

Abstract
Background:
Protein tyrosine phosphatase non-receptor type 1 is a therapeutic target for the type 2 diabetes mellitus. According to the International Diabetes Federation 2015 report, one out of 11 adults suffers from diabetes mellitus globally.
Objective:
Current anti-diabetic drugs can cause life-threatening side-effects. The present study proposes a pipeline for the development of effective and plant-derived anti-diabetic drugs that may be safer and better tolerated.
Methods:
Plant-derived protein tyrosine phosphatase non-receptor type 1 inhibitors possessing antidiabetic activity less than 10μM were used as a training set. A common feature pharmacophore model was generated. Pharmacophore-based screening of plant-derived compounds of the ZINC database was conducted using ZINCpharmer. Screened hits were assessed to evaluate their drug-likeness, pharmacokinetics, detailed binding behavior, and aggregator possibility based on their physiochemical properties and chemical similarity with reported aggregators.
Results:
Through virtual screening and in silico pharmacology protocol isosilybin (ZINC30731533) was identified as a lead compound with optimal properties. This compound can be recommended for laboratory tests and further analyses to confirm its activity as protein tyrosine phosphatase non-receptor type 1 inhibitor.
Conclusion:
The present study has identified plant-derived anti-diabetic virtual lead compound with the potential to inhibit protein tyrosine phosphatase non-receptor type 1, which may be helpful to enhance insulin production. This computer-aided study could facilitate the development of novel pharmacological inhibitors for diabetes treatment.
Keywords: Computer-aided drug design, diabetes mellitus, flavonoids, isosilybin, protein tyrosine phosphatase non-receptor type 1, common feature pharmacophore modeling, molecular docking, pharmacokinetics
1. INTRODUCTION
Plant-based medicine is a way to treat diabetes mellitus (DM). Traditional medicine has employed huge collection of plant-derived treatments effective in treatment of blood glucose imbalance and diabetes mellitus [1, 2]. Experimental studies have shown that the insulin-like growth factor domains of human insulin are common to the insulin sequence found in Canavalia ensiformis, Vigna unguiculata and Bauhinia purpurea [3]. Computer-aided molecular docking methods were applied to human insulin protein [4] and plant insulin present in Canavalia ensiformis to identify anti-diabetic compounds [5].
In our previous study, we discussed U.S. Food and Drug Administration (FDA) approved anti-DM medicines; insulin, biguanides, second generation sulfonylureas, alpha- glucosidase inhibitors, glinides, glucagon-like peptide-1 receptor agonists, thiazolidinediones, dipeptidyl peptidase-4 (DPP-4) inhibitors, bile acid sequestrants, dopamine agonists, amylin analogs, and sodium-dependent glucose cotransporter-2 inhibitors in detail [6]. However, currently available anti-DM drugs possess side effects such as headache, stomach upset, peripheral edema, increase in weight, and hypotension [7]. Therefore, compounds with ideal properties to stimulate insulin signaling pathway are required [8].
Molecular targets for pharmacological treatments of DM have been studied to develop unique anti-DM agents, including protein tyrosine phosphatase non-receptor type 1 (PTPN1) also known as protein tyrosine phosphatase 1B (PTP1B), peroxisome proliferator-activated receptor gamma, pyruvate dehydrogenase kinase, beta 3 adrenoceptors, glycogen synthase kinase 3, DPP-4, cannabinoid receptors, and fructose bisphosphatases enzymes [9, 10]. The protein tyrosine phosphatases are enzymes that catalyze protein tyrosine dephosphorylation in regulation of insulin action by dephosphorylation of activated auto phosphorylated insulin receptor and downstream substrate proteins [11]. The PTPN1 has been a target for treatment of diabetes and obesity [12], and PTPN1 knockout mice had insulin sensitivity and tolerance to diet-induced obesity [13, 14]. Recent technical advances in chemical synthesis have resulted in the design of potent synthetic PTPN1 inhibitors, but difficulties such as high polarity and low enzyme selectivity remain to be overcome [15]. The use of natural products has appreciated as an alternative source for discovery of PTPN1 inhibitors [16]. In vitro and in vivo methods confirmed that natural products are beneficial for discovery of new and potential PTPN1 inhibitors [11].
In the present study, we have discussed structural, biological and molecular activities of diverse plant-derived PTPN1 compounds reported in the last decades. We used computer-aided drug design (CADD) strategies for identification of novel compounds having PTPN1 inhibitory activity from the ZINC dataset of plant-derived compounds, which will be beneficial for medicinal chemist and pharmacologists to develop new PTPN1 inhibitors with anti-DM activity.
2. MaterialS and Method
2.1. Pharmacophore Modeling and Computer-based Screening of ZINC Database
In recent years, various experimental approaches have been developed to investigate flavonoids with PTPN1 inhibitory activity by incorporating novel approaches to previously tested models to improve their anti-DM activity. Botanical information, chemical structure and physicochemical properties of natural flavonoids with PTPN1 inhibitory activity were selected from reported data (Table 1) [17-22]. Eleven compounds were used as a training set based on their physiochemical properties, Lipinski’s filter, and IC50 values less than 10μM. These 11 compounds were used for pharmacophore modeling using LigandScout 4.1 [23]. ChemDraw Ultra 8.0 software [24] is used for sketching chemical structure of training dataset and saved in Protein Data Bank (PDB) format. Consequently, these files were used as input to LigandScout 4.1. A pharmacophore fit model was generated using the 11 compounds of training set and used for screening of plant-derived set of ZINC database. Table 2 shows pharmacophore features of the training set and common feature of a selected pharmacophore model. Pharmacophore features of the most appropriate model were also generated for each compound displayed in Table 3.
Table 1.
Selected compounds that possess Protein tyrosine phosphatase non receptor type 1 inhibitory activity used as a training set.
| Compounds | 2D Structure | Potency (IC50) | Physiochemical Properties | Source | Place of Origin | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 |

|
4.3 μM | MW: 424.491 cLogP: 5.5969 HBA: 6 HBD: 3 PSA: 96.22 RB: 3 |
Broussonetia Papyrifera (Extract of roots) |
China | [17] | ||||||||
| F2 |  |
2.6 μM | MW: 424.491 cLogP: 5.5969 HBA: 6 HBD: 3 PSA: 96.22 RB: 3 |
Erythrina addisoniae (EtOAc extract of the stem bark) |
West tropical Africa | [18] | ||||||||
| F3 |  |
4.1 μM | MW: 422.475 cLogP: 5.4331 HBA: 6 HBD: 3 PSA: 96.22 RB: 3 |
Erythrina addisoniae (EtOAc extract of the stem bark) |
West tropical Africa | [18] | ||||||||
| F4 |  |
7.6 μM | MW: 324.375 cLogP: 4.4262 HBA: 4 HBD: 1 PSA: 47.92 RB: 0 |
Erythrina abyssinica (Extract of stem bark) |
Africa (Nigeria) | [19] | ||||||||
| F5 |  |
8.8 μM | MW: 406.476 cLogP: 5.2725 HBA: 5 HBD: 2 PSA: 68.15 RB: 2 |
Erythrina abyssinica (Extract of stem bark) |
Africa (Nigeria) | [19] | ||||||||
| F6 |  |
6.0 μM | MW: 350.413 cLogP: 5.6154 HBA: 4 HBD: 2 PSA: 58.92 RB: 3 |
Erythrina abyssinica (Extract of stem bark) |
Africa (Nigeria) | [19] | ||||||||
| F7 |  |
9.7 μM | MW: 408.492 cLogP: 5.5966 HBA: 5 HBD: 2 PSA: 68.15 RB: 2 |
Erythrina abyssinica (Extract of stem bark) |
Africa (Nigeria) | [20, 21] |
||||||||
| F8 |  |
4.1 μM | MW: 336.386 cLogP: 4.9685 HBA: 4 HBD: 1 PSA: 51.83 RB: 3 |
Erythrina abyssinica (Extract of stem bark) |
Africa (Nigeria) | [20] | ||||||||
| F9 |  |
7.6 μM | MW: 324.375 cLogP: 4.9125 HBA: 4 HBD: 2 PSA: 58.92 RB: 2 |
Erythrina abyssinica (Extract of stem bark) |
Africa (Nigeria) | [20] | ||||||||
| F10 |  |
4.6 μM | MW: 354.401 cLogP: 4.6821 HBA: 5 HBD: 2 PSA: 75.99 RB: 4 |
Erythrina addisoniae (EtOAc extract of roots) |
West tropical Africa, Nigeria, Congo. | [21] | ||||||||
| F11 |  |
6.70 μM | MW: 288.254 cLogP: 1.81 HBA: 6 HBD: 4 PSA: 107.22 RB: 1 |
Abundantly present in various fruits and vegetables, e.g. Salvia tomentosa, Aiphanesaculeata. |
Various regions in the world | [22] | ||||||||
MW=molecular weight, cLogP= Partition coefficient, HBA=Hydrogen bond acceptor, HBD=hydrogen bond donor, PSA=polar surface area, and RB= rotatable bonds.
Table 2.
Pharmacophore features of the training set and common pharmacophore feature of a selected pharmacophore model.
| Compounds | HR | AR | HBA | HBD | Number of Confirmations | Common Pharmacophoric Feature | Pharmacophoric Fit |
|---|---|---|---|---|---|---|---|
| F1 | 5 | 2 | 7 | 5 | 54 | 5 | 56.30 |
| F2 | 5 | 2 | 6 | 3 | 65 | 6 | 64.01 |
| F3 | 6 | 2 | 6 | 2 | 28 | 6 | 65.02 |
| F4 | 3 | 2 | 4 | 1 | 4 | 6 | 65.37 |
| F5 | 6 | 2 | 5 | 2 | 21 | 6 | 65.34 |
| F6 | 5 | 2 | 4 | 2 | 23 | 6 | 64.62 |
| F7 | 5 | 2 | 5 | 2 | 49 | 6 | 65.34 |
| F8 | 4 | 3 | 2 | 1 | 5 | 5 | 48.49 |
| F9 | 3 | 2 | 4 | 2 | 19 | 6 | 65.34 |
| F10 | 3 | 2 | 4 | 2 | 106 | 6 | 65.07 |
| F11 | 1 | 2 | 6 | 4 | 6 | 5 | 56.62 |
Table 3.
Overlay of training set compounds upon the pharmacophore generated using LigandScout 4.1.

|
*Red spheres represent hydrogen bond acceptors, yellow spheres represent hydrophobic regions, and purple spheres represent aromatic rings.
Screening procedures were performed using a shared feature pharmacophore modeling approach for best flexible conformation exploration using ZINCpharmer [25]. The identified hits as outcome of the database search were subjected to drug-like filtration. Data Warrior [26] was used to calculate the physiochemical properties and toxicity estimation. Lipinski’s filter was applied to the screened dataset [27]. Partition coefficient logP values should be less than 5 or clogP values should be less than 6, molecular weight should be less than 500, hydrogen bond acceptors should be less 10, and hydrogen bond donors should be less than 5. Veber’s rule (rotatable bonds must be less than 10 while value of polar surface area must be less than 120 Å) was also considered because molecular flexibility of selected compounds is dependent on the number of rotatable bonds, an important property which influences bioavailability of drugs [28, 29]. Compounds were short-listed based on drug-likeness and were subsequently checked for toxicity using four criteria (mutagenicity, tumorigenicity, irritant and reproductive effects).
2.2. Molecular Docking and Interaction Analyses
The prerequisite of docking for the PTPN1 dataset is knowledge of the target protein structure. The target protein (ID: 3EAX) was downloaded from the PDB; its X-ray crystallographic structure demonstrated a high resolution of 1.9 Å [30]. The existing active site for ligand binding used in the nuclear magnetic resonance (NMR) study of 3EAX structure was used for the molecular docking study of screened hits. Plant-derived compounds from the ZINC database, after passing through drug-like filters, were docked into the binding pocket of the PTPN1 protein using the CLC drug discovery workbench tool [31]. A detail investigation of active site of target protein was performed to check if the significant residues which are responsible for activity were included in the binding site or not. The compound which holds the selected binding site was specified. After compound selection, docking was performed. The docking results summary was displayed in the project with dock scores. A 3D view was selected for manual inspection of the structural features of the docked complex involved in binding.
The docked complex in a mol format file was imported to chimera [32] and was saved in PDB format for interaction analysis in Ligplot. The ligand-protein interactions were predicted using Ligplot [33]. It generates 2D schematic diagrams of a docked complex to explain interactions with hydrophobic moieties and hydrogen bonds having a distance within 4 Å.
2.3. ADME Calculation and Aggregator Advisor Prediction
In silico ADME (absorption, distribution, metabolism and excretion) calculations are steadily gaining interest in computer aided drug discovery [34]. These methods are used here to shortlist data with suitable pharmacokinetic (ADME) and toxicity profiles in early phase of PTPN1 drug discovery. SwissADME [35] is used to calculate the pharmacological profile (drug action/effects within an organism) of selected 15 hits from the perspective of drug discovery. This online tool is also used to determine toxic structural moieties and synthetic accessibility of selected hits.
Aggregator possibility evaluation for selected 15 hits was conducted by Aggregator advisor [36]. This online tool compares chemical similarity of known aggregator compounds based on Tanimoto coefficient calculation and physiochemical properties. Input was provided using a SMILES file extension to obtain a reliable ADME prediction and information on already reported aggregates for each compound.
2.4. Lead Identification
After the systematic analyses of all compounds, lead compound was identified as a chemical compound that has the best pharmacological or biological activity against the PTPN1 therapeutic target. In detail, the lead compound was identified based on the best drug-likeness, pharmacokinetic properties, molecular docking and best binding interactions with the significant residues involved in binding the NMR structure of the target protein. The aggregator properties were used along with the other parameters to shortlist hits as lead compound.
3. Results
3.1. Training Set Selection
Flavonoids have extensive variety of biological activities, including anti-diabetic, anti-inflammatory, anti-oxidative, anti-allergic, anti-proliferative, anti-viral, anti-cancer activities, stomach and liver protection [37]. Initially, we selected 30 compounds reported in the literature in the last decade whose PTPN1 inhibitory activity is less than 10 μM. Drug-likeness filters were applied and 11 compounds fulfilled the criteria of drug-likeness. These are flavonoid compounds with PTPN1 inhibitory activity. Inhibitory activities against PTPN1 evaluated using experimental analyses, 2D structures, and physiochemical properties confirming drug-like properties, origin and botanical information concerning source plant species are listed in Table 1. This information is useful to understand the importance of these plants for anti-DM medicines.
3.2. Pharmacophore Generation
To achieve the goal of the ligand-based pharmacophore modeling using LigandScout 4.1, eleven compounds were used as input. Possible lowest energy conformations for each compound were generated (Table 2) and all conformations of least flexible compounds were then aligned. For a configurable number of best alignment solutions; common pharmacophoric features were interpolated and ten hypothetical pharmacophore models were created. The score generated for these models is shown in Fig. (1). Pharmacophore models were ranked using several adjustable scoring functions taking into account chemical feature overlap, steric overlap, or both. Pharmacophore models are set of common feature pharmacophores created by processing all compounds of the dataset. If minimum three common functional features can be identified by alignment and interpolation process, common feature pharmacophore generation is considered to be successful [23].
Fig. (1).

Ten pharmacophore hypothetical models (lower panel) were generated for eleven compounds using LigandScout 4.1. Six features are the best fit to generate the best pharmacophore model. The proposed pharmacophore model (model 1 shown in upper panel) used in this study contains three HBAs (red spheres), two ARs (purple spheres) and one HR (yellow spheres). (The color version of the figure is available in the electronic copy of the article).
The model 1 with the highest score is selected for a database search to retrieve similar hits from the plant-derived set of the ZINC database. Common features are important for the activity of compounds. The pharmacophore generated using 11 compounds contained four types of pharmacophore features: hydrogen bond acceptors (HBAs), hydrogen bond donors (HBDs), hydrophobic regions (HRs), and aromatic rings (ARs). Table 2 shows the total features present in each compound and the number of features which are common and the best fit to build the best model. Six features are the best fit to generate the best pharmacophore model. The best pharmacophore model (model 1) in this study contains three HBAs, two ARs and one HR, as shown in Fig. (1). Red spheres represent HBAs, purple spheres represent ARs and yellow spheres represent HRs for overlay of each compound of dataset upon the pharmacophore generated as shown in Table 3.
3.3. Computer-based Screening of the Plant-derived Set of the ZINC Database
To identify new plant-derived PTPN1 hits, pharmacophore based screening was conducted against 22,723,923 plant-derived compounds from the ZINC database and 6061 compounds fit with the pharmacophore query. Several filters were selected before screening by ZINCpharmer. These criteria dictated that the molecular mass of the compound should be less than 500 Daltons, the number of rotatable bonds should be less than 10 [38], the maximum hits per configuration should be one, the maximum hits per molecule should be one and the maximum root-mean-square deviation (RMSD) value for screened hits should be 1.5. The identified pharmacophore fit compounds from the databases were required to be drug-like. Therefore, Data Warrior was used for screening to calculate the physiochemical properties and toxicity estimation. Lipinski’s and Veber’s rules were applied to the screened dataset and reduced it to 4349 compounds. The dataset was subjected to a toxicity estimation using four criteria (mutagenicity, tumorigenicity, irritant and reproductive effects) and 2636 compounds with no risk of toxicity were retrieved. These compounds were used for molecular docking using the CLC drug discovery workbench and the 15 top scoring compounds were considered for interaction analyses using Ligplot. Then ADME calculations were performed for the selected 15 top scored hits to confirm their pharmacokinetic profile and medicinal parameters. Summary of drug-likeness and pharmacokinetic properties of the selected 15 selected hits are shown in Table 4. Many of the selected hits were shown to interact with the cytochrome P450 isoforms. Tyr46, Asp48, Ser216, Ala217 and Arg221 residues of target proteins are mostly involved in the binding of 15 hits as shown in Table 5. Summary of ligand-protein binding analyses of selected 15 hits is shown in Table 6. Synthetic accessibility score (SAS) based on the fragmental analyses of the structures of virtual hits is acceptable for easy synthesis in laboratories. The 11 hits demonstrated no drug safety alerts. The aggregator potential through comparison with previously reported aggregators is shown only for the four hits demonstrated in Table 7. A lead compound was identified after a series of filters based on a CADD scheme (Fig. 2).
Table 4.
Summary of drug-likeness and pharmacokinetic properties of the 15 selected virtual hits.
| Virtual Hits | Oral Bioavailability | Pharmacokinetic Properties |
Log Kp
(Skin Permeation) |
Water Solubility | Toxicity Estimation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MW | cLogP | HBA | HBD | RB | PSA | B-Score | |||||
| ZINC06137783 | 450.581 | 3.2659 | 7 | 2 | 5 | 73.91 | 0.55 | GI absorption BBB permeant P-gp substrate CYP2C9 inhibitor CYP2D6 inhibitor CYP3A4 inhibitor |
−6.19 | Moderately soluble | No risk |
| ZINC04259062 | 464.545 | 2.8826 | 8 | 2 | 4 | 119.22 | 0.55 | GI absorption P-gp substrate CYP1A2 inhibitor CYP2C9 inhibitor CYP2D6 inhibitor CYP3A4 inhibitor |
−7.34 | Moderately soluble | No risk |
| ZINC03841413 | 460.553 | 2.4036 | 9 | 2 | 5 | 116.43 | 0.55 | GI absorption P-gp substrate CYP3A4 inhibitor |
−8.02 | Soluble | No risk |
| ZINC04277683 | 458.516 | 3.016 | 8 | 2 | 4 | 90.98 | 0.55 | GI absorption P-gp substrate CYP2C9 inhibitor CYP2D6 inhibitor CYP3A4 inhibitor |
−7.31 | Moderately soluble | No risk |
| ZINC04259056 | 476.507 | 3.1168 | 8 | 2 | 4 | 90.98 | 0.55 | GI absorption P-gp substrate CYP1A2 inhibitor CYP2C9 inhibitor CYP2D6 inhibitor CYP3A4 inhibitor |
−7.35 | Moderately soluble | No risk |
| ZINC04259064 | 458.497 | 2.0691 | 9 | 2 | 4 | 103.87 | 0.55 | GI absorption P-gp substrate CYP2C9 inhibitor CYP2D6 inhibitor CYP3A4 inhibitor |
−7.84 | Soluble | No risk |
| ZINC05535232 | 415.448 | 3.7749 | 7 | 2 | 1 | 89.95 | 0.55 | GI absorption CYP2C19 inhibitor CYP2C9 inhibitor CYP3A4 inhibitor |
−6.90 | Moderately soluble | No risk |
| ZINC04237088 | 445.521 | 4.4229 | 5 | 0 | 2 | 64.41 | 0.55 | GI absorption BBB permeant CYP2C19 inhibitor CYP2C9 inhibitor CYP3A4 inhibitor |
−5.91 | Moderately soluble | No risk |
| ZINC13733603 | 421.448 | 3.5857 | 7 | 0 | 5 | 74.3 | 0.55 | GI absorption CYP1A2 inhibitor CYP2C19 inhibitor CYP2C9 inhibitor CYP2D6 inhibitor CYP3A4 inhibitor |
−5.97 | Moderately soluble | No risk |
| ZINC41585804 | 472.539 | 3.3751 | 7 | 1 | 6 | 71.47 | 0.55 | GI absorption BBB permeant P-gp substrate CYP2C19 inhibitor CYP2C9 inhibitor CYP2D6 inhibitor |
−6.20 | Moderately soluble | No risk |
| ZINC00004749 | 316.308 | 2.4978 | 6 | 4 | 1 | 107.22 | 0.55 | GI absorption BBB permeant P-gp substrate CYP2C19 inhibitor CYP2C9 inhibitor CYP2D6 inhibitor |
−6.20 | Moderately soluble | No risk |
| ZINC02093367 | 424.451 | 4.2868 | 5 | 0 | 4 | 61.83 | 0.55 | GI absorption CYP2C19 inhibitor CYP2C9 inhibitor CYP3A4 inhibitor |
−5.40 | Moderately soluble | No risk |
| ZINC30731533 | 482.44 | 2.1266 | 10 | 5 | 4 | 155.14 | 0.55 | GI absorption CYP3A4 inhibitor |
−7.89 | Moderately soluble | No risk |
| ZINC00968072 | 274.271 | 2.3608 | 5 | 4 | 1 | 90.15 | 0.55 | GI absorption P-gp substrate |
−7.02 | Soluble | No risk |
| ZINC13722309 | 485.512 | 2.4048 | 9 | 0 | 5 | 116.82 | 0.55 | GI absorption CYP2C19 inhibitor CYP2C9 inhibitor CYP3A4 inhibitor |
−7.30 | Moderately soluble | No risk |
* The toxicity estimation used four major criteria (mutagenicity, tumorigenicity, reproductive effects and irritant effects), Cytochrome p4501A2=CYP1A2, Cytochrome p4502C19=CYP2C19, Cytochrome p4502C9=CTP2C9, BBB permeant =blood brain barrier permeability, GI absorption= Gastrointestinal drug absorption and P-gp substrate= P-glycoprotein substrate.
Table 5.
Conserved interacting residues within the binding site of the target protein of the top scored 15 virtual hits.
| Virtual Hits | Tyr46 | Asp48 | Val49 | Ser216 | Ala217 | Gly220 | Arg221 | Gln262 | Gln266 |
|---|---|---|---|---|---|---|---|---|---|
| ZINC06137783 | + | + | − | + | + | + | + | − | + |
| ZINC04259062 | + | + | − | + | + | + | + | + | − |
| ZINC03841413 | + | − | − | + | + | + | + | + | + |
| ZINC04277683 | + | + | − | + | + | + | + | − | + |
| ZINC04259056 | + | + | − | + | + | + | + | + | + |
| ZINC04259064 | + | + | − | + | + | + | + | + | − |
| ZINC05535232 | + | + | + | + | + | + | + | − | − |
| ZINC04237088 | − | − | − | + | + | − | + | + | + |
| ZINC13733603 | + | + | − | + | + | + | + | − | − |
| ZINC41585804 | − | + | − | + | + | + | + | + | + |
| ZINC00004749 | + | + | − | + | − | − | + | + | + |
| ZINC02093367 | + | + | − | − | + | + | + | + | − |
| ZINC30731533 | + | + | − | + | + | − | + | − | − |
| ZINC00968072 | + | + | + | − | + | + | + | + | + |
| ZINC13722309 | + | + | − | + | + | − | + | + | − |
* If key residues are present in the binding interaction within 4Å of the binding site of the target protein, then this is represented by “+”. If the residues are not present this is represented by “−”.
Table 6.
Summary of molecular docking analyses of selected 15 virtual hits.
| Virtual Hits | RMSD | Score | Ligand Conformation Penalty | Interacting Residues in the Active Binding Site | No. of Hydrogen Bonds | No. of Hydrophobic Bonds | Total Number of Bonds |
|---|---|---|---|---|---|---|---|
| ZINC06137783 | 0.95 | −55.125 | 1.76 | Tyr46, Asp48, Phe182, Gly183, Cys215, Ser216, Ala217, Ile219, Gly220, Arg221, Gln266. |
0 | 29 | 29 |
| ZINC04259062 | 0.95 | −55.133 | 4.07 | Asp48, Lys116, Ala217, Ser216, Ile219, Gly220, Arg221, Gln262. |
0 | 28 | 28 |
| ZINC03841413 | 0.95 | −53.418 | 2.25 | Tyr48, Phe182, Gly183, Asp184, Cys215, Ser216, Ala217, Ile219, Gly220,Arg221,Gln262, Gln266. |
0 | 29 | 29 |
| ZINC04277683 | 0.95 | −52.773 | 4.26 | Tyr46, Asp48, Lys116, Phe182, Gly183, Cys215, Ser216, Ile219, Ala217,Gly220,Arg221, Gln266. |
0 | 29 | 29 |
| ZINC04259056 | 0.95 | −52.545 | 4.26 | Tyr46, Asp48, Lys116, Phe182, Ser216, Ala217, Ile219, Gly220, Arg221, Gln262, Gln266. |
0 | 28 | 28 |
| ZINC04259064 | 0.95 | −51.956 | 4.70 | Tyr46, Asp48, Lys116, Phe182, Ser216, Ala217, Ile219, Gly220, Arg221, Gln262. |
1 [Lys116: (2.88Å)] |
29 | 30 |
| ZINC05535232 | 1.03 | −51.241 | 0.35 | Tyr46, Asp48, Val49, Lys116, Lys120, Cys215,Ser216, Ala217, Gly220, Arg221. |
2 [Lys116: (3.00Å)] [Ser216: (3.24Å)] |
33 | 35 |
| ZINC04237088 | 1.11 | −51.215 | 6.53 | Phe182, Gly183,Cys215, Ser216, Ala217, Ile219, Gly220, Arg221, Gln262, Thr263, Gln266. | 0 | 29 | 29 |
| ZINC13733603 | 0.68 | −51.169 | 3.00 | Phe182, Gly183,Cys215, Ser216, Ala217, Ile219, Gly220, Arg221, Gln262, Thr263, Glu266. |
0 | 29 | 29 |
| ZINC41585804 | 0.57 | −50.542 | 5.69 | Tyr46, Asp48, Phe182, Gly183, Ser216, Ala217,Gly220, Arg221, Gln262, Thr263, Gln266. |
3 [Gln266: (3.01Å)] [Arg221: (3.12Å)] [Tyr46: (2.82Å)] |
28 | 31 |
| ZINC00004749 | 0.84 | −50.387 | 0.36 | Asp48, Tyr46, Trp179, Ser216, Arg221,Gln262,Thr263, Gln266. |
4 [Asp48: (2.63Å)] [Ser216: (2.99Å)] [Arg221: (2.73Å)] [Arg221: (2.64Å)] |
21 | 25 |
| ZINC02093367 | 0.94 | −50.372 | 3.44 | Tyr46, Asp48, Cys215, Ala217, Ile219, Gln262, Arg221, Gly220. |
3 [Arg47: (2.80Å)] [Arg47: (3.01Å)] [Arg47: (2.89Å)] |
20 | 23 |
| ZINC30731533 | 0.79 | −50.331 | 2.61 | Tyr46, Arg47, Asp48, Glu115, Lys120, Asp181,Ser216, Ala217, Arg221. |
7 [Asp48: (3.26Å)] [Arg47: (2.63Å)] [Arg47: (3.17Å)] [Asp181: (3.01Å)] [Ala217: (3.18Å)] [Glu115: (3.29Å)] [Arg221: (2.59Å)] |
30 | 37 |
| ZINC00968072 | 0.82 | −50.315 | 0.36 | Tyr46,Val49,Asp48,Cys215, Ala217,Arg221,Gly220, Gln266. |
5 [Gln266: (2.73Å)] [Arg221: (3.17Å)] [Arg221: (2.91Å)] [Cys215: (2.59Å)] [Asp48: (2.60Å)] |
20 | 25 |
| ZINC13722309 | 0.54 | −50.239 | 3.01 | Tyr46,Arg47,Asp48,Gln262, Ala217, Ile219, Cys215. | 3 [Arg221: (3.12Å)] [Ser216: (3.23Å)] [Ser216: (3.10Å)] |
37 | 40 |
Table 7.
Summary of aggregator advisor results and medicinal alerts for selected 15 virtual hits.
| Virtual Hits | Aggregator Likelihood | Medicinal Chemistry | |||||||
|---|---|---|---|---|---|---|---|---|---|
| LogP | TC | Structure of Similar Compound | Comments | SAS | PAINS | Brenk | |||
| ZINC06137783 | 4.4 | - | - | Not similar to any known aggregator in in-house database. | 5.13 | No alerts | No alerts | ||
| ZINC04259062 | 3.0 | - | - | Not similar to any known aggregator in in-house database. | 4.82 | No alerts | No alerts | ||
| ZINC03841413 | 2.3 | - | - | Has not been previously reported as an aggregator, or to be similar to an aggregator. | 5.00 | No alerts | No alerts | ||
| ZINC04277683 | 3.1 | - | - | Not similar to any known aggregator in in-house database. | 4.80 | No alerts | No alerts | ||
| ZINC04259056 | 3.3 | - | - | Not similar to any known aggregator in in-house database. | 4.82 | No alerts | No alerts | ||
| ZINC04259064 | 1.9 | - | - | Has not been previously reported as an aggregator, or to be similar to an aggregator. | 4.83 | No alerts | No alerts | ||
| ZINC05535232 | 3.5 | - | - | Not similar to any known aggregator in in-house database. | 4.51 | No alerts | No alerts | ||
| ZINC04237088 | 4.1 | - | - | Not similar to any known aggregator in in-house database. | 4.68 | No alerts | No alerts | ||
| ZINC13733603 | 4.7 | - | - | Not similar to any known aggregator in in-house database. | 4.28 | No alerts | No alerts | ||
| ZINC41585804 | 4.9 | 72% |
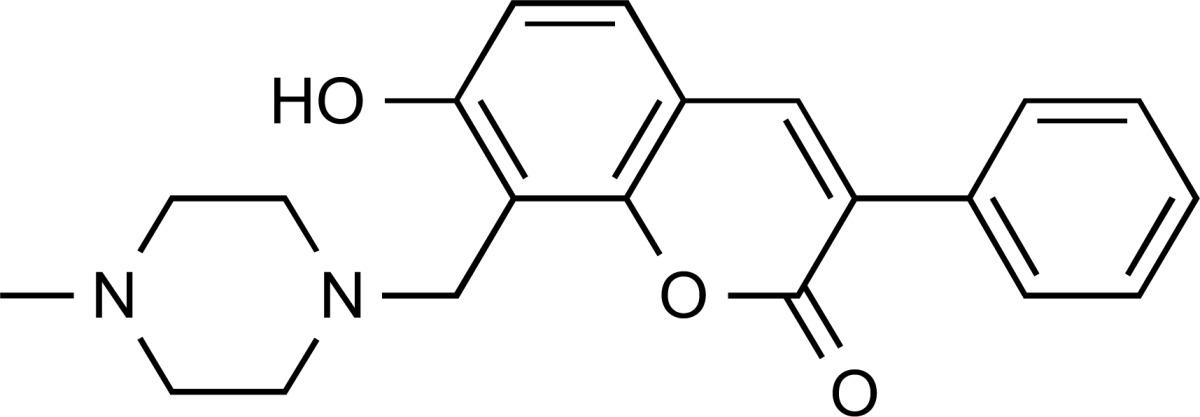
|
Reported as a colloidal aggregator. | 3.82 | Undesirable alerts | No alerts | ||
| ZINC00004749 | 2.4 | 78% | 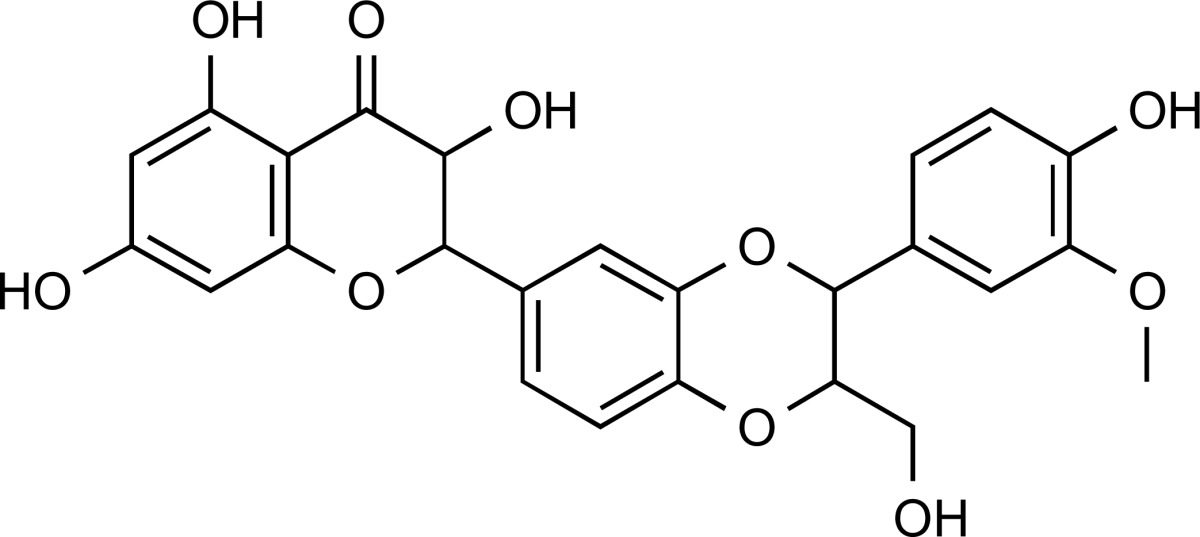 |
Reported as a colloidal aggregator. | 3.82 | Undesirable alerts | No alerts | ||
| ZINC02093367 | 6.0 | 71% | 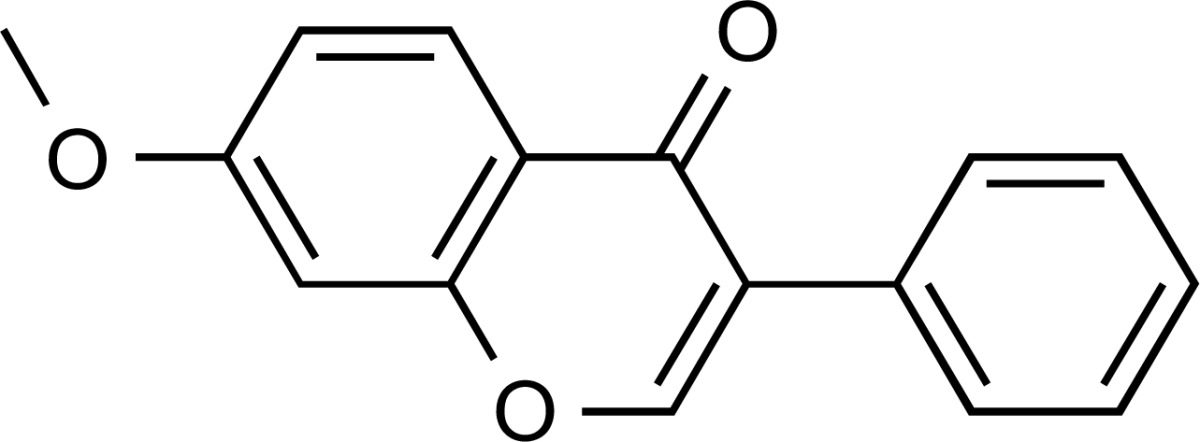 |
Reported as a colloidal aggregator. | 3.94 | No alerts | Undesirable alerts | ||
| ZINC30731533 | 1.5 | 100% | 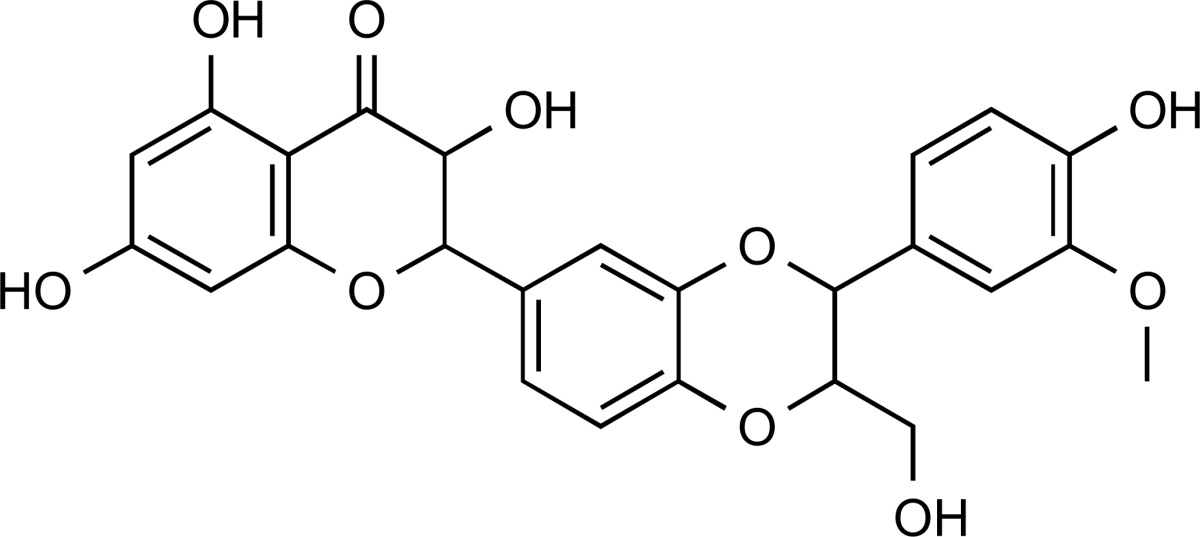 |
Reported as a colloidal aggregator. | 4.92 | No alerts | No alerts | ||
| ZINC00968072 | 2.5 | - | - | Has not been previously reported as an aggregator, or to be similar to an aggregator. | 3.07 | Undesirable alerts | Undesirable alerts | ||
| ZINC13722309 | 3.0 | - | - | Not similar to any known aggregator in in-house database. | 4.87 | No alerts | No alerts | ||
Fig. (2).

Schematic workflow summarizing the screening of Protein tyrosine phosphatase non receptor type 1 inhibitors using computer aided drug design.
3.4. Molecular Docking
Molecular docking study was conducted to evaluate the most favorable geometry of protein-ligand complex. We considered 2636 compounds for the molecular docking study. PTPN1 was used as a molecular target (PDB ID: 3EAX) [30].
We used the CLC drug discovery workbench for analyses of molecular binding of our screened dataset within the protein binding site. Comprehensive study of active site of PTPN1 X-rays crystal structure was conducted. We found that most of the protein binding site is hydrophobic. Fig. (3) shows the molecular surface recognition of 3EAX generated by using chimera software. Fig. (4) shows results of a detailed 2D interactions analysis of the target protein complex and three potential hits. Fig. (4) (A) shows that Tyr46, Asp48, Val49, Ser216, Ala217, Gly220, Arg221, Gln262, and Gln266 are interacting residues within 4 Å of the protein binding site of 3EAX. Molecular docking simulations identified hydrogen and hydrophobic bindings with significant residues of PTPN1 target protein as shown in Table 5.
Fig. (3).

Hydrophobic surface and the active binding site of the 3EAX protein showing LZP ligands, that is co-crystallized and overlaid at the active site, as generated using chimera.
Fig. (4).

Schematic representation of the binding mode of ligands with Protein tyrosine phosphatase non receptor type 1 protein (PDB ID: 3EAX). The protein site is hydrophobic and the NMR structure of the 3EAX protein complex bonded with LZP is shown in (A). Conserved interacting residues of the binding site of the target protein bonded with the virtual hits (B). ZINC04259056 shows only hydrophobic bonding (C). ZINC30731533 shows large network of hydrophobic and hydrogen bonding (D). ZINC00968072 also shows large network of hydrophobic and hydrogen bonding. Conserved interacting residues are displayed in red circles. (The color version of the figure is available in the electronic copy of the article).
On the basis of the best docking score, 15 hits were selected to identify a potent lead compound. The interactions of the active conformation of the best scoring 15 hits with the target protein were identified using Ligplot. The saved conformation for the docked complex was subjected to detailed interactions analyses. The docked files were uploaded to Ligplot to obtain its schematic representation of the hydrogen bonding and hydrophobic interactions. Detailed interactions of all docked complexes are shown in Table 6. These hits resulted from the hydrogen bonding and hydrophobic interactions with the significant residues within 4 Å, and the distance has been mentioned for hydrogen bonds.
The optimal binding mode of the three hits ZINC04259056, ZINC30731533 and ZINC00968072 having dock scores of −52.545, −50.331 and −50.315, is shown in Fig. (4B, C, D), respectively. The 2D analyses of these docked complexes revealed the significant residues involved in the binding interactions of the selected hits. Common binding residues are marked with red circles to highlight them (Fig. 4). The diagram provides a schematic representation of the docked complex. There are many hydrophobic interactions, so only residues are shown for clarity. Nine residues for the binding of the NMR protein (3EAX) to its ligand (LZP) are shown in Fig. (4) (A). While (B) ZINC04259056 and (D) ZINC00968072 show that eight interacting residues are common and (C) ZINC30731533 shows that five interacting residues are common to the 3EAX docked complex. Red circles are demonstrating similar binding residues (Fig. 4).
Seven compounds show prominent hydrophobic binding interactions and eight compounds also show hydrogen bonds. Hydrogens bonds are also of great importance in PTPN1 inhibitor design. In general, for most effective inhibition; inhibitory compounds should interact with the most possible surface residues of protein binding pocket. Therefore, the inhibitory compounds must have polar amino acid and be charged anionically at functional pH [39]. In addition, the inhibitory compound must be firmly anchored by the establishment of hydrogen bonds with particular amino acid residues and inhibitory functional groups in the protein binding pocket [39, 40]. However, interactions with polar amino acids will reduce the ability of PTPN1 inhibitors to cross cell membrane and to access cytosolic PTPN1 [39]. Hydrogen and hydrophobic interaction together contribute in PTPN1 inhibition. It contributes in strong bonding of selected hits with active binding site of respective protein. ZINC13722309 shows 40 binding interactions with the target protein, while ZINC30731533 shows 37 binding interactions and ZINC05535232 shows 35 binding interactions. ZINC30731533 shows the best binding mode with the hydrophobic moiety and the polar surface residues of the protein pocket. The dock scores of the top 15 hits are ≥ −50. The RMSD values are in the range of 0.5 to 1.1, which are considered to be acceptable values. The ligand conformation penalty is the conformational restriction energies that involve binding of flexible ligands in the protein pocket.
3.5. Aggregator Advisor Screening
Aggregator advisor was used for 15 selected hits to check their aggregator possibility (Table 7). While using this online tool, default affinity range (0.1 to 10 μM) was selected. On the basis of lipophilicity, LogP and chemical similarity thresholds (Tanimoto coefficient), three hits (ZINC03841413, ZINC04259064 and ZINC00968072) were not previously reported as aggregator and have not shown any similarity with known aggregator in the database. However, some hits were not similar to any known aggregators in the database and would require appropriate controls for possible aggregation if analyzed in vitro. Four hits (ZINC41585804, ZINC00004749, ZINC02093367, and ZINC30731533) with similarly threshold, LogP, and chemical structure of known aggregators are shown in Table 7 [41].
3.6. Lead Identification
Eleven compounds with acceptable physiochemical properties and without any expected toxicity or medicinal chemistry alerts were identified. The ZINC30731533 hit showed the best results in all the in silico protocols applied in the current study. It showed binding interactions, with a large network of hydrophobic interactions along with seven hydrogen bonds with the most important polar residues of the 3EAX NMR target protein structure. This compound did not show any toxicity risks like mutagenicity, tumorigenicity, reproductive effects and irritant effects. The pharmacokinetic calculations were also favorable. The aggregator likelihood with previous reported active compound was 100%, and it suggested ZINC30731533 as a potent lead compound through computational methods.
ZINC30731533 is known as isosilybin (major active constituent of silymarin), an abundant flavonolignans identified in milk thistle. Silymarin is famous as Chinese traditional medicine for over-indulgence of food or indigestion treatments. When an adjunct to oral diabetes therapy is used, it shows that level of fasting blood glucose reduces and HbA1c is maintained in animal models and in diabetic patients. It seems to increase insulin sensitivity. But more research is required to confirm its efficacy in management of diabetes mellitus [42]. Hence ZINC30731533 is suitable for in vivo studies to validate its PTPN1 inhibitory activity, with the potential for development of an antidiabetic drug.
4. DISCUSSION
To find potential plant-derived PTPN1 inhibitors and to deliver an idea for drug design, we have used both ligand and structure based methods. In our study, an integrated computational approach has been applied for ligand-based pharmacophore modeling of reported PTPN1 inhibitors and database screening to retrieve diverse plant-derived chemical scaffolds. Molecular docking has been applied in order to clarify the behavior of PTPN1 enzyme by binding plant-derived compounds with different binding affinities. It helps to identify significant residues involved in PTPN1 inhibition. We used integrated strategies for structural insights along with medicinal chemistry prospective, which will lead to robust bio-assay method for enabling the design of potential and selective PTPN1 inhibitors. The significance of physiochemical properties and ADME/toxicity filters for inhibitor design is also emphasized in the search of active plant-derived PTPN1 inhibitors. Completely similar reported aggregator compound is a useful reference point in designing inhibitor with better physiochemical properties and possibility of inhibition of PTPN1 enzyme for in vivo testing with a plant-derived lead compound.
We have discussed in our previous research that most of FDA approved anti-DM medicines now available cannot achieve a satisfactory level of glycemic control in DM patients, and have many side effects; therefore, new classes of anti-DM medicines are urgently needed. CADD techniques could be exploited to filter a large number of databases to produce efficient hits and to minimize the time and financial investments needed to discover novel anti-DM medications [6].
The merits of plant-based medicine have been demonstrated for various diseases. Metformin is mostly used medication for T2DM, derivative of guanidine which is obtained from Galegine Officinalis [43]. The potential advantages of plant-based medicines over synthetic medicines include less side-effects, increased efficacy, increased availability, and lower cost. Some of these plant-based medicines are better extracted and used in a crude form as it is the common practice in traditional anti-diabetic medicine [44]. It is mostly used as an adjunct to oral antidiabetic therapy to reach appropriate glycemic control.
PTPN1 is an acceptable therapeutic target that can be effectively targeted for the management of T2DM. Regardless of the accessibility of numerous synthetic PTPN1 inhibitors, their use often entails side-effects, some of which are life-threatening. However, to date, PTPN1 inhibitors are far from achieving regulatory approval from the FDA. Therefore, it is necessary to identify novel hits that have potential to evolve into effective inhibitors of PTPN1. The present study focuses on a pipeline for the development of effective plant-derived anti-DM drugs that are safer and better tolerated when compared with synthetic alternatives. To attain this target, compounds need to be designed with respect to the protein target. Discovering the substrate or inhibitors that successfully bind to the protein target has paved a path for numerous molecular docking strategies.
Significant research has been conducted on achieving PTPN1 inhibition and many developed compounds have reached stage I or II clinical trials only to be discarded because of bioavailability and toxicity issues. The development of PTPN1 inhibitors is challenging because of potency/affinity, selectivity, and cell permeability issues. We applied approximate filters and safety protocols to find a potent lead compound with sufficient oral bioavailability, highest docking score with favorable binding interactions, acceptable toxicity estimations, favorable pharmacokinetics, aggregation information and medicinal chemistry safety parameters.
In the present study, we selected 11 flavonoid compounds which have acceptable drug-like properties and have PTPN1 inhibitory activities, as a training set to use for pharmacophore modeling and generate a query to search similar compounds from the natural compounds listed in the ZINC database. Pharmacophore-based screening is applied to obtain and prioritize plant-derived compounds from ZINC databases for structure-based study. With correspondence to experimental drug discovery methods like high-throughput screening (HTS), computational methods [45, 46] like computer-based screening facilitate the selection of potential lead candidates in a cost-efficient manner by utilizing the available structural information on a protein. Here, we used PTPN1 target protein (PDB id: 3EAX) for molecular docking analyses. Based on the best dock score, 15 compounds were considered as virtual hits and satisfactory results. Optimal binding pose and interacting residues of selected hits analyses seem stable and have resulted in specific PTPN1 inhibition.
The selected hits showed acceptable bioavailability and pharmacokinetic profile. These virtual hits follow the rule of five and Veber’s rule of drug-likeness. A toxicity estimation based on four criteria was also acceptable for the 15 hits. There was no predicted mutagenic, tumorigenic, irritant and reproductive risk to a patient. The Log Kp (skin permeation) values were in an acceptable range, demonstrating that the cell permeability power of the selected candidates is sufficient. Water solubility was assessed because poor aqueous solubility of drug-like compounds limits their in vivo bioavailability because of less dissolution in the gastrointestinal fluids following oral administration [47]. The predicted solubility values for the selected hits were within a suitable range. In silico pharmacokinetic principles (rules that explain how a body affects/deals a drug with absorption, distribution, metabolism, and elimination) are used to assess if a drug-like compound is likely to be safe and effective for therapeutic management in patients. In addition, an estimation of cytochrome P450 (e.g. cytochrome p4501A2, cytochrome p4502C19 and cytochrome p4502C9) was performed for its most critical isoforms by SwissADME predictor shown in Table 4. The cytochrome P450 is a superfamily which performs important functions such as absorption, metabolism, and elimination of drug from liver [48]. If a drug does not metabolize and accumulate for long time in the body, it can cause toxicity. Many hits were shown to interact with the cytochrome P450 isoforms. In silico estimation for gastrointestinal (GI) drug absorption is acceptable; all selected hits have shown positive response towards oral administration. Selected 15 hits were also checked from a medicinal chemistry perspective, in terms of the presence of any toxic moieties, PAINS also known as pan assay interference compounds and Brenk filters, to determine an oral bioavailability and drug safety profile [35]. ZINC41585804, ZINC00004749, ZINC02093367, and ZINC00968072 showed high-risk structural alerts. Consequently, these four compounds should be discarded during the initial phase of drug development to avoid possible toxic effects. The remaining 11 compounds demonstrated no drug safety alerts and potential starting points for further studies. While ZINC30731533 showed complete similarity (100%) to the compound and it has been reported as an aggregator (Table 7). The aggregator likelihood assessment suggested a reference point for identified lead compound suitable for future study.
In this study, we focused on the identification of new candidates as anti-DM agents. By using structural information of already modeled PTPN1 structure (PDB id: 3EAX), detail binding behavior of PTPN1 was studied. Computational techniques were systematically used in this study to produce the results. It is worthy to note that we identified a lead compound (isosilybin; an active constituent of silymarin) as a PTPN1 inhibitor. Our recommendation is to test isosilybin in laboratories to confirm its activity as PTPN1 inhibitor. We highlighted the importance of PTPN1 enzyme in this study which is involved in many biological processes, however design and development of PTPN1 inhibitor is a hot research topic for treatment of obesity and cancer, as well as for T2DM [30]. The proposed virtual lead, isosilybin, is a flavonoid compound which shows extensive variety of biological activities, because flavonoids are beneficial for treatment of various diseases [37]. Furthermore, DM is a complicated disease, and DM patients usually have other complications along with the disease. In this regard, the proposed lead compound is expected to show multiple treatments for diabetic patients who have other complications.
CONCLUSION
By using computer-aided drug design methodologies, we successfully identified plant-derived therapeutic hits with the potential to inhibit the PTPN1 target, which may be helpful to enhance insulin production. The newly identified lead isosilybin (ZINC30731533) in this systematic study was without any predicted toxic effects and showed the best binding mode with the PTPN1 therapeutic target. Therefore, the lead compound is expected to function as an anti-diabetic drug after subsequent testing and validation. The present study could aid in the development of PTPN1 inhibitors for diabetes treatment.
CONSENT FOR PUBLICATION
Not applicable.
ACKNOWLEDGEMENTS
The authors thank the Otsuka Toshimi Scholarship Foundation, Japan, for financial assistance for this work, and Takumi Hidaka for editing data. Both authors had a significant role in the conceptualization and writing of this manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Li W.L., Zheng H.C., Bukuru J., DeKimpe N. Natural medicines used in the traditional Chinese medical system for therapy of diabetes mellitus. J. Ethnopharmacol. 2004;92(1):1–21. doi: 10.1016/j.jep.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 2.Potterat O., Hamburger M. Drug discovery and development with plant-derived compounds. Prog. Drug Res. 2008;65(45):47–118. doi: 10.1007/978-3-7643-8117-2_2. [DOI] [PubMed] [Google Scholar]
- 3.Koona S.J., Kudipudi S., Sridhar G.R., Rao S.B., Apparao A. Plant insulin: An in silico approach. Int. J. Diabetes Dev. Ctries. 2010;30(4):191–193. [Google Scholar]
- 4.Bibi S., Kulsoom S., Rashid H. In Silico Approach for lead identification and optimization of antidiabetic compounds. IOSR J. Pharm. Biol. Sci. 2013;7(3):36–46. [Google Scholar]
- 5.Bibi S., Kulsoom S., Rashid H., Sakata K. Lead identification and optimization of plant insulin-based antidiabetic drugs through molecular docking analyses. Int J. Pharm. Pharm. Sci. 2015;7(3):337–343. [Google Scholar]
- 6.Bibi S., Sakata K. Current status of computer-aided drug design for type 2 diabetes. Curr. Comput. Aided Drug Des. 2016;12(2):167–177. [PubMed] [Google Scholar]
- 7.Irons B.K., Minze M.G. Drug treatment of type 2 diabetes mellitus in patients for whom metformin is contraindicated. Diabetes Metab. Syndr. Obes. 2014;18(7):15–24. doi: 10.2147/DMSO.S38753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tiganis T. PTP1B and TCPTP-non redundant phosphatases in insulin signaling and glucose homeostasis. FEBS J. 2013;280(2):445–458. doi: 10.1111/j.1742-4658.2012.08563.x. [DOI] [PubMed] [Google Scholar]
- 9.Mohler M.L., He Y., Wu Z., Hwang D.J., Miller D.D. Recent and emerging anti-diabetes targets. Med. Res. Rev. 2009;29(1):125–195. doi: 10.1002/med.20142. [DOI] [PubMed] [Google Scholar]
- 10.Carpino P.A., Goodwin B. Diabetes area participation analysis: a review of companies and targets described in the 2008 - 2010 patent literature. Expert Opin. Ther. Pat. 2010;20(12):1627–1651. doi: 10.1517/13543776.2010.533171. [DOI] [PubMed] [Google Scholar]
- 11.Wang L.J., Jiang B., Wu N., Wang S.Y., Shi D.Y. Natural and semisynthetic protein tyrosine phosphatase 1B (PTP1B) inhibitors as anti-diabetic agents. RSC Advances. 2015;5(60):48822–48834. [Google Scholar]
- 12.Sun J., Qu C., Wang Y., Huang H., Zhang M., Li H., Zhang Y., Wang Y., Zou W. PTP1B, A potential target of type 2 diabetes mellitus. Mol. Biol. 2016;5(4):174. [Google Scholar]
- 13.Elchebly M., Payette P., Michaliszyn E., Cromlish W., Collins S., Loy A.L., Normandin D., Cheng A., Himms-Hagen J., Chan C.C., Ramachandran C., Gresser M.J., Tremblay M.L., Kennedy B.P. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283(5407):1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 14.Klaman L.D., Boss O., Peroni O.D., Kim J.K., Martino J.L., Zabolotny J.M., Moghal N., Lubkin M., Kim Y.B., Sharpe A.H., Stricker-Krongrad A., Shulman G.I., Neel B.G., Kahn B.B. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol. Cell. Biol. 2000;20(15):5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Combs A.P. Recent advances in the discovery of competitive protein tyrosine phosphatase 1B inhibitors for the treatment of diabetes, obesity, and cancer. J. Med. Chem. 2010;53(6):2333–2344. doi: 10.1021/jm901090b. [DOI] [PubMed] [Google Scholar]
- 16.Jiang C.S., Liang L.F., Guo Y.W. Natural products possessing protein tyrosine phosphatase 1B (PTP1B) inhibitory activity found in the last decades. Acta Pharmacol. Sin. 2012;33(10):1217–1245. doi: 10.1038/aps.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen R.M., Hu L.H., An T.Y., Li J., Shen Q. Natural PTP1B inhibitors from Broussonetia papyrifera. Bioorg. Med. Chem. Lett. 2002;12(23):3387–3390. doi: 10.1016/s0960-894x(02)00757-6. [DOI] [PubMed] [Google Scholar]
- 18.Bae E.Y., Na M., Njamen D., Mbafor J.T., Fomum Z.T., Cui L., Choung D.H., Kim B.Y., Oh W.K., Ahn J.S. Inhibition of protein tyrosine phosphatase 1B by prenylated iso flavonoids isolated from the stem bark of Erythrina addisoniae. Planta Med. 2006;72(10):945–948. doi: 10.1055/s-2006-946674. [DOI] [PubMed] [Google Scholar]
- 19.Nguyen P.H., Le T.V., Thuong P.T., Dao T.T., Ndinteh D.T., Mbafor J.T., Kang K.W., Oh W.K. Cytotoxic and PTP1B inhibitory activities from Erythrina abyssinica. Bioorg. Med. Chem. Lett. 2009;19(23):6745–6749. doi: 10.1016/j.bmcl.2009.09.108. [DOI] [PubMed] [Google Scholar]
- 20.Dao T.T., Nguyen P.H., Thuong P.T., Kang K.W., Na M., Ndinteh D.T., Mbafor J.T., Oh W.K. Pterocarpans with inhibitory effects on protein tyrosine phosphatase 1B from Erythrina lysistemon Hutch. Phytochemistry. 2009;70(17):2053–2057. doi: 10.1016/j.phytochem.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen P.H., Sharma G., Dao T.T., Uddin M.N., Kang K.W., Ndinteh D.T., Mbafor J.T., Oh W.K. New prenylated isoflavonoids as protein tyrosine phosphatase 1B (PTP1B) inhibitors from Erythrina addisoniae. Bioorg. Med. Chem. 2012;20(21):6459–6464. doi: 10.1016/j.bmc.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 22.Choi J.S., Islam M.N., Ali M.Y., Kim Y.M., Park H.J., Sohn H.S., Jung H.A. The effects of C-glycosylation of luteolin on its antioxidant, anti-Alzheimer’s disease, anti-diabetic, and anti-inflammatory activities. Pharmacol. Res. 2014;37(10):1354–1363. doi: 10.1007/s12272-014-0351-3. [DOI] [PubMed] [Google Scholar]
- 23.Wolber G., Langer T. Ligand Scout: 3-D pharmacophore derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005;45(1):160–169. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- 24. ChemDraw Ultra 8.0 package. Cambridge Soft Corp. USA. www.cambridgesoft.com
- 25.Koes D.R., Camacho C.J. ZINCPharmer: pharmacophore search of the ZINC database. Nucleic Acids Res. 2012;40:409–414. doi: 10.1093/nar/gks378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sander T., Freyss J., Korff M.V., Rufener C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015;55(2):460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- 27.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 28.Veber D.F., Johnson S.R., Cheng H., Smith B.R., Ward K.W., Kopple K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45(12):2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 29.Pajouhesh H., Lenz G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2(4):541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu S., Zeng L.F., Wu L., Yu X., Xue T., Gunawan A.M., Long Y.Q., Zhang Z.Y. Targeting inactive enzyme conformation: aryl diketoacid derivatives as a new class of PTP1B inhibitors. J. Am. Chem. Soc. 2008;130(50):17075–17084. doi: 10.1021/ja8068177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. CLC drug discovery work bench/Molegro Virtual Docker: CLC Inc A, Denmark. www.clcbio.com/products/clc-drug-discovery-work
- 32.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski R.A., Swindells M.B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011;51(10):2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 34.Ntie-Kang F., Lifongo L.L., Mbah J.A., Owono-Owono L.C., Megnassan E., Mbaze L.M., Judson P.N., Sippl W., Efange S.M. In silico drug metabolism and pharmacokinetic profiles of natural products from medicinal plants in the Congo basin. In Silico Pharmacol. 2013;1:12. doi: 10.1186/2193-9616-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daina A., Michielin O., Zoete V. iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model. 2014;54(12):3284–3301. doi: 10.1021/ci500467k. [DOI] [PubMed] [Google Scholar]
- 36.Aggregator Advisor Shoichet Laboratory at UCSF.University of California, San Francisco, USA. http://advisor.bkslab.org/
- 37.Yao L.H., Jiang Y.M., Shi J., Tomas-Barberan F.A., Datta N., Singanusong R., Chen S.S. Flavonoids in food and their health benefits. Plant Foods Hum. Nutr. 2004;59(3):113–122. doi: 10.1007/s11130-004-0049-7. [DOI] [PubMed] [Google Scholar]
- 38.Nogara P.A., Saraiva R.de.A., Bueno D.C., Lissner L.J., Corte C.L.D, Braga M.M., Rosemberg D.B., Rocha J.B.T. Virtual screening of acetylcholinesterase inhibitors using the lipinski’s rule of five and ZINC databank. Biomed. Res. Int., 2015 doi: 10.1155/2015/870389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Q., Gao J., Liu Y., Liu C. Molecular dynamics simulation of the interaction between protein tyrosine phosphatase 1B and aryl diketoacid derivatives. J. Mol. Graph. Model. 2012;38:186–193. doi: 10.1016/j.jmgm.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 40.Combs A.P., Yue E.W., Bower M., Ala P.J., Wayland B., Douty B., Takvorian A., Polam P., Wasserman Z., Zhu W., Crawley M.L., Pruitt J., Sparks R., Glass B., Modi D., McLaughlin E., Bostrom L., Li M., Galya L., Blom K., Hillman M., Gonneville L., Reid B.G., Wei M., Becker-Pasha M., Klabe R., Huber R., Li Y., Hollis G., Burn T.C., Wynn R., Liu P., Metcalf B. Structure-based design and structure-based design and discovery of protein tyrosine phosphatase inhibitors incorporating novel isothiazolidinone heterocyclic phosphotyrosine mimetics. J. Med. Chem. 2005;48(21):6544–6548. doi: 10.1021/jm0504555. [DOI] [PubMed] [Google Scholar]
- 41.Ferreira R.S., Simeonov A., Jadhav A., Eidam O., Mott B.T., Keiser M.J., McKerrow J.H., Maloney D.J., Irwin J.J., Shoichet B.K. Complementarity between a docking and a high-throughput screen in discovering new cruzain inhibitors. J. Med. Chem. 2010;53(13):4891–4905. doi: 10.1021/jm100488w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petrie M., McKay G., Fisher M. Silymarin. Drug notes. Pract. Diabetes. 2015;32(4):148–504. [Google Scholar]
- 43.Newman D.J., Cragg G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012;75(3):311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Osadebe P.O., Odoh E.U., Uzor P.F. Natural products as potential sources of antidiabetic drugs. Br. J. Pharm. Res. 2014;4(17):2075–2095. [Google Scholar]
- 45.Reddy A.S., Pati S.P., Kumar P.P., Pradeep H.N., Sastry G.N. Virtual screening in drug discovery – a computational perspective. Curr. Protein Pept. Sci. 2007;8(4):329–351. doi: 10.2174/138920307781369427. [DOI] [PubMed] [Google Scholar]
- 46.Thangapandian S., John S., Sakkiah S., Lee K.W. Potential virtual lead identification in the discovery of renin inhibitors: application of ligand and structure-based pharmacophore modeling approaches. Eur. J. Med. Chem. 2011;46(6):2469–2476. doi: 10.1016/j.ejmech.2011.03.035. [DOI] [PubMed] [Google Scholar]
- 47.Khadka P., Ro J., Kim H., Kim. I., Kim J.T., Kim H., Cho J.M., Yun G., Lee J. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J. Pharm. Sci. 2014;9(6):304–316. [Google Scholar]
- 48.Ogu C.C., Maxa J.L. Drug interactions due to cytochrome P450. Proc. Bayl. Univ. Med. Cent. 2000;13(4):421–423. doi: 10.1080/08998280.2000.11927719. [DOI] [PMC free article] [PubMed] [Google Scholar]