

We propose a promising new method of solar-driven ammonia synthesis with metal nanoparticles.
Abstract
Despite more than a century of advances in catalyst and production plant design, the Haber-Bosch process for industrial ammonia (NH3) synthesis still requires energy-intensive high temperatures and pressures. We propose taking advantage of sunlight conversion into surface plasmon resonances in Au nanoparticles to enhance the rate of the N2 dissociation reaction, which is the bottleneck in NH3 production. We predict that this can be achieved through Mo doping of the Au surface based on embedded multireference correlated wave function calculations. The Au component serves as a light-harvesting antenna funneling energy onto the Mo active site, whereby excited-state channels (requiring 1.4 to 1.45 eV, near-infrared–to–visible plasmon resonances) may be accessed. This effectively lowers the energy barriers to 0.44 to 0.77 eV/N2 (43 to 74 kJ/mol N2) from 3.5 eV/N2 (335 kJ/mol N2) in the ground state. The overall process requires three successive surface excitation events, which could be facilitated by amplified resonance energy transfer due to plasmon local field enhancement.
INTRODUCTION
Activation of the N2 bond—the second strongest chemical bond known—is of both fundamental and commercial interest. Despite its great abundance in the atmosphere, N2 must first be converted into a form that can be metabolized by living organisms [for example, ammonia (NH3), amines, and nitrates] to be agriculturally viable. The Haber-Bosch (HB) process was the “disruptive innovation” of its time that led to inexpensive, large-scale NH3 production from N2 and H2 (1). The process, which consumes ~5% of the world’s natural gas (2), requires high temperatures (~500°C) and pressures (~200 atm) to be kinetically feasible, despite the reaction being thermodynamically allowed at room temperature and ambient pressures (3, 4). Thus, NH3 production under milder conditions is a sensible trajectory toward a more energy-efficient HB process. The greatest hurdle in achieving this endeavor is the large (~10 eV) energy per molecule (~1000 kJ/mol) that has to be gained by the N2 molecule for it to dissociate in the gas phase (5). Therefore, the Herculean task of activating N2 is the primary rate-limiting step on most catalysts for the HB process. In catalytic N2 dissociation, the bond-breaking process is partly compensated by the favorable formation of metal–N bonds on the surface of a catalyst [for example, on Ru, Fe, Ir, and Mo (4)], and thus, together with high temperatures and pressures (as in the HB process), N–N bond scission becomes feasible. These metal–N bonds then are broken later via the formation of stronger or equally strong N–H bonds.
Recently, it has been demonstrated that catalysis on select single and tandem metal nanoparticles (MNPs) can be enhanced through excitation of the metal’s local surface plasmon resonances (LSPRs) (6–16). LSPRs involve the collective oscillation of the free-electron–like valence electrons of MNPs at incident light wavelengths (~400 to 1000 nm) that are much larger than the particles (~5 to 200 nm) (17). These excitations exhibit large extinction cross sections [much larger than the physical cross section of the MNP itself, with some that are ≥105 times greater than those of conventional chromophoric molecules (18, 19)] and local electric field enhancements at peak resonances (12, 17, 20). Perhaps, the most widely known application of LSPRs is in the field of surface-enhanced Raman spectroscopy (SERS) for molecules adsorbed on plasmonic MNPs, characterized by about a million-fold enhancement in the Raman signal (21, 22). LSPRs may generate energetic charge carriers or heat (through phonon coupling) upon decay (23) or may cause resonant surface excitations (20, 24), all potentially influencing the MNP surface’s chemical reactivity (fig. S1). Coupling of the few strongly plasmonic coinage metals (Cu, Ag, and Au) and Al with catalytically active metals, through decoration with small clusters of, for example, Pt and Pd, has been demonstrated experimentally to be successful in extending the library of reactions that may be plasmonically catalyzed (6–9). We previously proposed surface doping as an alternative configuration to enhance the catalytic properties of a plasmonic metal (25, 26). Here, we focus on a Mo-doped Au surface as a model catalyst, where the choice of the two components, Mo (the dopant and active site) and Au (the plasmonically active host), is due to their complementary properties. Mo is a known N2 dissociation catalyst that binds N very strongly (27). Au, on the other hand, is an excellent plasmonic material with an LSPR peak that can be modulated within the near-infrared (NIR) up to the ultraviolet (UV) range of the electromagnetic spectrum (18). Here, we theoretically predict, through a combination of periodic density functional theory (DFT) and a multireference second-order perturbation theory, that accessing multiple electronically excited states originating from Mo and the adsorbed N2 molecule, with energies at or near the plasmon resonance frequency of Au, is a potentially effective means to achieve a low-temperature HB process.
RESULTS AND DISCUSSION
Simulation of the catalyst surface
The (111) surface of Au with a face-centered cubic (fcc) structure was modeled with a very dilute Mo surface dopant concentration (see Methods). This model was chosen because of the known immiscibility of Mo and Au (28). Previously, we predicted theoretically that although doping of Au with Mo is thermodynamically unfavorable, oxidation with O2 could stabilize isolated Mo substituents on the surface of Au (25). Recovery of the active site can be achieved through reduction with, for example, H2(g), which is abundant during NH3 synthesis, at slightly elevated temperatures (25) or electrochemically. Furthermore, we predicted that lattice substitution is far more favorable than adsorption of single Mo atoms (25). Numerous experiments are consistent with our predictions. Substitutional surface alloying of Au(111) with Mo has been observed during physical vapor deposition of Mo on Au(111) at temperatures ≥250°C (29) and via molecular beam epitaxy at 500°C (30). It is noteworthy that well-mixed alloys of Au and first-row transition metals such as Ni, Co, and Fe, which are also immiscible in Au (31–33), can be synthesized electrochemically via simultaneous reduction of the dopants and Au erosion (34), and electrodeposition using salts of Au and of the dopants (35). Large-scale synthesis of dense Au–Ni alloy nanowires was achieved using an electrodeposition technique (36). Laser ablation was also successfully deployed to synthesize nanoalloys of Au and Fe (37). Both electrodeposition and laser ablation techniques could be potentially used to incorporate Mo into Au because these methods are strongly kinetically controlled. Although Au and Mo do not form thermodynamically stable alloys, such alloys were found to be kinetically accessible and stable, especially at the surface, and even argued to be thermodynamically favorable at the nanoscale (30). Thus, reducing the operating temperatures for NH3 synthesis [ideally far below the melting point of Au, 1064°C (30), to prevent atomic diffusion] is key to preventing eventual percolation of Mo, which we will show is possible via plasmonics.
Identification of the minimum energy pathway for N2 dissociation
The favorable adsorption configurations of N2 on the Mo active site and the intervening structures that connect them along the minimum energy pathway (MEP) toward dissociation are shown in Fig. 1A (see figs. S2 and S3 for the vibrational frequencies of the stationary and transition states, as well as changes in relevant structural parameters as a function of the reaction coordinate). The structures found at the energy minima and maxima along the MEP were identified using DFT with the climbing image–nudged elastic band method (CI-NEB) (38). As the N2 molecule approaches the Mo site, it orients such that it binds to the Mo via an end-on, η1-N2, configuration (Fig. 1, A and B, orange box and curve, respectively, henceforward referred to as path a). This binding is barrier-free and is exothermic with an associated ΔEa = −1.19 eV/N2 [−1.13 eV/N2 with zero-point energy (ZPE) correction] and a corresponding ΔGa = −0.69 eV/N2 at 298 K and 1 atm N2 (25).
Fig. 1. N2 dissociation trajectory and energetics on Mo-doped Au(111) surface.
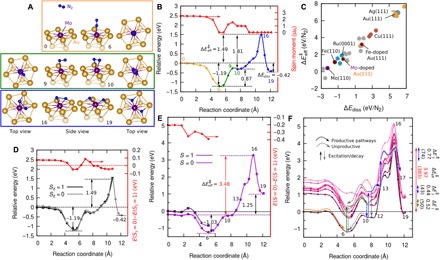
(A) Periodic slab DFT–predicted stationary and transition-state structures along the MEP for N2 dissociation (only the Au9Mo slab fragments are shown for clarity). (B) Corresponding spin-relaxed ground-state reaction energy curve. The upper red curve in (B) shows evolution of the net spin moment of the system (in μB). (C) Comparison of the DFT effective barrier for N2 dissociation over Mo-doped Au(111) versus over some other metallic surfaces investigated in the literature, plotted against the dissociative adsorption energy [Ru, Cu, Ag, Au, and others (39); Mo and Fe (27); Au-Fe (26)]. (D) Periodic slab DFT spin–constrained energetics, with Sz = 0 and 1. The upper red curve shows the energy splitting between the two spin structures. (E) Same type of curves in (D) for S = 0 and 1 obtained from emb-NEVPT2 (n-electron valence second-order perturbation theory). (F) emb-NEVPT2–predicted ground- and excited-state energy curves for S = 0, showing up to the sixth excited state. Possible lower-barrier trajectories are marked with arrows. Ground- and excited-state effective thermal barriers are shown on the right margin (in eV and kJ/mol).
The path toward dissociation requires a reorientation of the molecule until it lies parallel to the surface and binds side-on, η2-N2, onto the Mo (Fig. 1, A and B, green box and curve, respectively, path b). The η2-N2 configuration is a local minimum with a trivial, in-plane rotational degree of freedom (structure 10, denoted s:10). Here, ΔEb = 0.87 eV/N2 (0.83 eV/N2 ZPE-corrected) and ΔGb = 0.83 eV/N2 at 298 K, with a barrier of 0.92 eV/N2 (0.87 eV/N2 ZPE-corrected). Therefore, the barrier to go back to the η1-N2 configuration from this orientation is nearly nonexistent. From the η2-N2 configuration, the elongation of the N2 bond until complete dissociation may proceed (Fig. 1, A and B, blue box and curve, respectively, path c) with a relatively high reaction barrier of 1.81 eV/N2 (1.76 eV/N2 ZPE-corrected) because it is only slightly exothermic with ΔEc = −0.10 eV/N2 (−0.10 eV/N2 ZPE-corrected) and ΔGc = −0.08 eV/N2 at 298 K. Because the barrier for the bond elongation step is extremely high compared to the barriers of the two steps that precede it, the earlier two steps will be in a preequilibrium, and thus, the effective thermal ground-state barrier is simply = 1.49 eV (1.46 eV ZPE-corrected). The overall dissociation energy is ΔEdiss = ΔEa + ΔEb + ΔEc = −0.42 eV (−0.40 eV ZPE-corrected).
The calculated is comparable to the barriers predicted on the flat surfaces of the best HB catalysts, for example, 1.8 eV/N2 on the Ru(0001) surface (39) and 1.11 eV on the Fe(110) surface (27), which were calculated via DFT using a comparable method to that used here (see Fig. 1C). The barrier on the (110) surface of body-centered cubic Mo was previously calculated to be much smaller, 0.42 eV/N2, and very exothermic, ΔEdiss = −2.27 eV (27). However, strong binding is considered the reason behind the relatively poorer performance of Mo-based HB catalysts compared to Fe- and Ru-based catalysts (27). The stronger Mo–N bond results in the hydrogenation of N and the desorption of NH3 becoming rate-limiting (27). The effect of Au on Mo, Au being almost completely inert toward N2 adsorption (Fig. 1C), is thus favorable. The immediate Au environment has a lower reactivity than Mo; this enables AuMo to achieve optimal adsorption and dissociation energies for N2. Although DFT has clear quantitative limitations, comparing the relative activation and reaction energies within the same level of theory provides a qualitative measure (demonstrated by the linear trend in versus ΔEdiss in Fig. 1C) of how the new catalyst will behave relative to known active catalysts. If N2 dissociation remains as the rate-limiting elementary step for NH3 synthesis on the catalysts being compared, then the calculated ΔEdiss indicates that the Mo-doped Au surface should exhibit similar activity to the Ru- and Fe-based catalysts. However, the barrier is too large to allow N2 splitting at room temperature on Mo-doped Au. The advantage of Au over pure Fe and Ru is that Au is strongly plasmonic, which can be potentially used to enhance rates of surface reactions.
Corrections to the ground-state energy curves from many-body wave function methods
We use density functional embedding theory (DFET) (40) to examine electronic excited states and how they can be accessed to affect reaction kinetics, as well as to correct for the shortcomings of DFT in describing charge-transfer reactions (41–43). DFET is a divide-and-conquer approach in which the reaction center (here, an Au10Mo cluster; see fig. S4) is treated using, for example, a many-body multiconfigurational quantum mechanical method while embedded in an effective interaction potential (embedding potential) derived from a periodic DFT slab calculation (40). The embedding potential mimics the mutual interaction of the cluster and its environment (see Methods and fig. S4) (40). This method has been successfully used, for example, to corroborate the enhanced isotope exchange kinetics of H2 and D2 on Au and Al nanoparticles under mild conditions and illumination (15, 16). Here, we use the NEVPT2 (44) to solve for the corrected ground- and electronically excited–state reaction energies using the DFET approach. The complete active space self-consistent field method (CASSCF) (45, 46) was used to generate the reference wave functions. These calculations were done at the augmented triple-ζ basis set level (see Methods, tables S1 to S3, and fig. S5 for the definition of the basis set and conducted convergence tests). Henceforth, DFET with CASSCF and NEVPT2 will be referred to as emb-CASSCF and emb-NEVPT2, respectively.
Energy curves for two different spin manifolds, S = 0 and 1 (only on the early steps of the reaction), were also calculated. Note that DFT predicts Sz ~ 1 to be the ground-state spin upon N2 approach to the surface, which eventually quenches to Sz = 0 as the N2 adsorbs and dissociates (Fig. 1, B and D). However, emb-NEVPT2 predicts that the S = 0 state is lower energy than S = 1 all along the reaction path (Fig. 1E; see also fig. S6 and Supplementary Text). The reaction energy curves for paths a and b calculated from emb-NEVPT2 are comparable to the predictions of the periodic DFT calculations. However, for path c, the barrier and energy of the reaction markedly increase by ~2 and ~1.5 eV to 3.48 and 1.25 eV, respectively. DFT is known to underestimate dissociation barriers and energies of small molecules on metallic surfaces because of spurious electron overdelocalization due to electron self-interaction artifacts, originating from the approximation for the exchange interaction (41–43).
Figure 2 shows the evolution of the orbitals in the complete active space (CAS) used, defined by 12 electrons in 12 orbitals, CAS(12e,12o), composed of six N2 frontier states (5σ, 1π, 6σ*, and 2π*) and six Mo-Au 4d-6s–derived states. As the reaction progresses, the degree of mixing between the molecule and metal orbitals becomes increasingly apparent, which naturally arises from the eventual formation of Mo–N covalent bonds (see also fig. S7). The crossing of N2 1π and 2π* into the virtual and occupied spaces, respectively, and mixing with the Mo 4d states with proper symmetries, demonstrates the N–N bond breaking and Mo–N bond formation. π and δ bonds are shown to form near and at the transition state (s:13 to s:16). The changes in the residual charges of N2, that is, −0.62 e at 13 and −1.31 e at 16, accompany and reflect the degree of crossing of the N2 1π and 2π* orbitals (fig. S8).
Fig. 2. CAS natural orbitals from CASSCF.
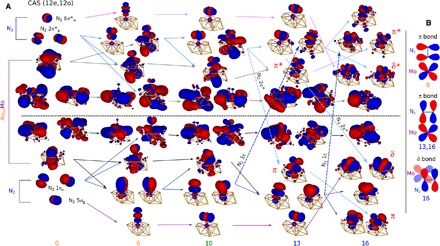
(A) Ground-state CASSCF natural orbitals demonstrating qualitative changes in the Mo-N2 bonding interactions in select structures along the dissociation trajectory (see orbital occupations in table S4). Orbitals above the black horizontal dashed line are nearly empty (virtual), and below are nearly completely filled (occupied). Arrows mark orbital lineage. Thick dashed arrows between s:10 and s:13 and s:13 and s:16 indicate crossing of an occupied orbital character (N2 π) into the virtual space or of a virtual (N2 π*) into the occupied space. Isosurface value: 0.02 atomic units (au); the Au10Mo-embedded cluster model + N2 are shown (see Fig. 1A for structural legend). (B) Schematic representation of the bonding orbital interactions between Mo 4d and N2 π or π* in s:6, s:13, and s:16.
Excited-state energy curves
We calculated several electronically excited states of the species along the MEP to identify the effects of electronic excitation on the kinetics of the reaction. In this section, we first discuss the ideal pathway that leads to barrier reduction and the necessary excitations to access it. We then evaluate the probability of these excitations and approximate their lifetimes. Finally, we discuss the mechanism of interaction between the surface species and the plasmon that leads to the energy transfer from the latter to the former.
We show the calculated emb-NEVPT2 ground- and excited-state curves (up to six excited states) for S = 0 in Fig. 1F. The reference wave functions were obtained from a seven-state state-averaged CASSCF (SA-CASSCF). We note that state-averaging on the CASSCF level led to lower relative energies for the ground states of s:17 to s:19 (by 0.2 to 1.25 eV) and higher energies for s:13 to s:16 (by ~0.5 eV). We compare SA-CASSCF/NEVPT2 to the more reliable single-state CASSCF (SS-CASSCF)/NEVPT2 (for predicting the ground-state curve) to find the optimal number of states with energies within the plasmon frequency of Au while also making sure that the ground-state curve of the pertinent region of the reaction coordinate is not severely compromised. The pertinent energies describing the kinetics of the dissociation are from s:0 to s:17. Within this range, the ground-state curve from the SA-CASSCF/NEVPT2 is nearly equivalent to the SS-CASSCF/NEVPT2 calculations (except for some up or down shifts by ~0.5 eV from s:10 to s:17; see fig. S9).
The pathways leading to reduced effective barriers involving both ground- and excited-state curves are traced out in Fig. 1F (orange, green, and blue traces). No energy input is required to adsorb a N2 molecule on the surface; thus, it can follow the ground-state curve (orange trace). The η1-N2 configuration is very stable such that energy input is required for desorption or the reorientation of the molecule into the η2-N2 configuration. Desorption can be thermally enhanced because the gas-phase N2 is entropically favored: desorption ΔS = 1.986 to 2.071 meV/K ~ S° of N2(g) for 298 to 700 K (5), corresponding to −TΔS of desorption = −0.60 eV at 298 K, which more than doubles to −1.45 eV at 700 K. High temperatures would be unfavorable for dissociation on the surface. Thus, to influence the reorientation without triggering desorption, an electronic excitation of 1.40 eV into the fifth excited state, e:5 (Fig. 1F, green vertical arrow), can provide the energy necessary to bring the molecule away from the ground-state energy. The molecule then only has to overcome a modest barrier of 0.52 eV to reorient, coupling through other lower index excited states along the pathway until it crosses the e:1 of s:9 (a transition state), which can then subsequently decay back into the ground state after evolving into the η2 configuration (Fig. 1F, green trace). Beginning at the η2 configuration, the large barrier for N–N bond dissociation can be overcome by a combination of two electronic excitations and thermally driven nuclear motion (Fig. 1F, blue traces). Excitation of s:10 from the ground (e:0) to e:5 (1.44 eV; Fig. 1F, blue vertical arrow) will provide enough energy to reach s:13 with an effective thermal barrier of only 0.44 eV. The molecule will follow the e:5 surface from 10 to 12, at which point it may then easily cross surfaces of lower state indices until it reaches the ground state at s:13 (blue trace). s:13 may be excited to its e:5 (1.41 eV; Fig. 1F, blue vertical arrow) under high intensity illumination or plasmonic field magnification before it decays back to a lower-energy structure, for example, s:10. The molecule will seek a pathway to relax at this excited state (e:5) of an unstable configuration (s:13). A relaxation pathway available is the molecular dissociation, with an effective barrier of only 0.77 eV (to reach the ground state of s:16; Fig. 1F, blue trace). Therefore, accounting for all of the steps, the highest thermal barrier for dissociation is 0.77 eV. Reactions with this barrier typically have reaction rates of about 6 × 10−1 to 2 × 106 molecules/s per active site at temperatures ranging from 25° to 300°C (298 to 573 K), derived assuming an Arrhenius rate expression with an approximate prefactor of ~ kBT/h = 0.6 × 1013 to 1.19 × 1013 s−1.
Although NH3 production from N2 and H2 is exergonic (spontaneous) under ambient conditions, the reaction is entropically unfavorable, primarily because of the consumption of two gas molecules ( N2 + H2) per NH3. For example, the values of ΔS for this reaction are −99.35, −110.03, −112.71, and −114.55 J/K-mol NH3 at 300, 500, 600, and 700 K, respectively, at 1 atm (4). The increase in temperature and decrease in the entropy of the reaction reduce the spontaneity of the reaction, from ΔG = −16.57 at 300 K to 4.44, 15.75, and 26.94 kJ/mol NH3 at 500, 600, and 700 K (4) at 1 atm. To offset the effect of high temperatures, which are necessary to improve the kinetics of the reaction, and reduced entropy, the total pressure = pN2 + pH2 + pNH3 (with pN2:pH2 = 1:3 and pNH3 = 20 atm) should be 33, 58, and 99 atm at 500, 600, and 700 K, respectively, to achieve at least an equilibrium. These pressures were calculated from ΔG = 0 = ΔG° + RTln (pNH3/[pN21/2pH23/2]), assuming weak dependence of the heat of reaction on pressure (4). These values illustrate that if the in operando temperature can be lowered from 427°C (700 K), which is the typical operating temperature of the HB process, to even 327°C (600 K), the operating pressure could be significantly reduced from ~100 to ~60 atm. The lowered pressure also improves the mole percent of NH3 in the gas-phase reactant-product mixture from 20 to 34%. These estimates are not far from the prediction of the most empirically consistent model, the Gillespie-Beattie equation of state (47), which are 19.4 and 30.3% NH3 at 430°C and 100 bar (98.7 atm) and 330°C and 50 bar (49.3 atm), respectively; see table 2 in the book by Appl (4). We therefore expect that this lower-temperature, plasmonic-based process will also improve the pressure requirements.
Excitation probabilities and excited-state lifetimes
Matching the relevant excitation energies along the pathway is the primary but not the only requirement for LSPRs to catalyze a reaction. The transition probabilities of these excitations must also be probed. To this end, we calculated the oscillator strengths of the excitations, f, which are proportional to the optical absorption cross sections of the transitions (see Methods) (48). f shows the relative “brightness” of an excitation, where the bright allowed transitions have f = 0.1 to 1 and forbidden transitions have f ~ 0 (48). Figure 3 shows the absorption spectrum for the critical structures (calculated from emb-SA-CASSCF transition dipole moments and emb-NEVPT2 excitation energies) as well as the transition-state character (shown via electron density difference plots from emb-SA-CASSCF) of the e:5 states, deemed as the relevant excitations based on their energies and relative oscillator strengths (see fig. S10 for the characters of other excited states). The embedded metal cluster on its own (s:0) has relatively bright absorptions between 1.2 and 1.7 eV, corresponding to the e:2, e:4, and e:5 states. The e:5 state is characterized by an Mo 4d (in-plane)–4d (out-of-plane) electronic transition, in which the surface facilitates symmetry-breaking between the Mo 4d orbitals. At s:6 (Mo–η1-N2), the aforementioned characteristic excitation now exhibits minor contribution from N2 (partial electron gain) and is lowered in energy but has a slightly smaller absorption strength than in s:0. From s:10 and up to s:12, the same characteristic excitation is present with similar transition energies (~1.4 V) and oscillator strengths (~0.02). At this state, the increase in electron density (blue) shares the same symmetry as the π interaction between Mo and N2. The aforementioned transition is no longer present at s:13, where (i) the N2 bond is considerably stretched, (ii) one of the π* orbitals of N2 has crossed into the occupied space, and (iii) significant charge is already transferred into the molecule (~0.6 e; fig. S8). The e:5 state is instead characterized as a d-d transition from a nonbonding Mo d orbital to a bonding d orbital, with symmetry appropriate for a δ-bond with N2 π*. This electronically excited state has the appropriate electronic redistribution for N2 to form the δ-bond with Mo, an interaction that emerges from s:14 to s:16 (transition state) (see orbital π*-4d δ in Fig. 2 in s:16). However, this excitation is much fainter with f = 0.002, which is an order of magnitude lower than previously described transitions. This is because no significant change in the 4d symmetry is associated with the transition, although it has some Mo d–to–N2 π* character (partial electron gain).
Fig. 3. Oscillator strengths and electron density differences.
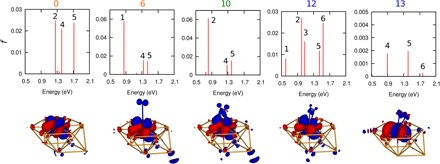
(Top) Absorption spectrum (oscillator strength, f, versus excitation energy) for select structures along the dissociation trajectory (Fig. 1F), calculated from emb-SA-CASSCF transition dipole moments and emb-NEVPT2 excitation energies. Excited-state index given at the top of each red vertical line. Note that vertical scale changes across the panels. (Bottom) Real-space electron density difference plots between the fifth excited state and the ground state for s:0, s:6, s:10, s:12, and s:13 from emb-SA-CASSCF. Red is electron loss, blue is electron gain, and isosurface value is ±0.002 au; the Au10Mo-embedded cluster model + N2 are shown (see Fig. 1A for structural legend).
Although it is desirable for the critical excitations to have large f, strong absorbers are also bright emitters (fluorescence). The excited state has to persist longer than it takes for the photo-activated reaction to occur. The emission lifetime of an excited state (τ in s) can be approximated from f according to classical theory via , where is the frequency of maximum absorption in cm−1 (48). Excitations at 1.40 eV (11,300 cm−1) will have τ of ~10−6 s for f = 0.01 and ~10−5 s for f = 0.001. Note that we approximated earlier the rate to be 2 × 106 molecules/s for a reaction with a barrier of 0.77 eV at 573 K (from [s:13,e:5] to [s:16,e:0]), which therefore requires an excited-state radiative lifetime to be longer than ~0.5 × 10−6 s so that the reaction can compete with the radiative decay. We note that [s:13,e:5] has an approximate radiative lifetime of ~0.6 × 10−5 s (f = 0.002), which is well within the abovementioned estimate.
Mechanism of plasmon energy transfer to the active site
It has been proposed recently that energetic charge carrier injection (“hot” electrons or holes) into an adsorbed molecule on MNPs is one of the primary mechanisms of plasmon-induced photochemical reactions (10, 11). This theory further supposes that a transient negative ion (TNI) state is formed, leading to the rapid turnovers of chemical reactions (10, 11), and even proposes to facilitate SERS (49). However, doubts on the existence of TNIs have been raised (22). Direct charge-transfer excitation from the metal Fermi level (EF) band into the π* of a physically adsorbed N2 molecule, the so-called 2π resonance state, is experimentally found to require energy of around 4 eV; for example, the 2π state of an adsorbed N2 on Ni(001) is 4.5 eV above the EF (50). We also showed with DFT that the 2π* state of η1-N2 on the AuMo surface is 3.3 eV above the EF, which is already expected to be underestimated by DFT (25). These relative energies provide clear evidence that a hot carrier injection leading to dissociation is unlikely for N2 at excitation energies within the NIR to visible spectrum (<3 eV) on at least high work function metals. Induced excitations within the surface defect due to plasmons are more likely mechanisms to enhance rates of reactions involving molecules with their lowest unoccupied molecular orbitals too high in energy for TNI states to exist.
The coupling between the LSPR of a plasmonic metal and an adjacent semiconducting material through a radiationless near-field dipole-dipole interaction via resonance energy transfer (Förster mechanism) was derived recently, coined as a plasmon-induced resonance energy transfer (PI-RET) (51). Extending this model to any acceptor on the surface of the MNP, one can obtain an expression for the PI-RET rate, wRET, using the methods of previous studies (17, 51, 52) (see the “PI-RET rate” section in the Supplementary Materials for the derivation):
| (1) |
where κ is an orientation factor between the plasmon and the acceptor transition dipoles and R is the distance between them, a is the MNP radius, c is the speed of light in vacuum, ω is the angular frequency of the incident light, Φsca is the LSPR scattering quantum yield, εd and εr are the complex and the real dielectric constants of the surrounding medium, εm is the complex dielectric constant of the metal, and f and IA(ω) are the oscillator strength and the normalized absorption spectrum of the acceptor. A is simply a proportionality constant relating f and the absorption cross section of the acceptor. |(εm − εd)/(εm + 2εd)|2 in Eq. 1 dictates the field enhancement |E/E0|2 outside the MNP. The electric field enhancements from LSPRs are on the order of |E/E0|2 ~ 102 for monomers and ≳ 104 between particles (hot spots) (6, 11, 12, 20) at plasmon resonance peaks (when εm ≈ −2εd for a spherical MNP), which therefore may facilitate transitions with f of even ~10−5 to 10−2. The magnified optical absorption of molecules on the surface of plasmonic MNPs due to electric field enhancements is the foundation of SERS (21, 22). The spectral overlap between the Au plasmon scattering and the surface absorption, defined by ∫|(εm − εd)/(εm + 2εd)|2ω2 IA(ω)dω, can be maximized by using nanorods with aspect ratios between 4 and 5 because they would have broad LSPR peaks centered around ~800 to 880 nm (1.40 to 1.5 eV) (18, 19).
Förster theory does not include a radiative term and applies to the weak electronic coupling limit: RET is incoherent and irreversible, that is, the energy transfer is only one way (52). In our case, the explicit quantum mechanical treatment of the mutual electronic effects of the surface and the molecule due to their chemical interaction takes care of the strong electronic coupling between the donor (metal) and acceptor (dopant + molecule), which is lacking in the Förster expression. Naturally, we assume that the dopant or molecule does not, in any quantitatively meaningful way, electronically affect the host metal Au. Although “lossy” metal dopants, such as Mo, can vary the spectral properties of Au (53), this may be easily accounted for in the variation in the complex dielectric function of the metal εm. In addition, Förster theory assumes that electron transfer does not occur or compete with RET, even at short distances between the acceptor and donor. This is shown to be a valid approximation in our system due to the absence of a clear charge-transfer excited state before the dissociation of N2 within the excitation energies probed (see Fig. 3 and fig. S8), which leads us to propose an alternative mechanism for energy transfer, such as RET.
The theoretically derived mechanistic picture provided here, where electronic excitations lower the activation barriers of a chemical reaction on an LSPR-active metal, is consistent with empirical evidence of the effect of LSPRs on chemical reactions in the literature. For example, Xiao and co-workers (9) showed that Au-Pd nanoalloys could lower the activation barriers of organic cross-coupling reactions by as much as ~50% with illumination: Sonogashira and Hiyama cross-coupling apparent barriers decrease from 217.3 and 150.4 kJ/mol to 120.6 and 50.8 kJ/mol, respectively, on the alloy under visible light illumination (0.45 W/cm2). Kim and co-workers (13) have shown wavelength-dependent lowering of the activation barrier to reduce Fe3+ to Fe2+ hexacyano complexes in the presence of spherical Au nanoparticles; the barrier is reduced by ~23 kJ/mol with 488-nm light illumination. Zhang and co-workers (14) found improved CO and CH4 production from CO2 and H2 on Rh nanocubes and Au spherical nanoparticles under UV illumination, with the activation barrier reduced from 78.6 (dark) to 50.4 kJ/mol for CH4 production on Rh and from 55.8 (dark) to 39.5 kJ/mol for CO production on Au.
It is clear from these experiments that certain reactions can have marked responses, whereas others can have modest responses, to LSPRs. It is worth pointing out that if the reactions are activated only by excited charge carriers, LSPRs should indiscriminately lower the barriers of reactions. However, instead, it is becoming clear that the nature of enhancement from LSPRs may depend strongly on the kind of reaction and the interaction of the adsorbate with the catalyst.
CONCLUSIONS
We showed that combining desirable properties of catalytic and plasmonic metals via surface doping of the former in the latter is a promising route to extend the library of reactions that can be plasmonically catalyzed. The advantages of plasmonic photocatalysts over other types of photocatalysts are their large extinction cross sections and their ability to locally enhance the incident electric field on the surface. The latter may augment the optical absorption of surface species, bringing them into their electronically excited states, and thus facilitate overcoming reaction barriers.
Specifically, we showed that, even if one considers the (generally too low) DFT barriers for N2 dissociation on flat [1.90 eV (39)] or stepped [0.78 eV (54)] Ru (0001) surfaces to be correct, the emb-NEVPT2 barriers on excited Mo-doped Au (highest barrier = 0.77 eV) suggest that the latter has comparable or even better (with respect to the flat surface) photochemical reactivity than the chemical reactivity of the best-in-class catalyst pure Ru in the dark. Experimentally, a barrier of 103 kJ/mol is measured for Fe, which is the main catalytic component in industrial HB catalysts (4, 55). Having only 74 kJ/mol as the highest barrier on the plasmon-active AuMo catalyst is a significant improvement over a known industrial catalyst. The LSPR response illustrated here shows how one may alter the surface chemistry of Mo-doped Au nanoparticles, potentially breaking the limitations of catalysts found in the ground state (54).
An NH3 synthesis catalyst should support H2 activation, as well as NH bond formation, and NH3 desorption, in addition to N2 activation. Plasmon-induced H2 dissociation on Au has been demonstrated recently (15). We expect that N–H bond formation and NH3 desorption would be highly favorable, considering that N and H atoms are only weakly adsorbed on the surface of Au in the ground state, although adsorption of N is strengthened by the presence of Mo. A rapid turnover of NH3 is expected, limited only by the rate at which atomic N is being produced on the surface, which should be controllable by light intensity.
Future iteration of the nanoplasmonic HB catalysts would require improvement in its synthesis for large-scale production. In parallel, research leading to discoveries of catalysts not involving precious metals should be conducted; in this regard, cheaper plasmonic metals such as Cu and Al may be considered. However, the interaction of the dopant and its efficacy as a catalyst may be different from when hosted by Au and therefore also would warrant detailed theoretical study. The key takeaway, though, is that surface doping is critical for activating these plasmonic metals to maximize the effect of LSPRs in catalysis.
A paradigm shift in how energy is delivered to drive reaction kinetics is required to develop low-temperature, energy-efficient HB processes, for example, from “low-quality” energy in the form of heat to “high-quality” and relatively more freely accessible energy in the form of visible light. Plasmonics can be one such avenue to drive the kinetics of N2 dissociation, overcoming limitations of thermal (ground-state) catalysts, as well as curtailing the unfavorable entropic effect of temperature on the thermodynamics of NH3 synthesis.
METHODS
Periodic DFT calculations
Periodic, spin-polarized, Kohn-Sham (KS) DFT calculations with the Perdew-Burke-Ernzerhof exchange-correlation functional (56) within the projector augmented-wave formalism (57) were performed using Vienna Ab initio Simulation Package (VASP) version 5.3.5 (58). A plane wave (PW) basis set with a kinetic energy cutoff of 500 eV was used for structure relaxations, in which all atom positions were relaxed until atomic forces were ≤0.01 eV/Å; a cutoff of 660 eV was used for single-point energy and vibrational frequency calculations (for an energy convergence of ≤1 meV/atom for the N2 molecule). A √21 × √21 five-layer periodic slab was used to model the surface, with one Au atom on one side of the slab substituted with Mo (Au104Mo) (25). The in-plane lattice constant was fixed to a = 4.168 Å, which was calculated to be optimal for the fcc Au crystal at this level of theory, and a vacuum of thickness ~14.5 Å normal to the surface was introduced. Brillouin zone sampling was conducted via 14 × 14 × 14 (Γ point–shifted) and 4 × 4 × 1 (Γ point–unshifted) k-point meshes within the Monkhorst-Pack method (59) for the bulk unit cell (four-atom basis cubic unit cell) and periodic slab, respectively. Electronic smearing following the Methfessel-Paxton method (60) at 0.09 eV was used to aid electronic convergence. van der Waals (vdW) interactions were included through Grimme’s semiempirical D3 with Becke-Johnson damping (61) to more adequately describe the interaction of N2 with the metal surface. Dipole corrections, in potential correction mode (62, 63) along the slab normal, were added to remove spurious dipole field interactions along this direction because of periodic images.
The vibrational modes of the adsorbate (N2) were calculated using the central difference method with ±0.015 Å atomic displacements from equilibrium. To manage computational cost, only the Au7Mo fragment of the surface was included in the construction of the Hessian, together with the molecule—suitable for calculating dopant- and adsorbate-derived local vibrational modes (see fig. S2).
The MEP was determined using the CI-NEB (38). In CI-NEB, a force constant of 3 eV/Å2 was used for the artificial spring connecting the images. The atomic forces were converged to ≤0.03 eV/Å, whereas the residual atomic forces on the transition state were converged to ≤0.01 eV/Å (similar to the stationary structures). The reaction coordinate, RC, is the length scale defined by cumulative distance between the images along the MEP
| (2) |
RC (M) is the RC at image M > 0, N is the total number of atoms per image, and (xi,j, yi,j, zi,j) are the coordinates of atom i at image j.
Constructing the embedding potential from DFET
A cluster with size Au10Mo (fig. S4A) was carved out from the slab, with the remaining 94 Au atoms designated as the environment. The embedding potential, Vemb, was determined from periodic PW-DFT using a modified VASP version 5.3.3 code via maximization of the extended Wu-Yang functional, W (64). W is a functional of the densities of the full system (represented by ∂Eref/∂Vemb), cluster (ρcl, via its energy ), and the environment (ρenv, via its energy )
| (3) |
The potential gradient is therefore defined as
| (4) |
resulting in the recovery of the ground-state slab electron density from the sum of the density of the fragments within DFT (40, 64). The embedded cluster and environment energies were obtained from modified one-electron KS Hamiltonians (H°)
| (5) |
Embedded cluster DFT, CASSCF, and NEVPT2 calculations
Once the optimized Vemb was derived (see fig. S4B), embedded cluster DFT () and correlated wave function (CW) () calculations, for different adsorbate configurations i, were calculated using Molpro version 2012.1 (65, 66). The molecular geometries were obtained from the MEP from the previous DFT slab NEB calculations. The internal coordinates of Mo and N2 were maintained along the reaction coordinate and placed/transplanted onto the fixed Au10Mo cluster.
All energies and calculated electronic properties reported for the embedded cluster were obtained using Gaussian-type orbital (GTO) basis sets, namely, Dunning’s augmented correlation-consistent polarized valence triple-ζ (aug-cc-pVTZ) for N (67) and Stuttgart/Cologne cc-pVTZ-PP with fully relativistic effective core potentials for both Mo (68) and Au (69, 70), unless otherwise stated (see table S1 for further details). Convergence of the predicted gas-phase N2 dissociation energy and surface reaction energies with respect to the basis set size is explored in tables S2 and S3 and fig. S5.
The Vemb, previously expressed on a real-space grid, is transformed into the GTO basis via an open-source embedding integral generation code (71), that is, ϵi,j = ∫gi*(r)Vembgj(r)dr were evaluated, where [gi(r)] are the GTO basis functions. The embedded cluster energies were then obtained using modified one-electron Hamiltonians, as described in the previous section, via the matrix manipulation feature in Molpro.
Accordingly, the energy of the system was evaluated following the DFET energy expression
| (6) |
is the energy of the full system from periodic PW-DFT (fully removing the empirical vdW correction to the energy) obtained from the CI-NEB calculations. Because of the multiconfigurational nature of metals and bond-breaking processes, here, the CW method we chose is the partially contracted NEVPT2 (44), using CAS(12e,12o) SS-CASSCF or seven-state SA-CASSCF to generate the reference wave functions (45, 46). All orbitals are optimized in the CASSCF calculations. The NEVPT2 procedure is essential to include (primarily dynamic) correlation originating from orbitals outside the CAS. The CAS(12e,12o) is composed of the (6e,6o) derived from molecular N2 (namely, the 5σ, the doubly degenerate 1π and 2π*, and the 6σ*) and the other (6e,6o) from the valence and low-lying virtual (unoccupied) orbitals of the Au10Mo cluster, derived from Mo 5d and Au 6s (Fig. 2 and fig. S7). In NEVPT2, the occupied N 2s and 2p; Mo 4p, 5s, and 4d; and Au 6s and 5d were explicitly correlated, whereas no additional correlation was included from the core N 1s, semicore Au 5s and 5p, and Mo 4s. Convergence of the energies with respect to the active space size is discussed in Supplementary Text, including fig. S6 and table S5.
The residual charges in N2 along the RC (fig. S8) were calculated using the Bader charge partitioning scheme (72). The electron densities of the embedded cluster + N2 were represented on a 180 × 180 × 320 real-space Cartesian grid, with uniform step size of 0.1 bohr, enough to contain the molecule.
The oscillator strengths of the electronic transitions (Fig. 3) were calculated using the equation
| (7) |
where ΔE is the excitation energy (calculated from emb-NEVPT2 in atomic units) and μs are the transition dipole moments of the excitation (from the reference emb-SA-CASSCF, also in atomic units) calculated with Molpro (48).
Supplementary Material
Acknowledgments
Funding: E.A.C. acknowledges financial support from the Air Force Office of Scientific Research via the Department of Defense Multidisciplinary University Research Initiative, under Award FA9550-15-1-0022. The High Performance Computing Modernization Program of the U.S. Department of Defense and Princeton University’s Terascale Infrastructure for Groundbreaking Research in Engineering and Science provided the computational resources. Author contributions: J.M.P.M. and E.A.C. designed and conceived the research. J.M.P.M. performed the calculations. J.M.P.M. and E.A.C. interpreted the data and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/12/eaao4710/DC1
Supplementary Text
fig. S1. Mechanisms of chemical enhancement on an MNP via an LSPR.
fig. S2. Adsorbate vibrational spectra.
fig. S3. Structural parameters versus reaction coordinate.
fig. S4. Metal cluster and embedding potential.
fig. S5. Reaction energy curve using a smaller basis set.
fig. S6. Singlet-triplet energy as a function of embedded metal cluster CAS size.
fig. S7. Additional CASSCF natural orbitals.
fig. S8. N2 charge versus reaction coordinate.
fig. S9. Comparison of the ground-state energy curves predicted by SA-CASSCF and SS-CASSCF.
fig. S10. Additional SA-CASSCF(12e,12o) electron difference density plots.
table S1. GTO basis sets.
table S2. Benchmark values for the gas-phase N2 dissociation energy (eV) with respect to the method and basis set used.
table S3. Dependence of reaction energies on basis set.
table S4. Ground-state CAS natural orbital occupations.
table S5. Dependence of reaction energies on CAS size.
movie S1. DFT + D3 CI-NEB–predicted structures along pathway a (physical adsorption).
movie S2. DFT + D3 CI-NEB–predicted structures along pathway b (reorientation).
movie S3. DFT + D3 CI-NEB–predicted structures along pathway c (dissociation).
data file S1. Atomic structure files.
REFERENCES AND NOTES
- 1.Smil V., Nitrogen and food production: Proteins for human diets. Ambio 31, 126–131 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Service R. F., New recipe produces ammonia from air, water, and sunlight. Science 345, 610 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Kandemir T., Schuster M. E., Senyshyn A., Behrens M., Schlögl R., The Haber-Bosch process revisited: On the real structure and stability of “ammonia iron” under working conditions. Angew. Chem. Int. Ed. 52, 12723–12726 (2013). [DOI] [PubMed] [Google Scholar]
- 4.M. Appl, Ammonia, in Ullmann’s Encyclopedia of Industrial Chemistry (Wiley-VCH Verlag GmbH & Co. KGaA, 2000), chap. 2. [Google Scholar]
- 5.M. W. Chase Jr., C. A. Davies, J. R. Downey Jr., D. J. Frurip, R. A. McDonald, A. N. Syverud, NIST JANAF Thermochemical Tables 1985 (National Institute of Standards and Technology, 1986), vol. 1–2. [Google Scholar]
- 6.Swearer D. F., Zhao H., Zhou L., Zhang C., Robatjazi H., Martirez J. M. P., Krauter C. M., Yazdi S., McClain M. J., Ringe E., Carter E. A., Nordlander P., Halas N. J., Heterometallic antenna–reactor complexes for photocatalysis. Proc. Natl. Acad. Sci. U.S.A. 113, 8916–8920 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng Z., Tachikawa T., Majima T., Plasmon-enhanced formic acid dehydrogenation using anisotropic Pd-Au nanorods studied at the single-particle level. J. Am. Chem. Soc. 137, 948–957 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Zheng Z., Tachikawa T., Majima T., Single-particle study of Pt-modified Au nanorods for plasmon-enhanced hydrogen generation in visible to near-infrared region. J. Am. Chem. Soc. 136, 6870–6873 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Xiao Q., Sarina S., Bo A., Jia J., Liu H., Arnold D. P., Huang Y., Wu H., Zhu H., Visible light-driven cross-coupling reactions at lower temperatures using a photocatalyst of palladium and gold alloy nanoparticles. ACS Catal. 4, 1725–1734 (2014). [Google Scholar]
- 10.Christopher P., Xin H., Linic S., Visible-light-enhanced catalytic oxidation reactions on plasmonic silver nanostructures. Nat. Chem. 3, 467–472 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Christopher P., Xin H., Marimuthu A., Linic S., Singular characteristics and unique chemical bond activation mechanisms of photocatalytic reactions on plasmonic nanostructures. Nat. Mater. 11, 1044–1050 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Linic S., Christopher P., Ingram D. B., Plasmonic-metal nanostructures for efficient conversion of solar to chemical energy. Nat. Mater. 10, 911–921 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Kim Y., Torres D. D., Jain P. K., Activation energies of plasmonic catalysts. Nano Lett. 16, 3399–3407 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Zhang X., Li X., Zhang D., Su N. Q., Yang W., Everitt H. O., Liu J., Product selectivity in plasmonic photocatalysis for carbon dioxide hydrogenation. Nat. Commun. 8, 14542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mukherjee S., Libisch F., Large N., Neumann O., Brown L. V., Cheng J., Lassiter J. B., Carter E. A., Nordlander P., Halas N. J., Hot electrons do the impossible: Plasmon-induced dissociation of H2 on Au. Nano Lett. 13, 240–247 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Zhou L., Zhang C., McClain M. J., Manjavacas A., Krauter C. M., Tian S., Berg F., Everitt H. O., Carter E. A., Nordlander P., Halas N. J., Aluminum nanocrystals as a plasmonic photocatalyst for hydrogen dissociation. Nano Lett. 16, 1478–1484 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Kelly K. L., Coronado E., Zhao L. L., Schatz G. C., The optical properties of metal nanoparticles: The influence of size, shape, and dielectric environment. J. Phys. Chem. B 107, 668–677 (2003). [Google Scholar]
- 18.Jain P. K., Huang X., El-Sayed I. H., El-Sayed M. A., Noble metals on the nanoscale: Optical and photothermal properties and some applications in imaging, sensing, biology, and medicine. Acc. Chem. Res. 41, 1578–1586 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Jain P. K., Lee K. S., El-Sayed I. H., El-Sayed M. A., Calculated absorption and scattering properties of gold nanoparticles of different size, shape, and composition: Applications in biological imaging and biomedicine. J. Phys. Chem. B 110, 7238–7248 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Boerigter C., Campana R., Morabito M., Linic S., Evidence and implications of direct charge excitation as the dominant mechanism in plasmon-mediated photocatalysis. Nat. Commun. 7, 10545 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moskovits M., Surface-enhanced spectroscopy. Rev. Mod. Phys. 57, 783–826 (1985). [Google Scholar]
- 22.Moskovits M., Persistent misconceptions regarding SERS. Phys. Chem. Chem. Phys. 15, 5301–5311 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Brongersma M. L., Halas N. J., Nordlander P., Plasmon-induced hot carrier science and technology. Nat. Nanotechnol. 10, 25–34 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Li J., Cushing S. K., Meng F., Senty T. R., Bristow A. D., Wu N., Plasmon-induced resonance energy transfer for solar energy conversion. Nat. Photonics 9, 601–607 (2015). [Google Scholar]
- 25.Martirez J. M. P., Carter E. A., Thermodynamic constraints in using AuM (M = Fe, Co, Ni, and Mo) alloys as N2 dissociation catalysts: Functionalizing a plasmon-active metal. ACS Nano 10, 2940–2949 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Martirez J. M. P., Carter E. A., Excited-state N2 dissociation pathway on Fe-functionalized Au. J. Am. Chem. Soc. 139, 4390–4398 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Nørskov J. K., Bligaard T., Logadottir A., Bahn S., Hansen L. B., Bollinger M., Bengaard H., Hammer B., Sljivancanin Z., Mavrikakis M., Xu Y., Dahl S., Jacobsen C. J. H., Universality in heterogeneous catalysis. J. Catal. 209, 275–278 (2002). [Google Scholar]
- 28.B. Predel, Au-W (gold-tungsten), in Ac-Au … Au-Zr, O. Madelung, Ed. (Springer, 1991), pp. 1–1. [Google Scholar]
- 29.Biener M. M., Biener J., Schalek R., Friend C. M., Surface alloying of immiscible metals: Mo on Au(111) studied by STM. Surf. Sci. 594, 221–230 (2005). [Google Scholar]
- 30.Li X. L., Ouyang G., Yang G. W., Surface alloying at the nanoscale: Mo on Au nanocrystalline films. Nanotechnology 19, 505303 (2008). [DOI] [PubMed] [Google Scholar]
- 31.T. Deutsch, A. Pasturel, Energetic effects in the Au-Ni system, in Stability of Materials, A. Gonis, P. E. A. Turchi, J. Kudrnovský, Eds. (Springer US, 1996), vol. 355, pp. 381–386. [Google Scholar]
- 32.Okamoto H., Massalski T. B., Nishizawa T., Hasebe M., The Au-Co (gold-cobalt) system. Bull. Alloy Phase Diagrams 6, 449–454 (1985). [Google Scholar]
- 33.Okamoto H., Massalski T. B., Swartzendruber L. J., Beck P. A., The Au–Fe (gold-iron) system. Bull. Alloy Phase Diagrams 5, 592–601 (1984). [Google Scholar]
- 34.Bratescu M. A., Takai O., Saito N., One-step synthesis of gold bimetallic nanoparticles with various metal-compositions. J. Alloys Compd. 562, 74–83 (2013). [Google Scholar]
- 35.Lu D.-l., Domen K., Tanaka K.-i., Electrodeposited Au-Fe, Au-Ni, and Au-Co alloy nanoparticles from aqueous electrolytes. Langmuir 18, 3226–3232 (2002). [Google Scholar]
- 36.Kumar S., Saini D., Large-scale synthesis of Au–Ni alloy nanowires using electrochemical deposition. Appl. Nanosci. 3, 101–107 (2013). [Google Scholar]
- 37.Amendola V., Scaramuzza S., Agnoli S., Polizzi S., Meneghetti M., Strong dependence of surface plasmon resonance and surface enhanced Raman scattering on the composition of Au-Fe nanoalloys. Nanoscale 6, 1423–1433 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Henkelman G., Uberuaga B. P., Jónsson H., A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000). [Google Scholar]
- 39.Falsig H., Shen J., Khan T. S., Guo W., Jones G., Dahl S., Bligaard T., On the structure sensitivity of direct NO decomposition over low-index transition metal facets. Top. Catal. 57, 80–88 (2014). [Google Scholar]
- 40.Huang C., Pavone M., Carter E. A., Quantum mechanical embedding theory based on a unique embedding potential. J. Chem. Phys. 134, 154110 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Libisch F., Huang C., Liao P., Pavone M., Carter E. A., Origin of the energy barrier to chemical reactions of O2 on Al(111): Evidence for charge transfer, not spin selection. Phys. Rev. Lett. 109, 198303 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Lundberg M., Siegbahn P. E. M., Quantifying the effects of the self-interaction error in DFT: When do the delocalized states appear? J. Chem. Phys. 122, 224103 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Cohen A. J., Mori-Sánchez P., Yang W., Insights into current limitations of density functional theory. Science 321, 792–794 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Angeli C., Cimiraglia R., Evangelisti S., Leininger T., Malrieu J. P., Introduction of n-electron valence states for multireference perturbation theory. J. Chem. Phys. 114, 10252–10264 (2001). [Google Scholar]
- 45.Roos B. O., The complete active space SCF method in a Fock-matrix-based super-CI formulation. Int. J. Quantum Chem. 18, 175–189 (1980). [Google Scholar]
- 46.Siegbahn P. E. M., Almlöf J., Heiberg A., Roos B. O., The complete active space SCF (CASSCF) method in a Newton–Raphson formulation with application to the HNO molecule. J. Chem. Phys. 74, 2384–2396 (1981). [Google Scholar]
- 47.Gillespie L. J., Beattie J. A., The thermodynamic treatment of chemical equilibria in systems composed of real gases. II. A relation for the heat of reaction applied to the ammonia synthesis reaction. The energy and entropy constants for ammonia. Phys. Rev. 36, 1008–1013 (1930). [Google Scholar]
- 48.N. J. Turro, Modern Molecular Photochemistry (University Science Books, 1991). [Google Scholar]
- 49.Michaels A. M., Nirmal M., Brus L. E., Surface enhanced Raman spectroscopy of individual rhodamine 6G molecules on large Ag nanocrystals. J. Am. Chem. Soc. 121, 9932–9939 (1999). [Google Scholar]
- 50.Johnson P. D., Hulbert S. L., Inverse-photoemission studies of adsorbed diatomic-molecules. Phys. Rev. B 35, 9427–9436 (1987). [DOI] [PubMed] [Google Scholar]
- 51.Cushing S. K., Li J., Meng F., Senty T. R., Suri S., Zhi M., Li M., Bristow A. D., Wu N., Photocatalytic activity enhanced by plasmonic resonant energy transfer from metal to semiconductor. J. Am. Chem. Soc. 134, 15033–15041 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Hsu L.-Y., Ding W., Schatz G. C., Plasmon-coupled resonance energy transfer. J. Phys. Chem. Lett. 8, 2357–2367 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Amendola V., Saija R., Maragò O. M., Iati M. A., Superior plasmon absorption in iron-doped gold nanoparticles. Nanoscale 7, 8782–8792 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Vojvodic A., Medford A. J., Studt F., Abild-Pedersen F., Khan T. S., Bligaard T., Nørskov J. K., Exploring the limits: A low-pressure, low-temperature Haber-Bosch process. Chem. Phys. Lett. 598, 108–112 (2014). [Google Scholar]
- 55.Brill R., Hensel J., Schaefer H., Über den einfluß der längsdiffusion auf die kinetik der ammoniakbildung. Ber. Bunsen. Phys. Chem. 73, 999–1003 (1969). [Google Scholar]
- 56.Perdew J. P., Burke K., Ernzerhof M., Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- 57.Blöchl P. E., Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994). [DOI] [PubMed] [Google Scholar]
- 58.Kresse G., Furthmüller J., Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996). [DOI] [PubMed] [Google Scholar]
- 59.Monkhorst H. J., Pack J. D., Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976). [Google Scholar]
- 60.Methfessel M., Paxton A. T., High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 40, 3616–3621 (1989). [DOI] [PubMed] [Google Scholar]
- 61.Grimme S., Ehrlich S., Goerigk L., Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011). [DOI] [PubMed] [Google Scholar]
- 62.Makov G., Payne M. C., Periodic boundary conditions in ab initio calculations. Phys. Rev. B 51, 4014–4022 (1995). [DOI] [PubMed] [Google Scholar]
- 63.Neugebauer J., Scheffler M., Adsorbate-substrate and adsorbate-adsorbate interactions of Na and K adlayers on Al(111). Phys. Rev. B 46, 16067–16080 (1992). [DOI] [PubMed] [Google Scholar]
- 64.Yu K., Libisch F., Carter E. A., Implementation of density functional embedding theory within the projector-augmented-wave method and applications to semiconductor defect states. J. Chem. Phys. 143, 102806 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Werner H.-J., Knowles P. J., Knizia G., Manby F. R., Schütz M., Molpro: A general-purpose quantum chemistry program package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2, 242–253 (2012). [Google Scholar]
- 66.P. J. Knowles, H.-J. Werner, G. Knizia, F. R. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklaß, D. P. O’Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang, Molpro Quantum Chemistry Software; www.molpro.net.
- 67.Dunning T. H., Jr., Gaussian-basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023 (1989). [Google Scholar]
- 68.Peterson K. A., Figgen D., Dolg M., Stoll H., Energy-consistent relativistic pseudopotentials and correlation consistent basis sets for the 4d elements Y-Pd. J. Chem. Phys. 126, 124101 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Figgen D., Rauhut G., Dolg M., Stoll H., Energy-consistent pseudopotentials for group 11 and 12 atoms: Adjustment to multi-configuration Dirac–Hartree–Fock data. Chem. Phys. 311, 227–244 (2005). [Google Scholar]
- 70.Peterson K. A., Puzzarini C., Systematically convergent basis sets for transition metals. II. Pseudopotential-based correlation consistent basis sets for the group 11 (Cu, Ag, Au) and 12 (Zn, Cd, Hg) elements. Theor. Chem. Acc. 114, 283–296 (2005). [Google Scholar]
- 71.C. M. Krauter, E. A. Carter, EmbeddingIntegralGenerator; https://github.com/EACcodes/EmbeddingIntegralGenerator.
- 72.Tang W., Sanville E., Henkelman G., A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 21, 084204 (2009). [DOI] [PubMed] [Google Scholar]
- 73.Malmqvist P. A., Rendell A., Roos B. O., The restricted active space self-consistent-field method, implemented with a split graph unitary group approach. J. Phys. Chem. 94, 5477–5482 (1990). [Google Scholar]
- 74.P. F. Bernath, Spectra of Atoms and Molecules (Oxford Univ. Press, ed. 2, 2005). [Google Scholar]
- 75.Su H., Zhong Y., Ming T., Wang J., Wong K. S., Extraordinary surface plasmon coupled emission using core/shell gold nanorods. J. Phys. Chem. C 116, 9259–9264 (2012). [Google Scholar]
- 76.Willets K. A., Van Duyne R. P., Localized surface plasmon resonance spectroscopy and sensing. Annu. Rev. Phys. Chem. 58, 267–297 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/12/eaao4710/DC1
Supplementary Text
fig. S1. Mechanisms of chemical enhancement on an MNP via an LSPR.
fig. S2. Adsorbate vibrational spectra.
fig. S3. Structural parameters versus reaction coordinate.
fig. S4. Metal cluster and embedding potential.
fig. S5. Reaction energy curve using a smaller basis set.
fig. S6. Singlet-triplet energy as a function of embedded metal cluster CAS size.
fig. S7. Additional CASSCF natural orbitals.
fig. S8. N2 charge versus reaction coordinate.
fig. S9. Comparison of the ground-state energy curves predicted by SA-CASSCF and SS-CASSCF.
fig. S10. Additional SA-CASSCF(12e,12o) electron difference density plots.
table S1. GTO basis sets.
table S2. Benchmark values for the gas-phase N2 dissociation energy (eV) with respect to the method and basis set used.
table S3. Dependence of reaction energies on basis set.
table S4. Ground-state CAS natural orbital occupations.
table S5. Dependence of reaction energies on CAS size.
movie S1. DFT + D3 CI-NEB–predicted structures along pathway a (physical adsorption).
movie S2. DFT + D3 CI-NEB–predicted structures along pathway b (reorientation).
movie S3. DFT + D3 CI-NEB–predicted structures along pathway c (dissociation).
data file S1. Atomic structure files.