

Abstract
Pseudogenes are fossil relatives of genes. Pseudogenes have long been thought of as “junk DNAs”, since they do not code proteins in normal tissues. Although most of the human pseudogenes do not have noticeable functions, ~20% of them exhibit transcriptional activity. There has been evidence showing that some pseudogenes adopted functions as lncRNAs and work as regulators of gene expression. Furthermore, pseudogenes can even be “reactivated” in some conditions, such as cancer initiation. Some pseudogenes are transcribed in specific cancer types, and some are even translated into proteins as observed in several cancer cell lines. All the above have shown that pseudogenes could have functional roles or potentials in the genome. Evaluating the relationships between pseudogenes and their gene counterparts could help us reveal the evolutionary path of pseudogenes and associate pseudogenes with functional potentials. It also provides an insight into the regulatory networks involving pseudogenes with transcriptional and even translational activities.
In this study, we develop a novel approach integrating graph analysis, sequence alignment and functional analysis to evaluate pseudogene-gene relationships, and apply it to human gene homologs and pseudogenes. We generated a comprehensive set of 445 pseudogene-gene (PGG) families from the original 3,281 gene families (13.56%). Of these 438 (98.4% PGG, 13.3% total) were non-trivial (containing more than one pseudogene). Each PGG family contains multiple genes and pseudogenes with high sequence similarity. For each family, we generate a sequence alignment network and phylogenetic trees recapitulating the evolutionary paths. We find evidence supporting the evolution history of olfactory family (both genes and pseudogenes) in human, which also supports the validity of our analysis method. Next, we evaluate these networks in respect to the gene ontology from which we identify functions enriched in these pseudogene-gene families and infer functional impact of pseudogenes involved in the networks. This demonstrates the application of our PGG network database in the study of pseudogene function in disease context.
Keywords: Pseudogene-gene (PGG) relationship, Network analysis, Pseudogene function, PGG network database
1 Introduction
Pseudogenes have long been deemed “relics of evolution”, because they are homologous to protein-coding genes but lack protein products1. Recently this nonfunctional label has started to be revised. Although most of the human pseudogenes do not have noticeable functions, ~20% of them exhibit transcriptional activity2. Recent studies have shown that pseudogenes can modulate gene expression and thus may influence signaling pathways in cancer3. Acquired somatic mutations can create pseudogenes in cancer development4. Evidence of pseudogene transcription and translation have been observed in cancer cell lines5. Besides, transcriptomics analysis has shown that transcribed pseudogenes are differentially expressed in specific cancer subtypes and could potentially be used as both prognostic and diagnostic biomarkers6. Another area of interest is the role of pseudogene transcripts in regulating gene expression. Some pseudogenes generate RNA products that can competitively bind to microRNAs thus regulating the expression of their homologous gene counterparts (i.e. ceRNAs)7–9. They can also be oncomodulatory, such as pseudogene PTENP1 regulating the PTEN tumor suppressor gene. PTENP1 locus lost in the genome could lead to tumorigenesis10,11. Pseudogenes might also represent a genetic diversity reservoir12 and play a role in new gene generation3. Because of all this, understanding the relationships of pseudogenes and gene counterparts on a systems biology level is important in not only understanding evolution but also understanding diseases like cancer. However, the role(s) of pseudogenes are still not fully understood.
Efforts have been made to explore the roles of pseudogenes. A prominent example was Pseudogene.org, which compiled information on pseudogenes of various species. This database annotated pseudogenes and compared pseudogenes with their parent genes13. We attempt to complement this knowledge by focusing only on human and by comparing all pseudogenes to all gene families.
Processed pseudogenes and duplicated pseudogenes are two major types of pseudogenes. They are derived from functional genes through (retro-)duplication followed by accumulation of loss-of-function mutations5,14. Conventionally, a pseudogene is often paired with a homologous gene counterpart referred to as a “parent gene”14,15. This understanding of pseudogenes, though informative, does not encompass the entirety of genome-wide relationships, where multiple genes and pseudogenes can be homologous. Thus we instead compare pseudogenes with homologous gene families, such that pseudogenes are not only considered as a descendent of a single gene, but rather a relative of a group of homologous genes and pseudogenes. In this study, we develop a novel approach to generate the comprehensive set of pseudogene-gene families in human, and further characterize the networks and study the potential functions of pseudogenes and their associated networks in more detail using sequence alignment, network analysis, and functional annotation.
2 Materials and Methods
The simplified workflow is shown in Figure 1.
Figure 1.
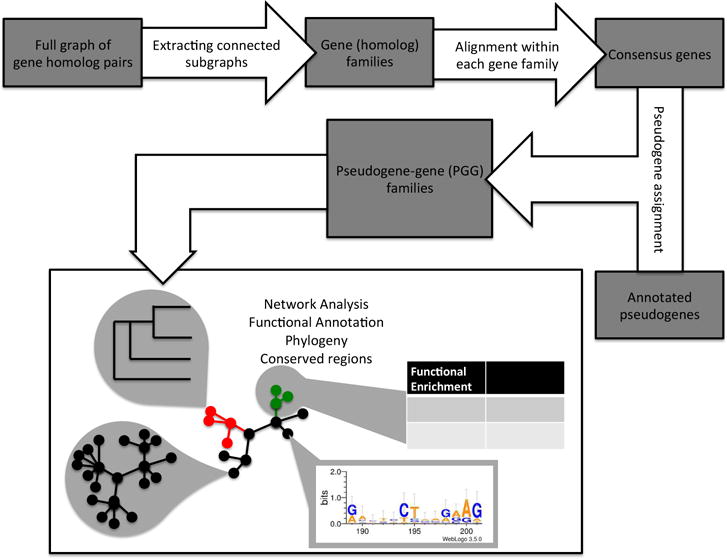
Simplified workflow from raw annotation through processing and analysis of the resulting pseudogene-gene (PGG) families.
2.1 Generating gene homolog families
We constructed the gene homolog families in which all the members are homologous genes. Specifically, all gene homolog pairs in human genome GRCh38 were downloaded from Ensembl BioMart. These gene homolog pairs were combined to generate a network of all homology relationships in the genome. In this network, each node represents a gene, and each edge indicates the existence of homology between a pair of genes. This basic structure of genes/pseudogenes as nodes and homology as edges was used throughout this project. Because not all genes share a common homolog, the full GRCh38 gene homology network is not a connected graph. Thus we performed an initial separation of the gene homolog network into connected subgraphs, denoted as gene families throughout this paper (Figure 1). In total we generated 3,281 gene families.
The human genome GRCh38 annotation was downloaded from Human GENCODE release 2416. Full length gene sequences were extracted using the gff2sequence tool17.
2.2 Mapping pseudogenes to gene homolog families
We extracted pseudogene sequences annotated in Human GENCODE release 24 using cufflinks gffread18. In total we obtained 14,548 pseudogenes. We assigned these pseudogenes to homologous gene families, by aligning every pseudogene sequence to a consensus sequence from each gene family. Consensus sequences were used to reduce the computational complexity and reduce bias associated to gene family size. The consensus sequence representing a gene family was selected from the set of all sequences within a gene family, by performing a multiple sequence alignment and selecting the gene with the highest sum of all pairwise alignment scores. To limit errors associated with long runtime, only sequences with less than 10,000 bp were retained for this alignment step. After a consensus gene was selected from each gene family, all pseudogenes were aligned to that consensus sequence using pairwise ClustalW19, which was installed on the high-performance computing cluster and has the ability to perform pairwise and multi-sequence alignment. The DataCutter framework20 was used to parallelize the alignments producing a 14,548 by 3,281 matrix of alignment scores between each pseudogene and every consensus sequence for the gene families. Each pseudogene was then subsequently assigned to the gene family with the highest alignment score. Not all gene families have pseudogenes assigned to them. Those gene families with assigned pseudogenes, i.e. pseudogene-gene (PGG) families, were examined further (Figure 1).
2.3 Network analysis of individual PGG families
The resulting PGG families were used to generate PGG networks based on the pairwise local alignment scores between all members of the family. Local alignment was used so that shorter sequences could still have high alignment scores when they match to a short segment of a larger sequence. The pairwise alignments were performed with a GPU parallelized local alignment tool CUDA-align21 in order to boost alignment performance for this large-scale computing. Using the resultant within-PGG family alignment matrix, a minimum spanning tree (MST) was generated for each PGG family. The alignment matrix for each PGG family consists of a complete network where all pseudogenes/genes within that family were nodes and the pairwise alignment scores edges. The MST was calculated from the alignment matrix producing a network in which bottlenecks had high sequence similarity to other nodes.
One facet of interest was identifying bottleneck nodes (gene or pseudogene) in the PGG family networks. As a measure of importance, betweenness centrality (BC) was calculated for all genes and pseudogenes contained within each network. A node with high non-zero BC is more likely to be a bottleneck in the network. (The smaller the proportion of zero-BC nodes is in a network, the more bottlenecks there are in the network.) Thus we record the proportion of zero-BC nodes in pseudogenes and genes respectively, and compare the number of bottlenecks in pseudogenes and genes. The distribution of BC for pseudogenes and genes were also plotted to evaluate the importance of pseudogene and gene bottlenecks.
2.4 Functional annotation of PGG families
Next we evaluated the functional enrichment of genes contained within pseudogene-gene (PGG) families (i.e. gene families that were assigned at least one pseudogene). A list of all genes (excluding the assigned pseudogenes) contained within PGG families were extracted and submitted to the DAVID Functional Annotation Tool22,23. DAVID functional annotation clusters (at High stringency) were evaluated for overrepresented annotations.
2.5 Identifying phylogenetic relationships and conserved regions
For each PGG family multiple sequence alignment (MSA) was performed with MUSCLE aligner24. Phylogenetic trees were created based on these alignments using FastTree25. The resulting MSAs and trees were used as input for the PhastCons26 program to identify conserved regions within each PGG family. We used the two-step approach outlined in their user manual in which the first step trains the Hidden Markov Model (HMM) transition model and the second identifies conserved regions (CRs). We then evaluated gene families (with no aligned pseudogenes) and PGG families (with at least one aligned pseudogene) for differences in their likelihood of containing conserved regions using a Fisher’s exact test. The test was conducted such that the two rows in the contingency table consisted of whether a gene family contained a pseudogene (row 1) or not (row 2). The columns consisted of whether a conserved region was identified in a gene family by PhastCons (column 1) or not (column 2).
2.6 Identifying GO networks associated with PGG families
The PGG networks identified (in-house PGG family IDs: 1149,1152,1235) were individually used to generate GO term networks using the BiNGO tool27 in cytoscape28,29. GO term annotations and hierarchy are used to generate a network from the members of each PGG family. Within each PGG family we use these GO networks to view the possible functional impact of the pseudogenes assigned to each PGG family. Each pseudogene that is contained within the PGG families could have an effect on the functions detected by the BiNGO tool. Specifically, functional impact (functional roles) of the pseudogenes of interest are interpretable from the proximity of pseudogenes to their gene counterparts in the PGG networks.
3 Results
3.1 Generating gene homolog families
In total, 3,281 exclusive subgraphs were generated by separating all connected subgraphs in the full GRCh38 gene homolog graph. These subgraphs represent 3,281 gene families that varied greatly in size with most having relatively few genes. The larger gene families had important structural features that included hub and bottleneck genes that connected to the entire family through homology. These gene family networks could take different forms containing a single or multiple hubs and bottlenecks (Figure 2A–C). These network structures indicate that genes in the same family can vary greatly in sequence, and help us understand how new genes arose and evolved through sequence changes.
Figure 2.
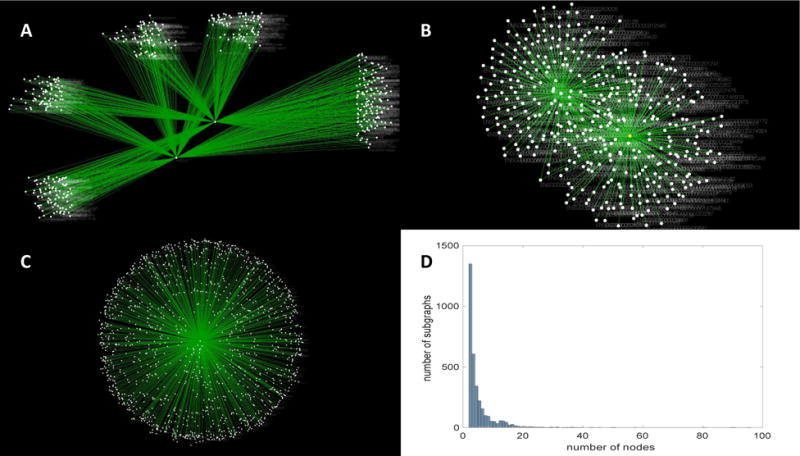
Subgraphs separated from gene homolog network. A: Gene family 6, B: Gene family 18, C: Gene family 32, D: Histogram of gene family sizes. Outliers were removed past 100 nodes so that the distribution of the common (smaller) sizes could be seen.
3.2 Mapping pseudogenes to gene homolog families
Through mapping pseudogenes to gene homolog families, we generated the comprehensive set of pseudogene-gene (PGG) families. Pseudogenes are relatives of genes and other pseudogenes in the same PGG family. The alignment scores between the pseudogenes and the consensus genes representing gene homolog families varied greatly between different pairs (i.e. alignments between pseudogenes and consensus genes) (Figure 3). Some had high conservation of sequences thus sequences are closely aligned with high scores. While some others had negative alignment scores indicating no relationship in sequence – the alignment incurred many more penalties than matches (these pairs would not be combined into PGG families). Pseudogenes were assigned to gene families with the highest alignment score for that pseudogene across all gene families. Thus each pseudogene was assigned to one unique gene family, and each gene family could accept multiple pseudogenes.
Figure 3.
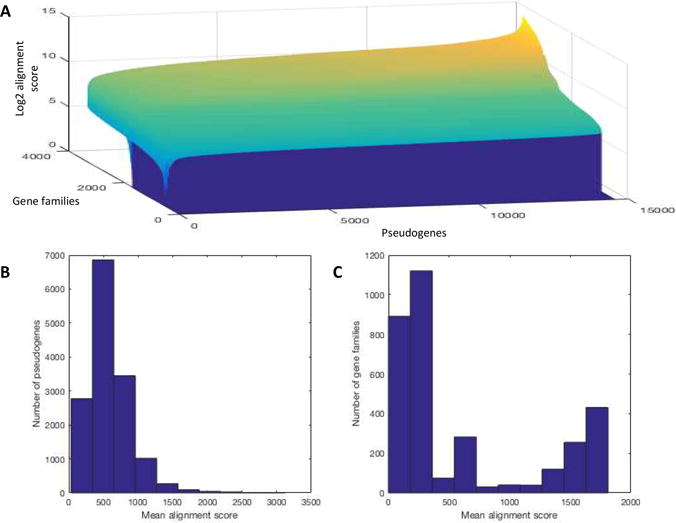
A) The distribution of pairwise pseudogene to consensus gene alignment scores for all pseudogenes and consensus genes (color also signifies Log2 alignment scores, lighter is higher and darker is lower). B) Distribution of mean pseudogene alignment score. C) Distribution of mean gene family alignment score.
3.3 Network analysis of individual PGG families
PGG networks were transformed into MSTs and graphed to view the relationship structure between the genes and pseudogenes in PGG families. The MSTs also highlight the bottlenecks. Three PGG families were selected as examples due to the large number of possible PGG families all of which could not be displayed. We also provide data matrices to generate all PGG family MSTs in Supplementary Materials which can be downloaded from GitHub (https://github.com/yanzhanglab/PGG_DB). Figure 4 shows the MSTs for three PGG families of interest. PGG family 1152 is of interest because it contains multiple genes that are related to chemokine receptors, olfactory receptors, and taste receptors. In this family a large portion of the nodes are pseudogenes, which is consistent with the knowledge of olfactory family reduction in primates with the greatest reduction of olfactory genes in hominoids (e.g. mice have more olfactory genes than primates of which humans have the least)30,31. There are even some examples of chemokine receptors becoming pseudogenes in Humans from other primates. An example is CCR5 which has a known pseudogene polymorphism in human that is known for reduced risk of HIV infection in exposed individuals32,33. PGG family 1149 is of interest because it contains the proto-oncogene RELA which has been implicated in pseudogene regulatory activity34,35. Another family of interest is 1235 that contains SOX2, which has been identified as a member of ceRNA networks36,37.
Figure 4.
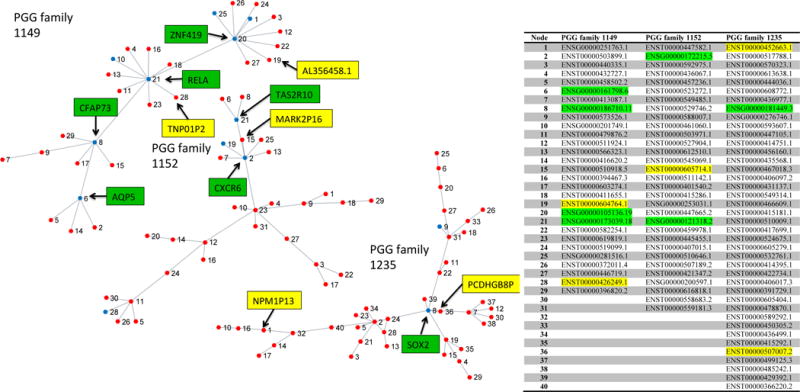
Minimum spanning trees of PGG families 1149, 1152 and 1235 with pseudogenes (red nodes) and genes (blue nodes). Genes of interest (with GO annotation or bottleneck node) are highlighted in green and pseudogenes of interest (with possible functional relationship to gene family) are highlighted in yellow.
The betweenness centrality (BC) of the PGG networks (Figure 5) showed that there were a higher proportion of genes with non-zero BC (17.29%) than pseudogenes (13.82%) with an odds ratio of 1.304 95% CI (1.056, 1.610) and p-value of 0.017. Higher non-zero BC implicates higher importance of the node in the network. After removing nodes with zero BC, it was found that the BC across all genes and pseudogenes was skewed higher in genes (Kolmogorov-Smirnov p-value = 2.856×10−8). These observations implicates that more genes than pseudogenes work as bottlenecks in the networks. Both gene and pseudogene BC followed exponential distributions with λ= 0.081 95% CI (0.075, 0.088) and λ=0.217 95% CI (0.199,0.235) respectively (Figure 5A–B).
Figure 5.
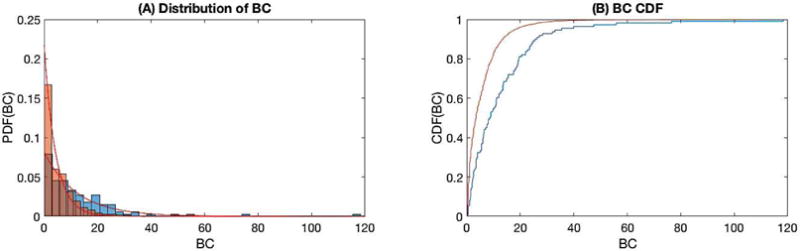
BC of PGG families. A) BC distributions and associated exponential empirical PDF for pseudogenes (red) and genes (blue). B) Empirical CDF for pseudogenes (red) and genes (blue).
3.4 Functional annotation of PGG families
Functional annotation of all genes contained in PGG families showed that there was enrichment in olfactory receptor and sensory receptor terms (Table 1). This supports the validity of our method since the most enriched function (Annotation Cluster 1) recapitulated the high number of known pseudogenes related to olfactory and other senses in human (Figure 5). Cluster 3 is also of interest due to the increasing evidence of the regulatory role of pseudogenes. DNA binding could be indicative of some forms of RNA regulation, which is further supported by the over-represented GO terms (GO:0045893:positive regulation of transcription, DNA-dependent and GO:0045944~positive regulation of transcription from RNA polymerase II promoter). Also, there is a growing body of evidence that proteins do not exclusively bind to DNA or RNA38 and a presence of over-represented ribonucleotide binding GO terms in our DAVID functional enrichment which could be indicative of ceRNA networks in which pseudogenes compete with genes for regulatory binding elements.
Table 1.
DAVID functional annotation of the genes contained within PGG families (High stringency).
| Category | Term | P-value | Fold Enrichment | FDR |
|---|---|---|---|---|
| Annotation Cluster 1 | Enrichment Score: 12.91 | |||
|
| ||||
| SP_PIR_KEYWORDS | olfaction | 2.89E-16 | 7.33 | 4.33E-13 |
| GOTERM_BP_FAT | GO:0007608~sensory perception of smell | 4.38E-15 | 6.45 | 6.98E-12 |
| INTERPRO | IPR000725:Olfactory receptor | 5.28E-15 | 6.52 | 7.15E-12 |
|
| ||||
| Annotation Cluster 2 | Enrichment Score: 12.36 | |||
|
| ||||
| SP_PIR_KEYWORDS | g-protein coupled receptor | 1.69E-19 | 5.45 | 2.20E-16 |
| INTERPRO | IPR017452:GPCR, rhodopsin-like superfamily | 1.15E-18 | 5.44 | 1.54E-15 |
| INTERPRO | IPR000276:7TM GPCR, rhodopsin-like | 1.20E-18 | 5.43 | 1.61E-15 |
|
| ||||
| Annotation Cluster 3 | Enrichment Score: 2.64 | |||
|
| ||||
| SMART | SM00389:HOX | 8.08E-04 | 4.46 | 0.83 |
| SP_PIR_KEYWORDS | Homeobox | 2.42E-03 | 3.85 | 3.11 |
| UP_SEQ_FEATURE | DNA-binding region:Homeobox | 2.56E-03 | 4.33 | 3.61 |
We extracted olfactory genes from the first functional annotation cluster using the full DAVID clustering table and labeled PGG networks as Olfactory if they contained at least one of the extracted olfactory genes (27 Olfactory and 418 Not Olfactory). Based on this stratification we found that olfactory related PGG families were more likely to contain both gene and pseudogene bottlenecks – olfactory related PGG families were more likely to have both genes and pseudogenes with non-zero BC (OR = 3.641 95% CI: (1.650,8.035), p-value = 0.002).
3.5 Identifying phylogenetic relationships and conserved regions
The phylogenetic tree (PGG family 1149) shows that one lineage consists purely of genes while the other consists of both genes and pseudogenes (Figure 6A). Not surprisingly these same genes are the bottlenecks in the MST graph (Figure 4), which could be indicative of the pseudogenes being generated from the bottleneck genes. Another result of note was the lack of conserved regions (CRs) found in the majority of PGG families. PGG family 1149 had an identified CR but PGG families 1152 and 1235 did not have identified CRs. We tested whether this was related to the containment of pseudogenes within PGG families and found that PGG families that have assigned pseudogenes are more likely to contain CRs with an odds ratio of 13.79 95% CI (10.70, 17.78) and p-value 3.169×10−95.
Figure 6.
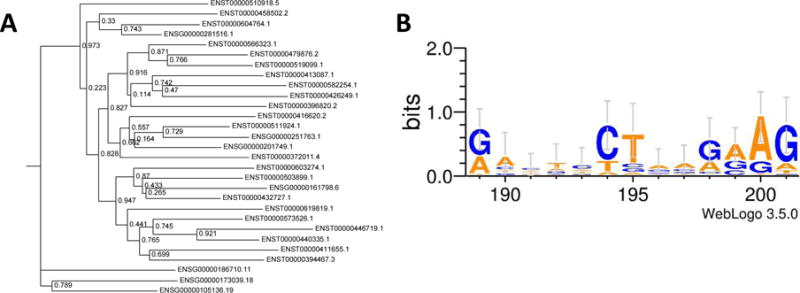
Phylogenetic trees and CRs, A) phylogenetic tree for PGG family 1149, B) CR for PGG family 1149 identified by PhastCons. IDs starting with ENSG constitute genes and ENST constitute pseudogenes.
3.6 Functional analysis of pseudogenes within PGG networks
Using BiNGO, we can evaluate the GO terms in each of the PGG networks. From PGG families 1149, 1152, 1235 we produced the following GO term networks (Figure 7), which can be used to evaluate the pseudogene functions included in the networks. PGG family 1149 had significant terms related to neurogeneration (Figure 7A). Pseudogene AL356458.1 is contained in PGG family 1149 and has copy number variations in oral carcinogenesis39. Pseudogene TNPO1P2 is also in PGG family 1149 and has implications in neurodegeneration in the frontotemporal lobe40. PGG family 1152 included multiple significant sensory related GO terms (Figure 7B). The pseudogene MARK2P16 is present in PGG family 1152 and its related gene MARK2 is needed for the migration of postnatal neuroblasts in the olfactory bulb41. PGG family 1235 included both NP1P13 and PCDHGB8P pseudogenes. PCDHGB8P is a protocadherin pseudogene with high sequence homology to other protocaderins such as PCDHGB3 and PCDHGB4 that have implications in multiple forms of cancer. The PGG family 1235 GO network includes significant proliferation terms (Figure 7C). PCDHGB3 and PCDHGB4 have implications in various cancer including lymphoma42 and PCDHBG4 has implications in metastatic breast cancer43. Analysis of Wilms’ tumors has shown frequent hypermethelated down-regulation of protocaderins (including PCDHGB4) in the tumor samples. NPM1P13 is implicated in a neurodevelopmental disorder, Saethre-Chotzen syndrome44. Significant GO terms in PGG family 1235 related to neurological development are also present (Figure 7C). PGG families 1149, 1152, and 1235 all have interesting functional relationships. These functional relationships (Figure 7) share documented functional similarities to either pseudogene members of the PGG family or the pseudogene members gene counterparts.
Figure 7.
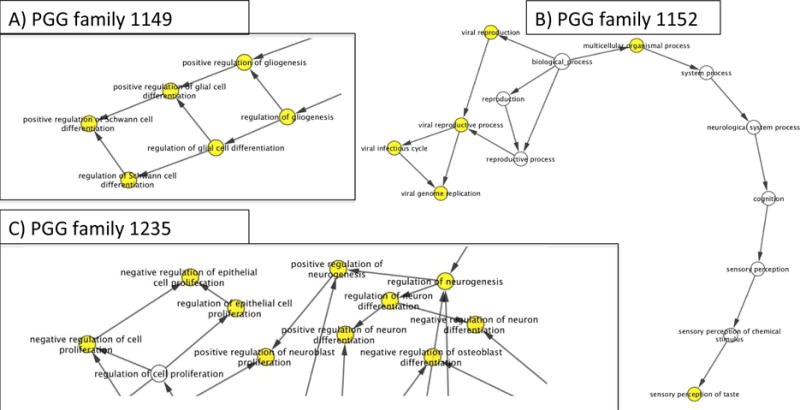
GO term networks from example PGG families. Each node denotes a GO term. Yellow nodes designate GO terms that are significant at p-value= 0.05. A) is a partial GO term BiNGO network for PGG family 1149 highlighting cell differentiation and glial cell development. B) is the full GO term BiNGO network from for PGG family 1152. C) is a partial BiNGO GO term network for PGG family 1235 highlighting cell proliferation GO terms and neuron development.
4 Discussion
4.1 Insights
Aside from the evolutionary relationships of pseudogenes to the genome we also find proto-oncogenes in PGG families containing pseudogenes. In PGG family 1149 we observed a network inclusive of a possible proto-oncogene RELA and small regulatory RNAs. RELA over-expression in mice was shown to delay the appearance of tumors and reduce proliferation in vitro45. The mapping of large numbers of pseudogenes to the RELA homolog family supports a possible regulatory relationship. Within these pseudogenes we find TNPO1P2 and AL356458.1 with some evidence in the literature of possible relationships to the BiNGO functional networks generated from the PGG families. Aside from RELA, another PGG family 1235 contained SOX2, a gene that has been implicated in ceRNA networks7. Within this network we identify PCDHGB8P and NPM1P13 that have literature supporting potential functions related to those identified in the BiNGO functional network. These findings support the hypothesis that pseudogenes may play a regulatory role in the genome, and the networks of interest are worth further investigation. Within the olfactory related PGG network 1152 we find olfactory related function in both the BiNGO functional network and literature supporting olfactory function for MARK2 the gene precursor to MARK2P16 pseudogene.
Genes and pseudogenes with a high non-zero BC are important to the structure of the PGG network. Directly it means these genes/pseudogenes constitute bridges between more dissimilar sequences within a PGG network. Biologically this could imply that that a gene/pseudogene likely contains key mutation signatures that triggers the change of function (e.g. silencing an ancient gene, or re-activating a pseudogene) or contributes to the gene/pseudogene family expansion (i.e. generating large number of descendants in the PGG network). The distribution of non-zero BC for genes and pseudogenes were also altered where genes tend to have higher BC values. Evolutionarily this could imply that genes are more likely to be bottlenecks in PGG families and are more important bottlenecks than pseudogenes.
We also find that gene families that were assigned pseudogenes were more likely to contain CRs. This could be examined further to evaluate what function this conservation could have. This difference in enrichment level of CRs may be related to family size, or biased by the sequence similarity threshold defined by computational tools used for identifying pseudogenes.
Of the gene families with many aligned pseudogenes, there was enriched annotation of olfactory receptor genes, as shown in DAVID results. This is in congruence with previous findings that the olfactory receptor family in humans has large numbers of pseudogenes46,47.
Another important note is that we generate PGG families through alignment of pseudogenes to gene families. Especially in the case of processed pseudogenes this unbiased approach should be taken into account when examining the potential of competing endogenous RNA. Since the sequences by definition must closely related to the assigned gene family, many of these pseudogenes could be candidates for ceRNA networks and evaluated further.
In the following work we will integrate all of the aspects of this project into an online query tool, which will return PGG families and functional prediction for specific novel pseudogene sequences of interest. The functional enrichment included in existing GO annotation tools (e.g. BiNGO) do not take into account the proximity of pseudogenes and genes in the same network. In the future we will try to overlay these weights in our GO inference method to improve the functional predictions given by our tool. Aside from the user interface and refined functional prediction, the tool itself due to its network structure could easily be integrated with other experimental methodologies (e.g. ChiP-Seq and Competition-ChiP) paired by gene/pseudogene IDs. Our goal is to use our current database as a baseline functional prediction for pseudogenes that can be easily augmented by the improved methodologies both currently available and developed in the future. This makes our database immensely scalable as new and improved features are added to aid in functional prediction of pseudogenes.
4.2 Limitations
One limitation affecting this approach includes the ambiguous definition of pseudogenes in available annotations. There are DNA segments annotated as genes but do not code proteins. There are also gene-like DNAs generating regulatory RNAs and are not annotated as pseudogenes. This ambiguity in annotation could introduce noise to the alignment steps.
In our current approach, we treat different pseudogene biotypes (processed, duplicated, or unitary pseudogenes) uniformly. The full gene sequences were used to make this approach more computationally tractable. Because there were intronic regions in gene homologs, whereas processed pseudogenes do not contain introns, the alignments of gene families to processed pseudogenes are not as accurate as aligning to duplicated or unitary pseudogenes. However, the effects of using full length gene sequences is mitigated by the use of local alignment. In our future studies, we will fine-tune our approach to treat different pseudogene biotypes respectively.
A large portion of the genes within the PGG families did not have associated functional annotation within DAVID, which implies annotation bias where not all genes are equally well-studied. Because the number of genes in some networks was small, some families had few genes to study functional enrichment from.
5 Conclusion
In this study, we investigate the functional relationships between pseudogene and gene homolog families, by integrating graph analysis, sequence alignment and functional analysis, and generate the comprehensive set of pseudogene-gene families in human. By studying the network structure of these pseudogene-gene networks, we find that there is an over-representation of olfaction related PGG families, differential BC between genes and pseudogenes, and structural patterns that can be used to differentiate PGG networks. These patterns in network structure also can be used to differentiate different classes of networks. Olfactory PGG families were associated with a network structure in which both genes and pseudogenes had bottleneck qualities (measurable BC). Similarly we view these networks as important tools in predicting function for pseudogenes, similar to previous methods that infer gene ontologies for under-annotated genes48. We use our PGG families to associate GO terms to under-documented pseudogenes and describe the utility of this database to query new pseudogene sequences to infer functional potential. In summary, here we have proposed a novel, comprehensive, and scalable evaluation of pseudogenes at the gene homolog level and showed that network structure can be related to functionality.
We also propose in future work a refined pipeline that i) treat different pseudogene biotypes respectively, ii) preprocesses the gene annotation prior to analysis to reduce ambiguity, iii) can identify possible ceRNA networks algorithmically, iv) provide a search utility to query novel pseudogene sequences against our network database to predict pseudogene function and v) use network weights to more accurately associate known and novel pseudogene sequences to GO terms within the assigned PGG family. Aside from these immediate improvements that are under development currently we also plan to make this database scalable in the different types of data that can be integrated (e.g. ChiP-Seq and Competition-ChiP) via pairing nodes via gene names or interactions.
Supplementary Material
Acknowledgments
The work is supported by NIH-NLM MIDAs Training Fellowship (4T15LM011270-05) to Travis Johnson, and The Ohio State University Startup Funds to Yan Zhang. The authors also thank the Ohio Supercomputer Center for providing computing resources.
Contributor Information
Travis S Johnson, Dept. Biomedical Informatics, Ohio State University, 5000 HITS, 410 W. 10th St. Indianapolis, Indiana, 46202.
Sihong Li, Dept. Biomedical Informatics, Ohio State University, 250 Lincoln Tower, 1800 Cannon Dr. Columbus, Ohio, 43210.
Jonathan R Kho, Dept. Computational Science and Engineering, Georgia Institute of Technology, Klaus Advanced Computing Building, 266 Ferst Dr. Atlanta, Georgia, 30332.
Kun Huang, Dept. Hematology Oncology, Indiana University, 335 Regenstrief Institute, 1101 W 10th St. Indianapolis, Indiana, 46202.
Yan Zhang, Dept. Biomedical Informatics, Ohio State University, 310-B Lincoln Tower, 1800 Cannon Dr. Columbus, Ohio, 43210.
References
- 1.Jacq C, Miller JR, Brownlee GG. A pseudogene structure in 5S DNA of Xenopus laevis. Cell. 1977;12(1):109–120. doi: 10.1016/0092-8674(77)90189-1. [DOI] [PubMed] [Google Scholar]
- 2.Zhang ZZ, Deyou Pseudogene evolution in the Human Genome. eLS. 2014 [Google Scholar]
- 3.Poliseno L. Pseudogenes: newly discovered players in human cancer. Sci Signal. 2012;5(242):re5. doi: 10.1126/scisignal.2002858. [DOI] [PubMed] [Google Scholar]
- 4.Cooke SL, Shlien A, Marshall J, et al. Processed pseudogenes acquired somatically during cancer development. Nat Commun. 2014;5:3644. doi: 10.1038/ncomms4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sisu C, Pei B, Leng J, et al. Comparative analysis of pseudogenes across three phyla. Proc Natl Acad Sci U S A. 2014;111(37):13361–13366. doi: 10.1073/pnas.1407293111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poliseno L, Marranci A, Pandolfi PP. Pseudogenes in Human Cancer. Front Med (Lausanne) 2015;2:68. doi: 10.3389/fmed.2015.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng DL, Xiang YY, Ji LJ, Lu XJ. Competing endogenous RNA interplay in cancer: mechanism, methodology, and perspectives. Tumour Biol. 2015;36(2):479–488. doi: 10.1007/s13277-015-3093-z. [DOI] [PubMed] [Google Scholar]
- 8.Poliseno L, Pandolfi PP. PTEN ceRNA networks in human cancer. Methods. 2015;77–78:41–50. doi: 10.1016/j.ymeth.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 9.Sanchez-Mejias A, Tay Y. Competing endogenous RNA networks: tying the essential knots for cancer biology and therapeutics. J Hematol Oncol. 2015;8:30. doi: 10.1186/s13045-015-0129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465(7301):1033–1038. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tay Y, Kats L, Salmena L, et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147(2):344–357. doi: 10.1016/j.cell.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Li S, Abyzov A, Gerstein MB. Landscape and variation of novel retroduplications in 26 human populations. PLoS Comput Biol. 2017;13(6):e1005567. doi: 10.1371/journal.pcbi.1005567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karro JE, Yan Y, Zheng D, et al. Pseudogene.org: a comprehensive database and comparison platform for pseudogene annotation. Nucleic Acids Res. 2007;35:D55–60. doi: 10.1093/nar/gkl851. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z, Harrison PM, Liu Y, Gerstein M. Millions of years of evolution preserved: a comprehensive catalog of the processed pseudogenes in the human genome. Genome Res. 2003;13(12):2541–2558. doi: 10.1101/gr.1429003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pink RC, Wicks K, Caley DP, Punch EK, Jacobs L, Carter DR. Pseudogenes: pseudo-functional or key regulators in health and disease? RNA. 2011;17(5):792–798. doi: 10.1261/rna.2658311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrow J, Frankish A, Gonzalez JM, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22(9):1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Camiolo S, Porceddu A. gff2sequence, a new user friendly tool for the generation of genomic sequences. BioData Min. 2013;6(1):15. doi: 10.1186/1756-0381-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 20.Beynon Michael DKT, Umit Catalyurek, Chialin Chang, Alan Sussman, Joel Saltz. Distributed processing of very large datasets with DataCutter. Parallel Computing. 2001;27(11):1457–1478. [Google Scholar]
- 21.Chirag Jain SK. Fine-grained GPU parallelization of pairwise local sequence alignment. 21st International Conference on High Performance Computing (HiPC. 2014 [Google Scholar]
- 22.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 23.Huang DW, Sherman BT, Tan Q, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–175. doi: 10.1093/nar/gkm415. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26(7):1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siepel A, Haussler D. Combining phylogenetic and hidden Markov models in biosequence analysis. J Comput Biol. 2004;11(2–3):413–428. doi: 10.1089/1066527041410472. [DOI] [PubMed] [Google Scholar]
- 27.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 28.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilad Y, Przeworski M, Lancet D. Loss of olfactory receptor genes coincides with the acquisition of full trichromatic vision in primates. PLoS Biol. 2004;2(1):E5. doi: 10.1371/journal.pbio.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rouquier S, Blancher A, Giorgi D. The olfactory receptor gene repertoire in primates and mouse: evidence for reduction of the functional fraction in primates. Proc Natl Acad Sci U S A. 2000;97(6):2870–2874. doi: 10.1073/pnas.040580197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dean M, Carrington M, Winkler C, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science. 1996;273(5283):1856–1862. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- 33.Zhang ZD, Frankish A, Hunt T, Harrow J, Gerstein M. Identification and analysis of unitary pseudogenes: historic and contemporary gene losses in humans and other primates. Genome Biol. 2010;11(3):R26. doi: 10.1186/gb-2010-11-3-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Porter KA, Duffy EB, Nyland P, Atianand MK, Sharifi H, Harton JA. The CLRX.1/NOD24 (NLRP2P) pseudogene codes a functional negative regulator of NF-kappaB, pyrin-only protein 4. Genes Immun. 2014;15(6):392–403. doi: 10.1038/gene.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rapicavoli NA, Qu K, Zhang J, Mikhail M, Laberge RM, Chang HY. A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. Elife. 2013;2:e00762. doi: 10.7554/eLife.00762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arancio W, Carina V, Pizzolanti G, et al. Anaplastic Thyroid Carcinoma: A ceRNA Analysis Pointed to a Crosstalk between SOX2, TP53, and microRNA Biogenesis. Int J Endocrinol. 2015;2015:439370. doi: 10.1155/2015/439370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu J, Feng L, Han Z, et al. Extensive ceRNA-ceRNA interaction networks mediated by miRNAs regulate development in multiple rhesus tissues. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkw587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hudson WH, Ortlund EA. The structure, function and evolution of proteins that bind DNA and RNA. Nat Rev Mol Cell Biol. 2014;15(11):749–760. doi: 10.1038/nrm3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vincent-Chong VK, Salahshourifar I, Razali R, Anwar A, Zain RB. Immortalization of epithelial cells in oral carcinogenesis as revealed by genome-wide array comparative genomic hybridization: A meta-analysis. Head Neck. 2016;38(Suppl 1):E783–797. doi: 10.1002/hed.24102. [DOI] [PubMed] [Google Scholar]
- 40.Troakes C, Hortobagyi T, Vance C, Al-Sarraj S, Rogelj B, Shaw CE. Transportin 1 colocalization with Fused in Sarcoma (FUS) inclusions is not characteristic for amyotrophic lateral sclerosis-FUS confirming disrupted nuclear import of mutant FUS and distinguishing it from frontotemporal lobar degeneration with FUS inclusions. Neuropathol Appl Neurobiol. 2013;39(5):553–561. doi: 10.1111/j.1365-2990.2012.01300.x. [DOI] [PubMed] [Google Scholar]
- 41.Mejia-Gervacio S, Murray K, Sapir T, Belvindrah R, Reiner O, Lledo PM. MARK2/Par-1 guides the directionality of neuroblasts migrating to the olfactory bulb. Mol Cell Neurosci. 2012;49(2):97–103. doi: 10.1016/j.mcn.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 42.Takata K, Tanino M, Ennishi D, et al. Duodenal follicular lymphoma: comprehensive gene expression analysis with insights into pathogenesis. Cancer Sci. 2014;105(5):608–615. doi: 10.1111/cas.12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shima J, Delaney J, Umesh A, et al. Disruption of protocadherin function and correlation with metastasis and cancer progression in TCGA patients. Journal of Clinical Oncology. 2012;30(30_suppl):70–70. [Google Scholar]
- 44.Shimbo H, Oyoshi T, Kurosawa K. Contiguous gene deletion neighboring TWIST1 identified in a patient with Saethre-Chotzen syndrome associated with neurodevelopmental delay: Possible contribution of HDAC9. Congenit Anom (Kyoto) 2017 doi: 10.1111/cga.12216. [DOI] [PubMed] [Google Scholar]
- 45.Ricca A, Biroccio A, Trisciuoglio D, Cippitelli M, Zupi G, Del Bufalo D. relA over-expression reduces tumorigenicity and activates apoptosis in human cancer cells. Br J Cancer. 2001;85(12):1914–1921. doi: 10.1054/bjoc.2001.2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niimura Y. Olfactory receptor multigene family in vertebrates: from the viewpoint of evolutionary genomics. Curr Genomics. 2012;13(2):103–114. doi: 10.2174/138920212799860706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prieto-Godino LL, Rytz R, Bargeton B, et al. Olfactory receptor pseudo-pseudogenes. Nature. 2016 doi: 10.1038/nature19824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dutkowski J, Kramer M, Surma MA, et al. A gene ontology inferred from molecular networks. Nat Biotechnol. 2013;31(1):38–45. doi: 10.1038/nbt.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.