increase glutamate reuptake and improved the phenotype of ALS mice,68 a clinical trial of 8 in ALS was unsuccessful. Compound 8 was evaluated in severe SMA mice and showed a modest extended survival.69 Treated animals demonstrated a modestly improved neuromuscular phenotype and increased survival. The neuroprotective effect appears to be mediated by increased levels of the glutamate transporter Glt1, the transcription factor Nrf2, and SMN protein. This study provided the first evidence of a potential positive effect of β-lactam antibiotics as a treatment for SMA, but further studies are needed.
2.3.2 Albuterol
Another FDA-approved compound repurposed for SMA is albuterol (9).70 Compound 9 is a β2 adrenergic receptor agonist used to relieve the symptoms of asthma and chronic obstructive pulmonary disease and increase skeletal muscle strength.70 Compound 9 has been shown to increase full-length SMN transcript levels in severe SMA patient-derived cell lines. In SMA-derived fibroblasts, 9 stimulated SMN2 mRNA and SMN protein production.71
Compound 9 has poor brain penetration in rat (e.g., brain to plasma ratio of 1:20). The very low lipophilicity (e.g., cLogP <1) and the 4 H-bond donors likely hinder passive diffusion across the BBB.72
A small, uncontrolled pilot study was performed to determine the clinical benefit of 9 in Type II and Type III SMA patients, and demonstrated a statistically significant increase in muscle function.73 An additional open-label trial using daily 9 treatments in Type II SMA patients reported possible functional improvement on the Hammersmith Motor Functional Scale. However, the data for this study should be cautiously interpreted because of the relatively small sample size and possible placebo effect.74 A sufficiently powered, randomized, double-blinded, placebo-controlled trial needs to be performed using 9 in SMA patients as none has been published to date.
2.3.3 Aclarubicin
Aclarubicin (10) is a member of the anthracycline drug class approved for the treatment of acute non-lymphocytic leukemia that was identified in a cell-based screen for compounds that correct SMN2 splicing.75 Compound 10 was shown to increase both total and exon 7-included SMN2 transcripts as well as SMN protein in primary SMA-derived fibroblasts. Compound 10 interacts with and antagonizes the activity of Type II DNA topoisomerases.5 Compound 10 can also intercalate into DNA and inhibit DNA replication, DNA repair, and RNA protein synthesis.
Despite observations of its activity on SMN expression by two independent groups,75,76 the use of 10 is restricted due to intrinsic toxicity. However, other related anthracycline derivatives did not affect SMN2 splicing, suggesting that the mechanism underlying compound 10-modulated splicing is different from other “anthracycline–like” activities. If the mechanism can be identified, it may be possible to design new analogs with less toxic effects.
3. SMALL MOLECULES IN CLINICAL TRIALS
3.1 NON-SPECIFIC TREATMENTS FOR SMA
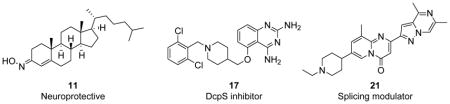
There are five small molecules currently being tested in Phase I to Phase III clinical trials for SMA. Two of these five compounds, neuroprotective olesoxime (11)77 and muscle protective CK-2127107 (structure not disclosed),78 are non-specific to SMA, meaning that the mechanism targets the CNS and muscles in general, and neither molecule directly affects the pathway of SMA. Each of these non-specific SMA small molecules will be discussed in terms of identification, rationale, mechanism of action, progression to date, and medicinal chemistry properties.
3.1.1 Olesoxime
Compound 11 is a novel neuroprotective compound that is non-specific to the disease mechanism of SMA (Table 2). This compound was identified in a cell-based screen as an agent protective against motor neuron death induced by neurotrophic factor withdrawal.79,80 This agent acts to preserve the integrity of mitochondria in neurons under cellular stress by preventing mitochondrial permeability transition pore openings. There is evidence that 11 binds to a translocator protein (18 kDa) and voltage-dependent anion channel, two proteins located on the outer mitochondrial membrane.80 Compound 11 is closely related to cholesterol and exists as a stable mixture of syn- and anti- isomers at the 3-position. Compound 11 is highly lipophilic (cLogP = 10) and is able to cross the BBB, although it has poor aqueous solubility at pH 7.4 and must be dosed orally in an oily excipient.77
Table 2.
Chemical properties of small molecules in clinical development for SMA.
| Name | Structure | Mode of Action | MWa | cLogPb | PSAc | HBAd | HBDe | Reff |
|---|---|---|---|---|---|---|---|---|
| Olesoxime (11) |
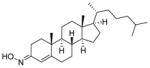
|
Neuro-protective | 400 | 10 | 33 | 2 | 1 | 77, 79–81 |
| CK-2127107 |
 Representative structure: 14 |
Fast skeletal troponin activator | 368 | 2.2 | 88 | 6 | 3 | 78, 82–85 |
| 17 |
|
DcpS inhibitor | 432 | 5.3 | 89 | 6 | 4 | 86–96 |
| RG7800 |
 Representative structure: 21 |
Sequence-specific splicing modulator | 416 | 3.7 | 64 | 7 | 0 | 97, 98 |
| LMI070 |
 Representative structure: 22 |
Sequence-specific splicing modulator | 393 | 3.2 | 91 | 3 | 1 | 100, 101 |
MW, molecular weight;
cLogP, calculated LogP;
PSA, polar surface area;
HBA, hydrogen-bond acceptor;
HBD, hydrogen-bond donor;
Ref, reference number
Compound 11 was developed as a neuroprotective therapy for ALS and SMA. In vitro neuronal cell death studies demonstrated a dose-dependent increase in cell survival with the use of 11 in trophic factor deprivation assays. When dosed at 3 mg/kg in SOD1G93A transgenic mouse models of ALS, 11 resulted in the prevention of weight loss, a delay in severe muscle function decline, and a significant increase in lifespan. In another study, 11 was subcutaneously administered (30 mg/kg) daily to 21-day old SMNF7/F7 SMA mice and resulted in a significant increase in life span. Only 15% of vehicle-treated mice survived longer than 40 days, whereas 45% of compound 11-treated mice lived beyond 40 days.77
Compound 11 is brain penetrant and is not a P-gp substrate. In mice, daily doses of 11 (3 mg/kg) resulted in a brain to plasma ratio of 0.35:1 after 7 days of treatment, and 0.51:1 after 42 days of treatment. Indeed, in a mouse nerve crush study, repeated dosing demonstrated drug accumulation in brain tissue over time, while the drug concentrations in plasma remained constant.77
Compound 11 did not provide significant treatment benefits during a Phase III clinical trial in patients with ALS, and it did not receive FDA approval. Although the compound showed no major effect on the survival of ALS patients, 11 was known to be safe in healthy individuals and was advanced directly into Phase II clinical trial studies for SMA.80,81 A randomized, double-blind Phase II clinical trial study was conducted to assess the efficacy and safety of 11 in SMA Type II and Type III non-ambulant patients ranging in age from 3 to 25 years old. Although no results were reported, a second Phase II clinical trial is underway. This trial, which is currently in the recruitment process, will focus on the nature, frequency, and severity of adverse events, as well as the effects on laboratory values, vital signs, and electrocardiography parameters.
3.1.2 Compound CK-2127107
In collaboration with Astellas Pharma, Cytokinetics has developed CK-2127107, which is believed to be a 2-aminoalkyl-5-N-heteroarylpyrimidine derivative that slows calcium release from fast skeletal muscle troponin and sensitizes the sarcomere to calcium (Table 2). The sarcomere is the contractile unit of skeletal muscle in the body. Within the sarcomere, calcium ions (Ca2+) are the key regulatory ions in the signaling process. The binding of Ca2+ to troponin C in the troponin complex of the muscle allows tropomyosin movement, actin-myosin interaction, and muscle contraction.78 As the magnitude of Ca2+ release is proportional to the level of neuromuscular transmission and the force of muscle contraction, control of this ion provides a therapeutic target of interest to treat SMA and other skeletal muscle-related diseases.82 With this mechanism of action, CK-2127107 helps increase skeletal muscle contractility and enhances physical performance in patients with neuromuscular diseases.78
Cytokinetics also developed an earlier small-molecule fast skeletal troponin activator, Tirasemtiv (12), and reported the first positive results from this mechanism of action by increasing skeletal muscle force and exercise performance in healthy and neuromuscular-diseased rats.78 Compound 12 is structurally distinct from CK-2127107, but it demonstrates efficacy in improving muscle strength and produces a significantly higher force output at submaximal levels of motor nerve stimulation (Figure 2). Compound 12 is currently in Phase III clinical trials for ALS treatment and provides proof-of-concept for this mechanism of action. Through the same mechanism, CK-2127107 has displayed multiple areas of effectiveness, improving exercise tolerance in vivo in a rate model of heart failure-mediated skeletal myopathy, increasing sarcomere Ca2+ sensitivity in left anterior descending coronary artery heart failure (LAD-HF) skeletal fibers in vitro, increasing force output at submaximal stimulation frequencies in the diaphragm and hind-limb muscles, and improving performance in LAD-HF rats.78 Importantly, CK-2127107 also has been shown to selectively activate fast skeletal myofibrils (EC50 = 3.4 umol/L and maximal activation = 3.6-fold increase) with no effect on slow skeletal or cardiac myofibrils. Although this study showed the effects of CK-2127107 in a heart failure rat model, there is a correlation between the effects in this mouse model and those of SMA mouse models, as both display significant muscle atrophy. CK-2127107 may decrease the severity of symptoms and allow for an increase in patient muscle function in SMA.78 The unique mechanism of CK-2127107 may allow combination therapy with agents that specifically increase levels of the SMN protein.
Figure 2.

A number of patents on the use of amino-pyridazines for the treatment of neuromuscular disorders (e.g., Markush 13, Figure 2) have published and the 2-aminopyridine (14) is shown as a representative analog.
Phase I clinical trials tested the safety, pharmacokinetics, and pharmacodynamics of CK-2127107 in healthy individuals using three randomized, placebo-controlled studies. Single and repeated doses of the compound were well-tolerated and increased the response of muscle to nerve activation.85 With positive safety results in the first phase of clinical trials, the drug is now moving into Phase II studies. Cytokinetics has opened enrollment for this double-blind, randomized, placebo-controlled clinical trial to test the pharmacodynamics effect of CK-2127107 on skeletal muscle function and fatigability in teens and adults with SMA Types II, III, and IV.
3.2 SPECIFIC TREATMENT FOR SMA
Treatment concepts shifted from non-specific to specifically targeting the SMN2 gene after this gene was discovered to produce normal SMN protein. The SMN2-derived SMN protein can be directly or indirectly upregulated through (i) increased transcription of SMN2, (ii) increased inclusion of exon 7 SMN2 (iii) stabilization of full-length exon 7-included SMN2 mRNA, and (iv) stabilization of the SMN protein. In this section, the identification, rationale, mechanism of action, progression to date, and medicinal chemistry properties of each small molecule specific to SMA-targeted therapy in clinical trials will be discussed.
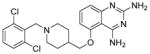
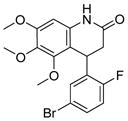
3.2.1 Compound RG3039 (17)
To identify compounds that increase the transcription of SMN2, a high-throughput, cell-based screen was performed by Aurora Biosciences (which was subsequently acquired by Vertex Pharmaceuticals) under the sponsorship of Families of SMA.86 The screen identified a class of 2,4-diaminoquinazolines (DAQ) (Figure 3; 15 and 16).86,87 The DAQ series of compounds was shown to increase total SMN2 mRNA with a proportional increase in exon 7-included SMN2 mRNA transcripts and SMN protein in SMA patient-derived primary fibroblasts. Medicinal chemistry optimization of the DAQ series focused on modifications to the 5-position and ultimately led to the discovery of the O-linked 2,6-dichlorobenzyl piperidine analog 17 (Table 2).88
Figure 3.

Discovery of clinical compound 17 and target ID probe compound 18.89
Following a single oral dose (10 mg/kg) in adult mice, 17 resulted in low plasma exposure (e.g., AUClast (plasma) = 157 ng.h/mL), but it displayed significant partitioning into the brain (AUClast (brain) = 6762 ng.h/mL) and consequently resulted in a very high brain to plasma ratio of 16:1.
Next, 17 was tested in three SMA mouse models. In the Taiwanese mouse model of SMA, in which the transgenic mice only have 2–4 copies of human SMN2 and no murine SMN (generated to be similar in survival to the SMAΔ7 by Li et al.), Compound 17-treated mice had improved motor abilities and a 38% increase in lifespan compared to vehicle-treated SMA mice.90 When tested in the intermediate 2B/– SMA mouse model (average survival 18.5 to 21 days), 17 extended survival by 100% relative to vehicle. When the study ended on day 112, 27% of compound 17-treated mice were still alive. In addition to improved lifespan, the muscle function and weight gain of the compound 17-treated mice improved as well.88
In order to identify the binding target of the DAQ series, a suitably active 125I radiolabeled compound was identified (Figure 3; 18) and used to identify potential binders in human protein microarrays.89 Compound 18 was found to bind the scaverger decapping enzyme (DcpS), and binding was confirmed using X-ray crystal structure determination.89,91–93 DcpS participates in 3′-5′ mRNA degradation, in which the protein targets the 5′ cap structure following 3′ end mRNA decay to release m7Gp and ppN products. In a separate pathway, DcpS also was shown to be a cofactor for Xrn1, a 5′-3′ exoribonuclease, and to regulate RNA stability through a transcript-specific mode of action. Thus, small molecules in the quazoline series, such as 17, appear to stabilize RNA levels through inhibition of DcpS.94
In the SMAΔ7 mouse model, 17 was administered intraperitoneally at 10 mg/kg for 10 days and resulted in 90% DcpS inhibition within 2 h of the last dose. DcpS levels remained 80% inhibited 72 h after dosing. To examine the effects of 17 on survival in the SMAΔ7 mouse model, mice were treated with daily doses (10 mg/kg) until death. Drug-treated mice showed a 26% increase in median survival in comparison to vehicle-treated mice and displayed an overall increase in weight gain and motor function. A modest increase in full-length SMN transcript levels was detected; however, no increase of the SMN protein in the CNS was detected.95,96 Notably, 17 improved survival, muscle function, and weight gain in all three mouse models, which varied in SMA severity.
A Phase I clinical trial to evaluate the safety profile of 17 in healthy volunteers began in 2012. Repligen, Inc. has released several reports about the success of this trial, detailing the effectiveness of 17 and its minor side effects. Pfizer Inc. acquired rights to this compound but subsequently terminated the project and further development of 17 as a treatment for SMA is questionable.
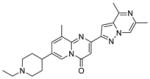
3.2.2 Compound RG7800
PTC Therapeutics aimed to identify compounds that selectively modulate SMN2 inclusion of exon 7. Through the use of a HEK293 human embryonic kidney cell line harboring an SMN2 mini-gene reporter assay, a library of proprietary small molecules was screened, and several chemical classes of compounds that induced exon 7 inclusion were identified. Deep sequencing of all mRNAs revealed that altered exon processing was highly specific to SMN2 transcripts. Following medicinal chemistry optimization in a collaborative project with Roche, three orally available compounds (i.e., the coumarins SMN-C1 (19) and SMN-C2 (20) and the pyrido[1,2-a]pyrimidin-4-one SMN-C3 (21)) with nanomolar potency were identified.97 These compounds increased full-length SMN2 mRNA levels and concomitantly reduced Δ7 mRNA levels in SMA Type I patient fibroblasts after 24 h of treatment.97
In RNA sequence studies, 21 was determined to be specific to SMA, displaying affinity for SMN2 exon 5 and 7 splice junctions. When dosed orally in the mild-type SMA mouse model, 21 increased SMN2 mRNA and SMN protein in the brain and muscle tissues by 90% and 50–70%, respectively. Compound 21 also showed an excellent PK profile; after oral administration (10 mg/kg), adult wild-type mice demonstrated a 3:1 brain penetration ratio and a plasma concentration peak of compound at approximately 2 to 2.5 h. In support of these findings, intraperitoneal injection of 21 in the severe SMNΔ7 mouse model resulted in increased SMN protein levels in the brain and muscle tissue by up to 150% and 90%, respectively. Dosing the animals daily with 21 (0.3 mg/kg) allowed 50% of the mice to survive past 40 days. Another study dosed Δ7 mice daily (1 or 3 mg/kg) and resulted in 90% survival until the end of the study or 65 days.97
PTC Therapeutics, in partnership with Roche, advanced RG7800 (structure not disclosed) as its clinical candidate and began a Phase I single-ascending dose study.98 The study revealed that the compound was safe and well-tolerated in healthy volunteers. Following this success, adult and pediatric SMA patients began to enroll in a randomized, placebo-controlled Phase Ib/IIa clinical trial. However, PTC/Roche suspended this clinical trial over concerns regarding an eye condition that developed in long-term animal studies. A Phase I trial began recruiting healthy volunteers in January 2016 to evaluate the safety profile and pharmacokinetic tolerability of an alternative candidate, RG7916 (structure not disclosed). Dosing regimens in the study will include a combination of RG7916 and itraconazole, an antifungal, as well as a placebo.
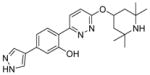
3.2.3 Compound LMI070
Novartis performed a high-throughput screen of over 1.4 million compounds using a minor modification of the SMN2 splicing reporter construct described by Cherry et al.99 In this screening assay, the reporter was introduced into the NSC-34 mouse neuroblastoma/spinal cord hybrid cell line.100 With a hit rate of <1%, a pyridazine class of orally active small molecules was identified and shown to elevate levels of the full-length SMN protein via the inclusion of exon 7. After substantial lead optimization and drug discovery efforts, two compounds from this series, NVS-SM1 (22) and NVS-SM2 (23), showed increased exon 7 inclusion and upregulated SMN protein expression in both SMA patient fibroblasts and SMNΔ7 5025-mouse myoblasts (Figure 5).100 The related analog NVS-SM3 (24) was inactive, indicating that the un-substituted pyrrole common to 22 and 23 is important for activity.100
Figure 5.

Examples of active and inactive pyridazines reported by Novartis.
Short-term dosing of 22 (1 mg/kg and 3 mg/kg) in the severe SMNΔ7 mouse model demonstrated an increase in both SMN protein levels in the brain and survival of the animals by 50% and 62% relative to the doses. In a long-term SMNΔ7 mouse model study, one cohort received daily doses of 22 (1 mg/kg) for an additional two weeks until day 49, and another received vehicle between days 36 and 49. Body weight and survival measurements between cohorts were nearly identical, supporting the duration effectiveness of 22 and suggesting that termination of treatment after 28 days allows sustained rescue from SMA phenotypic symptoms.100
Unexpected effects of alternative splicing prompted RNA sequencing studies to determine if 22 changed global gene expression or splice site selection. Following 22 treatment, 175 genes were altered by >2-fold with little difference in splicing factors or RNA binding proteins. Splice site analysis detected 39 22 specific events in 35 genes, indicating a very selective modulation of splicing. The mechanism of 22 exon 7 recognition was investigated with a series of chimeric splicing cassettes. This analysis revealed that the 21 nucleotides surrounding the 5′ splice site in intron 7 of SMN2 were enough to promote 22 specific changes in exon 7 inclusion. Surface plasmon resonance established changes in the disassociation kinetics between the SMN2 mRNA and the U1 snRNP in the presence of 22, suggesting that it interacts with the 5′ splice site of SMN2 intron 7 and stabilizes its interaction with the U1 snRNP complex. Combined, these results provide the first mechanistic insights for a sequence-specific, small molecule splicing modulator.100
The clinical lead compound in this series, LMI070 (structure not disclosed),101 has begun Phase II clinical trials to evaluate safety, tolerability, and efficacy in SMA patients. An open-label, multi-part, first-in-human clinical trial is now recruiting in Europe. During the trial, LMI070 will be orally administered to Type I SMA infants to determine the maximum tolerated dose and optimal dosing regimen.
4. PRECLINICAL SMALL MOLECULES
There are six small molecules currently in preclinical development for the treatment of SMA. All of these compounds are reported to be specific to the SMA-disease pathway, although not all of the companies and academic laboratories have published data to support these claims. Without published data, many of these compounds cannot be assessed for medicinal chemistry properties and their progress remains unknown.
4.1 Indiana University and the Laboratory for Drug Discovery in Neurodegeneration
The Androphy group at Indiana University and the Laboratory for Drug Discovery in Neurodegeneration (LDDN) at Brigham and Women’s Hospital combined the SMN promoter with exons 1–6 and an exon 7 splicing cassette in a single construct with a luciferase reporter to identify compounds that increase SMN transcription, increase exon 7 inclusion, or potentially stabilize the SMN mRNA or protein. Stable clonal HEK293 cell lines that expressed SMN1 or SMN2 reporters were isolated to reduce variation due to factors such as plasmid number. Using this SMN2 reporter assay, over 115,000 compounds were screened at the LDDN, and two hit compounds (LDN-75654 (25) and LDN-76070 (28)) were discovered to increase SMN expression from the SMN2 gene (Table 3).99
Table 3.
Preclinical small molecules in development for SMA.
| # | Structure | Mode of Action | MWa | cLogPb | PSAc | HBAd | HBDe | Reff |
|---|---|---|---|---|---|---|---|---|
| 25 |
|
Unknown (Post-transcriptional) | 282 | 3.5 | 51 | 4 | 1 | 99, 103 |
| 28 |
|
Unknown (Transcriptional) | 410 | 3.2 | 57 | 5 | 1 | 99 |
| 31 |
|
Unknown (Post-transcriptional) | 360 | 2.7 | 71 | 7 | 1 | 105 |
| 34 |
|
Glycogen synthase kinase 3-β inhibitor | 422 | 3.9 | 59 | 5 | 1 | 108 |
| 35 |
|
Unknown (Transcriptional) | 605 | 1.3 | 194 | 12 | 7 | 109 |
| 36 |
|
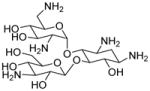
Read-through inducing | 483 | −7.9 | 288 | 15 | 16 | 111 |
MW, molecular weight;
cLogP, calculated LogP;
PSA, polar surface area;
HBA, hydrogen-bond acceptor;
HBD, hydrogen-bond donor;
Ref, reference number
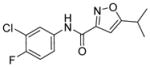
Compound 25 produced a >240% or 3.4-fold increase in the luciferase activity of the assay with an EC50 of 2 μM (Figure 6). Compound 25 is a 5-isopropyl isoxazole bearing a 3-chloro-4-fluorophenyl carboxamide at the 3-position. Compound 25 is compliant with Lipinski’s rules for drug-like molecules, has physicochemical properties that are considered suitable for BBB penetration102 (MW = 283 amu, cLogP = 3.4, PSA = 51 Å2, HBD = 1, HBA = 3), and promotes increases in SMN expression in SMA-derived fibroblasts. However, 25 was inactive for SMN protein induction in vivo. Administration to SMNΔ7 mice revealed that 25 did not consistently elevate levels of SMN protein in the brain or spinal tissue of the animals. Compound 25 had poor aqueous solubility (6 μM at pH 7.4) and a short microsomal half-life in mouse microsomes (T1/2 = 5 min). The lack of metabolic stability and poor solubility presumably accounts for its poor pharmacokinetics and disappointing performance in vivo. Modifications made to the aryl ring of 25 revealed that the 3-chloro-4-fluorophenyl could be replaced by a phenyl ring (Figure 6; LDN-5021 (26)) with a significant improvement in both potency (EC50 = 0.28 μM) and solubility (59 μM at pH 7.4), although the mouse microsomal stability was still poor (T1/2 = 5 min).103,104 The introduction of the 2-pyridyl group (Figure 6; LDN-5065 (27)) to replace the phenyl group gave a compound of similar potency and solubility to 26 but with significantly improved stability in mouse microsomes.
Figure 6.

Preliminary optimization of 25.
The mechanism of action of 25 was studied in SMN2-luciferase reporter cell lines using qRT-PCR. No change was detected in the mRNA levels, but concentration-dependent increases were observed in luciferase activity, suggesting that 25 and its analogs function through a post-transcriptional mechanism.99
Another distinct hit, compound 28, produced a >180% or 2.8-fold increase in the luciferase activity of the assay, with an average EC50 of 8.3 μM (Figure 7).99 Compound 28 is a 3,4-dihydroquinolinone and structurally distinct from the first lead, 25. Further, 28 follows Lipinski’s rules and had acceptable physicochemical properties for potential brain penetration (MW = 410 amu, cLogP = 3.3, PSA = 57 Å2, HBD = 1, HBA = 5). Using qRT-PCR, 28 was found to increase SMN transcription in the SMN2-luciferase reporter cell line.99
Figure 7.

Discovery of 30 and 31.
Additional in vitro studies showed 28 had good stability in mouse liver microsomes (T1/2 = 40 min), but had poor aqueous solubility (4 μM at pH 7.4). In a preliminary efficacy study, the un-optimized lead 28 was found to be active in vivo. Daily treatment of SMNΔ7 mice with 28 (20 mg/kg) increased SMN protein levels and survival. Compound 28-treated mice had a mean increase of SMN protein levels in the spinal cord up to 6-fold greater than untreated or vehicle-treated animals. Additionally, median survival increased from 7 days (vehicle-treated) to 16 days (compound 28-treated), a 2.4-fold increase in lifespan.99
4.2 NIH Chemical Genomics Center
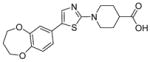
Using the cell-based SMN2-luciferase reporter assay previously described by Cherry et al., the NIH Chemical Genomics Center (NCGC) screened a library of 210,386 compounds to identify those that increased expression of full-length SMN transcripts.105 Following hit validation and prioritization, 1-(4-(4-bromophenyl) thiazol-2-yl)piperidine-4-carboxamide (29) was identified as a promising hit that increased SMN protein production in a dose-dependent manner (Figure 7).105
The hit, 29 (EC50 = 1.22 μM, 268% increase in SMN2) underwent medicinal chemistry optimization, including piperidine modification, thiazole replacement, and phenyl substitutions. Switching the location of the aryl substituent from the 4- to the 5-thiazole position and replacing the 4-bromo phenyl with the electron-donating 4-thiomethyl phenyl (e.g., ML200 (30), EC50 = 31 nM, 576% increase in SMN2) conferred increased potency and efficacy (Figure 7).105 Unfortunately, 30 had a very short half-life in mouse liver microsomes (T1/2 = 2 min) and low permeability in Caco-2 cells (Papp A to B = 1.6 × 10−6 cms−1 and Papp B to A = 0.2 × 10−6 cms−1) with a poor efflux ratio of 0.125.
Replacement of a potential metabolic liability, the 4-thiomethyl phenyl group, by 3,4-dihydro-2H-benzo[b][1,4]dioxepin-7-yl and replacement of the amide by carboxylic acid produced ML372 (31) (EC50 = 12 nM, 325% increase in SMN2).105 Compound 31 possessed a more stable half-life in mouse liver microsomes (T1/2 >60 min) and high permeability in Caco-2 cells (Papp A to B = 33.8 × 10−6 cms−1 and Papp B to A = 44.1 × 10−6 cms−1) with an efflux ratio close to unity (i.e., 1.3).
Pharmacokinetic studies in mice (oral dosing at 30 mg/kg) demonstrated that 31 reached high plasma levels (plasma Cmax = 44 μM) with a long plasma half-life (T1/2 >11 h). However, there was significant resistance to brain penetration (brain Cmax = 1.2 μM), and half-life in the brain was >13.7 h. In addition, 31 demonstrated high plasma protein binding (94.9%). Although 31 showed good oral absorption, its poor CNS penetration and high plasma protein binding indicated that free levels of 31 in brain were likely to be low following higher oral dosing (30 mg/kg).105
To determine their mechanism of action, 30 and 31 were studied using qRT-PCR. RNA expression and exon 7 inclusion levels were measured within a SMA reporter cell line, in which there was no increase in exon 7 mRNA. These compounds may, therefore, act post-transcriptionally to increase the stability of SMN protein by inhibition of its degradation.105
4.3 Kozikowski and Rubin Laboratories
An image-based screen of annotated chemical libraries revealed that small molecule inhibitors of the glycogen synthase kinase (GSK-3β) (e.g., alsterpaullone (32) and AR-A014418 (33)) could elevate SMN levels (Figure 8).106 In general, GSK-3β is known to regulate protein stability. More specific to SMA, there is a consensus GSK-3β phosphorylation site on serine 4 of the SMN protein.106 When this residue becomes phosphorylated, SMN protein degrades at a faster rate. Compound 32 is a known GSK-3β inhibitor, and treatment of mouse motor neurons with 32 resulted in increased amounts of SMN protein levels.106,107 This evidence suggests that the inhibition of GSK-3β is a potential therapeutic approach to treat SMA.
Figure 8.

GSK-3β inhibitors: 32, 33, and 34.
Kozikowski and Rubin recently described a potent ATP-competitive GSK-3β inhibitor, BIP-135 (34) (GSK-3β IC50 = 21 nm).108 In SMA patient fibroblasts, 34 (25 μM, 72 h exposure) stimulated a 7-fold increase in SMN protein levels and displayed neuroprotective properties in an SMA-related, cell-based model of oxidative stress-induced neurodegeneration. Subsequently, 34 was administered daily by intraperitoneal injection to SMNΔ7 neonatal mice in multiple doses (25, 75, and 125 mg/kg) in 100% DMSO. Compound 34 did not increase motor function or levels of SMN mRNA or protein, but it did improve median survival in the SMNΔ7 mouse model at the 75 mg/kg dose, increasing the lifespan by 2 days in comparison to untreated mice.108
Pharmacokinetic data (e.g., brain levels) were not reported for 34, and the choice of 100% DMSO as the vehicle for the in vivo study suggests poor compound solubility. Nevertheless, the modest extension in survival demonstrates potential for GSK-3β inhibitors as therapeutics; however, more potent and selective inhibitors with better drug-like properties and pharmacokinetics are required.
4.4 Paratek Pharmaceuticals
As previously mentioned, 10 was identified in a cell-based screen specifically designed to detect SMN protein upregulation via increased inclusion of exon 7 in SMN2. Although the precise mechanism in which 10 increases SMN expression is unclear, it was demonstrated to change the splicing ratio between exon 7-included and exon 7-excluded SMN2 transcripts.75 More importantly, 10 was found to be toxic in SMA Type I fibroblast 3061 cells dosed at concentrations >10 nM. Although toxic, the distinct mechanism of 10 was unlike any other tetracycline derivatives. Therefore, it is predicted that structure-activity relationship (SAR) studies can provide similarly structured compounds without the toxic effects.
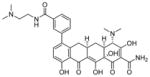
Paratek Pharmaceuticals reported that a lead compound from its novel series of tetracycline-like analogs, PTK-SMA1 (35) (Table 3), alters the splicing pattern of exon 7 and has shown progress in in vitro and in vivo studies.109 A dose of 35 (10 μM) increased the levels of SMN protein in SMA patient fibroblasts by about 40%, relative to vehicle-treated cells. Compound 35 (MW = 605 amu, cLogP = 1.3, PSA = 194 Å2) is a tetracycline derivative, modified at the C7 position of the tetracycline backbone.109 Due to its large molecular size and polarity, the compound does not demonstrate good CNS drug-like properties and is hindered in its ability to penetrate the brain tissue in vivo. When 35 was administered via intraperitoneal injection (25 and 50 mg/kg) in a mild SMA Type III mouse model over 6 days, a 1.5-fold increase of exon 7 inclusion within liver cells was found. Only data from liver was collected because the compound is unable to cross the BBB.109 Additional SAR studies must focus on BBB penetration to increase SMN expression in the CNS. Otherwise, the novel treatment may not be an effective therapy. No further information has been published on the progress of this preclinical phase project.
4.5 BioBlast Pharma
BBrm2 is the repurposed FDA-approved drug, azithromycin, redesigned in a proprietary intrathecal formulation. Azithromycin is an antibiotic approved to treat acute bacterial exacerbation of chronic obstructive pulmonary disease, acute bacterial sinusitis, and pneumonia, among other uses. Read-through agents cause skipping or read-through of the translational termination codon at the end of exon 7 and result in a protein that has an extended C-terminus.110 Aminoglycoside antibiotics (from a class of FDA-approved drugs including tobramycin, geneticin, amikacin, and TC-007 (36)),111 were found to induce higher levels of functional SMN protein after administration at the stop codon of exon 7 in the SMN Δ7 protein; however, toxicity limited any attempts to translate these agents into therapeutic candidates. With a safe profile, BBrm2 was shown to increase SMN protein expression and function in SMA patient cell lines, as well as increase SMN expression of the spinal cord, body weight, motor function, and survival in a SMNRT transgenic mouse model dosed via intracerebroventricular (ICV) administration. BioBlast Pharma reported that testing of the small molecule has been extended to additional studies in the Burgheron mouse model, where significant increases in tail length, body weight, and tissue SMN levels were observed.112
5. DISCUSSION
Repurposed drugs offer significant savings in discovery and development time and costs. However, many repurposed drugs were not originally developed for diseases that required brain penetration, limiting their utility in CNS diseases. While many of the compounds described in the preceding section demonstrated promising in vitro activity (e.g., increased SMN protein levels or neuroprotection), often there was no evidence of adequate brain penetration or target engagement.102,113,114 Indeed, a number of the compounds studied in vitro and in vivo for SMA have very poor brain penetration. When repurposing an approved drug for a CNS disease, there is a risk that negative information about a particular target or disease mechanism may be generated in the rush to achieve Phase II efficacy studies. For example, although a potent, HDAC-enzyme-specific, brain-penetrant molecule may have utility in the treatment of SMA, the non-specific mechanism through which HDAC inhibitors such as 2 and 3 act means these drugs are likely to cause side effects with long term usage, decreasing interest for developing this class of compound. Furthermore, many of the clinical trials of repurposed drugs in SMA were open label and ineffectively powered in trial design, so that it is difficult to know whether the therapeutic strategy or target had validity. Nonetheless, much was learned on how to develop a clinical trial and evaluate outcome measures. For instance, transport of a severely ill SMA Type 1 patient to a distant medical center could itself have deleterious effects that complicate trial result interpretations. There are also ethical issues in withholding or limiting palliative treatments during trials. Although repurposing small molecules can expedite clinical testing, opportunities were missed to develop new SAR and discovery molecules with suitable brain penetration, level of effect on the SMA target, off-target toxicity and specificity.
Clinical trials with agents specifically designed to target SMA and SMN2 have been initiated. Evidence shows that increasing the amount of SMN protein has the potential to decrease disease severity and symptoms. Because the SMN2 gene was discovered to produce some SMN protein, it has become a specific therapeutic target for SMA even though the threshold level of SMN protein required to decrease SMA symptoms remains undetermined.115 Depending on their mechanism of action, small molecules can be administered independently or in combination to help decrease SMA symptoms. Cherry et al. demonstrated the use and success of small molecule combination therapy targeted to SMA. The study used 25 (a small molecule that functions at the post-transcriptional level) and 28 (a small molecule that affects the transcription of the SMN2 gene) individually and in combination with the pan-HDAC inhibitor 4 in the SMN2-luciferase reporter assay.99 The combination of 25 with either 28 or 4 produced a greater-than-additive stimulation of the SMN2-luciferase reporter. Not only does this complementation confirm the separate and distinct mechanisms in which the two compounds work, but the results also suggest a synergistic relationship. On the other hand, 4, when combined with 28, appeared to overwhelm the transcriptional machinery and obscure the effect of 28. These results suggest that the compounds stimulate SMN2-luciferase expression similarly.99 Therapeutic approaches that combine modalities acting though different pathways may maximally increase SMN protein production and be of greatest clinical benefit.
Combination therapies can be applied to lead compounds when the mechanism of action is known and distinct for each molecule. Compound 11 and CK-2127107 might be paired with any SMA-specific treatment because the neuroprotective and muscular protective mechanisms provide them with a unique therapeutic opportunity compared to other treatments in the pipeline. Analyzing the mechanisms of more specific treatments, 17 has been shown to bind DcpS, an enzyme that participates in mRNA degradation.94 In comparison, LMI070 was determined to be a selective splicing module that works to increase the stabilization between U1 snRNP binding and the 5′ splice site.100 Compound 17 and LMI070 work to stabilize SMN2 mRNA levels and increase exon 7 inclusion of SMN2 mRNA, effectively working through two distinctive SMN2-targeted pathways. Importantly, some evidence suggests that RG7800 works through a similar mechanism as LMI070. The exact mechanism of RG7800 has not been reported, but affinity studies demonstrated that the compound shows moderate specificity for SMN2 exon 5 and 7 splice junctions.97 Based on their structural similarities, LMI070 and RG7800 may bind to the same protein or RNA structure in SMN2 intron 7. Expanding to other treatment types, the combination of IONIS-SMNRx, which increases exon 7 inclusion, with a drug that stabilizes the half-life of the SMN protein may be a more effective in clinical trials. With multiple therapeutic targets in the SMA disease pathway, there is the potential to manage the symptoms of SMA with a variety of treatment options and combinations.
6. CONCLUSION
Despite the lack of FDA-approved treatments for SMA, the last fifteen years have seen dramatic progress in the development of therapeutic strategies. These advances have widened our understanding of the disease process and helped researchers explore new options for treatment. Progress in the field of SMA therapy is attributed to understanding SMN function, SMN2 exon 7 splicing regulation, and the therapeutic opportunities that stem from SMN production defects. Thus far, the current use of repurposed drugs has not provided a short cut to disease treatment, due in large part to non-specific mechanistic pathways and lack of CNS-penetration. However, neuroprotective compounds, such as 11, and muscle targeting agents, such as CK-2127107, continue to progress through clinical trials due to their ability to target general atrophy symptoms. Very novel approaches that appear to specifically enhance the inclusion of SMN2 exon represent strong evidence that drug-like molecules can target RNA splicing. The field of SMA continues to advance upon the construction of extensive biology findings, sophisticated drug screening technologies and medicinal chemistry efforts that have translated to numerous therapeutic opportunities for SMA.
Figure 1.

SMA is caused by a loss of SMN1. The two SMA genes, SMN1 and SMN2, are found on chromosome 5q11-13 and encode for SMN protein. In most patients with SMA, complete lack of an SMN1 gene results in all SMN protein being derived from SMN2 expression. Although all patients retain one or more copies of the SMN2 gene, it is unable to fully compensate for loss of SMN1. Only about 15% of SMN2 transcripts encode for normal, full-length SMN protein.5
Figure 4.

Examples of PTC/Roche small molecules.
Acknowledgments
We thank the individuals with SMA, their families, and the foundations whose continued support has driven drug discovery and development. We acknowledge the many people in the field of SMA, who are involved in therapeutic development, and who have contributed to advancements in the field. The authors are supported by the National Institute of Health (R21 NS088522).
Abbreviations
- SMA
spinal muscular atrophy
- SMN
survival motor neuron
- MNs
motor neurons
- CSF
cerebral spinal fluid
- NMJ
neuromuscular junction
- SMNRT
SMN read through
- scAAV-9
self-complementary adeno-associated virus
- BBB
blood-brain barrier
- HDAC
histone deacetylase
- qRT-PCR
quantitative reverse transcriptase PCR
- ALS
amyotrophic lateral sclerosis
- CNS
central nervous system
- P-gp
P-glycoprotein
- HBD
hydrogen-bond donor
- HBA
hydrogen-bond acceptor
- MW
molecular weight
- cLogP
calculated LogP
- PSA
polar surface area
- LAD-HF
left anterior descending coronary artery heart failure
- DAQ
diaminoquinazoline
- DcpS
scavenger decapping enzyme
- LDDN
Laboratory for Drug Discovery in Neurodegeneration
- NCGC
NIH Chemical Genomics Center
- GSK
glycogen synthase kinase
- SAR
structure-activity relationship
Biographies
Alyssa N. Calder received her B.S. (summa cum laude) degree in Chemistry from Northeastern University in 2015. She performed undergraduate research at Northeastern University under Professor Graham Jones on the development of fluoro-pharmaceuticals and PET ligands utilizing continuous flow micro-reactor technology as well as at Genzyme Corporation, working on peptide chemistry for immune mediated disease discovery. Alyssa joined the LDDN in 2014, where she has focused her efforts on synthesizing and optimizing small molecules for the SMA project as the laboratory closes in on identifying a developmental candidate.
Elliot Androphy, M.D. is the Kampen-Norins Chair of the Department of Dermatology at Indiana University School of Medicine. After a post-doctoral fellowship at the National Cancer Institute/NIH, he began his SMA research at Tufts Medical Center, where his lab identified the critical difference between SMN1 and SMN2 exon 7. He served as Vice Chair for Research of the Department of Medicine and Director of the MD/PhD Program at the University of Massachusetts Medical School, where his research team focused on the mechanisms of exon 7 recognition and developed the first cell-based high-throughput screens to identify drug-like compounds that increase levels of the SMN protein. His lab continues to develop novel therapeutics and investigates the functions of the SMN protein in neurons.
Kevin Hodgetts, Ph.D. is a medicinal chemist, Director of the LDDN and Assistant Professor of Neurology at Brigham and Women’s Hospital and Harvard Medical School. He works in collaboration with researchers to translate discoveries in basic biology into opportunities for drug discovery.
References
- 1.Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- 2.McAndrew PE, Parsons DW, Simard LR, Rochette C, Ray RN, Mendell JR, Prior TW, Burghes AH. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet. 1997;60:1411–1422. doi: 10.1086/515465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearn J. Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J Med Genet. 1978;15:409–413. doi: 10.1136/jmg.15.6.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.d’Ydewalle C, Sumner CJ. Spinal muscular atrophy therapeutics: where do we stand? Neurotherapeutics. 2015;12:303–316. doi: 10.1007/s13311-015-0337-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cherry JJ, Androphy EJ. Therapeutic strategies for the treatment of spinal muscular atrophy. Future Med Chem. 2012;4:1733–1750. doi: 10.4155/fmc.12.107. [DOI] [PubMed] [Google Scholar]
- 6.Van Meerbeke JP, Sumner CJ. Progress and promise: the current status of spinal muscular atrophy therapeutics. Discovery Med. 2011;12:291–305. [PubMed] [Google Scholar]
- 7.DiDonato CJ, Ingraham SE, Mendell JR, Prior TW, Lenard S, Moxley RT, III, Florence J, Burghes AHM. Deletion and conversion in spinal muscular atrophy patients: is there a relationship to severity? Ann Neurol. 1997;41:230–237. doi: 10.1002/ana.410410214. [DOI] [PubMed] [Google Scholar]
- 8.Wirth B, Schmidt T, Hahnen E, Rudnik-Schöneborn S, Krawczak M, Müller-Myhsok B, Schönling J, Zerres K. De novo rearrangements found in 2% of index patients with spinal muscular atrophy: mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am J Hum Genet. 1997;61:1102–1111. doi: 10.1086/301608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Faravelli I, Nizzardo M, Comi GP, Corti S. Spinal muscular atrophy—recent therapeutic advances for an old challenge. Nat Rev Neurol. 2015;11:351–359. doi: 10.1038/nrneurol.2015.77. [DOI] [PubMed] [Google Scholar]; (b) Werdnig G. Two early infantile hereditary cases of progressive muscular atrophy simulating dystrophy, but on a neural basis. Arch Neurol. 1971;25:276–278. doi: 10.1001/archneur.1971.00490030102014. [DOI] [PubMed] [Google Scholar]
- 10.Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, Le Paslier D, Frézal J, Cohen D, Weissenbach J, Munnich A, Melki J. Identification and characterization of a spinal muscular atrophy determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 11.Echaniz-Laguna A, Miniou P, Bartholdi D, Melki J. The promoters of the survival motor neuron gene (SMN) and its copy (SMNc) share common regulatory elements. Am J Hum Genet. 1999;64:1365–1370. doi: 10.1086/302372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coovert DD, Le TT, McAndrew PE, Strasswimmer J, Crawford TO, Mendell JR, Coulson SE, Androphy EJ, Prior TW, Burghes AH. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet. 1997;6:1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 13.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci US A. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AHM, McPherson JD. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 15.Gennarelli M, Lucarelli M, Capon F, Pizzuti A, Merlini L, Angelini C, Novelli G, Dallapiccola B. Survival motor neuron gene transcript analysis in muscles from spinal muscular atrophy patients. Biochem Biophys Res Commun. 1995;213:342–348. doi: 10.1006/bbrc.1995.2135. [DOI] [PubMed] [Google Scholar]
- 16.Gavrilov DK, Shi XY, Das K, Gilliam TC, Wang CH. Differential SMN2 expression associated with SMA severity. Nat Genet. 1998;20:230–231. doi: 10.1038/3030. [DOI] [PubMed] [Google Scholar]
- 17.Lorson CL, Androphy EJ. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum Mol Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- 18.Lorson CL, Strasswimmer J, Yao JM, Baleja JD, Hahnen E, Wirth B, Le T, Burghes AH, Androphy EJ. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet. 1998;19:63–66. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- 19.Bunett BG, Munoz E, Tandon A, Kwon DY, Sumner CJ, Fischbeck KH. Regulation of SMN protein stability. Mol Cell Biol. 2009;29:1107–1115. doi: 10.1128/MCB.01262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Echaniz-Laguna A, Miniou P, Bartholdi D, Melki J. The promoters of the survival motor neuron gene (SMN) and its copy (SMNc) share common regulatory elements. Am J Hum Genet. 1999;64:1365–1370. doi: 10.1086/302372. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Melki J. Spinal muscular atrophy. Curr Opin Neurol. 1997;10:381–385. doi: 10.1097/00019052-199710000-00005. [DOI] [PubMed] [Google Scholar]
- 21.Arnold WD, Kassar D, Kissel JT. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve. 2015;51:157–167. doi: 10.1002/mus.24497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas NH, Dubowitz V. The natural history of type I (severe) spinal muscular atrophy. Neuromuscul Disord. 1994;4:497–502. doi: 10.1016/0960-8966(94)90090-6. [DOI] [PubMed] [Google Scholar]
- 23.Wang CH, Finkel RS, Bertini ES, Schroth M, Simonds A, Wong B, Aloysius A, Morrison L, Main M, Crawford TO, Trela A. Participants of the International Conference on SMA Standard Care. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22:1027–1049. doi: 10.1177/0883073807305788. [DOI] [PubMed] [Google Scholar]
- 24.Kaufmann P, McDermott MP, Darras BT, Finkel R, Kang P, Oskoui M, Constantinescu A, Sproule DM, Foley AR, Yang M, Tawil R, Chung W, Martens B, Montes J, O’Hagen J, Dunaway S, Flickinger JM, Quigley J, Riley S, Glanzman AM, Benton M, Ryan PA, Irvine C, Annis CL, Butler H, Caracciolo J, Montgomery M, Marra J, Koo B, De Vivo DC Muscle Study Group. The Pediatric Neuromuscular Clinical Research Network for Spinal Muscular Atrophy. Observational study of spinal muscular atrophy type 2 and 3: functional outcomes over 1 year. Arch Neurol. 2011;68:779–786. doi: 10.1001/archneurol.2010.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harada Y, Sutomo R, Sadewa AH, Akutsu T, Takeshima Y, Wada H, Matsuo M, Nishio H. Correlation between SMN2 copy number and clinical phenotype of spinal muscular atrophy: three SMN2 copies fail to rescue some patients from the disease severity. J Neurol. 2002;249:1211–1219. doi: 10.1007/s00415-002-0811-4. [DOI] [PubMed] [Google Scholar]
- 26.Wirth B, Herz M, Wetter A, Moskau S, Hahnen E, Rudnik-Schöneborn S, Wienker T, Zerres K. Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am J Hum Genet. 1999;64:1340–1356. doi: 10.1086/302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Kariya S, Park GH, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, Landmesser LT, Monani UR. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:2552–2569. doi: 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wong PC, Cai H, Borchelt DR, Price DL. Genetically engineered mouse models of neurodegenerative diseases. Nat Neurosci. 2002;5:633–639. doi: 10.1038/nn0702-633. [DOI] [PubMed] [Google Scholar]; (c) Seo J, Howell MD, Singh NN, Singh RN. Spinal muscular atrophy: an update on therapeutic progress. Biochim Biophys Acta. 2013;1832:2180–2190. doi: 10.1016/j.bbadis.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]; (b) Le TT, Pham LT, Butchbach MER, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AHM. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 29.(a) Bowerman M, Murray LM, Beauvais A, Pinheiro B, Kothary R. A critical Smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. NeuromusculDisord. 2012;22:263–276. doi: 10.1016/j.nmd.2011.09.007. [DOI] [PubMed] [Google Scholar]; (b) Cobb MS, Rose FF, Rindt H, Glascock JJ, Shababi M, Miller MR, Osman EY, Yen P-F, Garcia ML, Martin BR, Wetz MJ, Mazzasette C, Feng Z, Ko C-P, Lorson CL. Development and characterization of an SMN2-based intermediate mouse model of spinal muscular atrophy. Hum Mol Genet. 2013;22:1843–1855. doi: 10.1093/hmg/ddt037. [DOI] [PubMed] [Google Scholar]
- 30.Osborne M, Gomez D, Feng Z, McEwen C, Beltran J, Cirillo K, El-Khodor B, Lin MY, Li Y, Knowlton WM, McKemy DD, Bogdanik L, Butts-Dehm K, Martens K, Davis C, Doty R, Wardwell K, Ghavami A, Kobayashi D, Ko CP, Ramboz S, Lutz C. Characterization of behavioral and neuromuscular junction phenotypes in a novel allelic series of SMA mouse models. Hum Mol Genet. 2012;21:4431–4447. doi: 10.1093/hmg/dds285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bogdanik LP, Osborne MA, Davis C, Martin WP, Austin A, Rigo F, Bennett CF, Lutz CM. Systemic, postsymptomatic antisense oligonucleotide rescues motor unit maturation delay in a new mouse model for type II/III spinal muscular atrophy. Proc Natl Acad Sci US A. 2015;112:E5863–E5872. doi: 10.1073/pnas.1509758112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.(a) Tisdale S, Pellizzoni L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J Neurosci. 2015;35:8691–8700. doi: 10.1523/JNEUROSCI.0417-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dominguez E, Marais T, Chatauret N, Benkhelifa-Ziyyat S, Duque S, Ravassard P, Carcenac R, Astord S, Pereira de Moura A, Voit T, Barkats M. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMN mice. Hum Mol Genet. 2011;20:681–693. doi: 10.1093/hmg/ddq514. [DOI] [PubMed] [Google Scholar]
- 33.Staropoli JF, Li H, Chun SJ, Allaire N, Cullen P, Thai A, Fleet CM, Hua Y, Bennett CF, Krainer AR, Kerr D, McCampbell A, Rigo F, Carulli JP. Rescue of gene-expression changes in an induced mouse model of spinal muscular atrophy by an antisense oligonucleotide that promotes inclusion of SMN2 exon 7. Genomics. 2015;105:220–228. doi: 10.1016/j.ygeno.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 34.DiMasi JA. Innovating by developing new uses of already-approved drugs: trends in the marketing approval of supplemental indications. Clinical Therapeutics. 2013;35:808–818. doi: 10.1016/j.clinthera.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 35.DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J Health Econ. 2003;151:180–83. doi: 10.1016/S0167-6296(02)00126-1. [DOI] [PubMed] [Google Scholar]
- 36.Thayer AM. Drug repurposing. Chem Eng News. 2012;90:15–25. [Google Scholar]
- 37.O’Connor DJ. Orphan drug designation - Europe, the USA and Japan. Exp Opin Orphan Drugs. 2013;1:255–259. [Google Scholar]
- 38.Lunke S, El-Osta A. Applicability of histone deacetylase inhibition for the treatment of spinal muscular atrophy. Neurotherapeutics. 2013;10:677–687. doi: 10.1007/s13311-013-0209-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang JG, Hsieh-Li HM, Jong YJ, Wang NM, Tsai CH, Li H. Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci US A. 2001;98:9808–9813. doi: 10.1073/pnas.171105098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andreassi C, Angelozzi C, Tiziano FD, Vitali T, De Vincenzi E, Boninsegna A, Villanova M, Bertini E, Pini A, Neri G, Brahe C. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet. 2004;12:59–65. doi: 10.1038/sj.ejhg.5201102. [DOI] [PubMed] [Google Scholar]
- 41.Brichta L, Hofmann Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, Eyupoglu IY, Wirth B. Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet. 2003;12:2481–2489. doi: 10.1093/hmg/ddg256. [DOI] [PubMed] [Google Scholar]
- 42.Hahnen E, Eyupoglu IY, Brichta L, Haastert K, Trankle C, Siebzehnrubl FA, Riessland M, Holker I, Claus P, Romstock J, Buslei R, Wirth B, Blumcke I. In vitro and ex vivo evaluation of second-generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy. J Neurochem. 2006;98:193–202. doi: 10.1111/j.1471-4159.2006.03868.x. [DOI] [PubMed] [Google Scholar]
- 43.Riessland M, Ackermann B, Forster A, Jakubik M, Hauke J, Garbes L, Fritzsche I, Mende Y, Blumcke I, Hahnen E, Wirth B. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum Mol Genet. 2010;19:1492–1506. doi: 10.1093/hmg/ddq023. [DOI] [PubMed] [Google Scholar]
- 44.Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, Cizman Z, Di Prospero NA, Pellizzoni L, Fischbeck KH, Sumner CJ. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gilbert J, Baker SD, Bowling MK, Grochow L, Figg WD, Zabelina Y, Donehower RC, Carducci MA. A phase I dose escalation and bioavailability study of oral sodium phenylbutyrate in patients with refractory solid tumor malignancies. Clin Cancer Res. 2001;7:2292–2300. [PubMed] [Google Scholar]
- 46.Kemp S, Wei HM, Lu JF, Braiterman LT, McGuinness MC, Moser AB, Watkins PA, Smith KD. Gene redundancy and pharmacological gene therapy: Implications for X-linked adrenoleukodystrophy. Nat Med. 1998;4:1261–1268. doi: 10.1038/3242. [DOI] [PubMed] [Google Scholar]
- 47.Berg S, Serabe B, Aleksic A, Bomgaars L, McGuffrey L, Dauser R, Durfee J, Nuchtern J, Blaney S. Pharmacokinetics and cerebrospinal fluid penetration of phenylacetate and phenylbutyrate in the nonhuman primate. Cancer Chemother Pharmocol. 2001;47:385–390. doi: 10.1007/s002800000256. [DOI] [PubMed] [Google Scholar]
- 48.Mercuri E, Bertini E, Messina S, Pelliccioni M, D’Amico A, Colitto F, Mirabella M, Tiziano FD, Vitali T, Angelozzi C, Kinali M, Main M, Braheg C. Pilot trial of phenylbutyrate in spinal muscular atrophy. Neuromuscular Disorders. 2004;14:130–135. doi: 10.1016/j.nmd.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 49.Iannitti T, Palmieri B. Clinical and experimental applications of sodium phenylbutyrate. Drugs R D. 2011;11:227–249. doi: 10.2165/11591280-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rak K, Lechner BD, Schneider C, Drexl H, Sendtner M, Jablonka S. Valproic acid blocks excitability in SMA type I mouse motor neurons. Neurobiol Dis. 2009;36:477–487. doi: 10.1016/j.nbd.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 51.Sumner CJ, Huynh TN, Markowitz JA, Perhac JS, Hill B, Coovert DD, Schussler K, Chen X, Jarecki J, Burghes AHM, Taylor JP, Fischbeck KH. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann Neurol. 2003;54:647–654. doi: 10.1002/ana.10743. [DOI] [PubMed] [Google Scholar]
- 52.Harahap ISK, Saito T, San LP, Sasaki N, Gunadi, Nurputra DKP, Yusoff S, Yamamoto T, Morikawa S, Nishimura N, Lee MJ, Takeshima Y, Matsuo M, Nishio H. Valproic acid increases SMN2 expression and modulates SF2/ASF and hnRNPA1 expression in SMA fibroblast cell lines. Brain Dev. 2012;34:213–222. doi: 10.1016/j.braindev.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 53.Tsai LK, Tsai MS, Ting CH, Li H. Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice. J Mol Med. 2008;86:1243–1254. doi: 10.1007/s00109-008-0388-1. [DOI] [PubMed] [Google Scholar]
- 54.Kissel JT, Scott CB, Reyna SP, Crawford TO, Simard LR, Krosschell KJ, Acsadi G, Elsheik B, Schroth MK, D’Anjou G, LaSalle B, Prior TW, Sorenson S, Maczulski JA, Bromberg MB, Chan GM, Swoboda KJ Project Cure Spinal Muscular Atrophy Investigators’ Network. SMA carni-val trial part II: A prospective single-armed trial of L-carnitine and valproic acid in ambulatory children with spinal muscular atrophy. Neurology. 2011;76:A367–A367. doi: 10.1371/journal.pone.0021296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Narver HL, Kong LL, Burnett BG, Choe DW, Bosch-Mare M, Taye AA, Eckhaus MA, Sumner CJ. Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition. Ann Neurol. 2008;64:465–470. doi: 10.1002/ana.21449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu H, Yazdani A, Murray LM, Beauvais A, Kothary R. The Smn-independent beneficial effects of trichostatin A on an intermediate mouse model of spinal muscular atrophy. PLoS One. 2014;9:e101225. doi: 10.1371/journal.pone.0101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Doble A. The pharmacology and mechanism of action of riluzole. Neurology. 1996;47:S233–S241. doi: 10.1212/wnl.47.6_suppl_4.233s. [DOI] [PubMed] [Google Scholar]
- 58.Zarate CA, Manji HK. Riluzole in psychiatry: a systematic review of the literature. Expert Opin Drug Metab Toxicol. 2008;4:1223–1234. doi: 10.1517/17425255.4.9.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milane A, Fernandez C, Vautier S, Bensimon G, Meininger V, Farinotti R. Minocycline and riluzole brain disposition: interactions with p-glycoprotein at the blood-brain barrier. J Neurochem. 2007;103:164–173. doi: 10.1111/j.1471-4159.2007.04772.x. [DOI] [PubMed] [Google Scholar]
- 60.Jablonski MR, Jacob DA, Campos C, Miller DS, Maragakis NJ, Pasinelli P, Trotti D. Selective increase of two ABC drug efflux transporters at the blood-spinal cord barrier suggests induced pharmacoresistance in ALS. Neurobiol Dis. 2012;47:194–200. doi: 10.1016/j.nbd.2012.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Russman BS, Iannaccone ST, Samaha FJ. A phase 1 trial of riluzole in spinal muscular atrophy. Arch Neurol. 2003;60:1601–1603. doi: 10.1001/archneur.60.11.1601. [DOI] [PubMed] [Google Scholar]
- 62.Abbara C, Estournet B, Lacomblez L, Lelievre B, Ouslimani A, Lehmann B, Viollet L, Barois A, Diquet B. Riluzole pharmacokinetics in young patients with spinal muscular atrophy. Br J Clin Pharmacol. 2011;71:403–410. doi: 10.1111/j.1365-2125.2010.03843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grzeschik SM, Ganta M, Prior TW, Heavlin WD, Wang CH. Hydroxyurea enhances SMN2 gene expression in spinal muscular atrophy cells. Ann Neurol. 2005;58:194–202. doi: 10.1002/ana.20548. [DOI] [PubMed] [Google Scholar]
- 64.Xu C, Chen X, Grzeschik SM, Ganta M, Wang CH. Hydroxyurea enhances SMN2 gene expression through nitric oxide release. Neurogenetics. 2011;12:19–24. doi: 10.1007/s10048-010-0268-z. [DOI] [PubMed] [Google Scholar]
- 65.Chen TH, Chang JG, Yang YH, Mai HH, Liang WC, Wu YC, Wang HY, Huang YB, Wu SM, Chen YC, Yang SN, Jong YJ. Randomized, double-blind, placebo-controlled trial of hydroxyurea in spinal muscular atrophy. Neurology. 2010;75:2190–2197. doi: 10.1212/WNL.0b013e3182020332. [DOI] [PubMed] [Google Scholar]
- 66.Syvänen S, Barletta J, Blomquist G, Långström B, Bergström M. PET-evaluated transport of [11C]hydroxyurea across the rate blood-brain barrier—lack of influence of cyclosporine and probenecid. Drug Metab Lett. 2007;1:189–194. doi: 10.2174/187231207781369799. [DOI] [PubMed] [Google Scholar]
- 67.Spector R. Ceftriaxone transport through the blood-brain barrier. J Infect Dis. 1987;156:209–211. doi: 10.1093/infdis/156.1.209. [DOI] [PubMed] [Google Scholar]
- 68.Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Hoberg MD, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. β-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- 69.Nizzardo M, Nardini M, Ronchi D, Salani S, Donadoni C, Fortunato F, Colciago G, Falcone M, Simone C, Riboldi G, Govoni A, Bresolin N, Comi GP, Corti S. Beta-lactam antibiotic offers neuroprotection in a spinal muscular atrophy model by multiple mechanisms. Exp Neurol. 2011;229:214–225. doi: 10.1016/j.expneurol.2011.01.017. [DOI] [PubMed] [Google Scholar]
- 70.Martineau L, Horan MA, Rothwell NJ, Little RA. Salbutamol, a β2-adrenoceptor agonist, increases skeletal-muscle strength in young men. Clin Sci. 1992;83:615–621. doi: 10.1042/cs0830615. [DOI] [PubMed] [Google Scholar]
- 71.Angelozzi C, Borgo F, Tiziano FD, Martella A, Neri G, Brahe C. Salbutamol increases SMN mRNA and protein levels in spinal muscular atrophy cells. J Med Genet. 2008;45:29–31. doi: 10.1136/jmg.2007.051177. [DOI] [PubMed] [Google Scholar]
- 72.Albuterol. PDR electronic library. 2010 Retrieved from http://www.pdrnetwork.net/
- 73.Kinali M, Mercuri E, Main M, De Biasia F, Karatza A, Higgins R, Banks LM, Manzur AY, Muntoni F. Pilot trial of albuterol in spinal muscular atrophy. Neurology. 2002;59:609–610. doi: 10.1212/wnl.59.4.609. [DOI] [PubMed] [Google Scholar]
- 74.Pane MP, Staccioli SS, Messina SM, d’Amico AD, Pelliccioni MP, Mazzone EM, Cuttini MC, Alfieri PA, Battini RB, Main MM, Muntoni FM, Bertini EB, Villanova MV, Mercuri EM. Daily salbutamol in young patients with SMA type II. Neuromuscul Disord. 2008;18:762–762. doi: 10.1016/j.nmd.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 75.Andreassi C, Jarecki J, Zhou JH, Coovert DD, Monani UR, Chen XC, Whitney M, Pollok B, Zhang ML, Androphy E, Burghes AHM. Aclarubicin treatment restores SMN levels to cells derived from type I spinal muscular atrophy patients. Hum Mol Genet. 2001;10:2841–2849. doi: 10.1093/hmg/10.24.2841. [DOI] [PubMed] [Google Scholar]
- 76.Ting CH, Lin CW, Wen SL, Hsieh-Li HM, Li H. Stat5 constitutive activation rescues defects in spinal muscular atrophy. Hum Mol Genet. 2007;16:499–514. doi: 10.1093/hmg/ddl482. [DOI] [PubMed] [Google Scholar]
- 77.Bordet T, Berna P, Abitbol JL, Pruss RM. Olesoxime (TRO19622): A novel mitochondrial-targeted neuroprotective compound. Pharmaceuticals. 2010;3:345–368. doi: 10.3390/ph3020345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hwee DT, Kennedy AR, Hartman JJ, Ryans J, Durham N, Malik FI, Jasper JR. The small-molecule fast skeletal troponin activator, CK-2127107, improves exercise tolerance in a rat model of heart failure. J Pharmacol Exp Ther. 2015;353:159–168. doi: 10.1124/jpet.114.222224. [DOI] [PubMed] [Google Scholar]
- 79.Tisdale S, Pellizzoni L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J Neurosci. 2015;35:8691–8700. doi: 10.1523/JNEUROSCI.0417-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bordet T, Buisson B, Michaud M, Drouot C, Galéa P, Delaage P, Akentieva NP, Evers AS, Covey DF, Ostuni MA, Lacapere JJ, Massaad C, Schumacher M, Steidl EM, Maux D, Delaage M, Henderson CE, Pruss RM. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J Pharmacol Exp Ther. 2007;322:709–720. doi: 10.1124/jpet.107.123000. [DOI] [PubMed] [Google Scholar]
- 81.Lenglet T, Lacomblez L, Abitbol JL, Ludolph A, Mora JS, Robberecht W, Shaw PJ, Pruss RM, Cuvier V, Meininger V Mitotarget study group. A phase II-III trial of olesoxime in subjects with amyotrophic lateral sclerosis. Eur J Neurol. 2014;21:529–536. doi: 10.1111/ene.12344. [DOI] [PubMed] [Google Scholar]
- 82.Chin ER. Intracellular Ca2+ signaling in skeletal muscle: decoding a complex message. Exerc Sport Sci Rev. 2010;38:76–85. doi: 10.1097/JES.0b013e3181d495d2. [DOI] [PubMed] [Google Scholar]
- 83.Ashcraft LW, Bergnes G, Collibee S, Chuang C, Gardina J, Morgan BP, Muci AR, Qian X, Warrington J, Yang Z, Lu PP, Romero A. Certain Amino-Pyridazines, Compositions Thereof, and Methods of Their Use. WO Patent. 2011;133:882. [Google Scholar]
- 84.Jasper JR, Malik F, Hwee DT. Methods for Improving Diaphragm Function. WO Patent. 2013;034:824. [Google Scholar]
- 85.Andrews J, Vijayakumar V, James J, Lee J, Kulke S, Bian A, Meng L, Malik F, Wolff A. Pharmacokinetics and pharmacodynamics of the selective fast skeletal muscle troponin activator, CK-2127107. Muscle & Nerve. 2015;52:S11–S11. [Google Scholar]
- 86.Jarecki J, Chen X, Bernardino A, Coovert DD, Whitney M, Burghes A, Stack J, Pollok BA. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: early leads towards a therapeutic for spinal muscular atrophy. Hum Mol Genet. 2005;14:2003–2018. doi: 10.1093/hmg/ddi205. [DOI] [PubMed] [Google Scholar]
- 87.Thurmond J, Butchbach MER, Palomo M, Pease B, Rao M, Bedell L, Keyvan M, Pai G, Mishra R, Haraldsson M, Andrésson T, Bragason G, Thosteinsdottir M, Bjornsson JM, Coovert DD, Burghes AHM, Gurney ME, Singh J. Synthesis and biological evaluation of novel 2,4-diaminoquinazoline derivatives as SMN2 promoter activators for the potential treatment of spinal muscular atrophy. J Med Chem. 2008;51:449–469. doi: 10.1021/jm061475p. [DOI] [PubMed] [Google Scholar]
- 88.Gogliotti RG, Cardona H, Singh J, Bail S, Emery C, Kuntz N, Jorgensen M, Durens M, Xia B, Barlow C, Heier CR, Plasterer HL, Jacques V, Kiledjian M, Jarecki J, Rusche J, DiDonato CJ. The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum Mol Genet. 2013;22:4084–4101. doi: 10.1093/hmg/ddt258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Singh J, Salcius M, Liu SW, Staker BL, Mishra R, Thurmond J, Michaud G, Mattoon DR, Printen J, Christensen J, Bjornsson JM, Pollok BA, Kiledjian M, Stewart L, Jarecki J, Gurney ME. DcpS as a therapeutic target for spinal muscular atrophy. ACS Chem Biol. 2008;3:711–722. doi: 10.1021/cb800120t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 91.Milac A, Bojarska E, Wypijewska del Nogal A. Decapping Scavenger (DcpS) enzyme: advances in its structure, activity and roles in the cap-dependent mRNA metabolism. Biochim Biophys Acta. 2014;1839:452–462. doi: 10.1016/j.bbagrm.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 92.Sinturel F, Brechemier-Baey D, Kiledjian M, Condon C, Benard L. Activation of 5′-3′ exoribonuclease Xrn1 by cofactor Dcs1 is essential for mitochondrial function in yeast. Proc Natl Acad Sci USA. 2012;109:8264–8269. doi: 10.1073/pnas.1120090109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meziane O, Piquet S, Bossé GD, Gagné D, Paquet E, Robert C, Tones MA, Simard MJ. The human decapping scavenger enzyme DcpS modulates microRNA turnover. Sci Rep. 2015;5:16688. doi: 10.1038/srep16688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou M, Bail S, Plasterer HL, Rusche J, Kiledjian M. DcpS is a transcript-specific modulator of RNA in mammalian cells. RNA. 2015;21:1306–1312. doi: 10.1261/rna.051573.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Van Meerbeke JP, Gibbs RM, Plasterer HL, Miao W, Feng Z, Lin M, Rucki AA, Wee CD, Xia B, Sharma S, Jacques V, Li DK, Pellizzoni L, Rusche JR, Ko C, Sumner CJ. The DcpS inhibitor RG3039 improves motor function in SMA mice. Hum Mol Genet. 2013;22:4074–4083. doi: 10.1093/hmg/ddt257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Butchbach MER, Singh J, Thorsteinsdottir M, Luciano S, Slominski E, Thurmond J, Andrésson T, Zhang J, Edwards JD, Simard LR, Pellizzoni L, Jarecki J, Burghes AHM, Gurney ME. Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum Mol Genet. 2010;19:454–467. doi: 10.1093/hmg/ddp510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Naryshkin NA, Weetall M, Dakka A, Narasimhan J, Zhao X, Feng Z, Ling KKY, Karp GM, Qi H, Woll MG, Chen G, Zhang N, Gabbeta V, Vazirani P, Bhattacharyya A, Furia B, Risher N, Sheedy J, Kong R, Ma J, Turnpoff A, Lee C, Zhang X, Moon Y, Trifillis P, Welch EM, Colacino JM, Babiak J, Almstead NG, Peltz SW, Eng LA, Chen KS, Mull JL, Lynes MS, Rubin LL, Fontoura P, Santarelli L, Haehnke D, McCarthy KD, Schmucki R, Ebeling M, Sivaramakrishnan M, Ko C, Paushkin SV, Ratni H, Gerlach I, Ghosh A, Metzger F. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 2014;345:688–693. doi: 10.1126/science.1250127. [DOI] [PubMed] [Google Scholar]
- 98.Woll MG, Chen G, Choi S, Dakka A, Huang S, Karp GM, Lee CS, Li C, Narasimhan J, Naryshkin N, Paushkin S, Qi H, Turpoff AA, Weetall ML, Welch E, Yang T, Zhang N, Zhang X, Zhao X, Pinard E, Patni H. Compounds for Treating Spinal Muscular Atrophy. WO Patent. 2013;101:974. [Google Scholar]
- 99.Cherry JJ, Osman EY, Evans MC, Sungwoon C, Xing X, Cuny GD, Glicksman MA, Lorson CL, Androphy EJ. Enhancement of SMN protein levels in a mouse model of spinal muscular atrophy using novel drug-like compounds. EMBO Mol Med. 2013;5:1103–1118. doi: 10.1002/emmm.201202305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Palacino J, Swalley SE, Song C, Cheung AK, Shu L, Zhang X, Hoosear MV, Shin Y, Chin DN, Keller CG, Beibel M, Renaud NA, Smith TM, Salcius M, Shi X, Hild M, Servais R, Jain M, Deng L, Bullock C, McLellan M, Schuierer S, Murphy L, Blommers MJJ, Blaustein C, Berenshteyn F, Lacoste A, Thomas JR, Roma G, Michaud GA, Tseng BS, Porter JA, Myer VE, Tallarico JA, Hamann LG, Curtis D, Fishman MC, Dietrich WF, Dales NA, Sivasankaran R. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat Chem Biol. 2015;11:511–517. doi: 10.1038/nchembio.1837. [DOI] [PubMed] [Google Scholar]
- 101.Cheung A, Chin DN, Dales N, Fazal A, Hurley TB, Kerrigan J, O’Brien G, Shu L, Sun R, Snug M. 1,4-Disubstituted Pyridazine Analogs and Methods for Treating SMN-Deficiency-Related Conditions. WO Patent. 2014;028:459. [Google Scholar]
- 102.Hodgetts KJ. Case studies of CNS drug optimization—medicinal chemistry and CNS biology perspectives. In: Kerns EH, Di L, editors. Blood-Brain Barrier in Drug Discovery: Optimizng Brain Exposure of CNS Drugs and Minimizing Brain Side Effects for Peripheral Drugs. 1. John Wiley & Sons, Inc; Hoboken, NJ: 2015. pp. 425–446. [Google Scholar]
- 103.Androphy EJ, Cherry JJ, Cuny GD, Glicksman MA. Compounds for Treatment of Spinal Muscular Atrophy. WO Patent. 2014;012:050. [Google Scholar]
- 104.Reitz A, Kern NL, Cherry JJ, Choi S, Li H, Calder AN, Fritsche M, Xing X, Glicksman MA, Cuny GD, Androphy EJ, Hodgetts KJ. Unpublished results [Google Scholar]
- 105.Xiao J, Marugan JJ, Zheng W, Titus S, Southall N, Cherry JJ, Evans M, Androphy EJ, Austin CP. Discovery, synthesis and biological evaluation of novel SMN protein modulators. J Med Chem. 2011;54:6215–6233. doi: 10.1021/jm200497t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Makhortova NR, Hayhurst M, Cerqueira A, Sinor-Anderson AD, Zhao WN, Heiser PW, Arvanites AC, Davidow LS, Waldon ZO, Steen JA, Lam K, Ngo HD, Rubin LL. A screen for regulators of survival of motor neuron protein levels. Nat Chem Biol. 2011;7:544–552. doi: 10.1038/nchembio.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xu C, Kim NG, Gumbiner BM. Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle. 2009;8:4032–4039. doi: 10.4161/cc.8.24.10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen PC, Gaisina IN, El-Khodor BF, Ramboz S, Makhortova NR, Rubin LL, Kozikowski AP. Identification of a maleimide-based glycogen synthase kinase-3 (GSK-3) inhibitor, BIP-135, that prolongs the median survival time of Δ7 SMA KO mouse model of spinal muscular atrophy. ACS Chem Neurosci. 2012;3:5–11. doi: 10.1021/cn200085z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hastings ML, Berniac J, Hsiu Liu Y, Abato P, Jodelka FM, Barthel L, Kumar S, Dudley C, Nelson M, Larson K, Edmonds J, Bowser T, Draper M, Higgins P, Krainer AR. Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy. Sci Transl Med. 2009;1:5ra12. doi: 10.1126/scitranslmed.3000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cobb MS, Rose FF, Rindt H, Glascock JJ, Shababi M, Miller MR, Osman EY, Yen PF, Garcia ML, Martin BR, Wetz MJ, Mazzasette C, Feng Z, Ko CP, Lorson CL. Development and characterization of an SMN2-based intermediate mouse model of spinal muscular atrophy. Hum Mol Genet. 2013;22:1843–1855. doi: 10.1093/hmg/ddt037. [DOI] [PubMed] [Google Scholar]
- 111.Mattis VB, Ebert AD, Fosso MY, Chang CW, Lorson CL. Delivery of a read-through inducing compound, TC007, lessens the severity of a spinal muscular atrophy animal model. Hum Mol Genet. 2009;18:3906–3913. doi: 10.1093/hmg/ddp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Greif H, Rosin-Arbesfeld R, Megiddo D. BBrm2, a read-through repurposed drug, shows proof of efficacy in SMA treatment. Presentation presented at: Cure SMA. 19th Annual Spinal Muscular Atrophy Researcher Meeting; 2015 June 18–20; Kansas City, MO. [Google Scholar]
- 113.Rankovic Z. Designing CNS drugs for optimal brain exposures. In: Kerns EH, Di L, editors. Blood-Brain Barrier in Drug Discovery Optimizng Brain Exposure of CNS Drugs and Minimizing Brain Side Effects for Peripheral Drugs. 1. John Wiley & Sons, Inc; Hoboken, NJ: 2015. pp. 387–425. [Google Scholar]
- 114.Bunnage ME, Piatnitski Chekler EL, Jones LH. Target validation using chemical probes. Nat Chem Biol. 2013;9:195–199. doi: 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- 115.Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997;16:265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]