

Abstract
Here we examine the relationship between resolving power (Rp), resolution (Rpp), and collision cross section (CCS) for compounds analyzed in previous ion mobility (IM) experiments representing a wide variety of instrument platforms and IM techniques. Our previous work indicated these three variables effectively describe and predict separation efficiency for drift tube ion mobility spectrometry (DTIMS) experiments. In this work we seek to determine if our previous findings are a general reflection of IM behavior applicable to various instrument platforms and mobility techniques. Results suggest IM distributions are well characterized by a Gaussian model and separation efficiency can be predicted based on empirical difference in the gas-phase CCS and a CCS-based resolving power definition (CCS/ΔCCS). Notably traveling wave (TWIMS) was found to operate at substantially higher resolutions than a single-peak resolving power suggested. When a CCS-based Rp definition was utilized, TWIMS was found to operate at a resolving power of between 40 and 50, confirming the previous observations by Giles and coworkers. After converting the separation axis (and corresponding resolving power) to cross section space it is possible to effectively predict separation behavior for all mobility techniques evaluated (i.e. uniform field, trapped ion mobility, traveling wave, cyclic and overtone instruments) using the equations described in this work. Finally, we are able to establish for the first time that the current state-of-the-art ion mobility separations benchmark at a CCS-based resolving powers above 300 which is sufficient to differentiate analyte ions with CCS differences as small as 0.5%.
Graphical abstract
Ion mobility combined with mass spectrometry (IM-MS) is now an important and established analytical technique for characterizing chemical compounds simultaneously by molecular size (collision cross section, CCS) and molecular weight (mass-to-charge ratio, m/z).1–4 In the past decade, a myriad of ion mobility technologies have been developed and interfaced with MS (Table 1), including traveling wave drift cells (TWIMS),5,6 uniform field and confining RF drift tubes (DTIMS and rf-DTIMS),7,8 mobility separators (FAIMS/DMS),9,10 field-flow dispersive devices (DMA TIMS, and Transversal Modulation IMS, TMIMS),11–14 and confining 2-dimensional ion conveyors (SLIM),15,16 among others. While each of these IM technologies utilize different mobility dispersive fields to generate an ion mobility spectrum, all operate on a common basis of separating molecules based upon differences in their gas-phase ion mobility behavior. Thus, it should be possible to relate the separation efficiency of all IM techniques to a common, normalized parameter such as the reduced ion mobility coefficient (K0), or the gas-specific CCS value.
Table 1.
Various IM techniques and the respective dispersion dimension commonly reported for each technique.
| IM Technique | Abbreviation | Dispersion Dimension |
|---|---|---|
| Drift Tube Ion Mobility Spectrometry | DTIMS | Time |
| Confining RF DTIMS | rf-DTIMS | Time |
| Ambient Pressure DTIMS | ap-DTIMS | Time |
| Traveling Wave Ion Mobility Spectrometry | TWIMS | Time |
| Cyclic Traveling Wave Ion Mobility Spectrometry | Cyclic TWIMS | Time |
| Trapped Ion Mobility Spectrometry | TIMS | Time |
| Structures for Lossless Ion Manipulations | SLIM (DTIMS and TWIMS) | Time |
| Asymmetric Field Ion Mobility Spectrometry | FAIMS | Voltage |
| Differential Mobility Spectrometry | DMS | Voltage |
| Differential Mobility Analyzer | DMA | Voltage |
| Overtone Mobility Spectrometry | OMS | Frequency |
| Cyclic Ion Mobility Spectrometry | Cyclic IMS | Frequency |
In ion mobility and mass spectrometry, the resolving power (Rp) is defined quantitatively from a single peak as a ratio of the location of the peak divided by its width (eqn. 1).17
| (1) |
Here, x is the dimensional location of the measurement, which is technique-specific, and Δx is commonly defined as the full width of the peak at half its maximum height (fwhm). In mass spectrometry, the dimension of x is mass-to-charge (m/z) which is an intrinsic property of the ion. As all mass spectrometers report separations in m/z space, mass resolving power provides a convenient and reliable common basis for comparing results from different MS techniques (e.g., quadrupole, time-of-flight, and Fourier transform ion trap instruments). For modern mass spectrometers, mass separation efficiency can range from a few thousand (quadrupoles and electrodynamic ion traps), to tens of thousands (time-of-flight), to upwards of one million resolving power (Fourier transform MS). In contrast, IM resolving power is commonly calculated from the technique-specific mobility dispersion dimension (e.g., drift time or dispersion voltage) and resolving powers are rarely reported above 100.
While the time-based definition of resolving power (t/Δt) has been utilized for over two decades to report the separation efficiency of DTIMS instruments, the emergence of other IM techniques has complicated the interpretation of the time-based IM resolving power. For example, TWIMS ion mobility dispersion occurs on a timescale that is about an order of magnitude faster than many DTIMS platforms, resulting in erroneously low resolving power value when the time-based definition is utilized. The shortcomings of a time-based resolving power definition led Giles and coworkers to report a CCS-based resolving power (CCS/ΔCCS) when quantifying the separation efficiency of a second-generation TWIMS device.6 For some IM techniques, mobility separations do not directly occur in the time domain, such as with TIMS and FAIMS/DMS where a scanned electric field is utilized to generate a time-dependent IM spectrum. As recently underscored by Glish and coworkers, FAIMS/DMS is capable of achieving very high resolving power numbers (e.g., 7903), however, the corresponding 2-peak separation is lower than this high resolving power suggested.18 Hence, a technique-specific resolving power definition cannot give a comparable description of the analytical selectivity of different IM instruments.
While a single peak resolving power definition is a convenient metric for assessing IM instrument performance, practical separation efficiency is also often defined in terms of two-peak resolution (Rpp), which is a definition commonly used in condensed phase chromatography to quantify separation efficiency. IM shares similarities with chromatography, namely that analyte separations in both techniques are based on an extrinsic property (retention time and CCS, respectively) which can be altered to enhance the analytical selectivity of the separation. Two-peak resolution in IM is defined as separation of two closely-spaced Gaussian peaks (e.g. compounds A and B) via eqn 3.19,20
| (2) |
Conventionally for IM, eqn 2 is defined in the time-do-main (drift time for x) and the fwhm of each peak is used. Our previous study indicated a direct relationship between two-peak resolution (Rpp), single-peak resolving power (Rp) and the ion’s gas-phase collision cross section (CCS), through eqn. 3.21
| (3) |
Here the resolution is denoted as “predicted” to indicate that this value can be obtained theoretically. For a mobility separation of two analytes, Rp should be calculated as the average resolving power of both peaks, whereas the percent difference in cross section (ΔCCS%) is based on the average CCS, as defined in eqn 4.
| (4) |
We emphasize here that the ΔCCS% is a measure of how different two compounds are in cross section space, calculated as a percentage, and in this manner ΔCCS% is size-independent. For example, two molecules with CCS measurements of 100 and 101 Å2 (Δ 1 Å2) have the same ΔCCS% (1.0% difference) as a separate pair of compounds measuring 200 and 202 Å2 (Δ 2 Å2). These two separations should be equally challenging, and hence require the same measure of resolving power to separate as their percent difference in CCS space is equal. It is also important to note that when resolving power is measured as CCS/ΔCCS, that ΔCCS is the FWHM of a given IMS peak, and is not related to the percent difference in cross section of two peaks (ΔCCS%).
The validity of eqn 4 was previously established based on a Gaussian fit to uniform field IM data and provided the basis for predicting the IM separation of two analytes in a hypothetical mixture given that both their CCS values and the instrument resolving power was known.21
In this present study, we attempt to ascertain whether the relationship in eqn. 3 reflects a general observation of mobility behavior across various instrumentation and techniques. Thus this work endeavors to unify the various methods of mobility separation under a single descriptor of analytical efficiency in order to predict analyte separation for cross systems assessment.
Experimental Methods
Chemical Standards
L-leucine (61819) and L-isoleucine (I2752) were obtained from Sigma-Aldrich (St. Louis, MO, USA). The protein digest standard (MassPrep Mix 1) was obtained from Waters Co. (Milford, MA, USA) and consists of four tryptically-digested proteins (yeast enolase, rabbit phophorylase b, yeast alcohol dehydrogenase, and bovine serum albumin). The L-leucine and L-isoleucine standards were reconstituted to a final concentration of 10 μg/mL in high purity water (18 MΩ, Milli-Q, EMD Millipore, Billerica, MA, USA) buffered with 10 mM ammonium acetate (Sigma-Aldrich) to a pH of 6.5 (SevenEasy pH Meter, Mettler-Toledo, Columbus, OH, USA).
Instrumentation and Methods
A commercial DTIMS instrument (6560, Agilent Technologies) was used for all empirical CCS measurements performed in this study. Details of the instrumentation and CCS method have been previously described.7,19,22 Briefly, chemical standards were directly infused into the electrospray ionization source (Jet Stream, Agilent) at a flow rate of 5 μL/min using a syringe pump (KDS 101, KD Scientific, Inc.). Ion mobility separations were conducted in a uniform field drift tube operated with high purity nitrogen drift gas at 3.95 Torr and room temperature (ca. 298 K). A seven-frame stepped electric field method was utilized in the range of 10.9 to 18.5 V/cm, which provided the necessary data to perform a linear regression analysis used to determine the non-mobility ion transit times. This DTIMS CCS method was previously optimized based on the results of an interlaboratory study.23
Selection of Published Spectra
Previously published IM spectra were selected for this current study based on the quality of the published spectra, the inclusion of two well-defined ion mobility distributions (either partially or fully resolved), and the requirement that measurements were obtained from pure chemical standards. A diverse set of spectra were selected representing different IM instrumentation and techniques. The applicability of a Gaussian-based model for separation was assessed using a protocol described in the Supporting Information (Figure S1). Briefly, theoretical Gaussian distributions were generated and overlaid onto IM spectra obtained from the literature (see Figure 1 A–F). The important assumption made here is that the published IM spectra represent near optimal separations for each IM technique, as these spectra were obtained from experts in their respective fields. While not comprehensive, the spectra chosen here are representative of many well-conducted studies across the field. A comprehensive and annotated list of references to each spectrum used in this assessment is provided in the Supporting Information (Appendix S1).
Figure 1.
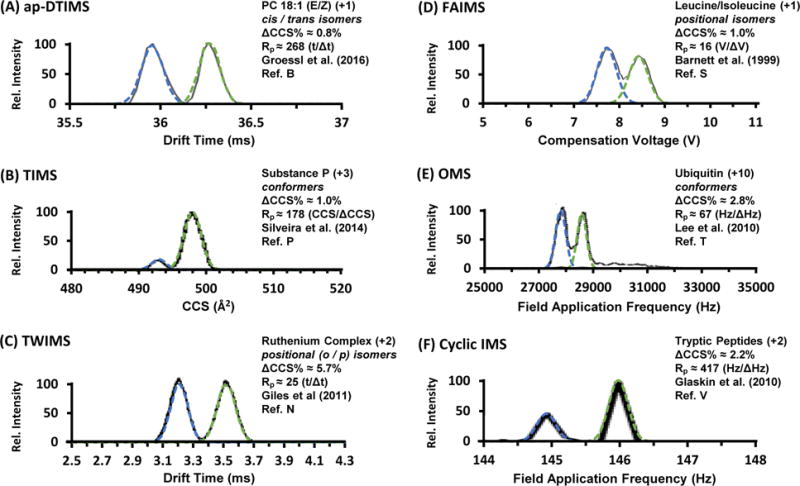
Selected IM spectra obtained from the literature representing challenging analyte separations using various IM techniques and instrumentation. Gaussian distribution overlays are shown as dotted traces. The corresponding single-peak resolving powers and percent differences in CCS as determined in the current analysis are provided for each example. (A) Reproduced/Adapted with permission from Groessl and coworkers, 2016 (see Supporting Information). (B) Reproduced/Adapted with permission from Ref. 30. American Chemical Society, 2014. (C) Reproduced/Adapted with permission from Ref. 6. Wiley and Sons, 2011. (D) Reproduced/Adapted with permission from Ref. 52. Elsevier, 1999. (E) Reproduced/Adapted with permission from Ref. 32. American Chemical Society, 2010. (F) Reproduced/Adapted with permission from Ref. 36. American Chemical Society, 2010.
Evaluation of Separation Efficiency
A total of 22 published ion mobility separations were examined from multiple sources including peer-reviewed literature, conference posters, and instrument vendor white papers, which represent various IM techniques and platforms (Table 2) and a broad range of analyte masses (131 to 8566 Da). For each separation, CCS values were used as reported from the cited source and the percent difference in CCS was calculated through eqn 4. Average resolving power and resolution were calculated via eqns 1 and 2, respectively, using the dimension of the reported separation. Utilizing the average resolving power and the calculated percent difference in CCS, the predicted resolution (Rpp) is subsequently calculated via eqn 3. In order to compare the theoretical resolution predicted by eqn 4 to the observed experimental two-peak resolution, we calculate the percent error in our prediction through eqn 5.
| (5) |
Table 2.
Separation parameters reported for various ion mobility platforms. The “Reference Point” column references the annotations in Figure 4 and in Appendix S1 of the supporting information.
| First Author | Ref. Point | Percent Difference in CCS (%)1 | Reported Resolving Power (Rp) 2 | Calculated Resolving Power (Rp)3 | Experimental Resolution (Rpp)4 | Predicted Resolution (Rpp)5 | Percent Error in Resolution (%)6 | Dispersion Axis Projected in IM Spectra | |
|---|---|---|---|---|---|---|---|---|---|
| DTIMS | Groessl, M. | A | 0.4 | >250 | 332 | 0.84 | 0.78 | 7.3 | td (ms) |
| Groessl, M. | B | 0.8 | 250 | 268 | 1.38 | 1.26 | 8.3 | td (ms) | |
| Groessl, M. | C | [1.2] | [251] | 251 | 1.77 | 1.77 | −0.3 | td (ms) | |
| Asbury, G. R. | D | [1.2] | [130] | 131 | 0.76 | 0.93 | −22.2 | td (ms) | |
| Groessl, M. | E | 1.3 | [187] | 187 | 1.31 | 1.46 | −11.8 | td (ms) | |
| Pierson, G. U. | F | 1.7 | [66] | 66 | 0.65 | 0.65 | 0.1 | CCS (Å2) | |
| Tang, G. R. | G | 1.7 | [62] | 63 | 0.58 | 0.63 | −8.0 | td (ms) | |
| Dodds, J. N. | H | 2.3 | 58 | 58 | 0.79 | 0.79 | 0.1 | CCS (Å2) | |
| Gaye, M. M. | I | 2.6 | [83] | 83 | 1.26 | 1.26 | 0.0 | CCS (Å2) | |
| Adamov, A. | J | 3.2 | 77 | 72 | 1.46 | 1.46 | −0.2 | K 2 0 (cm/Vs) | |
|
| |||||||||
| TWIMS | Deng, L. | K | 0.4 | 124 | 342 | 0.71 | 0.26 | 63.1 | td (ms) |
| Giles, K. | L | 1.5 | – | [208] | 4.34 | 1.90 | 56.3 | td (ms) | |
| Giles, K. | M | 5.1 | 18 | 40 | 1.21 | 0.55 | 55.0 | td (ms) | |
| Giles, K. | N | 5.7 | 25 | 41 | 1.36 | 0.83 | 38.7 | td (ms) | |
| Hofmann, J. | O | 5.9 | 26 | 43 | 1.51 | 0.88 | 41.4 | td (ms) | |
|
| |||||||||
| TIMS | Silveira, J. A. | P | 1.0 | 154–183 | 178 | 1.05 | 1.06 | −0.3 | CCS (Å2) |
| Bruker | Q | 1.0 | [113] | 113 | 0.68 | 0.69 | −0.6 | K0 (cm2/Vs) | |
| Bruker | R | 1.7 | 185 | 177 | 1.74 | 1.74 | 0.1 | K0 (cm2/Vs) | |
|
| |||||||||
| OTHER | Barnett, D. A. | S | [1.0] | – | 130 | 0.83 | – | – | Voltage (V) |
| Lee, S. | T | 2.8 | [66] | 67 | 1.09 | 1.09 | 0.1 | Frequency (Hz) | |
| Glaskin, R. S. | U | 2.7 | 121 | 85 | 1.42 | 1.33 | 6.3 | Frequency (Hz) | |
| Glaskin, R. S. | V | 2.2 | 417 | 145 | 1.90 | 5.72 | 185 | Frequency (Hz) | |
Calculated from equation 4. Bracketed values were determined using CCS values obtained in the PI’s laboratory
Bracketed values were calculated from equation 1 using the dispersion axis provided.
Calculated from equation 1 using the CCS-based definition for Rp.
Calculated from equation 2 using the dispersion axis provided.
Calculated from equation 3 using the dispersion axis provided.
Calculated from equation 5.
Results and Discussion
Gaussian Distributions
While the mechanics of ion diffusion in drift tube instruments are well characterized and can be described as Gaussian to a good approximation,24–26 the band-broadening mechanisms for other IM separation methods cannot be easily described by the first principles established in the kinetic theory of gases. To determine if other mobility techniques exhibit peak shapes that can modeled with a normal distribution, the IM spectra selected for this study were examined using a protocol described in the Supporting Information (Figure S1). Based on the quality of correlation observed between the published IM spectra and the theoretical Gaussian distributions, it was concluded here that the spectra from a wide distribution of IM techniques exhibit peak shapes accurately described by a normal distribution in standard operating conditions (e.g. no secondary conformers or peak saturation is observed (Figure 1A–F). This observation, in turn, justifies the use of a Gaussian-based mathematical description of ion mobility separation efficiency.
CCS-Based Resolving Power
Obtaining CCS from different IM experiments can be challenging as the fundamental ion mobility equation is only applicable for uniform field instruments with well-characterized gas compositions, e.g., DTIMS and DMA. Other IM techniques, namely TWIMS, OMS27–29 and TIMS12,30,31 have established protocols for converting the corresponding transmission frequency of analytes into CCS or Ko,31,32 which is of particular utility in relation to eqns 3 and 4. Defining the FAIMS and DMS separations in terms of CCS is more challenging as the mobility spectra are reported in terms of the compensation voltage that transmits the ion of interest. Depending on the particular experiment setup, nominal resolving power in FAIMS (V/ΔV) can be artificially low (Figure 1D), or uncharacteristically high, as noted by a recent report.18 It is not currently possible to translate FAIMS or DMS measurements directly into cross section space using a fundamental relationship. In addition, many FAIMS and DMS experiments are carried out in a mixture of drift gases in order to enhance selectivity,33,34 making comparisons to published CCS values (which are gas-specific) challenging.35 For the purposes of this study, chemical systems with known CCS values are selected, which allows each chosen spectra to be evaluated in terms of the percent difference in CCS.
Cross-Platform Assessment
For the 22 ion mobility separations surveyed in this study, both the observed experimental resolution (eqn 2) and predicted resolution (eqn 3), were calculated and the corresponding percent error between these calculations was determined via eqn 5. The percent error is a reflection of the ability of eqn. 3 to predict the level of separation efficiency for two analytes possessing a characterized difference in cross section at a given level of resolving power. Results are summarized in Table 2. Mobility separations for DTIMS, TIMS, and OMS instruments32,36–41 (Data Points A to J, and T) show sufficient agreement with eqn 3, with typically less than 10% error between the experimental and predicted resolution. We consider this good agreement as experimental single-peak resolving power can vary by as much as 11% between consecutive measurements on a DTIMS instrument.22 This good correlation suggests for DTIMS, TIMS, and OMS, the separation efficiency as determined from each corresponding dispersion dimension (time and frequency, respectively) correlate closely with the CCS-based Rp definition developed in this work. However, FAIMS/DMS and cyclic IMS separations are currently reported based on dispersion voltages or field application frequency, respectively, which yield Rp values that do not correlate to their respective CCS-based resolving powers. In some cases, for FAIMS/DMS, the dispersion axis is reported with negative values which cannot be used to determine resolving power. For cyclic IMS, the frequency based Rp values are higher than the CCS-based Rp, whereas utilizing the voltage axis in FAIMS yields Rp values that are lower than their corresponding CCS-based Rp. An erroneously low Rp was also found for TWIMS when using the time-domain definition of resolving power, and this result is discussed in detail in the following section.
Traveling Wave Resolving Power
Large deviations from eq 4 in terms of percent error (typically 40% or larger) were found for traveling wave instruments, which are utilized extensively by the IM-MS community. This limitation in time-based Rp calculations for TWIMS has been previously reported. For example, in their well-documented separation of reverse peptides (SDGRG and GRGDS, point M) Giles and coworkers were able to separate these two sequence isomers with ca. 5.1% difference in CCS to near baseline resolution using a second generation TWIMS geometry (Synapt G2).6 Using their experimental time-based resolving power of ca. 18 (td/Δtd) results in ca. 55% error through the prediction given by eqn. 4 (Figure S2A). The interpretation of this discrepancy is that although both drift tube and traveling wave experiments are time-dispersive separations, traveling wave devices operate at a higher level of selectivity than would be expected for their corresponding time based resolving power.6,42,43 Interestingly, using the established protocols for converting analyte drift time to CCS in TWIMS,44,45 Giles and coworkers also calculated resolving power in cross section space (CCS/ΔCCS). Their resulting experimental CCS-based Rp are nearly identical to what is found in this current work (ca. 40 CCS/ΔCCS) and show much more agreement to the predicted Rp (eqn. 3) than the time-based Rp, with 1% versus 55% error, respectively, based on eqn. 5 (Figure S2B). Following this example, we converted TWIMS resolving power from the time domain (td/Δtd) to CCS space (CCS/ΔCCS) for five different TWIMS separations reported in the literature, and the results are summarized in Table S1 (also Figures S3, S4, and Table S1). The five selected TWIMS separations include 3 studies of isomer separations obtained on the Synapt G2 (Points M, N, and O) where time based Rp is ca. 20-25,6,46 however once the CCS-based definition of eqn. 1 is used, the Rp is approximately doubled (ca. 40 CCS/ΔCCS). These larger Rp values better-reflect the analytical selectivity of TWIMS, with a corresponding low percent error predicted by eqn 5. Conversion to CCS-based Rp is also necessary for the recently developed cyclic TWIMS4,47 (Point L) which indicates that this device operates with a resolving power of ca. 480 for 50 cycles (c.f., Table S1). Additionally, the SLIM-based TWIMS instrument currently being developed by Smith and coworkers at Pacific Northwest National Laboratory (Point K),48 has shown very high analytical separation capabilities, and using the CCS-based definition, we can quantify for the first time the resolving power of current SLIM-based TWIMS devices as benchmarking around 340 (Figure S4). We note that the SLIM technology was initially developed for DTIMS-based separations.49,50
The discrepancy between CCS and time based Rp in TWIMS is related to the nonlinear relationship between voltage and analyte drift time in these devices, which has been discussed previously.42,43,51
Cross-Platform Assessment of Separation Capabilities
Unlike the TWIMS instruments, both time and CCS based Rp are nominally very similar for drift tube instruments. For example, in our previous work the separations related to isomers of leucine/isoleucine indicate the same level of nominal resolving power in both the time dimension and CCS space (ca. 60 td/Δtd and CCS/ΔCCS).21 Other DTIMS conversions from time to CCS-based Rp also indicated negligible differences in resolving power, typically less than 5%. This correlation of Rp in both the time and CCS dimension is a result of the linear relationship between drift time and CCS in uniform field instruments. Other IM techniques in Figure 1 measure Rp in terms of reduced mobility (Ko/ΔKo) and also exhibit low percent error. As FAIMS is not able to empirically measure CCS, the only FAIMS spectrum used in this work (Point S, Table 2),52 is included for comparison using previously measured nitrogen-based CCS values in our laboratory. Also, it should be noted that Barnett et. al. utilized ambient air (compressed) as the buffer gas instead of pure nitrogen, which will yield a slightly different CCS than what is used in this comparison.52
With a common frame of reference, we can compare the separation abilities of various IM techniques. The plot in Figure 2 depicts boundary regions representing various levels of separation efficiency calculated through eqn 3 covering a wide range of percent difference in CCS and resolving power. Numerical relationships between Rp and ΔCCS% are tabulated in Tables 2 and S1. The location of a given data point in Figure 2 corresponds to the percent difference in CCS of the specific compounds analyzed, the experimental resolving power (determined as described previously), and the calculated two-peak resolution of the published separation. The light shaded region at the top left section of the graph corresponds to all combinations of ΔCCS% and Rp which will yield baseline or greater separation (Rpp ≥ 1.23). For example, reverse peptides (SDGRG/GRGDS (+1), ΔCCS = 1.5%) were baseline separated by Giles et. al. using a prototype cyclic TWIMS instrument with ca. 480 resolving power (CCS/ΔCCS) (Point L, Rpp = 4.34).53 The darker shaded regions at the bottom right of Figure 2 indicate regions of greater than half-height separation (Rpp ≥ 1.23), equal to half height separation (Rpp = 0.83) and minimum resolution (Rp-p ≤ 0.61, or 10% separation), respectively. Note the current state-of-the art in IM performs with Rp over 300, which enables separation of ions differing by as little as ca. 0.5% in CCS (e.g., 1 Å2 at 200 Å2).54,55 In the ap-DTIMS examples chosen for this work, high resolution is achieved by operating the instrument at greater than atmospheric pressures (ca. 1050 Torr) and utilizing Hadamard transform multiplexing.56,57
Figure 2.
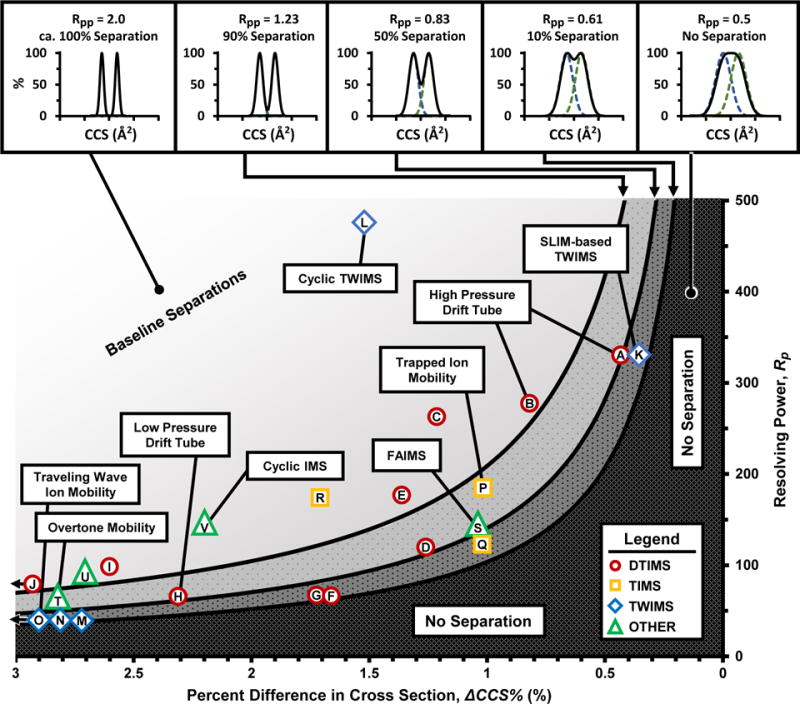
Plot depicting the required resolving power to separate two compounds in ion mobility with a known percent difference in cross section. Various levels of separation efficiency are indicated along the top panels both in terms of two-peak resolution (Rpp) and visually by means of percent separation. Previously published ion mobility separations (c.f., Table 2) are referenced in the plot and represent various ion mobility techniques. The techniques labeled “Other” (green triangles) include FAIMS, OMS, and cyclic IMS. Resolving powers for traveling wave and cyclic IMS instruments are reported here in the ion cross section domain (CCS/ΔCCS).
Figure 2 reveals several important analytical trends for the field of ion mobility. First, despite the wide range of CCS values represented here (ca. 100 to 500 Å2), the percent difference in CCS and the CCS-based Rp represents robust parameters for comparing the relative separation capabilities of different IM instrumentation. Second, various IM instruments operate across a very wide range of separation efficiencies, with the majority of commercially-available IM-MS platforms accessing IM resolving powers of 80 or less. The highest separation efficiencies represent ap-DTIMS and long path length TWIMS devices (both cyclic and serpentine), which have demonstrated resolving powers (CCS/ΔCCS) in excess of 300.
How Much Resolving Power is Necessary?
In order to assess the amount of resolving power required for routine ion mobility separation in biological applications, a protein digest was evaluated, which yielded a total of 100 CCS measurements for +1 and +2 protonated tryptic peptides. Identifications were made on the basis of mass measurement accuracy, which was less than 5 ppm for all peptides used in the subsequent analysis.
Mass Analysis
In order to describe the separation of the digested peptides by IM-MS, first we examine the mass dimension of the separation (Figure 3A). Of the 204 possible peptides (no missed cleavages), 10 peptides (ca. 5%) are constituents of isomeric pairs generated by permuted amino acid sequences (e.g., LAK and ALK) or isomeric amino acid substitutions (e.g. leucine/isoleucine) and hence are not resolvable by single-dimensional MS alone. To separate all 204 possible peptides in the digest (excluding the 5 isomer pairs) from the nearest neighboring peak (i.e. 203 separations) would require ca. 100,000 mass resolving power (Figure 3A and 3C), which is currently obtainable by FTMS (Orbitraps and ion cyclotron resonance).58–60 The smallest observable difference in the mass dimension was 0.062 Δm/z, which requires greater than 10,000 mass Rp to separate at half height. Interestingly, by noting all of the possible peptides produced in the digest, Figure 3A shows that a mass resolving power of 10,000 should be able to resolve ca. 95% of the possible peptides in the mixture at half height. If the mass Rp was increased by an order of magnitude to 100,000 (e.g., an Orbitrap mass analyzer) peptide coverage only increases by 3% (198/203 peptide pairs separable). Thus, for this proteomics example, high resolution MS is the primary analytical dimension in which most analytes are resolved.
Figure 3.
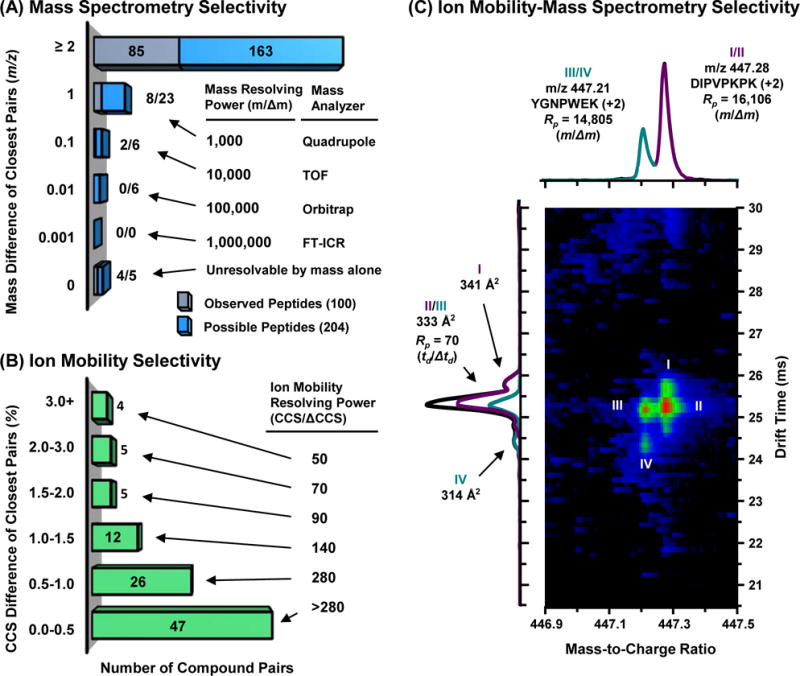
Comparison of mass spectrometry and ion mobility data collected from a tryptic peptide mixture originating from four proteins. (A) Difference in m/z between nearest neighbors for all possible peptides in the digest mixture (light blue) and from those peptides observed experimentally in this study (gray). (B) Bar graph of the percent difference in CCS between nearest-neighboring peptides for the 99 analyte pairs observed. (C) Separation of two doubly charged peptides by mass spectrometry and suspected conformers of each peptide noted through ion mobility.
Ion Mobility Analysis
To examine the potential of separating the peptides based on differences in mobility, the 100 observed analytes were sorted in order of increasing CCS and the percent difference in CCS from the nearest neighbor peak was calculated (Figure 3B). The results of this pair-wise CCS analysis indicate that more than half of the compound pairs analyzed (ca. 53%) have a percent difference in CCS of at least 0.5% from their nearest neighbor. Separating these compounds based purely on mobility alone would require 280 resolving power or less, which is currently obtainable (Table S2). 30,57,61 To separate ca. 95% (CCS ≥ 0.02%) of the peptides by IM alone would require about 7,000 mobility resolving power. Interestingly, this level of mobility resolving power is on the same order of magnitude as a moderate resolution mass analyzer (c.f., Figure 3A). However, this magnitude of resolving power is far beyond the capabilities of current IM instrumentation. Also, if the sample size was increased (i.e. N>100 compounds), the probability of concomitant IM peaks would be high and thus decreases the likelihood of discrete analytes being resolved. This indicates that IM selectivity is supplemental to the superior separation capabilities of MS and the best analytical performance is achieved when both techniques are used in concert (IM-MS).
Ion Mobility-Mass Spectrometry
Clearly, the full analytical utility of ion mobility is accessible only when directly coupled to a mass spectrometer.4,62,63 Current mass analyzers are highly selective (Rp > 100,000), and, in many cases, accurate mass measurement when combined with tandem MS/MS capabilities can elucidate an analyte identification. However, when investigating analytical mixtures that possess isomers or investigating proteins which often express many conformers, ion mobility analysis is chemically insightful. For example, Figure 3C illustrates a closer examination of two peaks noted in the IM-MS experiment for the protein digest. Two different peptides (from two different protein precursors) have an exact mass difference of 0.063 m/z and are resolvable by TOF mass analysis (features I/II and III/IV). However, the mass spectrometer cannot distinguish that DIPVPKPK (+2) has two distinct isobaric features (I and II) that are discernible in the ion mobility dimension at 70 resolving power (td/Δtd) for this +2 ion. Likewise, YGNPWEK (+2) also possesses secondary features when observed in the IM dimension (III/IV). However, the primary features (II and III) possess near identical CCS values and are unresolvable by ion mobility alone whereas the mass spectrometer resolves these features as two distinct peaks. Hence, the utility of IM-MS is evident in the analysis of complex samples that require identification of both distinct molecules (MS) and potential isomers, conformers or multimeric species of these compounds (IM), illustrating the well-known advantages of hyphenated separations.64,65
Conclusions
Here we develop a straightforward theoretical framework for comparing the separation efficiency of different IM techniques by defining the instrument resolving power in terms of the gas-phase CCS. We note that defining resolving power in this manner is particularly critical for obtaining meaningful metrics of separation capabilities for TWIMS techniques.
Based on the analysis developed in this work, the separation capabilities of various IM instrumentation can, for the first time, be compared relative to differences in the gas-phase CCS. The results of this study indicate that current ion mobility instruments operate across a broad range of separation efficiencies between 50 and 300 resolving power (CCS/ΔCCS) and the current state-of-the-art IM instruments are now demonstrating Rp in excess of 300 and thus are capable of separating compounds with CCS differences as low as 0.5% (c.f., Table 2). While this high level of structural selectivity enables IM to resolve constitutional isomers and conformers (typically 0.5% difference in CCS or greater), we hypothesize to resolve the majority of the components in a biological mixture using ion mobility alone would require resolving powers on the order of several thousand, which is far beyond the capabilities of current instrumentation and may not be achievable due to fundamental peak broadening limits imposed by ion diffusion. Ion mobility experiments therefore provide the greatest analytical benefits when combined with mass spectrometry, as well as other analytical dimensions (e.g. LC-IM-MS), which collectively function to broaden the analytical selectivity of the chemical separation. Nevertheless, routine ion mobility resolving powers in excess of 300 which are now being demonstrated will be essential to addressing chemical separations in highly challenging studies, such as in synthetic biology, medicine, and the omics sciences.
Finally, the guidance as illustrated in this manuscript suggest a potential criterion for reporting ion mobility resolving power in the future, similar to guidelines also reported in other scientific contexts, such as standards set forth for microarray (MIAME)”,66 proteomics (MIAPE)”,67 and glycomics experiments (MIRAGE).68
Supplementary Material
Acknowledgments
Financial support for aspects of this research was provided by The National Institutes of Health (NIH Grant R01GM099218) and under Assistance Agreement No. 83573601 awarded by the U. S. Environmental Protection Agency. This work has not been formally reviewed by EPA. The views expressed in this document are solely those of the authors and do not necessarily reflect those of the Agency. EPA does not endorse any products or commercial services mentioned in this publication. Furthermore, the content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies and organizations.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Process for extracting resolving power and resolution from previously published manuscripts, additional TWIMS spectra for comparison; extended appendix of cited spectra.
ORCID
John A. McLean: 0000-0001-8918-6419
Jody C. May: 0000-0003-4871-5024
James N. Dodds: 0000-0002-9702-2294
Notes
The authors declare no competing financial interest.
References
- 1.Zhong Y, Hyung S-J, Ruotolo BT. Expert Rev Proteomic. 2012;9:47–58. doi: 10.1586/epr.11.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lapthorn C, Pullen F, Chowdhry BZ. Mass Spectrom Rev. 2012;32:43–71. doi: 10.1002/mas.21349. [DOI] [PubMed] [Google Scholar]
- 3.Maurer MM, Donohoe GC, Valentine SJ. Analyst. 2015;140:6782–6798. doi: 10.1039/c5an00922g. [DOI] [PubMed] [Google Scholar]
- 4.May JC, McLean JA. Anal Chem. 2015;87:1422–1436. doi: 10.1021/ac504720m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giles K, Pringle SD, Worthington KR, Little D, L WJ, Bateman RH. Rapid Commun Mass Spectrom. 2004;18:2401–2414. doi: 10.1002/rcm.1641. [DOI] [PubMed] [Google Scholar]
- 6.Giles K, Williams JP, Campuzano I. Rapid Commun Mass Spectrom. 2011;25:1559–1566. doi: 10.1002/rcm.5013. [DOI] [PubMed] [Google Scholar]
- 7.May JC, Goodwin CR, Lareau NM, Leaptrot KL, Morris CB, Kurulugama RT, Mordehai A, Klein C, Barry W, Darland E, Overney G, Imatani K, Stafford GC, Fjeldsted JC, McLean JA. Anal Chem. 2014;86:2107–2116. doi: 10.1021/ac4038448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allen SJ, Giles K, Gilbert T, Bush MF. Analyst. 2016;141:884–891. doi: 10.1039/c5an02107c. [DOI] [PubMed] [Google Scholar]
- 9.Shvartsburg AA. Differential Ion Mobility Spectrometry: Nonlinear Ion Transport and Fundamentals of FAIMS. CRC Press; Boca Raton, FL: 2009. [Google Scholar]
- 10.Purves RW. Anal Bioanal Chem. 2013;405:35–42. doi: 10.1007/s00216-012-6496-3. [DOI] [PubMed] [Google Scholar]
- 11.Amo-Gonzalez M, Fernandez de la Mora J. J Am Soc Mass Spectrom. 2017;8:1506–1517. doi: 10.1007/s13361-017-1630-2. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez-Lima FA, Kaplan DA, Suetering J, Park MA. Int J Ion Mobil Spectrom. 2011;14:93–98. doi: 10.1007/s12127-011-0067-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michelmann K, Silveira JA, Ridgeway ME, Park MA. J Am Soc Mass Spectrom. 2015;26:14–24. doi: 10.1007/s13361-014-0999-4. [DOI] [PubMed] [Google Scholar]
- 14.Vidal-de-Miguel G, Macía M, Cuevas J. Anal Chem. 2012;84:7831–7837. doi: 10.1021/ac301127u. [DOI] [PubMed] [Google Scholar]
- 15.Deng L, Ibrahim YM, Hamid AM, Garimella SV, Webb IK, Zheng X, Prost SA, Sandoval JA, Norheim RV, Anderson GA. Anal Chem. 2016;88:8957–8964. doi: 10.1021/acs.analchem.6b01915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng L, Webb IK, Garimella SV, Hamid AM, Zheng X, Norheim RV, Prost SA, Anderson GA, Sandoval JA, Baker ES. Anal Chem. 2017;89:4628–4634. doi: 10.1021/acs.analchem.7b00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siems WF, Wu C, Tarver EE, Hill HH, Jr, Larsen PR, McMinn DG. Anal Chem. 1994;66:4195–4201. [Google Scholar]
- 18.Santiago BG, Harris RA, Isenberg SL, Glish GL. Analyst. 2015;140:6871–6878. doi: 10.1039/c5an00845j. [DOI] [PubMed] [Google Scholar]
- 19.Kurulugama RT, Darland E, Kuhlmann F, Stafford G, Fjeldsted J. Analyst. 2015;14:6834–6844. doi: 10.1039/c5an00991j. [DOI] [PubMed] [Google Scholar]
- 20.Tabrizchi M, Rouholahnejad F. Talanta. 2006;69:87–90. doi: 10.1016/j.talanta.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 21.Dodds JN, May JC, McLean JA. Anal Chem. 2016;89:952–959. doi: 10.1021/acs.analchem.6b04171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.May JC, Dodds JN, Kurulugama RT, Stafford GC, Fjeldsted JC, McLean JA. Analyst. 2015;140:6824–6833. doi: 10.1039/c5an00923e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stow SM, Causon TJ, Zheng X, Kurulugama RT, Mairinger T, May JC, Rennie EE, Baker ES, Smith RD, McLean JA, Hann S, Fjeldsted JC. Anal Chem. 2017;89:9048–9055. doi: 10.1021/acs.analchem.7b01729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spangler GE, Collins CI. Anal Chem. 1975;47:403–407. [Google Scholar]
- 25.Edelson D, Morrison J, McKnight L, Sipler D. Phys Rev. 1967;164:71–75. [Google Scholar]
- 26.Revercomb HE, Mason EA. Anal Chem. 1975;47:970–983. [Google Scholar]
- 27.Kurulugama RT, Nachtigall FM, Lee S, Valentine SJ, Clemmer DE. J Am Soc Mass Spectrom. 2009;20:729–737. doi: 10.1016/j.jasms.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valentine SJ, Stokes ST, Kurulugama RT, Nachtigall FM, Clemmer DE. J Am Soc Mass Spectrom. 2008;20:738–750. doi: 10.1016/j.jasms.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valentine SJ, Kurulugama RT, Clemmer DE. J Am Soc Mass Spectrom. 2011;22:804–816. doi: 10.1007/s13361-011-0087-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silveira JA, Ridgeway ME, Park MA. Anal Chem. 2014;86:5624–5627. doi: 10.1021/ac501261h. [DOI] [PubMed] [Google Scholar]
- 31.Hernandez DR, DeBord JD, Ridgeway ME, Kaplan DA, Park MA, Fernandez-Lima FA. Analyst. 2014;139:1913–1921. doi: 10.1039/c3an02174b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee S, Ewing MA, Nachtigall FM, Kurulugama RT, Valentine SJ, Clemmer DE. J Phys Chem B. 2010;114:12406–12415. doi: 10.1021/jp1060123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shvartsburg AA, Tang K, Smith RD. Anal Chem. 2004;76:7366–7374. doi: 10.1021/ac049299k. [DOI] [PubMed] [Google Scholar]
- 34.Santiago BG, Harris RA, Isenberg SL, Ridgeway ME, Pilo AL, Kaplan DA, Glish GL. J Am Soc Mass Spectrom. 2015;26:1746–1753. doi: 10.1007/s13361-015-1208-9. [DOI] [PubMed] [Google Scholar]
- 35.May JC, Morris CB, McLean JA. Anal Chem. 2017;89:1032–1044. doi: 10.1021/acs.analchem.6b04905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Glaskin RS, Valentine SJ, Clemmer DE. Anal Chem. 2010;82:8266–8271. doi: 10.1021/ac1017474. [DOI] [PubMed] [Google Scholar]
- 37.Reid Asbury G, Hill HH., Jr J Microcolumn Sep. 2000;12:172–178. [Google Scholar]
- 38.Pierson NA, Chen L, Valentine SJ, Russell DH, Clemmer DE. J Am Chem Soc. 2011;133:13810–13813. doi: 10.1021/ja203895j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang X, Bruce JE, Hill HH., Jr Rapid Commun Mass Spectrom. 2007;21:1115–1122. doi: 10.1002/rcm.2928. [DOI] [PubMed] [Google Scholar]
- 40.Gaye M, Nagy G, Clemmer D, Pohl N. Anal Chem. 2016;88:2335–2344. doi: 10.1021/acs.analchem.5b04280. [DOI] [PubMed] [Google Scholar]
- 41.Adamov A, Mauriala T, Teplov V, Laakia J, Pedersen CS, Kotiaho T, Sysoev AA. Int J Mass Spectrom. 2010;298:24–29. [Google Scholar]
- 42.Shvartsburg AA, Smith RD. Anal Chem. 2008;80:9689–9699. doi: 10.1021/ac8016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.May JC, McLean JA. Int J Ion Mobil Spectrom. 2013;16:85–94. doi: 10.1007/s12127-013-0123-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruotolo BT, Giles K, Campuzano I, Sandercock AM, Bateman RH, Robinson CV. Science. 2005;310:1658–1661. doi: 10.1126/science.1120177. [DOI] [PubMed] [Google Scholar]
- 45.Bush MF, Hall Z, Giles K, Hoyes J, Robinson CV, Ruotolo BT. Anal Chem. 2010;82:9557–9565. doi: 10.1021/ac1022953. [DOI] [PubMed] [Google Scholar]
- 46.Hofmann J, Hahm HS, Seeberger PH, Pagel K. Nature. 2015;526:241–244. doi: 10.1038/nature15388. [DOI] [PubMed] [Google Scholar]
- 47.Giles K, Ujma J, Wildgoose JL, Green MR, Richardson K, Langridge DJ, Tomczyk N. 65th Annual American Society for Mass Spectrometry Conference. Indianapolis, IN: 2017. [Google Scholar]
- 48.Deng L, Ibrahim YM, Baker ES, Aly NA, Hamid AM, Zhang X, Zheng X, Garimella SV, Webb IK, Prost SA. ChemistrySelect. 2016;1:2396–2399. doi: 10.1002/slct.201600460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Webb IK, Garimella SVB, Tolmachev AV, Chen T-C, Zhang X, Norheim RV, Prost SA, LaMarche B, Anderson GA, Ibrahim YM, Smith RD. Anal Chem. 2014;86:9169–9176. doi: 10.1021/ac502055e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tolmachev AV, Webb IK, Ibrahim YM, Garimella SVB, Zhang X, Anderson GA, Smith RD. Anal Chem. 2014;86:9162–9168. doi: 10.1021/ac502054p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhong Y, Hyung S-J, Ruotolo BT. Analyst. 2011;136:3534–3541. doi: 10.1039/c0an00987c. [DOI] [PubMed] [Google Scholar]
- 52.Barnett DA, Ells B, Guevremont R, Purves RW. J Am Soc Mass Spectrom. 1999;10:1279–1284. [Google Scholar]
- 53.Giles K, Wildgoose JL, Pringle S, Langridge DJ, Nixon P, Garside J, Carney P. 63rd Annual American Society for Mass Spectrometry Conference. St Louis, MO: 2015. [Google Scholar]
- 54.Kirk AT, Zimmermann S. Int J Ion Mobil Spectrom. 2015;18:17–22. [Google Scholar]
- 55.Kirk AT, Raddatz C-R, Zimmermann S. Anal Chem. 2017;89:1509–1515. doi: 10.1021/acs.analchem.6b03300. [DOI] [PubMed] [Google Scholar]
- 56.Zhang X, Knochenmuss R, Siems WF, Liu W, Graf S, Hill HH., Jr Anal Chem. 2013;86:1661–1670. doi: 10.1021/ac403435p. [DOI] [PubMed] [Google Scholar]
- 57.Groessl M, Graf S, Knochenmuss R. Analyst. 2015;140:6904–6911. doi: 10.1039/c5an00838g. [DOI] [PubMed] [Google Scholar]
- 58.Scheltema RA, Hauschild J-P, Lange O, Hornburg D, Denisov E, Damoc E, Kuehn A, Makarov A, Mann M. Mol Cell Proteomics. 2014;13:3698–3708. doi: 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nagornov KO, Gorshkov MV, Kozhinov AN, Tsybin YO. Anal Chem. 2014;86:9020–9028. doi: 10.1021/ac501579h. [DOI] [PubMed] [Google Scholar]
- 60.Shaw JB, Lin T-Y, Leach FE, Tolmachev AV, Tolić N, Robinson EW, Koppenaal DW, Paša-Tolić L. J Am Soc Mass Spectrom. 2016;27:1929–1936. doi: 10.1007/s13361-016-1507-9. [DOI] [PubMed] [Google Scholar]
- 61.Hamid AM, Garimella SVB, Ibrahim YM, Deng L, Zheng X, Webb IK, Anderson GA, Prost SA, Norheim RV, Tolmachev AV, Baker ES, Smith RD. Anal Chem. 2016;88:8949–8956. doi: 10.1021/acs.analchem.6b01914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoaglund CS, Valentine SJ, Sporleder CR, Reilly JP, Clemmer DE. Anal Chem. 1998;70:2236–2242. doi: 10.1021/ac980059c. [DOI] [PubMed] [Google Scholar]
- 63.McLean JA. J Am Soc Mass Spectrom. 2009;20:1775–1781. doi: 10.1016/j.jasms.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 64.Giddings JC. Anal Chem. 1984;56:1258A–1270A. doi: 10.1021/ac00276a003. [DOI] [PubMed] [Google Scholar]
- 65.May JC, McLean JA. Ann Rev Anal Chem. 2016;9:387–409. doi: 10.1146/annurev-anchem-071015-041734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, Gaasterland T, Glenisson P, Holstege FCP, Kim IF, Markowitz V, Matese JC, Parkinson H, Robinson A, Sarkans U, Schulze-Kremer S, Stewart J, Taylor R, Vilo J, Vingron M. Nat Genet. 2001;29:365–371. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- 67.Taylor CF, Paton NW, Lilley KS, Binz P-A, Julian RK, Jones AR, Zhu W, Apweiler R, Aebersold R, Deutsch EW, Dunn MJ, Heck AJR, Leitner A, Macht M, Mann M, Martens L, Neubert TA, Patterson SD, Ping P, Seymour SL, Souda P, Tsugita A, Vandekerckhove J, Vondriska TM, Whitelegge JP, Wilkins MR, Xenarios I, Yates JR, Hermjakob H. Nat Biotech. 2007;25:887–893. doi: 10.1038/nbt1329. [DOI] [PubMed] [Google Scholar]
- 68.Kolarich D, Rapp E, Struwe WB, Haslam SM, Zaia J, McBride R, Agravat S, Campbell MP, Kato M, Ranzinger R. Mol Cell Proteomics. 2013;12:991–995. doi: 10.1074/mcp.O112.026492. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.