

Abstract
Retinoblastoma is a rare cancer of the infant retina, which forms when both RB1 alleles mutate in a susceptible retinal cell, likely a cone photoreceptor precursor. Loss of the tumour suppressor functions of the retinoblastoma protein, pRB, leads to uncontrolled cell division and recurrent genomic changes during tumour progression. Although pRB is expressed in virtually all tissues, cone precursors have biochemical and molecular features that may sensitize to RB1 loss to enable tumourigenesis. Retinoblastoma is diagnosed in ~8,000 children each year worldwide. Patient survival is >95% in high-income countries, but <30% globally. However, outcomes are improving through increasing awareness for earlier diagnosis, new guidelines and sharing of expertise. Intra-arterial and intravitreal chemotherapy have emerged as promising methods to salvage eyes. Ongoing international collaborations will replace the multiple different classifications of eye involvement with standardized definitions to consistently assess eligibility, efficacy and safety of treatment options. Life-long follow-up is warranted since survivors of heritable retinoblastoma are at risk for developing second cancers. Defining the molecular consequences of RB1 loss in diverse tissues may open new avenues for treatment and prevention of retinoblastoma as well as second cancers in patients with germline RB1 mutations.
Introduction
Retinoblastoma is a rare cancer usually initiated by biallelic mutation of the retinoblastoma gene (RB1) in a single susceptible developing retinal cell. Inheritance of one mutant RB1 allele strongly predisposes to retinoblastoma tumours that form after the second RB1 allele is mutated.1 The principles discovered by studying retinoblastomas have led to the recognition that inactivation of tumor suppressor genes broadly contributes to human tumorigenesis.
Retinoblastoma initiates after an RB1−/− retinal cell undergoes limited proliferation to form a non-malignant retinoma (Figure 1)2 An intraretinal tumour develops after genetic or epigenetic changes lead to uncontrolled proliferation. Most commonly, this is first detected when the white tumour is visible through the pupil (leukocoria) or blocks vision.3 When noticed early, prompt treatment can cure the cancer and save the eye(s). However, delayed diagnosis can lead to incurable invasion of the optic nerve and brain, or to metastases elsewhere in the body that are sometimes cured by extensive intervention.
Figure 1. Progression of retinoblastoma.

a) Anatomical features of a healthy eye. Genomic damage (orange lightning bolt) leads to mutation of RB1 resulting in biallelic functional loss of RB1 in a developing retinal cell (possibly a cone photoreceptor precursor cell that is dependent on pRB to stop proliferation). b) Genomic instability leads to a benign retinoma; only 5% of patients show retinoma without retinoblastoma. Inset shows a small retinoma/tumour not visible except by optical coherence tomography. c) Intraretinal retinoblastoma arises as additional genomic changes promote uncontrolled cell proliferation; the tumour grows and seeds become independent, floating under the retina and into vitreous. d) Retinoblastoma can invade adjacent tissues: into the optic nerve, uvea, or sclera to constitute high-risk pathologic features. e) Eventually, retinoblastoma can extend extraocularly into orbit and metastasize especially to the bone marrow, or into the brain (direct or via the cerebrospinal fluid).
Several questions related to our understanding of retinoblastoma and its optimal clinical management remain. The cell of origin of retinoblastoma has been difficult to pinpoint; new research we review in this Primer has potential to improve our understanding of why the infant retina is uniquely predisposed to malignancy upon RB1 loss. The evidence base for clinical management is weak. Rigorous randomized clinical trials (RCTs) in retinoblastoma are missing for multiple reasons: too few patients in countries with capacity to conduct clinical trials; often complex presentation (two eyes with differing severity); too few patients to interest the pharmaceutical industry; often-opposing schools of thought and difficulties in transdisciplinary collaborations; and high societal value on eyes and vision imposes considerations beyond curing the cancer. New technologies showing dramatic tumour responses have been quickly embraced, without rigorous clinical science. Differences in resource availability, medical expertise, and societal and health system structure vary between and within nations, making recommendations for optimal care complex and context-dependent.
To address these challenges, this Primer on retinoblastoma introduces unprecedented new science, ideas, therapies and global collaboration. The Internet has opened many avenues: parents diagnose retinoblastoma themselves; colleagues share patients around the globe; centres of excellence are mapped; and a common database for all children — no matter where they live — is within reach. These developments are set to empower a ‘learning health system’ that will build the evidence base to optimise care.
Epidemiology
Global incidence and outcomes
The expected number of patients with retinoblastoma annually per country is most accurately calculated by multiplying the global retinoblastoma incidence (1 in 16,000–18,000 live births) by number of forecast surviving infants (birth rate minus infant mortality rate) (Table 1).4–6 Using these formulas, approximately 8,000 new cases are predicted each year. The number of children predicted in each country are indicated in Supplementary Table 1. Calculations as in Table 1 accurately predict retinoblastoma in many countries.7–9 There is no validated evidence of geographical or racial variations of retinoblastoma incidence.6,10,11
Table 1.
Estimated global distribution of retinoblastoma.
| Category | Population, total | Birth rate, crude (per 1,000 people) | Mortality rate, infant (per 1,000 live births) | Forecast live birth total | Retinoblastoma incidence (high) 1:16,000 | Retinoblastoma incidence (low) 1:18000 | % of world retinoblastoma |
|---|---|---|---|---|---|---|---|
| World | 7 × 109 | 19.4 | 34.6 | 131 106 | 8,200 | 7,300 | 100% |
| High income | 1,299,489,520 | 11.6 | 5.5 | 14,941,320 | 934 | 830 | 11.4% |
| High income: non-OECD | 249,927,994 | 13.8 | 10.3 | 3,418,872 | 214 | 190 | not applicable |
| High income: OECD | 1,049,561,526 | 11.0 | 4.4 | 11,518,845 | 720 | 640 | not applicable |
| Low & middle income | 5,743,616,071 | 21.1 | 38.1 | 116,699,203 | 7,294 | 6,483 | 88.7% |
| Middle income | 4,913,580,797 | 19.2 | 33.7 | 91,300,546 | 5,706 | 5,072 | 69.4% |
| Upper middle income | 2,390,210,774 | 14.8 | 16.3 | 34,737,317 | 2,171 | 1,930 | 26.4% |
| Lower middle income | 2,523,370,023 | 23.4 | 45.3 | 56,490,388 | 3,531 | 3,138 | 42.9% |
| Low income | 830,035,274 | 32.3 | 54.5 | 25,373,890 | 1,586 | 1,410 | 19.3% |
Forecast births were calculated using most recent data (2012) on each World Bank Category for population, birth rate and mortality rate (World Bank (http://data.worldbank.org) accessed on 5 February 2015. Low (1:18,000 live births) and high estimates (1:16,000 live births) for retinoblastoma were calculated, following the example of Kivela.5 Since retinoblastoma arises before and near birth, the usual cancer incidence rate calculations (number of cases diagnosed in a given population, sometimes adjusted for the under-514 or under-1411 populations in children’s cancers) do not provide accurate data for retinoblastoma. OECD, Organisation for Economic Co-operation and Development
Of all affected children, 11% reside in high-income, 69% in middle-income and 20% in low-income countries.12 Although prevalence is higher in middle- to low-income countries (Figure 2), most retinoblastoma treatment centres are in middle- and high-income countries, creating a gap in healthcare access (Table 2). Consistent with income being a surrogate for non-economic measures of standard of living, retinoblastoma in low-income countries is associated with low patient survival (~30%13,14) compared with high-income countries (>95%8), but comprehensive nation-wide data are lacking.
Figure 2. Global retinoblastoma treatment centres and patient distribution relative to resources.
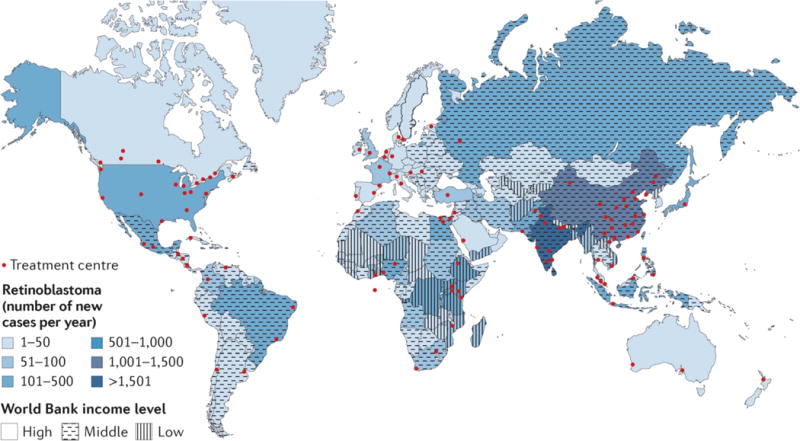
One Retinoblastoma World (www.1rbw.org) shows Retinoblastoma Centres of Excellence, providing a resource for affected families to access expert care. The majority of patients reside in low and middle income countries, while the majority of Retinoblastoma Centres are in high income countries. Images from www.1rbw.org.
Table 2.
Availability of treatment and imaging at retinoblastoma centres worldwide.
| Countries | Total number of centres | Enucleation (%) | Laser therapy (%) | Cryotherapy (%) | IVC (%) | Retinal imaging (%) |
|---|---|---|---|---|---|---|
| High income | 47 | 100% | 91% | 89% | 85% | 89% |
| Middle income | 75 | 100% | 91% | 91% | 69% | 63% |
| Low income | 10 | 100% | 50% | 20% | 5 (50%) | 30% |
| Total | 132 | 100% | 88% | 85% | 97 (73%) | 70% |
Retinoblastoma centres worldwide were surveyed and featured on the One Retinoblastoma World Map (1rbw.org, accessed on 25 March 2015). Information is available for 54 countries (22 high-income countries, 28 middle-income countries and 4 low-income countries, following World Bank Classification). The percentages depict the percentage of centres offering the specific treatment or diagnostic tools. IVC, intravenous chemotherapy.
Poor outcome correlates with late diagnosis, difficulty accessing retinoblastoma-specific health care, and socio-economic issues leading to poor compliance, including family refusal to remove the affected eye (enucleation) and abandonment of therapy.11,15,16 Without timely diagnosis and appropriate treatment, difficult-to-cure metastatic disease may develop. Fortunately, with early diagnosis,17 many eyes can be safely treated and support a lifetime of good vision, pointing to key elements for global focus: awareness, collaboration, and affordable expert care.
Solutions for global retinoblastoma
A number of initiatives address the inequality in retinoblastoma treatment between developing and developed countries. In 2009, the Canadian National Retinoblastoma Strategy (NRbS) published the first-ever retinoblastoma clinical practice guidelines,8 adapted by the Kenyan National Retinoblastoma Strategy (KNRbS) and published in 2014 in partnership with the Kenyan Ministry of Health.18 The KNRbS has supported the standardization of histopathologic examination of the enucleated eye to inform treatment decisions and discussion of prognosis with families. Adoption of upfront (first-line) enucleation with implants and immediate prosthetic eyes19 (sourced from India), parent-to-parent interactions to allay uninformed fears, and standardization of information provided to parents has reduced the rate of non-compliance with treatment.18,20 In 2013, the Paediatric Oncology in Developing Countries Committee of the SIOP (International Society of Paediatric Oncology) published a consensus guideline for the management of retinoblastoma in countries with limited resources21 with clear ideas that can shape resource development. The Mexican National Strategy22 and the Brazilian SOBOPE (Brazilian Society of Paediatric Oncology)23 guidelines are also applicable at a national level with governmental support for treatment.
Peer-to-peer collaborations and twinning programmes have built a framework for knowledge and expertise exchange, filling gaps in specialized training, and donating equipment and resources with the ultimate aim to ensure sustainable local capacity to deliver optimal health care.24 Retinoblastoma-specific twinning programmes include partnerships between St. Jude’s Children’s Research Hospital (USA) and the Middle-East,25 Central America26 and Mexico,22 and between the Institut Curie and a centre in Bamako, Mali.27 The Central American Association of Paediatric Hematology Oncology (AHOPCA) created a cooperative group and implemented multicentre protocols for retinoblastoma treatment,26,28,29 a major achievement, not yet paralleled in developed countries. The AHOPCA funding is now 90% local. Twinning programmes benefit from strong participation of both governmental and non-governmental organizations (NGO). However, where government may prove volatile and unpredictable, sustainable local capacity is a challenge. Less formal cooperation between developed and less developed countries can also result in highly efficient programmes.30,31
A successful national strategy has been developed in China. About 1,100 newly diagnosed cases are forecast annually, scattered over 32 provinces. Before 2005, enucleation was the only available treatment for most children. For better treatment options and follow-up, centres classified by expertise and resources were established in 28 hospitals covering 25 provinces (over 90% of the population), which profoundly reduced travel costs12. The improved efficiency and national collaboration from 2006 to 2014 has led to standardized classification and treatment of 2,097 newly diagnosed patients with retinoblastoma on common protocols, which has led to increased survival (unpublished data, J.Z.). In Argentina, a single centre with high expertise coordinates affiliated retinoblastoma clinics. This collaboration between governmental hospitals, local and international NGOs with prospective protocols has significantly improved survival in two decades, in addition to stimulating translational research and population-based incidence and survival studies.32,33,34
In a rapidly changing world, online strategies are facilitating global collaboration. The One Retinoblastoma World initiative (www.1rbw.org) leads families to the nearest centre with known expertise.12 The retinoblastoma-specific point-of-care database (eCancerCareRB, eCCRB) summarizes the medical record for retinoblastoma for each affected child. eCCRB is freely accessible online to every retinoblastoma centre with local ethics and privacy approvals. The Disease-specific, electronic Patient Illustrated Clinical Timeline (DePICT) is language-independent, is understandable by parents, and supports fully informed care choices.35 With guardian consent, identifiable data is accessible to those in the circle of care of each child. Ultimately, the de-identified data will be fuel for powering a global learning health system for retinoblastoma.
Mechanisms/pathophysiology
Genetic basis of retinoblastoma
The search for the genetic basis of retinoblastoma provided the two-hit model of tumour suppressor gene inactivation. In 1971, Knudson discovered that the ages of retinoblastoma diagnosis are consistent with one rate-limiting event in bilaterally affected patients (who have heritable disease) (median age at diagnosis 12 months), and with two rate-limiting events in unilaterally affected patients with no family history (who usually have the non-heritable form) (median age at diagnosis 24 months).36 This prompted the proposal that heritable retinoblastomas result from a germline mutation (‘first hit’, M1) and an acquired somatic mutation (‘second hit’, M2), whereas non-heritable retinoblastomas arise when two somatic mutations are present in the same transformation suppressor gene in a susceptible cell (Figure 3).37 Chromosomal deletions in some patients pointed to a chromosome (chr.) 13q14 locus. Loss of heterozygosity of chr. 13q14 polymorphic markers in 70% of retinoblastoma tumours38 implied that the second hit involved the same locus. The breakthrough came when one retinoblastoma was found to be missing the sequence of a chr. 13q DNA clone,39 that was shown to be a conserved exon sequence of RB1.39–42 Although RB1 loss consistently initiates cancer in the retina, RB1 function can be lost with progression in almost all cancer types.43
Figure 3. Genetic origins of retinoblastoma.
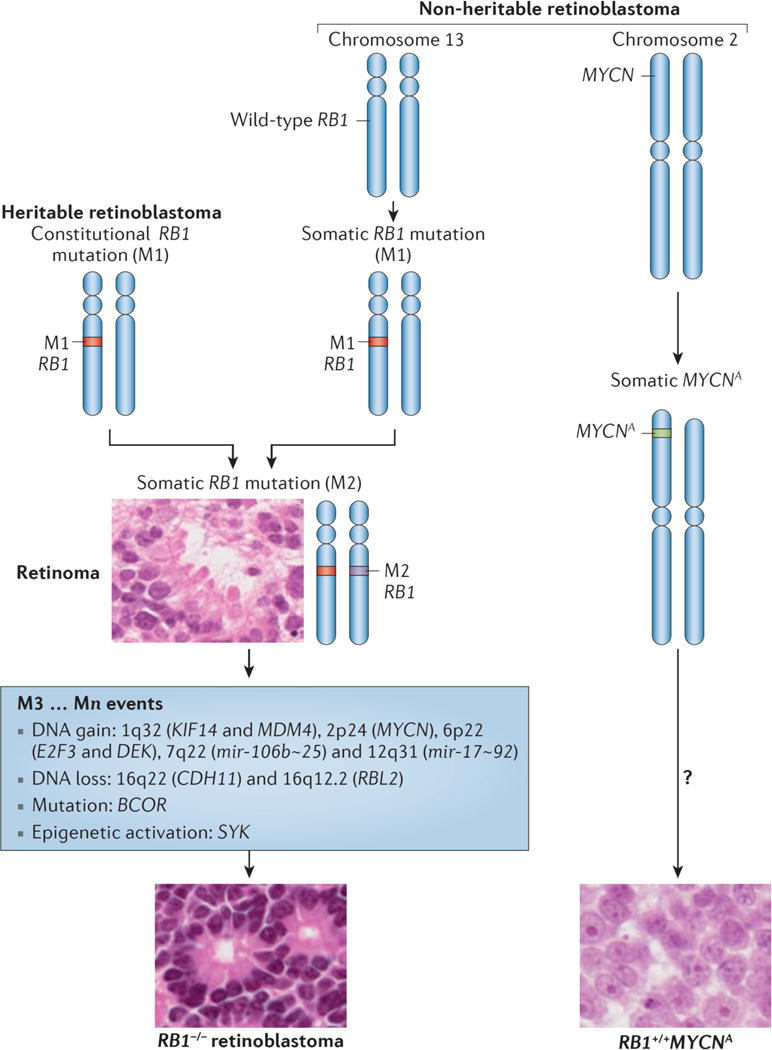
Three genetic subtypes of retinoblastoma are known. Heritable retinoblastoma patients have a constitutive inactivating mutation (M1) in the RB1tumor suppressor gene in all cells of their body. A second, somatic mutation (M2) in a susceptible retinal cell can lead to benign retinoma. Further genetic and/or epigenetic events (M3…Mn) are required to transform to retinoblastoma. Non-heritable, RB1−/− retinoblastomas progress similarly, except both M1 and M2 occur in one susceptible retinal cell. RB1+/+MYCN-amplified (RB1+/+MYCNA) retinoblastoma is a rare, non-heritable retinoblastoma subtype driven by amplification of MYCN with normal RB1; other changes in these tumours remain uncharacterized. Retinoma histology shows distinct photoreceptor-like fleurettes, whereas RB1−/− retinoblastoma can show Flexner-Wintersteiner (insert) and Homer Wright rosettes (not shown). RB1+/+MYCNA retinoblastoma have a distinct morphology with rounded nuclei and prominent nucleoli related to the high MYCN protein.
RB1 is a large (190 kb) gene with 27 exons, encoding a 4.7 kb mRNA that translates into a 928 amino acid protein, pRB. Many modifications impair pRB function, including point mutations, promoter methylation, and small and large deletions.44 Even chromothripsis, a “shattering” of a genomic region resulting in widespread rearrangements, has been shown to underlie a minority of cases.45 The A/B “pocket” region46 harbours most missense mutations.
pRB is best known as a cell cycle regulator that binds to E2F transcription factors to repress cell proliferation-related genes. Hyperphosphorylation of pRB by cyclin-dependent kinases (CDKs) in response to mitogenic signals normally relieves repression and promotes the G1 to S phase transition. pRB loss relieves this repression in the absence of mitogenic signals to enable cell cycle entry. It is tempting to surmise that pRB is primarily needed to suppress E2F transcription factors, and that loss of this function is the main cause of retinoblastoma formation.46 However, several “low penetrance” RB1 mutations encode proteins with minimal ability to bind E2F but unexpectedly these mutations predispose to far fewer tumours than RB1 null alleles.47,48 Thus, such defective E2F-binding alleles may function to substantially block retinoblastoma development through one or more of pRB’s E2F-independent functions.46 For example, pRB also up-regulates p27, and this effect has been implicated in cell differentiation, apoptosis, and genomic integrity.46 However, during cone precursor maturation increasing pRB is associated with decreasing p27 expression,49 arguing that p27 would not mediate retinoblastoma suppression in these cells.49 The loss of pRB N-terminus functions such as non-homologous end-joining may also be important but are not clearly implicated in retinoblastoma initiation.50,51 Thus, the exact mechanism by which pRB normally suppresses retinoblastoma remains to be identified.
Biallelic RB1 inactivation is necessary to initiate most retinoblastomas, but is not sufficient, since the benign retinal lesion, retinoma, has lost both RB1−/− alleles (Figures 1 and 3).2 Further genetic or epigenetic changes are likely needed for malignant transformation.52 Epigenetic alterations might drive retinoblastoma formation by inducing H3K4 trimethylation and H3K9 and H3K14 acetylation marks and expression of the SYK oncogene.53 Comparative genomic hybridization studies identified DNA copy number gains encompassing the candidate retinoblastoma oncogenes mitotic kinesin KIF14 and the p53 regulator MDM4 (chr. 1q32), transcription factors E2F3 and DEK (chr. 6p22), and the onco-miR clusters miR-106b~25 (chr. 7q22.1) and miR-17~92 (chr. 13q31, as well as losses encompassing the cadherin-11 (CDH11) (chr. 16q22) and pRB family member RBL2 (chr. 16q12.2) tumour suppressor genes.52 Whole-genome sequencing identified inactivating mutations in the transcriptional corepressor BCOR encoding the BCL-6 corepressor.53 Interestingly, some tumors had no detected alterations,53 consistent with acquisition of such changes during tumour progression.54
Many other genes and microRNAs also show altered expression in retinoblastoma compared to normal retina.52 Gene expression profiles were initially proposed to reveal only a single relatively homogeneous class of retinoblastoma55 and were subsequently proposed to segregate RB1−/− retinoblastomas into two subtypes.56 However, analyses of a larger cohort recently revealed a spectrum of phenotypes reflecting progression from a state with high photoreceptor differentiation and few cytogenetic changes to a state with lower differentiation, increased mRNA and ribosome biogenesis and mitosis gene expression signatures, and increased cytogenetic aberrations.54 Thus, RB1−/− retinoblastomas appear to progress from self-limiting retinomas to differentiated retinoblastoma to less differentiated malignancies.
Whereas both RB1 alleles are mutated in nearly all retinoblastomas, a subset of unilateral tumours (1.4%) show no evidence of RB1 mutation but have high-level amplification of the oncogene MYCN (amplified MYCN, MYCNA).57 These RB1+/+MYCNA tumours are always unilateral, are diagnosed in much younger children than unilateral RB1−/− tumours, and have a distinct morphology, reflecting a unique subtype. Although they also fall within the photoreceptor differentiation and mitosis/RNA biogenesis spectrum of RB−/− tumours, RB1+/+MYCNA tumors are distinct outliers in levels of expression of many genes,54 consistent with the idea that they originate in a earlier retinal precursor.57 Another 1.5% of unilateral non-familial retinoblastomas are unexplained and have apparently normal RB1 and MYCN genes.57
The retinoblastoma cell of origin
Since RB is expressed in most if not all cells, the retina’s unique sensitivity to RB loss has been perplexing. While the key to this sensitivity likely relates to the features of the cell of origin, the identity of this cell has been elusive. Studies addressing the cell of origin through analysis of tumours offered puzzling clues, as the tumours expressed diverse retinal cell type markers. Together with other evidence, the diverse cell marker expression suggested either a pluripotent cell of origin, aberrant co-expression of marker genes due to oncogenic transformation, or the presence of normal RB1+/+ retinal cells within largely cone-like tumours.55,58 Mouse models have not clarified the cell-of-origin, since such models have required an additional mutation of Rbl1, Rbl2, or Cdkn1b on top of Rb1 loss,59 and the resulting tumours express a different spectrum of retinal markers, raising the possibility that they might not extrapolate to humans.60
A clue to the cell of origin came from the finding that the topographic distribution of emerging retinoblastomas mimics the horizontal visual streak characteristic of red and green cones, which provided evidence that the tumours could derive from a cell in the cone photoreceptor lineage.48 Consistent with this view, RB1−/− retinoblastomas consistently express cone photoreceptor markers but not other retinal cell type-specific proteins. In addition, normal post-mitotic maturing cone precursors have unusually high expression of pRB as well as oncoproteins (MDM2 and MYCN) that could collaborate with RB1 loss to enable cone precursor proliferation (Figure 4a, b).46,61 More recently it was shown that experimental depletion of RB1 induces human cone precursor cell proliferation in vitro and pRB-depleted human cone precursors form tumours typical of differentiated retinoblastomas in orthotopic xenografts.62 The proliferation of these normally post-mitotic cells depends upon their intrinsically high levels of MYCN and MDM2, cone-specific transcription factors RXRγ and TRβ2, and down-regulation of p27, which is associated with normal cone precursor maturation.49,62 Thus, pRB may counter an oncogenic programme associated with cone precursor maturation.
Figure 4. Retinoblastomas originate in the retina.
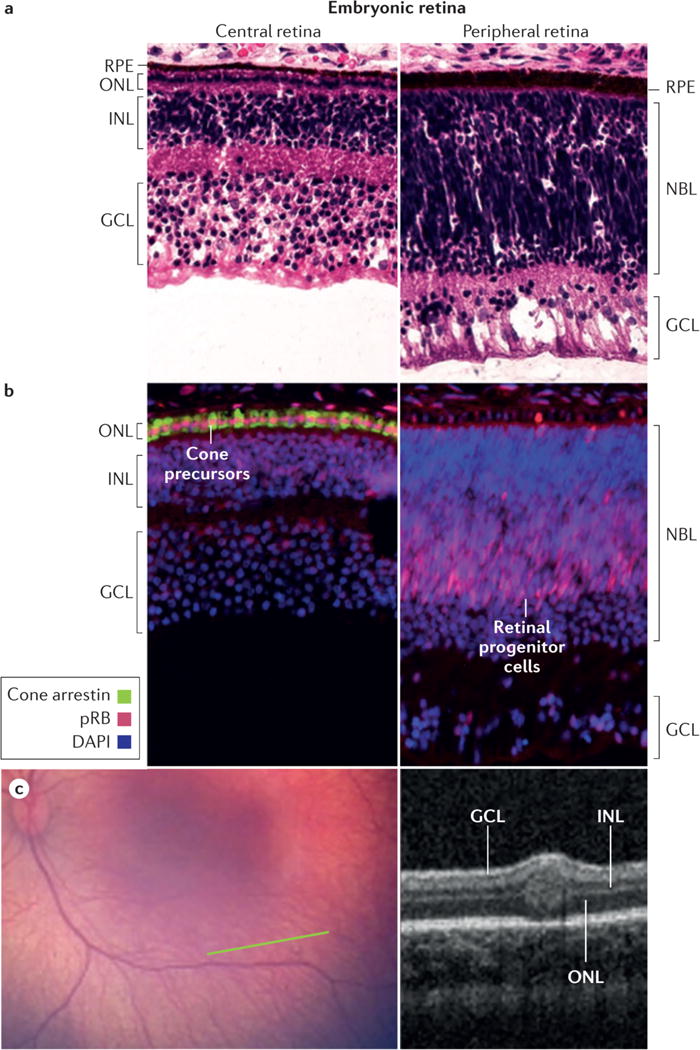
a, The retina has a complex structure and contains multiple cell types. Haematoxylin and eosin staining of post-fertilization week 19 retina shows three post-mitotic nuclear layers in the central retina near the fovea (left) and two nuclear layers in the less mature periphery of the same histologic section (right). Cell types in each layer are indicated. b, Cone precursors normally have high expression of the RB protein (pRB). Immunofluorescence staining of post-fertilization week 19 retina shows especially strong pRB signal (pink) in maturing cone precursors in the central retina (left, counterstained for cone arrestin, green) and in retinal progenitor cells in the peripheral retina of the same histologic section (right). pRB staining is less intense in DAPI-stained nuclei (blue) in other retinal cell types. c, Wide-angle retinal image (left image) shows no tumor. In the plane of the green line, optical coherence tomography (OCT; right image) shows a tiny intra-retinal tumor in a 2.5 month old infant, which seems to extend from the inner nuclear layer to the outer nuclear layer. The tumour has an uncertain epicentre making it difficult to infer the retinal layer from which the tumor arose. The layer-of-origin may be defined in the future with more images of higher resolution OCT of retina in very young children carrying an inherited germline RB1 mutation. Abbreviations: ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer; NBL, neuroblastic layer; RPE, retinal pigment epithelium.
Notably, the smallest tumours detected via optical coherence tomography (OCT) appear to be centered in the inner nuclear layer (INL) of the retina, not the outer nuclear layer (ONL) where mature cones reside (Figure 4c).63 Possibly, the cone precursor cell that loses pRB mislocalizes to the INL. However, as they grow, the small tumors also extend into ONL and other retinal layers (Figure 4c), making it difficult to infer the retinal layer in which tumors first form. Perhaps blood vessels and retinal astrocytes that are normally present in the inner retina promote retinoblastoma cell growth in the INL.64
Translating knowledge of pathogenesis
Understanding retinoblastoma molecular pathways could lead to treatment and prevention opportunities. Oncoproteins such as MYCN could be targeted65 in both RB1+/+MYCNA tumours57 and RB1−/− tumours that have MYCN-dependence.61 Furthermore, studies based on molecular discoveries showed that exposing retinoblastoma-prone murine foetuses to small molecule inhibitors of E2f or Cdk inhibited subsequent tumourigenesis without disrupting normal retinal development.59
These new translational opportunities require cell lines and animal models that accurately reflect retinoblastoma cell responses. Most in vitro studies have used two old cell lines, but many others are available.66 Primary xenografts in newborn rats and immunodeficient mice67 are useful53 but will not reflect the cell and immune environment of natural retinoblastoma, potentially limiting translation to patients. Genetically engineered mouse models can also be used to assess novel treatments.68 Whereas Rb1+/- mice (which are expected to lose the wild type Rb1 allele as in RB1+/- humans) do not develop retinoblastoma, retinal-specific deletion of both alleles of Rb1 (using Pax6a, Nes (Nestin), or Chx10 promoters) in Rbl1, Rbl2 or Cdkn1b deficient backgrounds achieves retinal tumour formation.60 Models such as overexpression microRNA cluster miR-17~92 have been used to examine genetic interactions in vivo.69 Mice expressing the viral oncoprotein Simian Virus 40 large T-antigen develop retinoblastoma specifically in precursors of Müller cells (a type of retinal glia cells), and have been used extensively for pre-clinical therapeutic studies.70,71 However, experimental murine tumours have different collaborating mutations and cell type origin, reducing their ability to predict human treatment responses. New models that more precisely recapitulate human retinoblastoma pathogenesis are needed to identify novel therapies that will target RB1−/− cancers.
Diagnosis, screening and prevention
Clinical diagnosis
Diagnosis of retinoblastoma is usually clear from presenting signs and clinical examination.3 The most common sign is leukocoria (white pupil) (Figure 5). When parents report a strange reflection in the child’s eye, retinoblastoma should be at the top of the differential diagnosis. The second most common sign is strabismus (misaligned eyes) when central vision is lost. Advanced disease may present with iris colour change, enlarged cornea and eye due to increased pressure, or non-infective orbital inflammation. Very late, the eye may bulge from the orbit, a common presentation where awareness and resources are inadequate.
Figure 5. Online diagnosis of retinoblastoma.
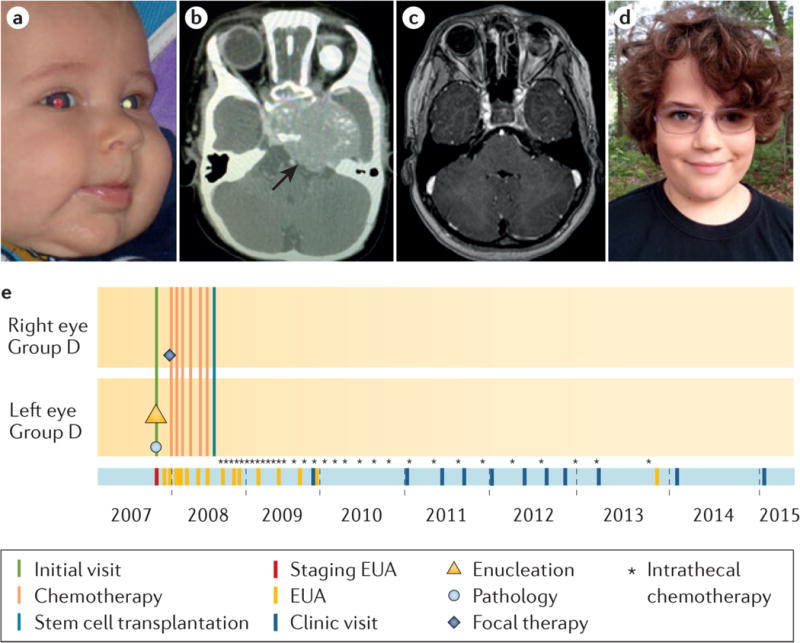
a, Detection of photoleukocoria (white pupil) on this digital image led the parents to the diagnosis of retinoblastoma. The left eye was determined by examination under anaesthetic to have IIRC78 Group D retinoblastoma and was enucleated two days later; CT scan was scheduled to check for trilateral tumour. (Today, MRI would be recommended to reduce radiation exposure.) The pathologic examination of the eye showed no high-risk features. The other eye was normal at diagnosis but on the next examination a small tumour was detected and treated with only laser. b, One month after surgery, the child presented with high intracranial pressure. A large intracranial midline tumour (trilateral disease) was diagnosed on CT scan, which was treated with chemotherapy, high-dose chemotherapy with hematopoietic rescue by stem cell transplant, and intrathecal chemotherapy injections (directly into the cerebro-spinal fluid) through an implanted intraventricular catheter. c, Follow-up MRI at age 8 years shows no residual disease and d, the child is well with one good-looking artificial eye and one eye with normal vision; he wears poly-carbonate lenses to protect his only eye. e, The eCCRB timeline of treatments; not all intrathecal chemotherapy injections (*) are shown.
Diagnosis of retinoblastoma does not rely on histopathologic examination since biopsy incurs risk of metastasis.72 Indirect ophthalmoscopy with the pupil pharmacologically dilated is usually sufficient for diagnosis by an eye specialist. Calcification, a characteristic of retinoblastoma, is detected by ocular ultrasonography (b-scan). MRI is used to assess invasion of the optic nerve73 and the presence of trilateral retinoblastoma (pinealoblastoma and primitive neuroectodermal intracranial tumours associated with RB1 mutations).73,74 CT scans are now avoided because radiation induces second primary cancers in people carrying RB1 mutations.75
Detailed retinal examination under general anaesthesia is required to distinguish the differential diagnoses (Coats’ disease, persistent foetal vasculature, and vitreous haemorrhage) and classify the severity of intraocular disease. Accurate fundus drawing is essential to map tumour burden and location, and in some low-resource settings is the only available form of imaging. Where resources allow, the wide-angle, hand-held fundus camera is used to view and record the whole retina, while an expert depresses the sclera to bring the most anterior retina into view. At the same time that the focus is on viewing the anterior retina, care must be taken that there is not excessive pressure on the eye that could cause ischemia and would be indicated by optic nerve arterial pulsations. Very useful are high frequency (50 MHz) ultrasound biomicroscopy,76 and OCT63 to discover invisible tumours in infants with familial disease. Good imaging and documentation of the whole retina supports eye classification and cancer staging, documents treatment responses, supports consultation with colleagues, and helps parents understand treatment options.
Classification and staging
Classification is necessary to determine appropriate management and predict outcome. The Reese-Ellsworth classification77 for intraocular retinoblastoma was used to predict outcomes of external beam radiation. When new therapies became available, an international collaboration led by Murphree developed the International Classification of Intraocular Retinoblastoma (IIRC)78 which better predicted responses to intravenous chemotherapy than the Reese-Ellsworth classification (Figure 6).78 The IIRC classifies eyes in groups A to E, with E being the most severe. However, subsequent modification79 resulted in a significantly different versions of IIRC such that severely involved (dangerous) eyes are classified with less affected eyes.80 Subsequently, the Children’s Oncology Group (COG) inserted a further minor variation.81 The TNM cancer classification system for all cancers (defined by primary tumour (T), lymph node extension (N), and distant metastasis (M) is yet another slightly different system for retinoblastoma.82 The use of multiple classification schemes seeds confusion and undermines research in retinoblastoma. A priority to advance management strategies in retinoblastoma is to collect outcome evidence to develop one universally accepted classification based on evidence of the most important features at presentation that predict success for life and eye salvage, that will allow comparison between studies.
Figure 6. Different classification schemes for intraocular retinoblastoma confound comparison of outcomes.
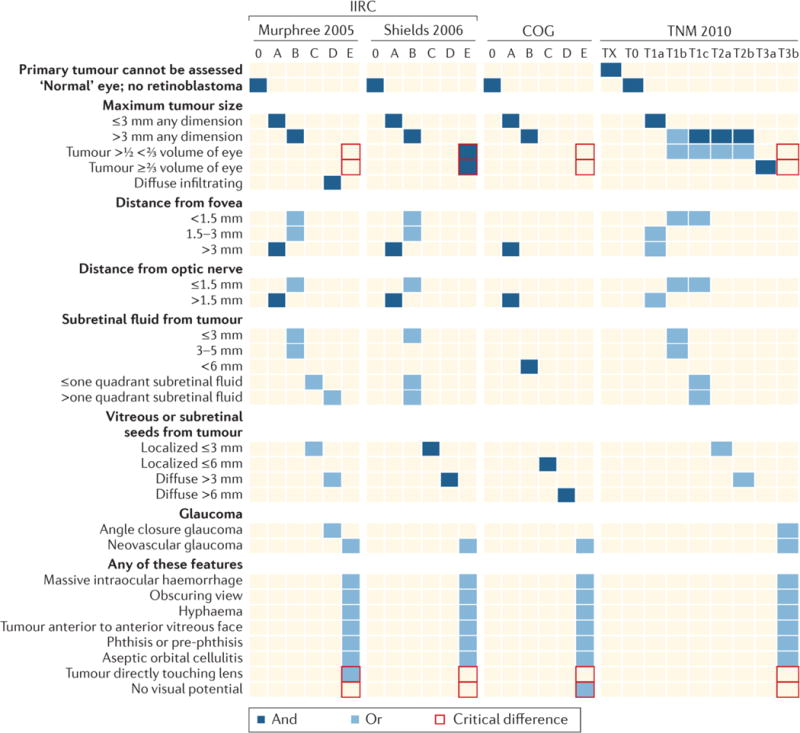
The features listed determine the overall classification, ranging from small tumours not threatening vision (“Group A”) to tumours clinically noted to have features suggesting potential spread outside the eye (“Group E”). Most importantly, size of tumour alone does not make an eye dangerous by Murphree,78 Children’s Oncology Group (COG),81 or TNM classification;82 but any eye with tumour >50% of eye volume is E (advanced-stage disease) by Shields classification.79 The consequence is widespread confusion in the literature undermining clinical research, since studies using the different classifications cannot be compared. The red boxes indicate the critical differences between the different classifications.
A number of staging systems also exist for extraocular retinoblastoma. In a coordinated action with the group that developed the original IIRC78, retinoblastoma clinicians developed the International Retinoblastoma Staging System (IRSS), focused on overall staging (different again from the standard TNM).83 The American Joint Committee on Cancer (AJCC) and the International Union Against Cancer (UICC) staging system developed the TNM classification for retinoblastoma82, which is currently under revision for the 2016 edition. Owing to its general acceptance in the oncology field, this scheme has the potential to achieve a single, collaborative, consensus classification of eyes and cancer staging, critical to clinical research.
Pathology
Histological examination of the retinoblastoma after enucleation of the affected eye (Figure 3) is the only way to evaluate high-risk features (tumour invasion into the optic nerve beyond the confines of the eye or to the cut end of nerve; more than 3 mm of invasion of the vascular layer (choroid) under the retina; invasion of sclera) and establish pathological staging.84–86 Emerging evidence indicates that high grade anaplasia (loss of differentiated characteristics) may be a risk factor for metastasis and death.87 High-risk features by the Murphree IIRC78 were observed in 15–33% of Group D and 50–61% of Group E eyes,85,88, but by the Shields IIRC (which classifies less affected D eyes as E), only 24–39%89 of Group E eyes showed high risk. Adjuvant systemic chemotherapy after high-risk features are observed in an enucleated eye may reduce risk of metastatic relapse.88–90
The preparation and examination of the enucleated eye have been optimized to evaluate all risk features.84,91 Pathology specimens are staged using the pathology TNM (pTNM) and the IRSS classification schemes.82,84 However, still too frequently retrospective examination finds a previously unnoticed risk after metastatic disease is diagnosed. Staining of specific markers for photoreceptor cells (the cone-rod homeobox transcription factor; CRX)92 and for tumour cells (N-glycosylated ganglioside; NeuGc-GM3)93 may facilitate detection of tumour cells otherwise missed or interpreted as artefact.
Genetic diagnosis
Many patients with retinoblastoma are the first individuals to be diagnosed in a family; only 6% of patients indicated when tested for RB1 mutations that a family member was previously diagnosed with retinoblastoma.94 However, a population-based study in England suggested that 35% of bilaterally affected patients had a family history.9 In three population-based studies9,11,95 representing 2,738 retinoblastoma patients on three continents, 30–37% of cases were bilateral. When we add the 15–18% of unilateral cases who also carry an RB1 mutant allele, approximately 45% of people with retinoblastoma carry one RB1 mutation in their constitutional cells. Of the remaining patients with unilateral retinoblastoma, 98% have somatic biallelic RB1 loss and 2% have normal RB1 genes in the tumour, but somatic amplification of the MYCN oncogene instead.57
Knowledge of the patient’s RB1 mutation profile enables precise screening of relatives and subsequent generations. Without genetic testing, all children at risk (already one eye affected and/or positive family history) should undergo multiple exams under anaesthesia in the first 3 years of life to enable detection of small, easily treatable tumours. When genetic testing reveals RB1 mutations in a family, children at risk can be tested for that mutation. Of unilateral patients, 85% will test negative with <1% residual risk for undetectable low level mosaicism, which reduces the intensity of surveillance for tumours in the normal eye and improves estimates of cancer risk of family members.8 In familial retinoblastoma, prenatal screening can be performed on DNA obtained from amniotic fluid. When such prenatal testing results in a positive finding, the retinoblastoma team works with specialists to safely induce labour near term, to facilitate detection and treatment of retinoblastoma tumours as early as possible.8 Pre-implantation genetic diagnosis for the known mutation of a parent offers the option for a family to have an unaffected child.96,97
Cerebrospinal fluid and bone marrow can be molecularly screened to as part of a surveillance strategy, when clinically indicated, to detect metastasis. Harvested stem cells to detect minimal residual disease. Markers that have been used are the RB1 tumour mutation (when mutation is not present or different in the germline alleles),98 the signature of post-RB1 genomic gains and losses99 and CRX expression.92
Best outcomes in familial retinoblastoma management depend on the effectiveness of the healthcare team to identify and counsel affected families. Parents need appropriate genetic counselling and understanding of the risks and actions for each at-risk pregnancy. Alarmingly, a study of retinoblastoma in several developing countries observed that familial cases were diagnosed later than non-familial probands.100 The authors inferred that the parents did not understand the risks to themselves and/or to their children, or were afraid that there was no treatment available. Alternatively, socioeconomic or geographic barriers may have reduced desired healthcare access. Clearly, study of social determinants of health, such as health seeking behaviour, perceptions of medical care, and sociocultural issues related to cancer inheritance would inform counselling approaches that meet the needs of families.101
Screening
Screening through eye examination
To detect retinoblastoma as early as possible, vision screening and eye examination have been developed. Childhood vision screening with effective training to detect signs of retinoblastoma will occasionally identify a child with retinoblastoma. Red-eye reflex examination is recommended and sometimes mandated for each doctor’s visit of babies and toddlers in many parts of the world, yet this does not often identify children with retinoblastoma. A negative childhood eye screening test does not mean this child will remain tumour-free since a tumour might appear later, or may have been in the periphery and not visible (false negative).
An effective way to achieve early diagnosis is for all health professionals to be aware of retinoblastoma and the common words that parent use universally to describe the white pupil (Figure 5) that they have observed, and know that this might mean a life-threatening condition in the child that needs urgent attention. Too often health workers fail to take the parent’s complaint seriously, due to lack of awareness. Photoleukocoria (the appearance of leukocoria on flash photographs, Figure 5) can bring retinoblastoma to the attention of parents, but is hindered by red-eye reduction in the camera, which constricts pupils with a pre-photograph flash, limiting photoleukocoria detection.3 Red-eye and pet-eye correction tools to make portraits prettier removes photoleukocoria from photos, which might delay diagnosis. Awareness campaigns bring retinoblastoma to attention, but lose effectiveness with time. An innovative study called PhotoRed in India trained healthcare professionals to use flash photography to identify childhood eye diseases, including retinoblastoma.102 Other pioneering efforts target software to enable cameras to detect photoleukocoria.103 The global imaging industry could play a role in early diagnosis of retinoblastoma.
Surveillance
Individuals with germline RB1 mutation who have been treated with radiotherapy have high risk to develop specific second cancers (50% risk to develop cancer at 50 years if they received external beam radiotherapy (EBRT)), including leiomyosarcoma, osteosarcoma, melanoma, lung and bladder cancer.104 Surveillance screening for second cancers with and without radiation is a pressing need in the opinion of retinoblastoma survivors.105,106 The first study to evaluate annual whole body MRI surveillance for individuals with predisposing RB1 mutation showed it was feasible to detect second cancers, however only with 67% sensitivity and one false negative test.107 Efforts to detect second cancers earlier are important to reduce mortality.
In addition, lifestyle counselling educates survivors on ways to mitigate their second cancer risk by reporting unexplained lesions, and avoiding an unnecessary radiation and carcinogens (e.g. cigarettes and excessive alcohol). The extent to which these ideas will prevent second cancers is unknown.
Management
Management of retinoblastoma depends on extent of disease at diagnosis (classification of intraocular disease, stage of systemic disease), status of the opposite eye, overall health of the child, socioeconomic circumstances108, and access to expert care.3,21,109
Intraocular retinoblastoma
Primary treatment
Choice of primary treatment is based on likelihood of cure (patient survival), eye salvage and ultimate vision, and the status of the other eye, weighed against short term and long term complications of treatment (Table 3).109 In order of approximate frequency of global use, primary treatments for intraocular disease include enucleation, intravenous chemotherapy (IVC) with focal therapy (laser therapy, cryotherapy), intra-arterial chemotherapy (IAC) with focal therapy, and focal therapy alone when tumours are small at diagnosis. External beam radiotherapy (EBRT) is no longer recommended for first line therapy for primary intraocular retinoblastoma, since radiation, especially in the first year of life, imposes a high risk of secondary cancers when the patient carries an RB1 mutation.104,110 Although there is no role for EBRT in primary treatment of intraocular retinoblastoma, for salvage of the only remaining eye that has failed other therapies, EBRT may still have a role.
Table 3.
Qualitative comparison of enucleation, IVC and IAC for unilateral retinoblastoma (Murphree IIRC Groups B, C, D).
| Enucleation | IVC and focal therapy | IAC and focal therapy | |
|---|---|---|---|
|
|
|||
| Treatment | Major muscles removed, optic nerve cut, eye removed; orbital volume reconstituted with implant; muscles sutured to gain motility of the artificial eye, held in place by the eyelids. | Chemotherapy (carboplatin, vincristine, and etoposide) is given intravenously, best through an implanted “port”, every 3-4 weeks; tumour response consolidated by focal therapy (laser and cryotherapy) every 1–4 months for 3 years. | Chemotherapy (melphalan, and/or carboplatin, and/or topotecan) infused via micro-catheter through femoral artery, near, or into the ophthalmic artery with radiographic confirmation; tumour response consolidated by focal therapy (laser and cryotherapy), every 1–4 months for 3 years. |
|
|
|
|
|
| Success rate to save eye (%) | 0% | 47% Group78 D; >90% Group B & C | 45129 to 95130% Group78 D*; >90% Group B & C |
| Acute toxicity | Loss of eye; parental distress waiting for artificial eye fitting; transient bruising, swelling and headache | Hair loss; low white cell count; fevers require IV antibiotics and hospital admission; protective isolation at a time of social development; parental distress over prolonged treatment | Groin haematoma; transient low white cell count; potential carotid vascular spasm and stroke; vascular compromise of the ophthalmic artery, retinal artery, or choroidal vessels; headache |
| No. of Procedures | 1 surgery | 2–8 chemotherapy cycles every 3 weeks | 2–8 treatments, every 4 weeks |
| No. of EUAs | 1 (unless patients are RB1+/−) | >20 for consolidation and monitoring | >10 since only 30% need consolidation |
| Patient survival | >99% | >98% | high unless undetected high risk features |
| High risk pathology detected | Yes | No | No |
| Hidden metastases treated | No | Yes | No |
| Identification of tumour genetics | Yes | No | No |
| Duration of treatment | 2 months (if not RB1+/−) | > 1 year | > 1 year |
| Invasive surgery | Loss of eye | 1 central venous catheter | 2–8 intracranial arterial canulations |
| Long term toxicity | Loss of eye | High tone hearing loss ~20% (Carboplatin); <1% AML (etoposide) | 4% severe vascular choroidal toxicity; long term unknown vision risk |
|
|
|
||
| psychological harms | Possibly low self-esteem due to loss of the eye or artificial eye; distress of fitting and maintenance of artificial eye | Years of social isolation for treatments; delay physical, emotional, social development; long term separations of families; parental relationship breakdown; hospital-associated anxiety that hinders future medical care; post-traumatic stress disorder; Developmental Trauma Disorder; parental anxiety over illnesses; PTSD in parents or siblings; financial distress | |
|
|
|
||
| Radiation | none | No Group78 D get EBRT; enucleation preferred | Fluoroscopy; No Group78 D to need EBRT |
| Socio-economic impact108 | minimal | profound | very profound |
| Disease-specific mortality | ≪1% | <1% | unknown |
| Resource | Anaesthesia; ophthalmic surgery, ocularist; artificial eye | Anaesthesia; retinoblastoma and oncology expertise; drugs; laser and cryotherapy equipment; imaging equipment; Child Life support;181 long term follow-up | Anaesthesia; retinoblastoma-specific interventional radiology and oncology expertise; drugs; laser and cryotherapy equipment; imaging equipment; Child Life support;181 long term follow-up |
| Long term follow-up | Minimal unless inherited | Every 2–6 months for 4 years after tumour control | Every 2–6 months for 4 years after tumour control |
| Availability: | |||
| Available in high income countries | Yes | Yes | Yes |
| Available in middle income countries | Yes | Yes | Limited |
| Available in low income countries | Yes | No | No |
AML, acute myeloid leukaemia; EUA, examination under anaesthesia; IV, intravenous; IVC, intravenous chemotherapy; IAC, intra-arterial chemotherapy; * very wide range reflects different classifications and different follow-up durations.
Comprehensive analysis of the published literature on eye salvage by any strategy is confounded by the use of five different classification systems77–79,81,82 in different studies (Figure 6), preventing a clear-cut comparison of results between centres, especially for Group D and E eyes. There is a general consensus that eyes with clinical features such as neovascular glaucoma, pthisis bulbi (a shrunken, non-functional eye), and anterior disease are not appropriate for any conservative therapy and require enucleation to know if further therapy is needed for high risk pathlogical features.79,85,88,89 This may be particularly problematic when IAC is offered for eyes with advanced disease, since only the eye receives the chemotherapy.79,85,88,89 Clinical variables strongly associated with high risk features included older age, longer lag from diagnosis to enucleation, anterior tumour or blood, scleral invasion, invasion of the post-laminar optic nerve and orbital cellulitis.89,111,112 Attempted salvage by any method of an eye with features indicating high risk for extraocular extension imposes the danger of undiagnosed disease with risk for metastasis. By the Murphree IIRC78 high-risk features were present in 15–33% of Group D and 50–61% of Group E eyes85,88, and by the Shields IIRC79 24–39%89 of Group E eyes showed high-risk. Enucleation of the high-risk eye alerts those in the circle of care consideration of adjuvant chemotherapy to reduce risk for metastatic disease.88–90 If enucleation of Group78 E eyes is delayed longer than 3 months, for example by prior systemic chemotherapy, disease-specific survival is significantly lower (p<0.001).113 Choices for primary bilateral advanced retinoblastoma are outlined in Figure 7. However, where there is lack of expertise, equipment, resources, and difficulties with close monitoring, bilateral retinoblastoma may still be best treated with bilateral enucleation.3,21 Many bilaterally enucleated retinoblastoma survivors lead active, productive and satisfying lives because they were treated by timely surgery as infants.
Figure 7. Primary treatment choices based on the Murphree IIRC.
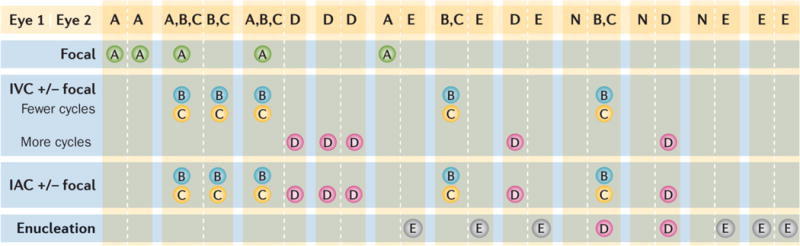
Treatment depends on the combined severity of each of the affected eyes (Eye 1 | Eye 2); the preferred option for each eye is depicted in the blue boxes. IIRC Group78 A eyes can be treated with only laser or cryotherapy (focal therapy consolidation). Group78 B and C eyes require several cycles of systemic intravenous chemotherapy (IVC) or intra-arterial chemotherapy (IAC) followed by focal therapy and intravitreal chemotherapy with melphalan and/or topotecan for residual or recurrent vitreous seeds. Isolated single tumours in Group78 B or C eyes may occasionally be appropriate for primary radioactive plaque therapy. Group78 D eyes require either IVC (with focal therapy consolidation) or IAC (with focal therapy consolidation). All eyes with features suggesting imminent extraocular extension (Group78 E) should be removed so that accurate pathological examination can be performed to determine risk of metastasis requiring adjuvant chemotherapy. IIRC, International Intraocular Retinoblastoma Classification.78
Second-line (salvage) therapy
Second line salvage means initiating a new management plan, in a second attempt to save an eye that has failed the first attempt. A range of modalities is appropriate, but each subsequent plan has a lower success rate114 and long drawn out attempts to salvage an eye incur high risks for the child and family, including metastasis and death.108,115,116
Second line treatment options include focal therapies, repeated systemic chemotherapy,117,118 IAC,114,119 brachytherapy (internal radiotherapy),120,121, EBRT122–124 and whole-eye121 or proton beam125 radiation. Criteria for secondary enucleation are not well defined but are dominated by refractory subretinal and vitreous seeding, vitreous haemorrhage and secondary neovascular glaucoma,117,126 and socio-economic and psychological fatigue to save an eye with poor vision.108
IVC and focal therapy could avoid EBRT and enucleation in 37–47% of Group D or Reese-Ellsworth Group V eyes in three studies with long-term follow-up.124,126,127 The three studies of IAC with the most patients128–130 suggest that IAC achieved eye salvage in 57% of more advanced eyes, and a higher % of less advanced eyes using less focal therapy than is required to consolidate primary care following IVC. A combination of repeat IAC and intravitreal chemotherapy may play an important part in saving eyes that have failed IVC, radiation or IAC.119,130,131 However, in general, extensive treatments over a long time to save an eye may increase family stress, decrease family resources,108 and risk metastases and death.114,116,129
Vitreous seeding is the major cause of failure to save or salvage an eye. Pharmacokinetic studies show poor vitreous levels of drugs administered by either IVC or IAC.132 The highest drug bioavailability in the vitreous is achieved by intravitreal chemotherapy using a safety-enhanced injection technique in carefully selected eligible eyes.133 Following control of the source of seeds, intravitreal chemotherapy achieved two-year 98.5% control of target seeds and 90.4% event-free ocular survival.133–135 The efficacy of intravitreal and IAC chemotherapy may ultimately eliminate the need for EBRT, even as second line therapy.136 A combination of repeat IAC and intravitreal chemotherapy may play an important part in saving eyes that have failed IVC, radiation or IAC119,130,131 However, in general, extensive treatments over a long time to save an eye may increase family stress, decrease family resources,108 and risk metastases and death.114,116,129
A combination of treatments can target salvage therapies to the site of relapse. For relapse confined to the retina and/or vitreous, focal therapy and/or intravitreal chemotherapy are recommended, as long as whole-eye therapy is not required. Recurrent tumours that touch the optic nerve head and/or vision-critical regions such as the maculo-papillary bundle, or have a diffuse shallow retinal or subretinal tumour represent good indications for IAC, which might achieve better visual outcome than focal treatments.114,119
Ocular therapies
Enucleation
Enucleation is a first-line therapy for the majority of eyes with retinoblastoma globally; it is the fastest and least costly treatment137,138. Since the majority of children without a family history of retinoblastoma have Group78 D or E disease at diagnosis, and more than 50% have unilateral disease with the other eye unaffected, cure can be achieved with enucleation (Table 3). Most Group78 E eyes require enucleation because they carry risk of extraocular spreading by definition which can only be confirmed by the identification of high-risk pathological features in the enucleated eye. Good cosmetic outcome is achieved by replacement of the volume of the eye with an implant deep in the orbit, and provision of a prosthetic eye, worn in the conjunctival sac behind the eyelids (Figure 8). Many different reconstruction techniques are used worldwide.139 Comparative studies have shown best artificial eye motility with a simple, cheap, polymethyl methacrylate implant and muscles sutured to the conjunctiva to form the pockets to hold the artificial eye (myoconjunctival approach), which is affordable world-wide.140,141 Porous implants that become vascularized with muscles sutured to the implant are commonly used but are susceptible to infection and extrusion, and are more costly. Provision of a temporary prosthetic eye at the time of enucleation has a positive psychological impact on families,19 to help the family accept enucleation for their child.
Figure 8. Triplets with retinoblastoma.

These triplets developed retinoblastomas in all six eyes illustrating the full expressivity and penetrance of RB1−/− germline mutations that completely abolish pRB expression. The pedigree indicates that the three children all have the same RB1 mutation, which is not detected in their parents or older brother. The eCCRB timelines reveal that all eyes had individualized therapy, with choices considering the overall impact on each child. Each child lost one eye, has a good-looking artificial eye and an eye with normal vision, and has disease control in less than one year. The number of eye exams under anaesthesia (EUA) was least for the child who had primary enucleation. OD, right eye; OS, left eye. Eye involvement at diagnosis is indicated in by the International Intraocular Retinoblastoma Classification;78 the eye labelled “Group 0” had no tumour at initial diagnosis but developed a tumour 6 weeks later.
Intravenous chemotherapy and focal therapy
Since 1996{Ferris, 1996 #7587} first-line therapy to control Groups78 B, C and D disease has been IVC with different combinations, doses, schedules, and durations of carboplatin, etoposide and vincristine (CEV) followed by focal therapy to consolidate chemotherapy responses (Figures 7, 9). {Chan, 1996 #10251;Gallie, 1996 #10578;Shields, 2002 #9137;Manjandavida, 2014 #21453} One initiated RCT142 includes high–dose, short-duration cyclosporine simultaneous with the chemotherapy to modulate multidrug resistance.142,143 Eyes in Groups78 B and C do well with CEV and focal therapy. In three studies with long-term follow-up of IVC and focal therapy, 47% of Group78 D eyes124, 47% of Reese-Ellsworth Group V,126 and 47% of Group{Shields, 2006 #21122} D eyes{Shields, 2006 #21122} avoided EBRT and enucleation (Figure 9).
Figure 9. Retinoblastoma treated with IVC.
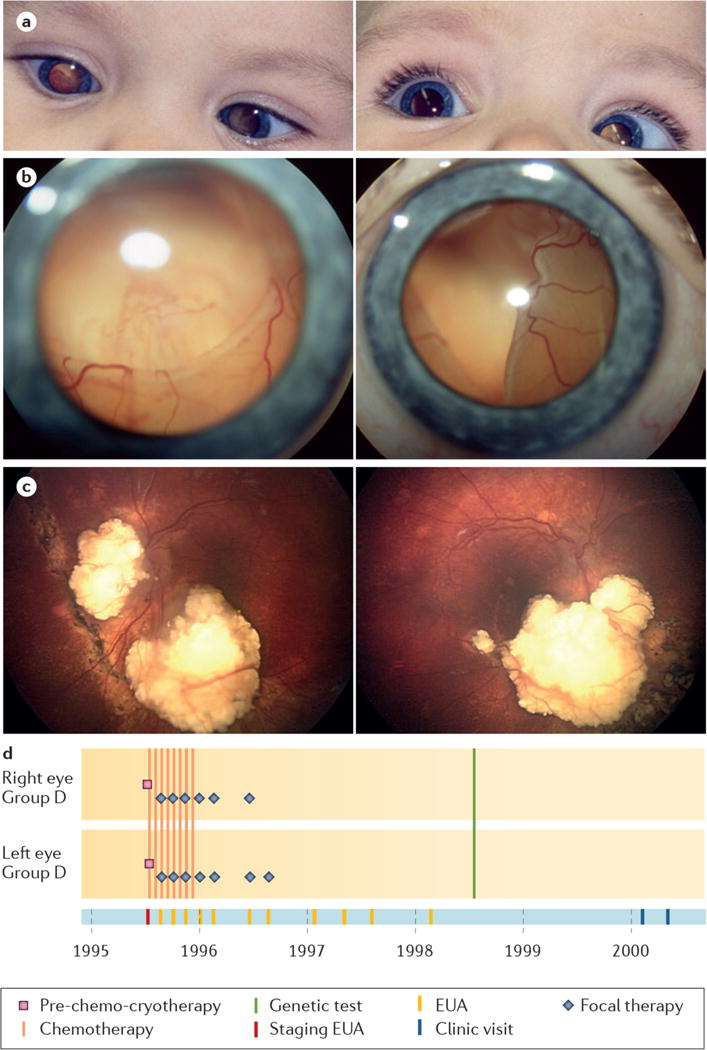
a, Detection of photoleukocoria in both eyes at age 8 months. b, Retinal photography at diagnosis showed total retinal detachment with underlying large tumours in the right (left image) and left eyes (right image). c, Both eyes have calcified tumor remnants and attached retinas 20 years after diagnosis, with 0.25 vision, right eye and 0.1 vision, left eye (Vision decimal system, 1 = normal, 0.1=legal blindness). d, eCCRB time line of shows treatment with IVC and focal therapy, then only follow-up visits.
Fundamental principles for systemic cancer therapy also apply to retinoblastoma: optimized outcomes are anticipated with high dose intensity and combination of several agents with complementary mechanisms of action and optimal delivery to the eye. This is illustrated by the reduced effectiveness of single agent, low dose carboplatin for retinoblastoma.144 Acute toxicities of IVC for retinoblastoma are as for other paediatric cancers, including short-term transient pancytopenia (reduction in red blood cells, white blood cells and platelets), hair loss, vincristine-induced neurotoxicity, and infections. Long term toxicities include carboplatin-induced ototoxicity (high tone hearing loss) in some patients,145 second non-ocular cancer risk with alkylating agents95,146 and secondary acute myeloid leukaemia following intense chemotherapy including topoisomerase inhibitors, doxorubricin and alkylating agents.147,148
IVC alone rarely eradicates the retinoblastoma entirely, and focal therapy with repeated examinations under general anaesthesia is very important (Figure 9).126,149,150 Tumours in the macular region, threatening vision, are particularly at risk of recurrence without focal therapy,151 perhaps because of caution to not damage vision results in under-treatment. Optical coherence tomography can be used to monitor macular tumours and scars for small recurrences, improving outcomes.152
Location of tumour is the most important predictor of final visual acuity (near-normal visual acuity ≥0.5 in 22% of eyes with macular tumour, 67% of eyes with extrafoveal tumour).153 In children with two eyes affected, patching of the better eye to force use-dependent development of the eye with macular involvement (occlusion therapy) achieved near-normal vision in 53% and useful vision in 73% of patients.154 There is no documented local toxicity of IVC to the eye (Table 3).
Intra-arterial chemotherapy
IAC has been used for eye salvage therapy for decades in Japan129 and has been recently refined to achieve a more selective delivery to the eye.128,155 Interventional radiologists pass a micro-catheter through the femoral artery up to the orifice of the ophthalmic artery of the eye with retinoblastoma, and chemotherapy (single drug or combination; melphalan, topotecan, carboplatin) is infused in a pulsatile fashion over 30 minutes. Different interventional approaches are used for specific anatomic situations.156
The three papers with mean follow-up 13 to 79 months and a total of 488 patients128–130 suggest that IAC achieved eye salvage for 65% of all eyes (Figure 10). Overall globe (eyeball) salvage was 74% when IAC was used as first-line treatment and 67% when used as second-line treatment.130 In the North American experience,130,157 globe salvage without needing EBRT (definition of “success”) was 96-100% for less advanced eyes (IIRC A-C or RE I-III) and 94% for Group79 D eyes and 36% of selected Group E eyes79 with a median follow-up time <24 months; 80% of RE V that received OAC as primary treatment could be saved (n=36). The longest follow-up study (median 74 months; range 0–252 months) for IAC salvaged the eye in 88% of Group B, 65% of Group C, and 45% of Group D eyes with vision ≥0.5 (Vision decimal system, 1 = normal, 0.1=legal blindness) in 50% of the eyes that had no foveal involvement.129 This evidence suggests that IAC is a reasonable approach to primary therapy for eye salvage, with good evidence for a role in secondary eye salvage.114,119
Figure 10. Retinoblastoma treated with IAC.
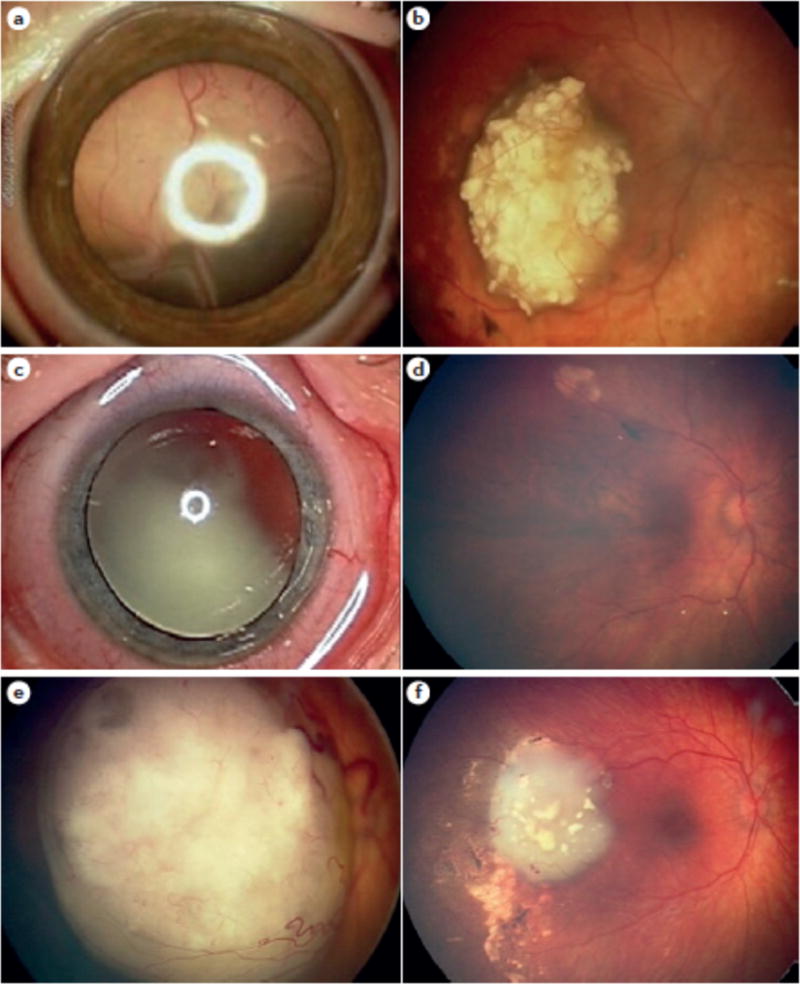
a, Before IAC for a massive exophytic (outward growing tumour) retinoblastoma with total retinal detachment; and b, after IAC, showing complete response and eye salvage, leaving a 6 mm calcified scar. c, Before IAC for an endophytic (inward growing tumour) retinoblastoma tumour with extensive vitreous seeding; and d, after IAC showing complete control with clinically intact fovea and visual acuity 20/25. e, Before IAC for an eye with macular retinoblastoma demonstrating excellent tumor regression away from the macula; and f, after IAC leaving intact fovea and hope for vision.
From the IAC publications, the total number of patients with metastases is unclear, but at least 14 are reported.116,128,129,158 Since many other treatments were given simultaneously with IAC, the 8 deaths from metastases of 343 patients with long follow-up129 are not clearly assigned to IAC alone. From the IAC publications with high patient number and longer follow-up, metastatic relapse occurred in <2% of the children receiving IAC, but the relapsed patients often had received extensive, multimodal therapies116,128,129,158. The long term Japanese experience129 and the more recent New York experience128 have not reported an increase in metastases or metastatic deaths to date.
Ocular complications of IAC included vitreous haemorrhage (2%), branch retinal artery obstruction (1%), ophthalmic artery spasm with reperfusion (2%), ophthalmic artery obstruction (2%), partial choroidal ischaemia (2%) and optic neuropathy (< 1%).130 At the time of IAC, minor transient complications included transient eyelid oedema (5%), blepharoptosis (droopy eyelid 5%), and forehead increased blood flow, 2%).130 Electroretinogram showed minor evidence of retinal toxicity which lasted up to 12 months, which was not considered clinically relevant.159
Focal therapy
Focal therapy (laser therapy, cryotherapy, local chemotherapy) is the local application of therapy to the eye, under direct visualization through the pharmacologically dilated pupil. Focal therapy is the primary treatment for Group78 A eyes and is used to consolidate responses of Group78 B, C, D eyes after IVC or IAC with the aim to physically destroy residual or recurrent small-volume, active retinoblastoma. In general, focal therapy is repeated monthly until the tumour is completely atrophic or calcified (Figures 8, 9).
Two types of laser treatments are applied monthly until the tumour is flat, atrophic or calcified. Transpupillary thermotherapy involves a 810 nm diode laser through the dilated pupil to heat tumour, rather than precisely coagulate the tumour for 3–5 minutes per spot. Photocoagulation lasers with 532 nm, 810 nm or continuous wave 1064 nm laser beams are directly applied by multiple short (0.7 s) burns to small volume active or suspicious tumour, gradually increasing power from sub-coagulation power intensity until the tumour is coagulated opaque white. Cryotherapy, on the other hand, involves freezing the tumour through the sclera with a nitrous oxide probe; the tumour is directly visualized and duration of freeze judged to completely eliminate the tumour. Since tumour cells die when thawing, one minute is allowed for each thaw between freezing cycles. Cryotherapy effectively destroys small primary tumour(s) or recurrences in the periphery of the retina. Plaque radiotherapy involves positioning a radioactive probe on the eye to deliver trans-scleral radiotherapy with an apex dose of 35–40 Gy over 4–7 days. Plaque focal radiation has not been associated with new primary tumours and is effective to treat a single primary or recurrent tumour in a location that will not compromise vision. Finally, paraocular chemotherapy has been useful for small volume recurrences and vitreous seeds.118,160–162
Intravitreal Chemotherapy
Primary treatments mainly fail due to the presence of vitreous seeds.118 Intravitreal chemotherapy is adjunctive to other treatments, and is only initiated after the source of the seeds is controlled. The use of intravitreal chemotherapy to control vitreous seeds — most difficult to control form of retinoblastoma — has been promising, especially in combination with a safety-enhanced injection technique and well defined eligibility criteria.133,135,136,163,164 Under anaesthesia, the intraocular pressure is lowered by removing a small volume of fluid from the anterior chamber or by digital massage. Melphalan (or combined with topotecan) is injected into the vitreous behind the lens, through the conjunctival, sclera, and pars plana with a small-gauge needle. On needle withdrawal, the injection site is sealed and sterilized with cryotherapy and the eye is shaken gently to distribute the drug throughout the vitreous. Three clinically defined classes of vitreous seeds have significantly different median times to regression, mean number of injections and cumulative and mean melphalan dose.135 Ultrasound biomicroscopy may be used to evaluate the otherwise hidden ciliary region behind the iris76 to confirm that the injection site is tumour-free prior to intravitreal chemotherapy treatment. The toxicity of intravitreal chemotherapy is limited to technique-dependent localized peripheral retinal toxicity.136
Extraocular retinoblastoma
Extraocular at presentation
Retinoblastoma may present with evident extraocular disease, especially in low-income countries. Orbital extension of an intraocular retinoblastoma may be detected clinically in children with proptosis or a fungating mass or occasionally by imaging studies. Treatment includes neo-adjuvant chemotherapy, including carboplatin, etoposide and vincristine, as for intra-ocular retinoblastoma; other agents that are useful include cisplatin, cyclophosphamide and anthracyclines.21 This is followed by enucleation and limited excision of affected tissues without mutilation, orbital radiation and adjuvant chemotherapy. Intrathecal chemotherapy (injection into the cerebrospinal fluid space) may also be used and high dose chemotherapy with stem cell rescue may be added in metastatic disease. Children with orbital disease or metastatic spread without central nervous system invasion may be cured with intensive therapy. Long term outcomes for retinoblastoma patients presenting with extraocular disease are complicated by late-developing effects, but overall they are moderate.165
Palliation
Palliation includes pain management, symptom relief, nutritional support, and psychosocial support for the child and families.21 Untreated retinoblastoma is highly sensitive to most chemotherapy agents. Children presenting with orbital retinoblastoma are usually in severe pain and discomfort that may be alleviated with judicious use of anticancer therapy, including conventional chemotherapy, even when no curative intent is pursued. These children often present with severe emaciation (severe weight loss) needing prompt medical treatment. Radiotherapy may also be helpful, especially for a central nervous system relapse or for the treatment of massive orbital extension, but it is often unavailable in low-income countries.166
Quality of life
Quality of life (QoL) describes the level of physical, emotional and psychological wellbeing experienced by an individual. Cancer, its treatment and effects on the developing body, brain and mind can significantly decrease QoL, which has implications for treatment decisions and supportive and long-term follow-up care.
Measured life-long impact
In one study, children with retinoblastoma followed from diagnosis to age 5 years showed decline in developmental functioning over time.167 However, survivors diagnosed under 1 year of age performed significantly better in short-term and long-term verbal memory, verbal learning and verbal reasoning abilities compared with those diagnosed at over 1 year of age.167,168 Surprisingly, verbal IQ was significantly above both age-matched sighted and non-retinoblastoma blind persons.169 This unexpected higher performance might relate to the heritable RB1 mutant allele in the younger children, since the opposite outcome is expected in this group.
Adults who lost one eye to retinoblastoma in early–life have altered development of visual, auditory and multisensory brain morphology, suggesting that the remaining eye acquired increased contralateral visual and other cortical connections, also measured as enhanced functioning of the remaining eye, if the other eye is removed at a young age.170–172 However, clinical and animal studies suggest that repeated anaesthesia of young children, such as is necessary for eye salvage and to discover and treat small tumours, may impair neurocognitive development.138,173,174 Clearly the long-term development of retinoblastoma survivors is an important area for future study, to both guide future care of newly diagnosed children and survivors, and learn about human development.
Direct insight from survivors
Social media have brought parents of patients with retinoblastoma and survivors into peer-support communities that share experiences and insights with each other. It is not yet clear how to reference and include the unique and rich data of social media in formal research.
Children’s perception of pain and medical interventions changes over time.175–177 Since children with retinoblastoma have to undergo many repeated procedures, normal anticipation anxiety around procedures might be magnified, resulting in intolerance of even minimally invasive experiences and mild pain. The child’s initial strong emotions may be suppressed as the child gives up, and re-emerge as depression, post-traumatic stress or developmental trauma disorder.115,178–180 Child Life interventions at any age and with any treatment help children cope and even thrive during treatment, reduce treatment costs, ease family stress and improve long-term mental health (Figure 11).5,75,181–184
Figure 11. Child Life promotes effective coping through play, preparation, education, positive-touch and self-expression activities, based on natural child development.

Child Life181 helps children and their families cope with challenging healthcare issues, hospitalization and therapeutic interventions. This child plays doctor and nurse, wearing gloves and stethoscopes to examine Puppet Kevin who had one eye removed for retinoblastoma. They also learn how to put in his artificial eye, to better understand their own artificial eye. Play (medical play and just general play) promotes a sense of control for children within their medical experience.
Retinoblastoma treatment is often the childrens’ only life experience and forms the centerpiece of their earliest memories. Adult survivors may not remember being anaesthetized, however, many describe acute fear of their mouth and nose being covered. Extended isolation during therapy may impact social functioning. Although most adult survivors perform well socially, many report low confidence and intense anxiety, especially in large groups and crowded environments. Most survivors are high cognitive performers.168 However, reduced vision causes some children to become frustrated by their inability to keep up with peers, damaging self-esteem and confidence.165,185,186
Late effects of radiotherapy
Although radiotherapy is nowadays rarely used for primary treatment of retinoblastoma, thousands of adult survivors live with its long-term effects. Many feel neglected and demoralized by lack of follow-up and prospective management. Facial deformity induced by radiation blocking tissue growth causes low self-confidence and social anxiety. Reconstructive surgery is a painful process that may impact remaining vision, but its cosmetic effects can dramatically improve QoL. Dry eye is very painful, and corneal vascularization reduces already limited vision. Use of ocular lubricants may prevent complications, if started early before pain and vision loss occur.
Second cancer risk
Individuals at risk of second primary cancers as a consequence of RB1 gene mutation require life-long follow-up.95,104,187 Lack of agreed-upon protocols causes confusion, frustration and fear as adults struggle to access informed care, compounded by primary doctors who are not aware of late effects and life-long implications of RB1 mutations. Looking forwad, full information about the patient’s cancer history, genetic status, life-long risks and action plan when concerns arise will empower survivors to be advocates for their own and their children’s health.188–191
Family planning
Many adult survivors have little knowledge of retinoblastoma genetics, genetic counselling or testing, their own status, or options for their baby. Genetic testing provides knowledge of inheritance of the parental RB1 mutant alleles, empowering several options.8 Pre-implantation genetic diagnosis is expensive but available for parents with retinoblastoma in some countries96,97. Choices for an affected foetus are complex and dependent on social contexts. Parents seek an agreed-upon screening protocol for at-risk babies, and the discussion and planning may cause, or remove anxiety among survivor-parents. Anger and guilt about somehow being responsible for the child’s cancer is amplified when diagnosis is delayed. Screening protocols for at-risk children will reduce survivor-parent anxiety and enhance early diagnosis to achieve minimally invasive therapy.
Outlook
Retinoblastoma is curable if diagnosed early. We propose that within 10 years retinoblastoma can be a ZERO-death cancer. Our anti-retinoblastoma arsenal will consist not only of curative treatments, but also measures to prevent development of disease in predisposed individuals. To achieve this vision, critical efforts are needed to build upon the current state-of-the-art for retinoblastoma as presented in this Primer. Here, we outline the key action items, respond to the immediate obvious targets for improvements, and set the stage for innovative discoveries in retinoblastoma (Box 1). We are confident that the 8,000 children that will be diagnosed each year will have optimized timely diagnosis, stage-appropriate care, and survive and have productive, happy lives. We are also inspired by the principles that define a “High Reliability Organization”192 such as decision support tools based on evidence; deep analysis of unexpected events (such as metastasis unexpected for the stage at presentation, technical errors, failure to achieve parental compliance) and action to prevent such events; and full, honest and open communication.
Box 1. Outstanding research questions.
Patient-centered, global research focus
Collect outcome evidence to develop one universally accepted classification based on clinical features of the eye at presentation that predict success for life and eye salvage, that will allow comparison between studies.
Address the reluctance of health care workers to explain the true risks (such as in Jordan where doctors do not want to destroy families), and the choice of families who know and understand the risks but choose not to follow through because of stigma.
Define treatment success: cure (life), salvage of vision (one eye), salvage of eye (vision and cosmetic), fewest examinations under anaesthesia (cumulative toxicity of anaesthesia).
Meta-analysis of all children treated primarily with intra-arterial chemotherapy to quickly establish that this treatment is both safe and effective.
Develop a “retinoblastoma index” to predict outcome and include levels of awareness, access to centres with the necessary resources and expertise, and application of evidence-based care.
Collect evidence about the quality of life following retinoblastoma, and develop action plans to address the issues that most affect survivors.
Determine what failures led to death of children from retinoblastoma, and focus global effects on these targets to make retinoblastoma a ZERO death cancer.
Implement a global retinoblastoma health record, such as eCCRB, to underpin a Learning Health System bringing real-time evidence to treatment choices.
Pathogenesis
What underlies the genomic progression post RB1 loss?
What molecular changes promote (early) metastasis?
Are there other RB1+/+ genetic subtypes beyond MYCN-amplified retinoblastoma?
What dictates the tissue specificity of RB1 loss leading to cancer?
What are the molecular features of second cancers in RB1 mutation carriers?
Develop drugs with low systemic toxicity and high effectiveness against retinoblastoma.
We propose to determine the most likely cause of every death from retinoblastoma. Similar in concept to the groundbreaking Million Death Study,193 each death will be an opportunity to avoid more deaths. The identified key risk factors will lead to systematic guidelines to avert preventable deaths. These guidelines will fall into categories such as public awareness (public and health system understanding of the urgency of leukocoria, use of social media), health services194 (infant screening, financial support for every child, regional retinoblastoma expertise, etc.), clinical research (co-operative groups for clinical trials, outcomes analysis from learning health records, etc.) and basic research (define in retinoblastoma tumour cells targets for new therapy, define molecular targetable pathways for treatment and prevention of RB1−/− cancers, starting with heritable retinoblastoma).
Patient-centered, global research
Retinoblastoma will benefit from clinical science at the level that has achieved dramatic change for paediatric cancer. In retinoblastoma, we have lagged behind in systematic accumulation of evidence on which to base therapy. Today we are faced with sweeping change in treatment of retinoblastoma using IAC and salvage of unresponsive vitreous seeds by intravitreal chemotherapy. However, we do not have full data on which to judge patient eligibility, long-term effectiveness or outcomes. In-depth systematic reviews and meta-analyses of therapies and outcomes for retinoblastoma can capture retrospective evidence on which to base present recommendations for therapy. Global collaboration will empower RCTs of many aspects of care for children, families and survivors of retinoblastoma.
We have opportunities now to learn from initiatives and innovations everywhere, and to listen to the patient’s voice to become an inclusive, participatory global network. Patients and families, empowered by social media, will be heard in the research approaches, making these approaches relevant, ethical, and appropriate. Stakeholders from low-and-middle income countries will be active innovators, assuring that protocols are feasible and acceptable. Equality for retinoblastoma will come by education (internet access), with shared collaborative care coordinated online. Retinoblastoma expertise will be developed locally (proportional to the local burden of retinoblastoma) but derived globally, with best practices drawing on rigorous molecular and clinical research. Broad inclusion in high quality studies of all aspects of care, including traditional, complementary and alternative medicine, will enhance outcomes.195 Retinoblastoma RCTs will be collaborative, high-enrolment studies. Effective innovations will include all outcomes (survival, eye salvage, short- and long-term toxicities, social impact, etc.) and sustainability (cost, uptake by health ministries, social acceptability of therapy, etc.). The global burden of retinoblastoma shows the inherent inequity in how patients are diagnosed and cared for around the world.5 Documentation of human and material resources required for optimal care, drawn from established clinical care guidelines, represents a first step towards health equity. National strategies in areas currently under represented in the retinoblastoma literature are poised to fill the knowledge gap.
We need a universally endorsed single classification in order to avoid confusion regarding eligibility and salvage rates for new treatments. Progress in therapy is critically dependent on achieving consensus on a standard classification of intraocular disease. The ongoing AJCC/UICC international survey of features of eyes at diagnosis, treatments given and outcomes will be used to derive one evidence-based classification to replace the current clinical staging schemes that confuse analysis, eligibility and outcomes.78–80,82
We are poised to collect uniform data on all children with retinoblastoma. The 1RBW.org map documents “staff, stuff systems and spaces”196 and provides an alternative system of global collaboration, perhaps unique to small neglected diseases, but possibly a fore-runner for future learning health systems. The eCancerCareRB (eCCRB) point-of-care database summarizes the health record from diagnosis (including features of eyes and patient, treatments, complications, and outcomes) and is available to all retinoblastoma centres.197,198 eCCRB also supports the caregivers and families by graphically displaying this information ensuring high motivation to collect good data quality by clinicians who directly benefit from efficient and accurate patient care (Figures 5, 8, 9). The next phase of this online project (aiming at 2020) will combine the 1RBW map (http://www.1rbw.org) with eCCRB de-identified patient data to develop a ‘learning health system” which will provide real-time analysis of outcomes and will guide clinicians to apply evidenced-based care.199
Targeted therapies
Our knowledge of the cell of origin of retinoblastoma and its unique sensitivity to RB1 inactivation is steadily increasing, and may reveal strategies to prevent tumourigenesis. Moreover, ongoing work to define the molecular signature of retinoblastoma will continue to yield novel therapeutic targets. Advances in therapeutic development and prevention depend on these molecular studies, but we do not yet have preclinical models that accurately reflect retinoblastoma pathogenesis and treatment.
One of the earliest targeted therapies envisioned for use in retinoblastoma, based on preclinical information generated in transgenic mice, was Nutlin-3.200 Nutlin-3 blocks the interaction of MDM2 and MDM4 with p53, resulting in stabilization of p53 and stimulation of apoptotic cell death. Nutlin-3 showed promising activity in combination with topotecan for retinoblastoma control in preclinical models. Although the SYK antagonist R406 was also considered a promising candidate based on preclinical studies,53 its ocular pharmacokinetic profile following peri-ocular administration rendered it unfavourable for clinical use.201 Preclinical studies further suggest that retinoblastoma initiation and tumor growth might be suppressed by inhibitors of MYCN, SKP2, E2F, or CDKs (Figure 12).
Figure 12. Proposed roles for cone precursor circuitry in retinoblastoma tumourigenesis and targeted therapy.
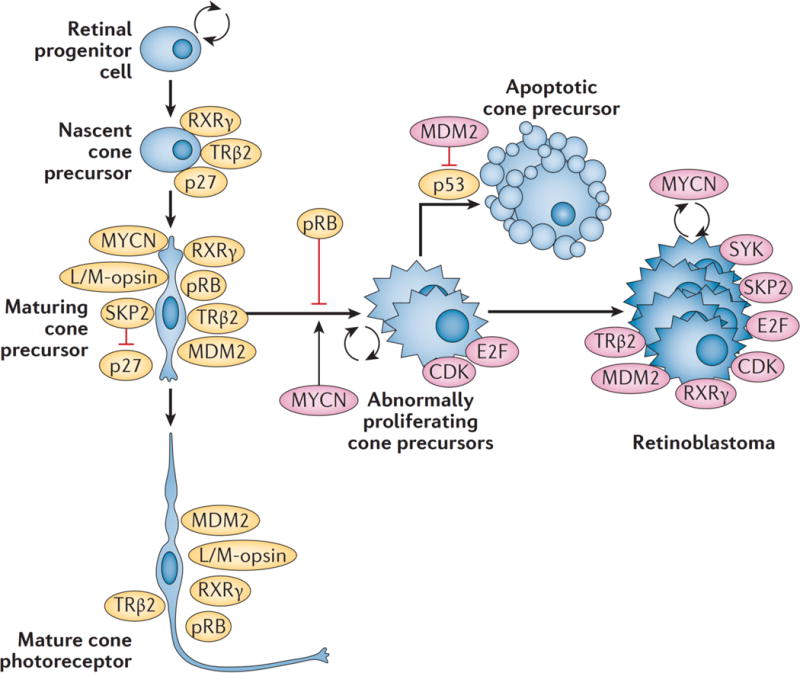
Signalling circuitry specific to maturing L/M-cone precursors is uniquely conducive to oncogenic transformation in response to pRB loss.49,61,62 Specifically, these cone precursors express the transcription factors RXRγ and thyroid hormone receptor (TR) β2, and high levels of MYCN, MDM2, and pRB, and down regulate p27 via a SKP2-related mechanism. pRB depletion in maturing cone precursors blocks cone photoreceptor differentiation and enables proliferation and survival dependent upon MYCN, SKP2, and MDM2. Mouse models suggest that E2Fs and CDKs contribute to the proliferative response to pRB loss59; these genes/proteins are also needed for proliferation and survival of human retinoblastoma cells.61,207 Identifying the molecular mechanism mediating the cone precursor proliferative response to pRB loss provides opportunities to prevent tumor initiation, at least in preclinical models. Potential drugs are inhibitors of MYCN expression (JQ1),208 inhibitors of MDM2-mediated p53 degradation (Nutlin 3a),200 inhibitors of SKP2-mediated p27 degradation (C25),209 and inhibitors of E2F and CDK activities (HLM006474 and R547, respectively)59. In addition, SYK was highly expressed in retinoblastoma but not normal retina.53 Additional targeted agents may be effective against growing tumours, such as improved versions of the SYK inhibitor R406.53,201
Preclinical studies have also sought to identify and evaluate pharmacokinetics of new agents alone or in combination202 before clinical use. A few translational studies have resulted in changes in retinoblastoma clinical practice.203 Innovative delivery systems to target small-volume subretinal tumours and vitreous seeding include episcleral implants204 and nanoparticles205. Approaches adjusting chemotherapy schedules and dose intensity (metronomic therapy)206 may particularly play an important role in palliative care for retinoblastoma. New biomarkers for molecular dissemination of retinoblastoma outside the eye may lead to earlier diagnosis of metastasis and targeted treatments.92,93
Quality of life
Without thinking about the lifelong outlook for patients, however, all of the above endeavours become moot. Quality of life begins in infancy, assisted by Child Life181 and respect for the child’s need to play. Patient-oriented research will develop and validate life-long survivor follow-up care protocols, including centralized specialist eye care, genetic testing, second cancer screening, ongoing psychological support and fertility care. The education of primary physicians and development of survivor clinics will reduce morbidity and facilitate research to inform patient management with a major outcome being good quality of life during treatment and throughout life.
Supplementary Material
Acknowledgments
T.W.C. acknowledges NIH NCATS KL2TR001106 and Research to Prevent Blindness, Inc.
D.C. acknowledges NIH R01CA137124 and support from the Larry and Celia Moh Foundation and the Margaret E. Early Medical Research Trust.
H.D. and B.L.G. acknowledge TUYF (Hong Kong).
Footnotes
Competing interests
There is NO competing interest.
Author contributions
Introduction (B.L.G., H.D., D.C., T.W.C.); Epidemiology (H.D., G.C., J.Z, F.N.); Mechanisms/pathophysiology (T.W.C., D.C., H.D., B.L.G.); Diagnosis, screening and prevention (H.D., G.C., J.Z., B.L.G.); Management (F.L.M., C.S., D.H.A., G.C., F.N., J.Z., B.L.G.); Quality of life (A.W., H.D., B.L.G.); Outlook (ALL); overview of Primer (B.L.G.).
References
- 1.Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1:157–162. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- 2.Dimaras H, et al. Loss of RB1 induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet. 2008;17:1363–1372. doi: 10.1093/hmg/ddn024. [DOI] [PubMed] [Google Scholar]
- 3.Dimaras H, et al. Retinoblastoma. Lancet. 2012;379:1436–1446. doi: 10.1016/S0140-6736(11)61137-9. [DOI] [PubMed] [Google Scholar]
- 4.Seregard S, Lundell G, Svedberg H, Kivela T. Incidence of retinoblastoma from 1958 to 1998 in Northern Europe: advantages of birth cohort analysis. Ophthalmology. 2004;111:1228–1232. doi: 10.1016/j.ophtha.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 5.Kivela T. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death. Br J Ophthalmol. 2009;93:1129–1131. doi: 10.1136/bjo.2008.150292. doi:93/9/1129 [pii]10.1136/bjo.2008.150292. [DOI] [PubMed] [Google Scholar]
- 6.Broaddus E, Topham A, Singh AD. Incidence of retinoblastoma in the USA: 1975–2004. Br J Ophthalmol. 2009;93:21–23. doi: 10.1136/bjo.2008.138750. [DOI] [PubMed] [Google Scholar]
- 7.Nyamori JM, Kimani K, Njuguna MW, Dimaras H. The incidence and distribution of retinoblastoma in Kenya. Br J Ophthalmol. 2012;96:141–143. doi: 10.1136/bjophthalmol-2011-300739. [DOI] [PubMed] [Google Scholar]
- 8.National Retinoblastoma Strategy Canadian Guidelines for Care/Stratégie thérapeutique du rétinoblastome guide clinique canadien. Canadian journal of ophthalmology. 2009;44:S1–88. doi: 10.3129/i09-194. [DOI] [PubMed] [Google Scholar]
- 9.MacCarthy A, et al. Retinoblastoma in Great Britain 1963-2002. Br J Ophthalmol. 2009;93:33–37. doi: 10.1136/bjo.2008.139618. [DOI] [PubMed] [Google Scholar]
- 10.Krishna SM, Yu GP, Finger PT. The effect of race on the incidence of retinoblastoma. J Pediatr Ophthalmol Strabismus. 2009;46:288–293. doi: 10.3928/01913913-20090903-06. [DOI] [PubMed] [Google Scholar]
- 11.Moreno F, et al. A population-based study of retinoblastoma incidence and survival in Argentine children. Pediatr Blood Cancer. 2014;61:1610–1615. doi: 10.1002/pbc.25048. [DOI] [PubMed] [Google Scholar]
- 12.One Retinoblastoma World. 2015 < http://1rbw.org/>.
- 13.Nyawira G, Kahaki K, Kariuki-Wanyoike M. Survival among retinoblastoma patients at the Kenyatta National Hospital, Kenya. Journal of Ophthalmology of Eastern Central and Southern Africa. 2013:15–19. [Google Scholar]
- 14.Dean M, et al. Increased incidence and disparity of diagnosis of retinoblastoma patients in Guatemala. Cancer letters. 2014;351:59–63. doi: 10.1016/j.canlet.2014.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar A, Moulik NR, Mishra RK, Kumar D. Causes, outcome and prevention of abandonment in retinoblastoma in India. Pediatr Blood Cancer. 2013;60:771–775. doi: 10.1002/pbc.24454. [DOI] [PubMed] [Google Scholar]
- 16.Gichigo EN, Kariuki-Wanyoike MM, Kimani K, Nentwich MM. Retinoblastoma in Kenya: Survival and prognostic factors. Ophthalmologe. 2014 doi: 10.1007/s00347-014-3123-z. [DOI] [PubMed] [Google Scholar]
- 17.Asencio-Lopez L, Torres-Ojeda AA, Isaac-Otero G, Rosales Lopez SL, Leal-Leal CA. Treating retinoblastoma in the first year of life in a national tertiary pediatric hospital in Mexico. Acta Paediatr. 2015 doi: 10.1111/apa.13033. [DOI] [PubMed] [Google Scholar]
- 18.Kenyan Ministry of Health. Kenya national retinoblastoma strategy: Best practice guidelines. 2014 < http://www.health.go.ke/images/downloads/Best-Practice-September-2014.pdf>.
- 19.Vincent AL, Webb MC, Gallie BL, Heon E. Prosthetic conformers: a step towards improved rehabilitation of enucleated children. Clin Experiment Ophthalmol. 2002;30:58–59. doi: 10.1046/j.1442-9071.2002.00472.x. [DOI] [PubMed] [Google Scholar]
- 20.Dimaras H, White A, Gallie BL. Ophthalmology Rounds. Vol. 6. Toronto: 2008. [Google Scholar]
- 21.Chantada G, et al. SIOP-PODC recommendations for graduated-intensity treatment of retinoblastoma in developing countries. Pediatr Blood Cancer. 2013;60:719–727. doi: 10.1002/pbc.24468. [DOI] [PubMed] [Google Scholar]
- 22.Perez-Cuevas R, et al. Scaling up cancer care for children without medical insurance in developing countries: The case of Mexico. Pediatr Blood Cancer. 2013;60:196–203. doi: 10.1002/pbc.24265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Castro Junior CG, Macedo CR. Brazilian Society of Pediatric Oncology - SOBOPE: 30 years of history, a lot in the present, full of the future. Revista brasileira de hematologia e hemoterapia. 2011;33:326–327. doi: 10.5581/1516-8484.20110089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naseripour M. “Retinoblastoma survival disparity”: The expanding horizon in developing countries. Saudi journal of ophthalmology: official journal of the Saudi Ophthalmological Society. 2012;26:157–161. doi: 10.1016/j.sjopt.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qaddoumi I, et al. Team management, twinning, and telemedicine in retinoblastoma: a 3-tier approach implemented in the first eye salvage program in Jordan. Pediatr Blood Cancer. 2008;51:241–244. doi: 10.1002/pbc.21489. [DOI] [PubMed] [Google Scholar]
- 26.Wilimas JA, et al. Development of retinoblastoma programs in Central America. Pediatr Blood Cancer. 2009;53:42–46. doi: 10.1002/pbc.21984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Traore F, et al. Retinoblastoma: inventory in Mali and program to develop early diagnosis, treatments and rehabilitation. Bull Cancer. 2013;100:161–165. doi: 10.1684/bdc.2013.1703. [DOI] [PubMed] [Google Scholar]
- 28.Luna-Fineman S, et al. Retinoblastoma in Central America: report from the Central American Association of Pediatric Hematology Oncology (AHOPCA) Pediatr Blood Cancer. 2012;58:545–550. doi: 10.1002/pbc.23307. [DOI] [PubMed] [Google Scholar]
- 29.Barr RD, et al. Asociacion de Hemato-Oncologia Pediatrica de Centro America (AHOPCA): a model for sustainable development in pediatric oncology. Pediatr Blood Cancer. 2014;61:345–354. doi: 10.1002/pbc.24802. [DOI] [PubMed] [Google Scholar]
- 30.Waddell KM, et al. Improving survival of retinoblastoma in Uganda. Br J Ophthalmol. 2015 doi: 10.1136/bjophthalmol-2014-306206. [DOI] [PubMed] [Google Scholar]
- 31.Waddell KM, et al. Clinical features and survival among children with retinoblastoma in Uganda. Br J Ophthalmol. 2015;99:387–390. doi: 10.1136/bjophthalmol-2014-305564. [DOI] [PubMed] [Google Scholar]
- 32.Schvartzman E, et al. Results of a stage-based protocol for the treatment of retinoblastoma. J Clin Oncol. 1996;14:1532–1536. doi: 10.1200/JCO.1996.14.5.1532. [DOI] [PubMed] [Google Scholar]
- 33.Chantada GL, et al. Results of a prospective study for the treatment of unilateral retinoblastoma. Pediatr Blood Cancer. 2010;55:60–66. doi: 10.1002/pbc.22503. [DOI] [PubMed] [Google Scholar]
- 34.Chantada GL, et al. Impact of chemoreduction for conservative therapy for retinoblastoma in Argentina. Pediatr Blood Cancer. 2014;61:821–826. doi: 10.1002/pbc.24857. [DOI] [PubMed] [Google Scholar]
- 35.Chiu HH, Dimaras H, Downie R, Gallie B. Breaking down barriers to communicating complex retinoblastoma information: can graphics be the solution? Canadian journal of ophthalmology. doi: 10.1016/j.jcjo.2015.02.003. In Press. [DOI] [PubMed] [Google Scholar]
- 36.Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Science, USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Comings DE. A general theory of carcinogenesis. Proc Natl Acad Sci U S A. 1973;70:3324–3328. doi: 10.1073/pnas.70.12.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cavenee WK, et al. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature. 1983;305:779–784. doi: 10.1038/305779a0. [DOI] [PubMed] [Google Scholar]
- 39.Dryja TP, Rapaport JM, Joyce JM, Petersen RA. Molecular detection of deletions involving band q14 of chromosome 13 in retinoblastomas. Proceedings of the National Academy of Science, USA. 1986;83:7391–7394. doi: 10.1073/pnas.83.19.7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Friend SH, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643–646. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- 41.Lee WH, et al. Human retinoblastoma susceptibility gene: cloning, identification, and sequence. Science. 1987;235:1394–1399. doi: 10.1126/science.3823889. [DOI] [PubMed] [Google Scholar]
- 42.Fung YK, et al. Structural evidence for the authenticity of the human retinoblastoma gene. Science (New York, NY. 1987;236:1657–1661. doi: 10.1126/science.2885916. [DOI] [PubMed] [Google Scholar]
- 43.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 44.Lohmann DR. RB1 gene mutations in retinoblastoma. Hum Mutat. 1999;14:283–288. doi: 10.1002/(SICI)1098-1004(199910)14:4<283::AID-HUMU2>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 45.McEvoy J, et al. RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget. 2014 doi: 10.18632/oncotarget.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14:297–306. doi: 10.1038/nrm3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lohmann DR, Brandt B, Hopping W, Passarge E, Horsthemke B. Distinct RB1 gene mutations with low penetrance in hereditary retinoblastoma. Human genetics. 1994;94:349–354. doi: 10.1007/BF00201591. [DOI] [PubMed] [Google Scholar]
- 48.Munier FL, Balmer A, van Melle G, Gailloud C. Radial asymmetry in the topography of retinoblastoma. Clues to the cell of origin. Ophthalmic genetics. 1994;15:101–106. doi: 10.3109/13816819409057835. [DOI] [PubMed] [Google Scholar]
- 49.Lee TC, Almeida D, Claros N, Abramson DH, Cobrinik D. Cell cycle-specific and cell type-specific expression of Rb in the developing human retina. Invest Ophthalmol Vis Sci. 2006;47:5590–5598. doi: 10.1167/iovs.06-0063. [DOI] [PubMed] [Google Scholar]
- 50.Goodrich DW. How the other half lives, the amino-terminal domain of the retinoblastoma tumor suppressor protein. J Cell Physiol. 2003;197:169–180. doi: 10.1002/jcp.10358. [DOI] [PubMed] [Google Scholar]
- 51.Cook R, et al. Direct Involvement of Retinoblastoma Family Proteins in DNA Repair by Non-homologous End-Joining. Cell reports. 2015;10:2006–2018. doi: 10.1016/j.celrep.2015.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Theriault BL, Dimaras H, Gallie BL, Corson TW. The genomic landscape of retinoblastoma: a review. Clin Experiment Ophthalmol. 2014;42:33–52. doi: 10.1111/ceo.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang J, et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012;481:329–334. doi: 10.1038/nature10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kooia IE, et al. Loss of photoreceptorness and gain of genomic alterations in retinoblastoma reveal tumor progression. doi: 10.1016/j.ebiom.2015.06.022. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McEvoy J, et al. Coexpression of normally incompatible developmental pathways in retinoblastoma genesis. Cancer Cell. 2011;20:260–275. doi: 10.1016/j.ccr.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kapatai G, et al. Gene expression profiling identifies different sub-types of retinoblastoma. Br J Cancer. 2013;109:512–525. doi: 10.1038/bjc.2013.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rushlow DE, et al. Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol. 2013;14:327–334. doi: 10.1016/S1470-2045(13)70045-7. [DOI] [PubMed] [Google Scholar]
- 58.Kyritsis AP, Tsokos M, Triche TJ, Chader GJ. Retinoblastoma–origin from a primitive neuroectodermal cell? Nature. 1984;307:471–473. doi: 10.1038/307471a0. [DOI] [PubMed] [Google Scholar]
- 59.Sangwan M, et al. Established and new mouse models reveal E2f1 and Cdk2 dependency of retinoblastoma, and expose effective strategies to block tumor initiation. Oncogene. 2012;31:5019–5028. doi: 10.1038/onc.2011.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cobrinik D. In: Animal Models of Brain Tumors. Martinez-Murillo R, Martinez A, editors. Springer; 2013. pp. 141–152. [Google Scholar]
- 61.Xu XL, et al. Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell. 2009;137:1018–1031. doi: 10.1016/j.cell.2009.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu XL, et al. Rb suppresses human cone-precursor-derived retinoblastoma tumours. Nature. 2014;514:385–388. doi: 10.1038/nature13813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rootman DB, et al. Hand-held high-resolution spectral domain optical coherence tomography in retinoblastoma: clinical and morphologic considerations. Br J Ophthalmol. 2013;97:59–65. doi: 10.1136/bjophthalmol-2012-302133. [DOI] [PubMed] [Google Scholar]
- 64.Xu XL, et al. Tumor-associated retinal astrocytes promote retinoblastoma cell proliferation through production of IGFBP-5. Am J Pathol. 2010;177:424–435. doi: 10.2353/ajpath.2010.090512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hook KE, et al. An integrated genomic approach to identify predictive biomarkers of response to the aurora kinase inhibitor PF-03814735. Mol Cancer Ther. 2012;11:710–719. doi: 10.1158/1535-7163.MCT-11-0184. [DOI] [PubMed] [Google Scholar]
- 66.Gallie BL, Trogadis J, Han L-P. In: Human Cell Culture. Masters JR, Palsson B, editors. Vol. 2. Springer; 1999. pp. 361–374. [Google Scholar]
- 67.Gallie BL, Albert DM, Wong JJ, Buyukmihci N, Puliafito CA. Heterotransplantation of retinoblastoma into the athymic “nude” mouse. Investigative ophthalmology & visual science. 1977;16:256–259. [PubMed] [Google Scholar]
- 68.Pacal M, Bremner R. Insights from animal models on the origins and progression of retinoblastoma. Curr Mol Med. 2006;6:759–781. doi: 10.2174/1566524010606070759. [DOI] [PubMed] [Google Scholar]
- 69.Conkrite K, et al. miR-17~92 cooperates with RB pathway mutations to promote retinoblastoma. Genes Dev. 2011;25:1734–1745. doi: 10.1101/gad.17027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Windle JJ, et al. Retinoblastoma in transgenic mice. Nature. 1990;343:665–669. doi: 10.1038/343665a0. [DOI] [PubMed] [Google Scholar]
- 71.Pajovic S, et al. The TAg-RB murine retinoblastoma cell of origin has immunohistochemical features of differentiated Müller glia with progenitor properties. Invest Ophthalmol Vis Sci. 2011;52:7618–7624. doi: 10.1167/iovs.11-7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karcioglu ZA. Fine needle aspiration biopsy (FNAB) for retinoblastoma. Retina (Philadelphia, Pa. 2002;22:707–710. doi: 10.1097/00006982-200212000-00004. [DOI] [PubMed] [Google Scholar]
- 73.de Jong MC, et al. Diagnostic performance of magnetic resonance imaging and computed tomography for advanced retinoblastoma: a systematic review and meta-analysis. Ophthalmology. 2014;121:1109–1118. doi: 10.1016/j.ophtha.2013.11.021. [DOI] [PubMed] [Google Scholar]
- 74.de Jong MC, et al. Trilateral retinoblastoma: a systematic review and meta-analysis. The lancet oncology. 2014;15:1157–1167. doi: 10.1016/S1470-2045(14)70336-5. [DOI] [PubMed] [Google Scholar]
- 75.McCarthy M, et al. Comfort First: an evaluation of a procedural pain management programme for children with cancer. Psychooncology. 2013;22:775–782. doi: 10.1002/pon.3061. [DOI] [PubMed] [Google Scholar]
- 76.Moulin AP, Gaillard MC, Balmer A, Munier FL. Ultrasound biomicroscopy evaluation of anterior extension in retinoblastoma: a clinicopathological study. Br J Ophthalmol. 2012;96:337–340. doi: 10.1136/bjophthalmol-2011-300051. [DOI] [PubMed] [Google Scholar]
- 77.Reese AB, Ellsworth RM. The evaluation and current concept of retinoblastoma therapy. Trans Am Acad Ophthalmol-Otolaryngol. 1963:164–172. [PubMed] [Google Scholar]
- 78.Murphree AL. Intraocular retinoblastoma: the case for a new group classification. Ophthalmology clinics of North America. 2005;18:41–53. doi: 10.1016/j.ohc.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 79.Shields CL, et al. The International Classification of Retinoblastoma predicts chemoreduction success. Ophthalmology. 2006;113:2276–2280. doi: 10.1016/j.ophtha.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 80.Novetsky DE, Abramson DH, Kim JW, Dunkel IJ. Published international classification of retinoblastoma (ICRB) definitions contain inconsistencies–an analysis of impact. Ophthalmic genetics. 2009;30:40–44. doi: 10.1080/13816810802452168. doi:908192764 [pii]10.1080/13816810802452168. [DOI] [PubMed] [Google Scholar]
- 81.Children’s Oncology Group. Newly diagnosed with retinoblastoma. 2011 < http://www.childrensoncologygroup.org/index.php/newlydiagnosedwithretinoblastoma>.
- 82.Finger PT, et al. In: AJCC Cancer Staging Manual. 7th. Edge SB, Byrd DR, Carducci MA, Compton CC, editors. Springer; 2010. pp. 561–568. Vol. Ch. 52. [Google Scholar]
- 83.Chantada G, et al. A proposal for an international retinoblastoma staging system. Pediatr Blood Cancer. 2006;47:801–805. doi: 10.1002/pbc.20606. [DOI] [PubMed] [Google Scholar]
- 84.Sastre X, et al. Proceedings of the consensus meetings from the International Retinoblastoma Staging Working Group on the pathology guidelines for the examination of enucleated eyes and evaluation of prognostic risk factors in retinoblastoma. Arch Pathol Lab Med. 2009;133:1199–1202. doi: 10.1043/1543-2165-133.8.1199. [DOI] [PubMed] [Google Scholar]
- 85.Wilson MW, Qaddoumi I, Billups C, Haik BG, Rodriguez-Galindo C. A clinicopathological correlation of 67 eyes primarily enucleated for advanced intraocular retinoblastoma. The British journal of ophthalmology. 2011;95:553–558. doi: 10.1136/bjo.2009.177444. [DOI] [PubMed] [Google Scholar]
- 86.Sullivan EM, et al. Pathologic risk-based adjuvant chemotherapy for unilateral retinoblastoma following enucleation. J Pediatr Hematol Oncol. 2014;36:e335–340. doi: 10.1097/MPH.0000000000000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mendoza PR, et al. Histopathologic grading of anaplasia in retinoblastoma. American journal of ophthalmology. 2015;159:764–776. e763. doi: 10.1016/j.ajo.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yousef YA, et al. Predictive Value of Tnm Classification, International Classification, and Reese-Ellsworth Staging of Retinoblastoma for the Likelihood of High-Risk Pathologic Features. Retina (Philadelphia, Pa. 2015 doi: 10.1097/IAE.0000000000000547. [DOI] [PubMed] [Google Scholar]
- 89.Kaliki S, Srinivasan V, Gupta A, Mishra DK, Naik MN. Clinical Features Predictive of High-Risk Retinoblastoma in 403 Asian Indian Patients: A Case-Control Study. Ophthalmology. 2015 doi: 10.1016/j.ophtha.2015.01.018. [DOI] [PubMed] [Google Scholar]
- 90.Kim JW. Retinoblastoma: evidence for postenucleation adjuvant chemotherapy. International ophthalmology clinics. 2015;55:77–96. doi: 10.1097/IIO.0000000000000048. [DOI] [PubMed] [Google Scholar]
- 91.Grossniklaus HE, Kivëla T, Harbour JW, Finger PT. Protocol for the Examination of Specimens From Patients With Retinoblastoma. 2011 < http://www.cap.org/apps/docs/committees/cancer/cancer_protocols/2011/Retinoblast_11protocol.pdf>.
- 92.Torbidoni AV, et al. Association of Cone-Rod Homeobox Transcription Factor Messenger RNA With Pediatric Metastatic Retinoblastoma. JAMA ophthalmology. 2015 doi: 10.1001/jamaophthalmol.2015.0900. [DOI] [PubMed] [Google Scholar]
- 93.Torbidoni AV, et al. Immunoreactivity of the 14F7 Mab raised against N-Glycolyl GM3 Ganglioside in retinoblastoma tumours. Acta Ophthalmol. 2014 doi: 10.1111/aos.12578. [DOI] [PubMed] [Google Scholar]
- 94.Genetics I. What is retinoblastoma? 2015 < http://impactgenetics.com/testing-services/retinoblastoma/what-is-rb/>.
- 95.Wong JR, et al. Risk of subsequent malignant neoplasms in long-term hereditary retinoblastoma survivors after chemotherapy and radiotherapy. J Clin Oncol. 2014;32:3284–3290. doi: 10.1200/JCO.2013.54.7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xu K, et al. Preimplantation genetic diagnosis for retinoblastoma: the first reported liveborn. American journal of ophthalmology. 2004;137:18–23. doi: 10.1016/s0002-9394(03)00872-9. [DOI] [PubMed] [Google Scholar]
- 97.Dommering CJ, Moll AC, Imhof SM, de Die-Smulders CE, Coonen E. Another liveborn after preimplantation genetic diagnosis for retinoblastoma. American journal of ophthalmology. 2004;138:1088–1089. doi: 10.1016/j.ajo.2004.07.048. [DOI] [PubMed] [Google Scholar]
- 98.Dimaras H, et al. Retinoblastoma CSF metastasis cured by multimodality chemotherapy without radiation. Ophthalmic genetics. 2009;30:121–126. doi: 10.1080/13816810902988780. [DOI] [PubMed] [Google Scholar]
- 99.Bowles E, et al. Profiling genomic copy number changes in retinoblastoma beyond loss of RB1. Genes Chromosomes Cancer. 2007;46:118–129. doi: 10.1002/gcc.20383. [DOI] [PubMed] [Google Scholar]
- 100.Chantada GL, et al. Familial retinoblastoma in developing countries. Pediatr Blood Cancer. 2009;53:338–342. doi: 10.1002/pbc.21970. [DOI] [PubMed] [Google Scholar]
- 101.He LQ, et al. Developing clinical cancer genetics services in resource-limited countries: the case of retinoblastoma in Kenya. Public health genomics. 2014;17:221–227. doi: 10.1159/000363645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Patel T, et al. Consumer Digital Cameras: A feasible strategy for the early detection of childhood blindness. Association for Research in Vision and Ophthalmology (ARVO) 2012 [Google Scholar]
- 103.Abdolvahabi A, et al. Colorimetric and longitudinal analysis of leukocoria in recreational photographs of children with retinoblastoma. PLoS One. 2013;8:e76677. doi: 10.1371/journal.pone.0076677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.MacCarthy A, et al. Second and subsequent tumours among 1927 retinoblastoma patients diagnosed in Britain 1951-2004. Br J Cancer. 2013;108:2455–2463. doi: 10.1038/bjc.2013.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Abramson DH, Beaverson KL, Chang ST, Dunkel IJ, McCormick B. Outcome following initial external beam radiotherapy in patients with Reese-Ellsworth group Vb retinoblastoma. Archives of ophthalmology. 2004;122:1316–1323. doi: 10.1001/archopht.122.9.1316. [DOI] [PubMed] [Google Scholar]
- 106.Fletcher O, et al. Lifetime risks of common cancers among retinoblastoma survivors. Journal of the National Cancer Institute. 2004;96:357–363. doi: 10.1093/jnci/djh058. [DOI] [PubMed] [Google Scholar]
- 107.Friedman DN, et al. Whole-body magnetic resonance imaging (WB-MRI) as surveillance for subsequent malignancies in survivors of hereditary retinoblastoma: a pilot study. Pediatr Blood Cancer. 2014;61:1440–1444. doi: 10.1002/pbc.24835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Soliman SE, Dimaras H, Souka AA, Ashry MH, Gallie BL. Socioeconomic and psychological impact of treatment for unilateral intraocular retinoblastoma. J Fr Ophtalmol. 2015;38:550–558. doi: 10.1016/j.jfo.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 109.Shields CL, et al. Retinoblastoma frontiers with intravenous, intra-arterial, periocular, and intravitreal chemotherapy. Eye (Lond) 2013;27:253–264. doi: 10.1038/eye.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Eng C, et al. Mortality from second tumors among long-term survivors of retinoblastoma. Journal of the National Cancer Institute. 1993;85:1121–1128. doi: 10.1093/jnci/85.14.1121. [DOI] [PubMed] [Google Scholar]
- 111.Chantada GL, et al. Treatment results in patients with retinoblastoma and invasion to the cut end of the optic nerve. Pediatr Blood Cancer. 2009;52:218–222. doi: 10.1002/pbc.21735. [DOI] [PubMed] [Google Scholar]
- 112.Kashyap S, et al. Clinical predictors of high risk histopathology in retinoblastoma. Pediatr Blood Cancer. 2012;58:356–361. doi: 10.1002/pbc.23239. [DOI] [PubMed] [Google Scholar]
- 113.Zhao J, et al. Pre-enucleation chemotherapy for eyes severely affected by retinoblastoma masks risk of tumor extension and increases death from metastasis. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:845–851. doi: 10.1200/JCO.2010.32.5332. [DOI] [PubMed] [Google Scholar]
- 114.Francis JH, et al. Efficacy and Toxicity of Second-Course Ophthalmic Artery Chemosurgery for Retinoblastoma. Ophthalmology. 2015 doi: 10.1016/j.ophtha.2014.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hamama-Raz Y, Rot I, Buchbinder E. The coping experience of parents of a child with retinoblastoma-malignant eye cancer. Journal of psychosocial oncology. 2012;30:21–40. doi: 10.1080/07347332.2011.633977. [DOI] [PubMed] [Google Scholar]
- 116.Mathew AA, Sachdev N, Staffieri SE, McKenzie JD, Elder JE. Superselective intra-arterial chemotherapy for advanced retinoblastoma complicated by metastatic disease. J AAPOS. 2015;19:72–74. doi: 10.1016/j.jaapos.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 117.Gunduz K, et al. Causes of chemoreduction failure in retinoblastoma and analysis of associated factors leading to eventual treatment with external beam radiotherapy and enucleation. Ophthalmology. 2004;111:1917–1924. doi: 10.1016/j.ophtha.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 118.Manjandavida FP, Honavar SG, Reddy VA, Khanna R. Management and outcome of retinoblastoma with vitreous seeds. Ophthalmology. 2014;121:517–524. doi: 10.1016/j.ophtha.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 119.Schaiquevich P, et al. Intra-arterial chemotherapy is more effective than sequential periocular and intravenous chemotherapy as salvage treatment for relapsed retinoblastoma. Pediatr Blood Cancer. 2013;60:766–770. doi: 10.1002/pbc.24356. [DOI] [PubMed] [Google Scholar]
- 120.Shields CL, et al. Iodine 125 plaque radiotherapy as salvage treatment for retinoblastoma recurrence after chemoreduction in 84 tumors. Ophthalmology. 2006;113:2087–2092. doi: 10.1016/j.ophtha.2006.04.032. [DOI] [PubMed] [Google Scholar]
- 121.Munier FL, et al. New developments in external beam radiotherapy for retinoblastoma: from lens to normal tissue-sparing techniques. Clin Experiment Ophthalmol. 2008;36:78–89. doi: 10.1111/j.1442-9071.2007.01602.x. [DOI] [PubMed] [Google Scholar]
- 122.Chan MP, Hungerford JL, Kingston JE, Plowman PN. Salvage external beam radiotherapy after failed primary chemotherapy for bilateral retinoblastoma: rate of eye and vision preservation. Br J Ophthalmol. 2009;93:891–894. doi: 10.1136/bjo.2007.129981. [DOI] [PubMed] [Google Scholar]
- 123.Pica A, et al. Preliminary experience in treatment of papillary and macular retinoblastoma: evaluation of local control and local complications after treatment with linear accelerator-based stereotactic radiotherapy with micromultileaf collimator as second-line or salvage treatment after chemotherapy. International journal of radiation oncology, biology, physics. 2011;81:1380–1386. doi: 10.1016/j.ijrobp.2010.07.033. [DOI] [PubMed] [Google Scholar]
- 124.Berry JL, et al. Long-term outcomes of Group D eyes in bilateral retinoblastoma patients treated with chemoreduction and low-dose IMRT salvage. Pediatr Blood Cancer. 2013;60:688–693. doi: 10.1002/pbc.24303. [DOI] [PubMed] [Google Scholar]
- 125.Mouw KW, et al. Proton radiation therapy for the treatment of retinoblastoma. International journal of radiation oncology, biology, physics. 2014;90:863–869. doi: 10.1016/j.ijrobp.2014.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shields CL, et al. Chemoreduction plus focal therapy for retinoblastoma: factors predictive of need for treatment with external beam radiotherapy or enucleation. American journal of ophthalmology. 2002;133:657–664. doi: 10.1016/s0002-9394(02)01348-x. [DOI] [PubMed] [Google Scholar]
- 127.Abramson DH, Schefler AC. Update on retinoblastoma. Retina. 2004;24:828–848. doi: 10.1097/00006982-200412000-00002. [DOI] [PubMed] [Google Scholar]
- 128.Gobin YP, Dunkel IJ, Marr BP, Brodie SE, Abramson DH. Intra-arterial chemotherapy for the management of retinoblastoma: four-year experience. Archives of ophthalmology. 2011;129:732–737. doi: 10.1001/archophthalmol.2011.5. [DOI] [PubMed] [Google Scholar]
- 129.Suzuki S, Yamane T, Mohri M, Kaneko A. Selective ophthalmic arterial injection therapy for intraocular retinoblastoma: the long-term prognosis. Ophthalmology. 2011;118:2081–2087. doi: 10.1016/j.ophtha.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 130.Shields CL, et al. Intra-arterial chemotherapy for retinoblastoma in 70 eyes: outcomes based on the international classification of retinoblastoma. Ophthalmology. 2014;121:1453–1460. doi: 10.1016/j.ophtha.2014.01.026. [DOI] [PubMed] [Google Scholar]
- 131.Abramson DH, et al. Intra-arterial chemotherapy for retinoblastoma in eyes with vitreous and/or subretinal seeding: 2-year results. Br J Ophthalmol. 2012;96:499–502. doi: 10.1136/bjophthalmol-2011-300498. [DOI] [PubMed] [Google Scholar]
- 132.Schaiquevich P, et al. Pharmacokinetic analysis of topotecan after superselective ophthalmic artery infusion and periocular administration in a porcine model. Retina (Philadelphia, Pa. 2012;32:387–395. doi: 10.1097/IAE.0b013e31821e9f8a. [DOI] [PubMed] [Google Scholar]
- 133.Munier FL, et al. Intravitreal chemotherapy for vitreous disease in retinoblastoma revisited: from prohibition to conditional indications. Br J Ophthalmol. 2012;96:1078–1083. doi: 10.1136/bjophthalmol-2011-301450. [DOI] [PubMed] [Google Scholar]
- 134.Shields CL, et al. Intravitreal melphalan for persistent or recurrent retinoblastoma vitreous seeds: preliminary results. JAMA ophthalmology. 2014;132:319–325. doi: 10.1001/jamaophthalmol.2013.7666. [DOI] [PubMed] [Google Scholar]
- 135.Francis JH, et al. The classification of vitreous seeds in retinoblastoma and response to intravitreal melphalan. Ophthalmology. 2015;122:1173–1179. doi: 10.1016/j.ophtha.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 136.Munier FL. Classification and management of seeds in retinoblastoma. Ellsworth Lecture Ghent August 24th 2013. Ophthalmic genetics. 2014;35:193–207. doi: 10.3109/13816810.2014.973045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mallipatna AC, Sutherland JE, Gallie BL, Chan H, Heon E. Management and outcome of unilateral retinoblastoma. J AAPOS. 2009;13:546–550. doi: 10.1016/j.jaapos.2009.09.004. doi:S1091-8531(09)00308-5 [pii]10.1016/j.jaapos.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 138.Rappaport BA, Suresh S, Hertz S, Evers AS, Orser BA. Anesthetic neurotoxicity–clinical implications of animal models. The New England journal of medicine. 2015;372:796–797. doi: 10.1056/NEJMp1414786. [DOI] [PubMed] [Google Scholar]
- 139.Mourits DL, Hartong DT, Bosscha MI, Kloos RJ, Moll AC. Worldwide enucleation techniques and materials for treatment of retinoblastoma: an international survey. PLoS One. 2015;10:e0121292. doi: 10.1371/journal.pone.0121292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yadava U, Sachdeva P, Arora V. Myoconjunctival enucleation for enhanced implant motility. result of a randomised prospective study. Indian J Ophthalmol. 2004;52:221–226. [PubMed] [Google Scholar]
- 141.Shome D, Honavar SG, Raizada K, Raizada D. Implant and prosthesis movement after enucleation: a randomized controlled trial. Ophthalmology. 2010;117:1638–1644. doi: 10.1016/j.ophtha.2009.12.035. [DOI] [PubMed] [Google Scholar]
- 142.Chan HSL. Combination Chemotherapy and Cyclosporine Followed by Focal Therapy for Bilateral Retinoblastoma NCT00110110. 2005 < https://clinicaltrials.gov/ct2/show/NCT00110110?term=retinoblastoma&intr=cyclosporin&rank=1>.
- 143.Chan HS, et al. Combining cyclosporin with chemotherapy controls intraocular retinoblastoma without requiring radiation. Clin Cancer Res. 1996;2:1499–1508. [PubMed] [Google Scholar]
- 144.Dunkel IJ, et al. A phase II trial of carboplatin for intraocular retinoblastoma. Pediatr Blood Cancer. 2007 doi: 10.1002/pbc.21163. [DOI] [PubMed] [Google Scholar]
- 145.Jehanne M, et al. Analysis of ototoxicity in young children receiving carboplatin in the context of conservative management of unilateral or bilateral retinoblastoma. Pediatr Blood Cancer. 2009;52:637–643. doi: 10.1002/pbc.21898. [DOI] [PubMed] [Google Scholar]
- 146.Draper GJ, Sanders BM, Kingston JE. Second primary neoplasms in patients with retinoblastoma. British journal of cancer. 1986;53:661–671. doi: 10.1038/bjc.1986.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Smith MA, et al. Secondary leukemia or myelodysplastic syndrome after treatment with epipodophyllotoxins. J Clin Oncol. 1999;17:569–577. doi: 10.1200/JCO.1999.17.2.569. [DOI] [PubMed] [Google Scholar]
- 148.Gombos DS, et al. Secondary acute myelogenous leukemia in patients with retinoblastoma: is chemotherapy a factor? Ophthalmology. 2007;114:1378–1383. doi: 10.1016/j.ophtha.2007.03.074. [DOI] [PubMed] [Google Scholar]
- 149.Gombos DS, Kelly A, Coen PG, Kingston JE, Hungerford JL. Retinoblastoma treated with primary chemotherapy alone: the significance of tumour size, location, and age. Br J Ophthalmol. 2002;86:80–83. doi: 10.1136/bjo.86.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee V, et al. Globe conserving treatment of the only eye in bilateral retinoblastoma. Br J Ophthalmol. 2003;87:1374–1380. doi: 10.1136/bjo.87.11.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shields CL, et al. Macular retinoblastoma managed with chemoreduction: analysis of tumor control with or without adjuvant thermotherapy in 68 tumors. Archives of ophthalmology. 2005;123:765–773. doi: 10.1001/archopht.123.6.765. doi:123/6/765 [pii]10.1001/archopht.123.6.765. [DOI] [PubMed] [Google Scholar]
- 152.Astudillo PP, Chan HS, Heon E, Gallie BL. Late-diagnosis retinoblastoma with germline mosaicism in an 8-year-old. J AAPOS. 2014;18:500–502. doi: 10.1016/j.jaapos.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 153.Narang S, Mashayekhi A, Rudich D, Shields CL. Predictors of long-term visual outcome after chemoreduction for management of intraocular retinoblastoma. Clin Experiment Ophthalmol. 2012;40:736–742. doi: 10.1111/j.1442-9071.2012.02757.x. [DOI] [PubMed] [Google Scholar]
- 154.Watts P, et al. Visual results in children treated for macular retinoblastoma. Eye. 2002;16:75–80. doi: 10.1038/sj.eye.6700070. [DOI] [PubMed] [Google Scholar]
- 155.Abramson DH, Dunkel IJ, Brodie SE, Kim JW, Gobin YP. A phase I/II study of direct intraarterial (ophthalmic artery) chemotherapy with melphalan for intraocular retinoblastoma initial results. Ophthalmology. 2008;1151404:1398–1404. e1391. doi: 10.1016/j.ophtha.2007.12.014. doi:S0161-6420(07)01359-0 [pii]10.1016/j.ophtha.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 156.Klufas MA, et al. Intra-arterial chemotherapy as a treatment for intraocular retinoblastoma: alternatives to direct ophthalmic artery catheterization. Ajnr. 2012;33:1608–1614. doi: 10.3174/ajnr.A3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Abramson DH, et al. Ophthalmic artery chemosurgery for less advanced intraocular retinoblastoma: five year review. PLoS ONE. 2012;7:e34120. doi: 10.1371/journal.pone.0034120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ong SJ, Chao AN, Wong HF, Liou KL, Kao LY. Selective ophthalmic arterial injection of melphalan for intraocular retinoblastoma: a 4-year review. Jpn J Ophthalmol. 2015;59:109–117. doi: 10.1007/s10384-014-0356-y. [DOI] [PubMed] [Google Scholar]
- 159.Francis JH, et al. Electroretinogram monitoring of dose-dependent toxicity after ophthalmic artery chemosurgery in retinoblastoma eyes: six year review. PLoS One. 2014;9:e84247. doi: 10.1371/journal.pone.0084247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Abramson DH, Frank CM, Dunkel IJ. A phase I/II study of subconjunctival carboplatin for intraocular retinoblastoma. Ophthalmology. 1999;106:1947–1950. doi: 10.1016/S0161-6420(99)90406-2. [DOI] [PubMed] [Google Scholar]
- 161.Mallipatna AC, Dimaras H, Chan HS, Heon E, Gallie BL. Periocular topotecan for intraocular retinoblastoma. Arch Ophthalmol. 2011;129:738–745. doi: 10.1001/archophthalmol.2011.130. [DOI] [PubMed] [Google Scholar]
- 162.Mulvihill A, et al. Ocular motility changes after subtenon carboplatin chemotherapy for retinoblastoma. Archives of ophthalmology. 2003;121:1120–1124. doi: 10.1001/archopht.121.8.1120. [DOI] [PubMed] [Google Scholar]
- 163.Ghassemi F, Amoli FA. Pathological findings in enucleated eyes after intravitreal melphalan injection. International ophthalmology. 2014;34:533–540. doi: 10.1007/s10792-013-9851-2. [DOI] [PubMed] [Google Scholar]
- 164.Ghassemi F, Shields CL, Ghadimi H, Khodabandeh A, Roohipoor R. Combined intravitreal melphalan and topotecan for refractory or recurrent vitreous seeding from retinoblastoma. JAMA ophthalmology. 2014;132:936–941. doi: 10.1001/jamaophthalmol.2014.414. [DOI] [PubMed] [Google Scholar]
- 165.Friedman DN, et al. Long-term medical outcomes in survivors of extra-ocular retinoblastoma: the Memorial Sloan-Kettering Cancer Center (MSKCC) experience. Pediatr Blood Cancer. 2013;60:694–699. doi: 10.1002/pbc.24280. [DOI] [PubMed] [Google Scholar]
- 166.Bansal M, Patel FD, Mohanti BK, Sharma SC. Setting up a palliative care clinic within a radiotherapy department: a model for developing countries. Supportive care in cancer: official journal of the Multinational Association of Supportive Care in Cancer. 2003;11:343–347. doi: 10.1007/s00520-002-0418-4. [DOI] [PubMed] [Google Scholar]
- 167.Willard VW, et al. Developmental and adaptive functioning in children with retinoblastoma: a longitudinal investigation. J Clin Oncol. 2014;32:2788–2793. doi: 10.1200/JCO.2013.53.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Brinkman TM, et al. Cognitive function and social attainment in adult survivors of retinoblastoma: a report from the St. Jude Lifetime Cohort Study. Cancer. 2015;121:123–131. doi: 10.1002/cncr.28924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tobin M, Hill E, Hill J. Retinoblastoma and superior verbal IQ scores? British Journal of Visual Impairment. 2010;28:7–18. [Google Scholar]
- 170.Kelly KR, McKetton L, Schneider KA, Gallie BL, Steeves JK. Altered anterior visual system development following early monocular enucleation. NeuroImage Clinical. 2014;4:72–81. doi: 10.1016/j.nicl.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kelly KR, DeSimone KD, Gallie BL, Steeves JK. Increased cortical surface area and gyrification following long-term survival from early monocular enucleation. NeuroImage Clinical. 2015;7:297–305. doi: 10.1016/j.nicl.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gonzalez EG, Lillakas L, Lam A, Gallie BL, Steinbach MJ. Horizontal saccade dynamics after childhood monocular enucleation. Investigative ophthalmology & visual science. 2013;54:6463–6471. doi: 10.1167/iovs.13-12481. [DOI] [PubMed] [Google Scholar]
- 173.Rappaport B, Mellon RD, Simone A, Woodcock J. Defining safe use of anesthesia in children. The New England journal of medicine. 2011;364:1387–1390. doi: 10.1056/NEJMp1102155. [DOI] [PubMed] [Google Scholar]
- 174.Ing C, et al. Long-term differences in language and cognitive function after childhood exposure to anesthesia. Pediatrics. 2012;130:e476–485. doi: 10.1542/peds.2011-3822. [DOI] [PubMed] [Google Scholar]
- 175.Khin Hla T, et al. Perception of pediatric pain: a comparison of postoperative pain assessments between child, parent, nurse, and independent observer. Paediatric anaesthesia. 2014;24:1127–1131. doi: 10.1111/pan.12484. [DOI] [PubMed] [Google Scholar]
- 176.Chen E, Zeltzer LK, Craske MG, Katz ER. Children’s memories for painful cancer treatment procedures: implications for distress. Child development. 2000;71:933–947. doi: 10.1111/1467-8624.00200. [DOI] [PubMed] [Google Scholar]
- 177.Dahlquist LM, Pendley JS. When distraction fails: parental anxiety and children’s responses to distraction during cancer procedures. J Pediatr Psychol. 2005;30:623–628. doi: 10.1093/jpepsy/jsi048. [DOI] [PubMed] [Google Scholar]
- 178.van Dijk J, et al. Behavioural functioning of retinoblastoma survivors. Psychooncology. 2009;18:87–95. doi: 10.1002/pon.1381. [DOI] [PubMed] [Google Scholar]
- 179.van Dijk J, et al. Coping strategies of retinoblastoma survivors in relation to behavioural problems. Psychooncology. 2009;18:1281–1289. doi: 10.1002/pon.1507. [DOI] [PubMed] [Google Scholar]
- 180.Gold JI. RESEARCHLA Blog. Vol. 2015. Children’s Hospital Los Angeles The Saban Research Institute; Los Angeles: [Google Scholar]
- 181.Committee on Hospital Care & Child Life Council. Child life services. Pediatrics. 2014;133:e1471–1478. doi: 10.1542/peds.2014-0556. [DOI] [PubMed] [Google Scholar]
- 182.O’Callaghan C, Sexton M, Wheeler G. Music therapy as a non-pharmacological anxiolytic for paediatric radiotherapy patients. Australasian radiology. 2007;51:159–162. doi: 10.1111/j.1440-1673.2007.01688.x. [DOI] [PubMed] [Google Scholar]
- 183.Post-White J, et al. Massage therapy for children with cancer. Journal of pediatric oncology nursing: official journal of the Association of Pediatric Oncology Nurses. 2009;26:16–28. doi: 10.1177/1043454208323295. [DOI] [PubMed] [Google Scholar]
- 184.Hildenbrand AK, Clawson KJ, Alderfer MA, Marsac ML. Coping with pediatric cancer: strategies employed by children and their parents to manage cancer-related stressors during treatment. Journal of pediatric oncology nursing: official journal of the Association of Pediatric Oncology Nurses. 2011;28:344–354. doi: 10.1177/1043454211430823. [DOI] [PubMed] [Google Scholar]
- 185.van Dijk J, et al. Restrictions in daily life after retinoblastoma from the perspective of the survivors. Pediatr Blood Cancer. 2010;54:110–115. doi: 10.1002/pbc.22230. [DOI] [PubMed] [Google Scholar]
- 186.van Dijk J, et al. Quality of life of adult retinoblastoma survivors in the Netherlands. Health and quality of life outcomes. 2007;5:30. doi: 10.1186/1477-7525-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Shinohara ET, DeWees T, Perkins SM. Subsequent malignancies and their effect on survival in patients with retinoblastoma. Pediatr Blood Cancer. 2014;61:116–119. doi: 10.1002/pbc.24714. [DOI] [PubMed] [Google Scholar]
- 188.Ford JS, Chou JF, Sklar CA. Attendance at a survivorship clinic: impact on knowledge and psychosocial adjustment. Journal of cancer survivorship: research and practice. 2013;7:535–543. doi: 10.1007/s11764-013-0291-9. [DOI] [PubMed] [Google Scholar]
- 189.Schulz CJ, Riddle MP, Valdimirsdottir HB, Abramson DH, Sklar CA. Impact on survivors of retinoblastoma when informed of study results on risk of second cancers. Medical and pediatric oncology. 2003;41:36–43. doi: 10.1002/mpo.10278. [DOI] [PubMed] [Google Scholar]
- 190.Rowland E, Metcalfe A. Communicating inherited genetic risk between parent and child: a meta-thematic synthesis. International journal of nursing studies. 2013;50:870–880. doi: 10.1016/j.ijnurstu.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 191.Clarke SA, Sheppard L, Eiser C. Mothers’ Explanations of Communicating Past Health and Future Risks to Survivors of Childhood Cancer. Clinical Child Psychology and Psychiatry. 2008;13:157–170. doi: 10.1177/1359104507080997. [DOI] [PubMed] [Google Scholar]
- 192.Weaver RR. Seeking high reliability in primary care: Leadership, tools, and organization. Health care management review. 2014 doi: 10.1097/HMR.0000000000000022. [DOI] [PubMed] [Google Scholar]
- 193.Jha P, et al. Prospective study of one million deaths in India: rationale, design, and validation results. PLoS Med. 2006;3:e18. doi: 10.1371/journal.pmed.0030018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Farmer P. Who Lives and Who Dies? Medical Examiner. 2015 [Google Scholar]
- 195.Ladas EJ, et al. Improving our understanding of the use of traditional complementary/alternative medicine in children with cancer. Cancer. 2015;121:1492–1498. doi: 10.1002/cncr.29212. [DOI] [PubMed] [Google Scholar]
- 196.Farmer P, Mukherjee J. Ebola: Countries Need ‘Staff, Stuff, and Systems’. 2014 < http://www.pih.org/blog/for-ebola-countries-need-tools-to-treat-patients-in-their-communities>.
- 197.Panton RL, et al. A visual approach to providing prognostic information to parents of children with retinoblastoma. Psychooncology. 2009;18:300–304. doi: 10.1002/pon.1397. [DOI] [PubMed] [Google Scholar]
- 198.Chiu HH, Dimaras H, Downie R, Gallie B. Breaking down barriers to communicating complex retinoblastoma information: can graphics be the solution? Can J Ophthalmol. 2015;50:230–235. doi: 10.1016/j.jcjo.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 199.Grossmann C, Sanders J, English RA. KNOWLEDGE GENERATION IN A LEARNING HEALTH SYSTEM: Workshop Summary. National Academies Press; 2013. [PubMed] [Google Scholar]
- 200.Laurie NA, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–66. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- 201.Pritchard EM, et al. Pharmacokinetics and efficacy of the spleen tyrosine kinase inhibitor r406 after ocular delivery for retinoblastoma. Pharmaceutical research. 2014;31:3060–3072. doi: 10.1007/s11095-014-1399-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Mahida JP, et al. A synergetic screening approach with companion effector for combination therapy: application to retinoblastoma. PLoS ONE. 2013;8:e59156. doi: 10.1371/journal.pone.0059156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Taich P, et al. Clinical pharmacokinetics of intra-arterial melphalan and topotecan combination in patients with retinoblastoma. Ophthalmology. 2014;121:889–897. doi: 10.1016/j.ophtha.2013.10.045. [DOI] [PubMed] [Google Scholar]
- 204.Carcaboso AM, et al. Episcleral implants for topotecan delivery to the posterior segment of the eye. Investigative ophthalmology & visual science. 2010;51:2126–2134. doi: 10.1167/iovs.09-4050. doi:iovs.09-4050 [pii]10.1167/iovs.09-4050. [DOI] [PubMed] [Google Scholar]
- 205.Kang SJ, Durairaj C, Kompella UB, O’Brien JM, Grossniklaus HE. Subconjunctival nanoparticle carboplatin in the treatment of murine retinoblastoma. Archives of ophthalmology. 2009;127:1043–1047. doi: 10.1001/archophthalmol.2009.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Oronsky B, et al. The war on cancer: a military perspective. Frontiers in oncology. 2014;4:387. doi: 10.3389/fonc.2014.00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wang H, et al. Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1+/- mice. Nat Genet. 2010;42:83–88. doi: 10.1038/ng.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Puissant A, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer discovery. 2013;3:308–323. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Chan CH, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154:556–568. doi: 10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.