

Abstract
Ion mobility (IM) is an important analytical technique for determining ion collision cross section (CCS) values in the gas-phase and gaining insight into molecular structures and conformations. However, limited instrument resolving powers for IM may restrict adequate characterization of conformationally similar ions, such as structural isomers, and reduce the accuracy of IM-based CCS calculations. Recently, we introduced an automated technique for extracting “pure” IM and collision-induced dissociation (CID) mass spectra of IM overlapping species using chemometric deconvolution of post-IM/CID mass spectrometry (MS) data [J. Am. Soc. Mass Spectrom., 2014, 25, 1810–1819]. Here we extend those capabilities to demonstrate how extracted IM profiles can be used to calculate accurate CCS values of peptide isomer ions which are not fully resolved by IM. We show that CCS values obtained from deconvoluted IM spectra match with CCS values measured from the individually analyzed corresponding peptides on uniform field IM instrumentation. We introduce an approach that utilizes experimentally determined IM arrival time (AT) “shift factors” to compensate for ion acceleration variations during post-IM/CID and significantly improve the accuracy of the calculated CCS values. Also, we discuss details of this IM deconvolution approach and compare empirical CCS values from traveling wave (TW)IM-MS and drift tube (DT)IM-MS with theoretically calculated CCS values using the projected superposition approximation (PSA). For example, experimentally measured deconvoluted TWIM-MS mean CCS values for doubly-protonated RYGGFM, RMFGYG, MFRYGG, and FRMYGG peptide isomers were 288.8 Å2, 295.1 Å2, 296.8 Å2, and 300.1 Å2; all four of these CCS values were within 1.5% of independently measured DTIM-MS values.
Keywords: peak deconvolution, chemometric analysis, resolving power, structural isomers, collision-induced dissociation (CID), ion mobility-mass spectrometry
TOC Image
1. INTRODUCTION
Ion mobility-mass spectrometry (IM-MS) is an important analytical technique for differentiating the structure(s) and conformation(s) of gas-phase ions.1,2 IM-MS can be used to rapidly separate ions based on their gas-phase sizes and shapes, while collision cross section (CCS) values (determined from the IM drift times) can aid molecular identification and be used to gain insight into the size and folding of molecules. There is compelling evidence that structural insights obtained from IM-MS studies may be relevant to solution-phase structures and chemistry.3–9 Several commercial IMS and IM-MS systems are available which are capable of determining CCSs; the most prevalent of these measurements are based on drift tube (DT)IM-MS and traveling wave (TW)IM-MS technologies.1,2 CCS measurements in DTIM-MS are relatively straightforward because ion drift times (tD) are directly proportional to their CCS.2,10 However, in TWIM-MS (where a non-uniform electric field is used to move ions) ions’ CCS values are proportional to tDx, where “x” is an empirically-derived parameter that must be calculated from the analysis of calibrants with known CCS values.11
The ability to resolve isomeric species is governed by the IM resolving power (i.e., arrival time (AT)/ΔAT50% or CCS/ΔCCS50%) of the instrument.10,12–15 Continuous improvements in achievable resolving power of contemporary instrumentation have extended the ability to differentiate isomeric systems. To improve IM resolving power, several groups have focused on different strategies including using alternative drift gases or gas modifiers,16–21 developing new instruments,22–27 or customizing existing systems.28 Additional improvements in software and post-data acquisition approaches can also enhance data interpretation for convoluted IM peaks. For instance, to circumvent instrumental limitations, some groups have used post-IM fragmentation and assigned IM profiles of diagnostic fragment ions to IM-unresolved precursor ions.29–35 For example, similar to spectral deconvolution of overlapping gas chromatography (GC)/MS peaks,36 Clemmer and coworkers demonstrated that isomers with unresolved drift time distributions could be differentiated by analyzing extracted ion drift time distributions (EIDTDs) of unique fragment ions generated from post-DTIM collision-induced dissociation (CID).31,32 Recently, Khakinejad et al. demonstrated that EIDTDs from post-DTIM/electron transfer dissociation data could be used to calculate CCS values of IM-unresolved peptide conformers.34 These post-IM/CID approaches demonstrated that fragment ions could be used to differentiate unresolved isomers present in a mixed AT; however, to date, there have not been any attempts at reconstructing pure AT profiles from convoluted IM spectra for determination of CCS values for IM-overlapping ions.
Previously, we introduced a chemometric data deconvolution technique37 based on SIMPLe-to-use Interactive Self-modeling Mixture Analysis (SIMPLISMA)38 which allowed the extraction of “pure” IM and post-IM/CID mass spectra for IM-unresolved species. Subsequently, we integrated SIMPLISMA into an automated IM deconvolution (AIMD) software, which improved the speed and accuracy of IM-MS analyses.39 Moreover, we showed that chemical rank determination techniques40,41 could be used to identify the number of IM-unresolved species without prior knowledge of the sample composition.37,39,42–44
In this manuscript, we demonstrate another unique utility of chemometric deconvolution of post-IM/CID MS data; specifically, we show that CCSs of IM-unresolved species, such as those for reverse sequence pentapeptide isomers (i.e., SDGRG and GRGDS) and scrambled sequence enkephalin hexapeptide isomers (i.e., RYGGFM, RMFGYG, MFRYGG, and FRMYGG), can be accurately calculated. In this method, we utilize AT “shift factors” to compensate for effects of electric field variations during post-IM/CID and reduce differences between mean calculated CCSs for IM deconvoluted profiles and CCSs obtained from individually measured analytes. Finally, we compare our deconvoluted TWIM-MS CCS values for enkephalin hexapeptide isomers to (a) experimental values obtained by using DTIM-MS and (b) theoretical values calculated by using the projected superposition approximation (PSA).
2. MATERIALS AND METHODS
2.1 Sample Preparation
Two reverse pentapeptide isomers (amino acid sequences: SDGRG and GRGDS) and poly-alanine (used as the CCS calibrant)11,45 were purchased from Sigma-Aldrich (St. Louis, MO, USA) and [Arg0] Met-enkephalin (amino acid sequence: RYGGFM) was purchased from American Peptide (Sunnyvale, CA, USA). Three sequence-scrambled enkephalin hexapeptides (amino acid sequences: RMFGYG, MFRYGG, and FRMYGG) were custom synthesized by Peptide 2.0, Inc. (Chantilly, VA, USA) and their identities were confirmed by CID MS analyses (supporting information, Figure S1). The pentapeptide mixture was chosen because of its previous use in characterizing the IM resolving powers of the Synapt HDMS TWIM-MS instruments.28 The four hexapeptide isomers were selected as model systems because of their overlapping IM profiles; moreover, to the best of our knowledge, CCS values of these enkephalin hexapeptides have not been previously characterized. Optima grade acetic acid and methanol were purchased from Fisher Scientific (Waltham, MA, USA). Water was purified using a Direct-Q 3 UV water purification system (EMD Millipore Corporation, Billerica, MA, USA). Peptides were prepared by reconstitution in a water: methanol: acetic acid (49.95: 49.95: 0.1) solvent composition at final analyte concentrations of ~1 μM. Peptide mixtures contained approximately equimolar concentrations of each isomer.
2.2 Ion Mobility-Mass Spectrometry
TWIM-MS experiments were performed as previously described37,39,42–44,46 using Synapt G1 (for the pentapeptide binary mixture) and G2-S (for the hexapeptide quaternary mixture) systems (Waters Corporation, Milford, MA, USA). Experimental details on collection of TWIM-MS data reported in this manuscript are included in the supporting information, Table S1. Drift tube CCS measurements were obtained on a commercial ESI-IM-QTOF Agilent 6560 (Agilent Technologies, Santa Clara, CA, USA) as previously described;47,48 briefly, buffer gas (i.e., nitrogen (N2)) was maintained at a pressure of ~4 Torr and drift voltages were varied to correct for the non-IM flight time of the ions through interfacing ion optics. Agilent tune mix was run with each hexapeptide sample as an internal CCS standard.
2.3 Data Analysis
Post-IM/CID MS data were deconvoluted using in-house developed automated IM deconvolution (AIMD) software39, which utilized the SIMPLISMA algorithm to extract “pure” IM and mass spectra. The nominal m/z values with the highest corresponding “purity value” (used to guide the deconvolution) for deconvoluted singly-protonated SDRGR and GRGDS (i.e., [SDGRG+H]+ and [GRGDS+H]+, respectively) were m/z 289 (i.e., y3+) and m/z 386 (i.e., b4+), respectively. Likewise, the nominal m/z values with the highest corresponding “purity values” for deconvoluted doubly-protonated RYGGFM, RMFGYG, MFRYGG, and FRMYGG (i.e., [RYGGFM + 2H]2+, [RMFGYG + 2H]2+, [MFRYGG + 2H]2+, and [FRMYGG + 2H]2+, respectively) were m/z 150 (i.e., y1+), m/z 492 (i.e., b4+), m/z 599 (i.e., y5+), and m/z 133 (i.e., y2+), respectively. All IM arrival times from the raw data (i.e., poly-DL-alanine, individually analyzed peptides, and peptide mixtures) correspond to the experimental values for the centroid m/z of the monoisotopic (viz.,12Call) peaks using a ±0.03 m/z mass ranges (e.g., m/z 491.22 ± 0.03 for singly- [SDGRG + H]+). Note that the term “arrival time” (AT) is used throughout this manuscript to denote “raw” times that have not been corrected for charge state, mass, or time spent outside of the IM cell (e.g., in the transfer cell or TOF). Post-IM/CID MS data were analyzed with AIMD software with averages of 10 min (588 scans; pentapeptides) or 1 min acquisition times (58 scans; enkephalin hexapeptides). All other TWIM data were collected for 1 min acquisition time (58 scans), unless otherwise noted. DTIM-MS experiments were collected for 2 min acquisition time (108 scans). All experiments were performed a minimum of three times; all reported errors are at the 95% confidence level (C.L.). Experimental TWIM-MS CCSs were calculated using the method proposed by Ruotolo and coworkers.11 The Mason-Schamp equation49 was used to determine experimental CCS values from ion drift times for all DTIM-MS experiments. Microsoft Excel (Office 2010, Microsoft Corp., Redmond, WA, USA) spreadsheets containing CCS calculations (including insignificant figures), AT corrections, and statistical tests can be found in the supporting information (viz., Spreadsheet S1 corresponds to the pentapeptides, Spreadsheet S2 corresponds to the TWIM-MS hexapeptide measurements, Spreadsheet S3 corresponds to the DTIM-MS hexapeptide measurements).
2.4 Theoretical CCS Calculations
Theoretical CCSs for the four enkephalin hexapeptide isomers (i.e., RYGGFM, RMFGYG, MFRYGG, and FRMYGG) were calculated using a simulated annealing protocol implemented with the AMBER software.50 The doubly-protonated hexapeptide isomers were constructed using standard amino acid templates in the xLEaP module such that protonation sites were at the arginine and N-termini. Because xLEaP contains amino acid templates that do not incorporate hydrogens on C-terminal residues, these were constructed manually and subsequently geometry optimized at the Hartree-Fock level with a 6–31G* basis set using Gaussian 09.51 The hexapeptide isomers were then energy minimized with the sander module,50 and subsequently heated to 700–800 K over a 10 ps molecular dynamics simulation. An extended molecular dynamic simulation was run at 700–800 K for 9,000 ps and structural snapshots were saved every 3 ps throughout the simulation, resulting in 3,000 unique structures. These structures were cooled to 300 K during 15 ps molecular dynamics simulations resulting in low-energy conformations that were used for theoretical CCS determination.52,53
A combination of MOBCAL54–57 and projected superposition approximation (PSA)58–61 were used to calculate the theoretical CCS values. Helium (He) values were obtained in MOBCAL using the projection approximation. PSA was then used to calculate nitrogen (N2) CCS values for a series of low-energy structures (~20) spanning the entire CCS range for which the structures were obtained. A linear function was determined from these values to convert the remaining He CCS values to N2 CCS values as previously demonstrated.52 PSA CCS values were calculated using the online service hosted by the University of California, Santa Barbara.62
3. RESULTS AND DISCUSSION
Figure 1 (black) shows the IM profile of a mixture of two isomeric reverse pentapeptides (i.e., [SDGRG + H]+ and [GRGDS + H]+ at m/z 491.22). As previously reported, the IM resolving power of the Synapt G1 is insufficient to separate these two peptides.28,39 Characterization of unknown samples with overlapping IM profiles (such as those shown in Figure 1, black IM profile) would be challenging, as IM-unresolved peaks could erroneously be assigned to a single analyte. Particularly in this example, the mixture exhibits a symmetric Gaussian-like IM profile indicative of what would be obtained from a single analyte. Presuming a single IM peak for this binary mixture would lead to measurement of a CCS corresponding to a mixture of two analytes. In the following sections, we show that chemometric deconvolution of post-IM/CID MS data can be used to extract ATs of IM-unresolved isomers that can be used to calculate accurate CCS values. Because of ion acceleration during post-IM/CID, ATs of deconvoluted IM profiles are shorter than ATs of individually analyzed species without post-IM/CID (i.e., obtained under low-energy conditions). Here, we demonstrate that corrected ATs can be used for deconvoluted IM spectra to improve experimentally calculated CCS values.
Figure 1.
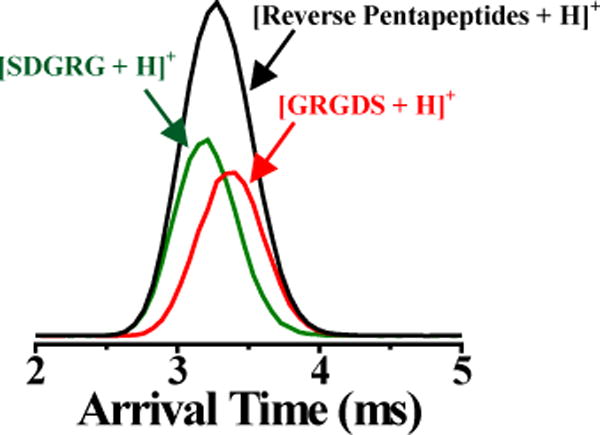
IM profiles of a binary mixture containing [SDGRG + H]+ (green) and [GRGDS + H]+ (red) before (black IM profile) and after (colored IM profiles) IM deconvolution using IM wave height voltage, wave velocity, and post-IM/CID acceleration voltage of 7.6 V, 300 m/s, and 40 V, respectively.
3.1 Chemometric Deconvolution
Using our previously reported AIMD software,39 the unresolved IM profile in Figure 1 (black IM profile) was deconvoluted to yield extracted IM profiles for [SDGRG + H]+ and [GRGDS + H]+ (green and red IM spectra in Figure 1, respectively). To check the validity of the deconvolution, we compared constructed CID mass spectra for each peptide from deconvolution of the data in Figure 1 to CID mass spectra of individually analyzed peptides (supporting information, Figure S2). The degree of similarity between deconvoluted and pure CID mass spectra were assessed using a previously described matching score (R) algorithm:46
| Equation 1 |
where n is the total number of fragment ion m/z values, m, with xm and x′m as fragment ion intensities (for a given value of m) for pure and deconvoluted spectra, respectively. R values of 0 and 1 indicate no correlation and a perfect match, respectively.46 An R value of ≥0.75 was considered a “successful” threshold for our deconvolution.46 Calculated R values for all deconvoluted reverse peptide isomer CID mass spectra were >0.86. Moreover, cross-validation matching scores (R′) were calculated by comparing the deconvoluted CID mass spectra for each peptide isomer with the reference spectrum for the non-matching isomer. The highest calculated R′ value was ~0.46 (i.e., indicating “unsuccessful match” for cases with wrong comparison spectra). Validation and cross-validation results suggested that, for each peptide isomer, deconvolution was successful both in terms of the fragment ions present and their relative abundances in each mass spectrum.
3.2 Arrival Time Correction Factor and CCS Calculations
Although match scores (i.e., R values) for all deconvoluted CID mass spectra (supporting information, Figure S2) for the pentapeptides suggested that MS deconvolution was successful, the degree of match between ATs of deconvoluted and pure isomers could not be discerned or quantified from the MS data alone. In-fact, as a result of electric potential difference used to induce post-IM/CID, ATs extracted from the deconvoluted post-IM/CID data in Figure 1 did not match ATs of individually run peptides (i.e., without post-IM/CID). The higher potential difference between the IM and transfer cells in post-IM/CID experiments results in faster acceleration of the ions as they exit the IM cell and pass through the transfer cell and thus leads to shorter measured ATs than what is obtained in low-energy (non-fragmentation) experiments. For example, the AT of deconvoluted (high-energy) [SDGRG + H]+ (3.18 ± 0.06 ms: green IM profile in Figure 2a) was approximately 0.22 ms shorter than the AT for the individually analyzed (low-energy) pure [SDGRG + H]+ (3.40 ± 0.10 ms: green IM profile in Figure 2b); these AT values are statistically different at the 95% C.L. and suggest that deconvoluted ATs should only be used for CCS calculations after applying an appropriate AT correction factor to the deconvoluted IM spectra.
Figure 2.
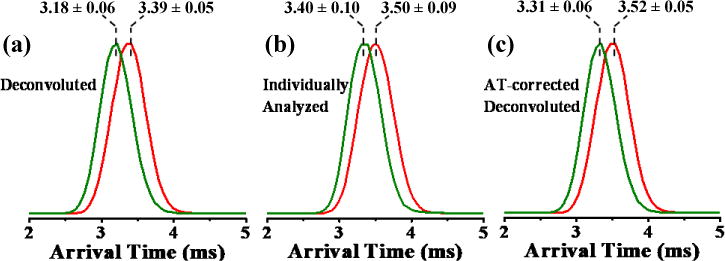
Normalized IM profiles for (a) deconvoluted, (b) individually analyzed pure, and (c) AT-corrected deconvoluted [SDGRG +H]+ (green) and [GRGDS + H]+ (red) using IM wave height voltage, wave velocity, and post-IM/CID acceleration voltage of 7.6 V, 300 m/s, and 40 V, respectively. AT errors are reported at the 95% C.L. (npure = 5 and ndecon = 4).
One solution to avoid AT variations would be to run the CCS calibration mixture at the same post-IM/CID energy as used for deconvolution. However, in cases where the CCS calibrant is a polymer (such as poly-alanine used here),42,45,63,64 post-IM/CID could generate several isomers with different and/or overlapping IM profiles (e.g., CID of Ala5 through Alan, where n > 5, can generate Ala1–5), or completely fragment the precursor ion (e.g., Ala5 is depleted while CID of Alan, where n > 5, still generates the Ala5 fragment ion), and complicate the extraction of pure IM ATs. Moreover, if post-IM/CID were used for a calibration mixture, then calibration plots would only be valid for that specific collision-energy. Therefore, multiple calibration profiles would have to be generated to calculate CCSs for IM-unresolved species deconvoluted using different collision-energies.46 Hence, it would be advantageous to correct ATs of deconvoluted IM profiles and fit them to a “conventional” low-energy CCS calibration plot.
To correct for change in ATs caused by post-IM/CID acceleration voltage, we ran the pentapeptide mixture at low collision-energy (i.e., 4 V; which is insufficient to induce CID) and high collision-energy (i.e., 40 V) and calculated the difference in AT (herein referred to as the AT “shift factor”). We then added this shift factor (of ~0.13 ms) to ATs of deconvoluted IM profiles to generate corrected-IM distributions (Figure 2c, discussed in-detail below); note that similar shift factors can be applied to all ions exiting the IM cell. Garmón-Lobato et al. presented a similar strategy for aligning low- and high-energy IM profiles;65 however, their approach was not focused on characterizing ions with similar CCSs, such as those presented in this manuscript, or calculating CCSs from the resulting aligned spectra.65
3.3 Estimation of Time Correction Factor
The magnitude of AT correction can be estimated from known parameters of the experiment. Experimentally measured ATs in the Synapt G1 (and G2-S) instruments are the summation of four ion transit times including: (1) IM drift time, (2) time spent in the transfer cell, (3) TOF MS flight time, and (4) time spent traveling across the interface between each of these instrument regions.11,28,66 IM drift times should not change significantly between low- and high-energy experiments because the electric fields (i.e., TW, entrance and exit lenses, and radio frequency confinement voltages) and IM pressure, which govern ions’ mobilities through the IM cell, are the same in both sets of experiments.28,67 Ion flight times within the TOF analyzer for precursor ions remain constant for both low-energy (no CID) and high-energy (post-IM/CID) experiments. However, due to larger potential difference between IM and transfer cells during a high-energy post-IM/CID experiment, molecular and potential fragment ions should spend less time in this interface region and exhibit mass-dependent time shifts.
In the Synapt G1, the center-to-center spacing between ring electrodes (e.g., between the IM and transfer cells) is ~1.5 mm and the length of the transfer cell is ~100 mm.66,67 Based on the physical geometry and electric fields of the instrument,66,67 ions will spend a considerably longer time in the transfer cell than in the region between the IM and transfer cells or in the TOF mass spectrometer. Therefore, we can approximate a maximum AT shift factor to be equal to the maximum amount of time an ion can spend in the transfer cell. The transfer cell is operated at lower pressure than the IM cell and ions therefore travel with the TW rather than falling over the wave as they might do in IM.14,21,66,67 Therefore, assuming an ion entering the transfer cell had insufficient kinetic energy to overtake the TW in the transfer cell, and ignoring phase effects of the TW, the maximum amount of time an ion could spend in the transfer cell (ttransfer) would be equal to the length of the cell (dt) divided by the transfer cell wave velocity (vt):11
| Equation 2 |
For a cell length of 100 mm (0.1 m) and transfer cell wave velocity of 248 m/s (as used in our experiments), the maximum time an ion should spend in the transfer cell (under our experimental conditions) is ~0.40 ms. Based on these simple estimations, under our experimental conditions, we should expect shift factors between ~0.10 × 10−2 ms (minimum TOF variation for fragment and precursor ions)68 and ~0.40 ms (maximum transfer cell time).
3.4 Empirical Shift Factors
Figure 3 shows the observed shift in IM AT for the binary pentapeptide mixture at 4 V (black IM profile) and 40 V (blue IM profile) post-IM/CID collision-energies. The measured shift factor for 7.6 V IM wave height was consistently 0.13 ms across eight trials (i.e., for four low-energy (4 V) and four high-energy (40 V) experiments). As expected, calculated shift factors varied as a function of wave height, ranging from 0.12 ms to 0.18 ms for wave heights of 7.0 V to 8.0 V (specific values can be found in the supporting information, Spreadsheet S1). Experimentally determined AT shift factors (i.e., 0.12 ms to 0.18 ms) are within the estimated ~0.10 × 10−2 ms to ~0.40 ms range (as discussed earlier in this section). Deconvoluted IM profiles were corrected by adding the AT shift factor time (i.e., 0.13 ms) to the IM profiles in Figure 2a to yield time-corrected IM profiles (Figure 2c). ATs for individually measured peptide isomers and AT-corrected deconvoluted IM profiles at wave height of 7.6 V were statistically indistinguishable at the 95% confidence level. Because of the approximations involved in the deconvolution process (e.g., peak centroid identification)39 and CCS dependent post-IM ion velocity, adding a single correction factor is not expected to improve all AT values identically. For example, the average corrected-AT value for GRGDS (3.52 ms, from the red IM profile in Figure 2c) is closer to its individually measured value (3.50 ms, from the red IM profile in Figure 2b) than the corrected-AT for SDGRG (3.31 ms, from the green IM profile in Figure 2c) is to its individually measured value (3.40 ms, from the green IM profile in Figure 2b). However, based on the aforementioned statistical justifications, corrected-deconvoluted ATs (as labeled in Figure 2c) were sufficient for use in CCS calculations (i.e., CCS results from AT-corrected deconvolution and pure IM peak analyses agreed at the 95% C.L.).
Figure 3.
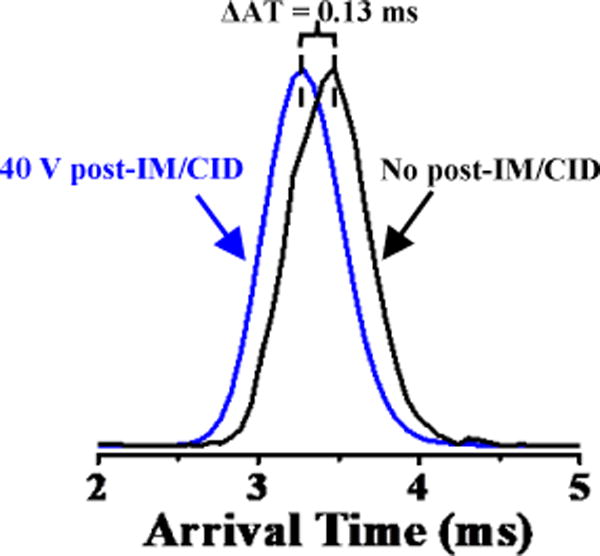
Unresolved IM profiles of an equimolar mixture of [SDGRG + H]+ and [GRGDS + H]+ (m/z 491.22) using IM wave height and velocity of 7.6 V and 300 m/s, respectively, without post-IM/CID (black IM profile) and with post-IM/CID at 40 V acceleration voltage (blue IM profile). The arrival time shifts (ΔAT = 0.13 ms) to a shorter value, due to post-IM/CID acceleration voltage.
Table 1 shows a summary of CCS calculations for the pentapeptide mixture. The calculated CCS of the pentapeptide mixture (i.e., 210.3 ± 3.2 Å2; where the subscript digit denotes the first insignificant figure) is comparable to the mathematical average CCS (~211 Å2) of individually measured [SDGRG +H]+ (209.1 ± 3.1 Å2) and [GRGDS + H]+ (212.7 ± 3.3 Å2) (Table 1). Our calculated CCSs for individual [SDGRG + H]+ and [GRGDS + H]+ (from pure samples) are within the 3% error range of the previously reported values for the same peptide samples calculated using DTIM-MS.69 Slight differences in CCS measurements may be due to several factors including (but not limited to) differences in IM cell humidity,70,71 mixture of gasses between the trap/transfer and IM cells (i.e., changing the reduced mass and average drift gas polarizability),45,69,72–74 accuracy and precision of calibrants’ CCSs,69 and/or other experimental condition variations.10 CCSs of deconvoluted [SDGRG + H]+ and [GRGDS + H]+ without AT corrections were 204.9 ± 3.0 Å2 and 209.5 ± 3.1 Å2, respectively (Table 1). Uncorrected CCSs were not statistically different than the individually measured values for both peptides at the 95% confidence level; however, at the 90% C.L., the uncorrected CCS for [SDGRG + H]+ was significantly different than the corresponding pure CCS. By comparison, calculated CCSs for the AT-corrected deconvoluted IM profiles for [SDGRG + H]+ and [GRGDS + H]+ were 208.3 ± 4.4 Å2 and 212.7 ± 4.6 Å2, respectively (Table 1); these calculated CCSs (after applying the AT correction approach) are not statistically different than the CCSs of individually measured peptide isomers at either the 90% or 95% confidence levels.
Table 1.
Experimental CCSs for Two Reverse Pentapeptide Isomers
| Identity | Mixture CCS (Å2)* |
Pure CCS (Å2)* |
Decon. CCS (Å2)* |
Corrected Decon. CCS (Å2)* |
|---|---|---|---|---|
| [Reverse Peptides + H]+ (mixture) | 210.3 ± 3.2 | — | — | — |
| [GRGDS + H]+ | — | 212.7 ± 3.3 | 209.5 ± 3.1 | 212. 7 ± 4.6 |
| [SDGRG + H]+ | — | 209.1 ± 3.1 | 204.9 ± 3.0 | 208.3 ± 4.4 |
Subscript digits denote the first insignificant figure. Errors are reported at the 95% C.L. (nmixtures = 24, npure = 25).
Our results indicate that CCSs of IM-unresolved reverse sequence peptide isomers (i.e., SDGRG and GRGDS) can be accurately calculated using chemometric deconvolution and AT correction using a Synapt G1 system. In the next section, we extend those results to demonstrate the utility of IM-unresolved CCS calculations on a four-component IM-unresolved enkephalin isomer mixture (i.e., RYGGFM, RMFGYG, MFRYGG, and FRMYGG) using a higher IM resolving power Synapt G2-S system.
3.5 Application of IM-unresolved CCS Calculations Using a Synapt G2-S System
Clemmer and coworkers demonstrated that by using LC-IM-MS they could identify ~82% of isomers in an approximately 4000-component combinatorial peptide library.75 For complex mixture analyses, such as performed by Clemmer,75 it is unlikely (although not impossible) to have more than two LC/IM unresolved isomers. However, to test our approach in a “worst case scenario” we analyzed a mixture of four constitutional isomeric enkephalin hexapeptides with unreported CCSs and limited our data collection times to one minute (as opposed to 10 minutes as used for the pentapeptides or 45 minutes as used for our previously reported four-component mixture results46). Results were compared to calculated theoretical CCSs (i.e., using PSA) and experimentally derived DTIM-MS values.
Figure 4 shows the IM profile for a doubly-protonated (m/z 365.67) four-component hexapeptide mixture before (black IM profile) and after (colored IM profiles where the orange, purple, cyan, and gray IM profiles correspond to RYGGFM, RMFGYG, MFRYGG, and FRMYGG, respectively) IM deconvolution at IM wave height of 22 V and wave velocity of 1300 m/s. The enkephalin isomer mixture (Figure 4, black IM profile) yielded a bimodal IM distribution which could erroneously be characterized as corresponding to two components if peak widths of the two overlapping Gaussian-like distributions were not considered. For example, the peak width of the later AT profile (which is generated from the additive sum of three individual IM profiles, as discussed in detail below) is much broader than the early AT conformer (which corresponds to a single isomer).76
Figure 4.
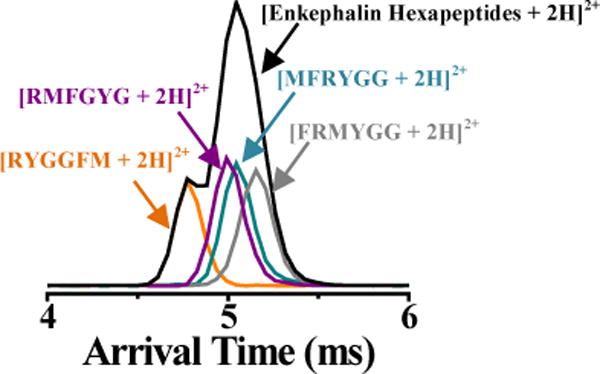
IM profiles of [RYGGFM + 2H]2+ (orange), [RMFGYG + 2H]2+ (purple), [MFRYGG + 2H]2+ (cyan), and [FRMYGG + 2H]2+ (gray) before (black IM profile) and after (colored IM profiles) IM deconvolution at IM wave height voltage, wave velocity, and post-IM/CID acceleration voltage of 40 V, 1300 m/s, and 22 V, respectively.
Deconvolution, R and R′ match scoring, and AT corrections were performed as discussed in the preceding sections. Calculated R and R′ values for all deconvoluted CID mass spectra for the hexapeptides were ≥0.83 and ≤0.37, respectively. For brevity, only final CCS values for the hexapeptides are discussed in this section. Examples of deconvoluted and pure CID mass spectra for hexapeptide isomers can be found in the supporting information, Figure S1.
Table 2 shows a summary of CCS calculations for the enkephalin isomer mixture. Similar to the pentapeptide mixture (Table 1), if the late-AT IM profile in Figure 4 was misidentified as a single component, then the calculated CCS (i.e., 296.9 ± 7.0 Å2) would be close to the average of the CCS values of the individual IM-unresolved isomers that contribute to the late-AT IM peak. Likewise, the calculated CCS for the early-AT profile in the hexapeptide mixture (i.e., without deconvolution; 288.8 ± 6.8 Å2) at the 95% C.L. is statistically indistinguishable from the CCS of the first eluting hexapeptide (i.e., [RYGGFM + 2H]2+; 292.7 ± 6.9 Å2).
Table 2.
Experimental CCSs for Four Enkephalin Hexapeptide Isomers
| Identity | Mixture CCS (Å2)* |
Pure CCS (Å2)* |
Decon. CCS (Å2)* |
Corrected Decon. CCS (Å2)* |
Drift Tube CCS (Å2)* |
|---|---|---|---|---|---|
| [Enkephalins + 2H]2+ (mixture; early AT Peak) | 288.8 ± 6.8 | — | — | — | — |
| [Enkephalins + 2H]2+ (mixture; late AT Peak) | 296.9 ± 7.0 | — | — | — | — |
| [RYGGFM + 2H]2+ | — | 292.7 ± 6.9 | 285.7 ± 6.7 | 288.8 ± 9.6 | 285.23 ± 0.53 |
| [RMFGYG + 2H]2+ | — | 299.6 ± 7.1 | 292.0 ± 6.9 | 295.1 ± 9.9 | 296.11 ± 0.38 |
| [MFRYGG + 2H]2+ | — | 299.2 ± 7.1 | 293.8 ± 6.9 | 296.8 ± 10.0 | 298.97 ± 0.48 |
| [FRMYGG + 2H]2+ | — | 301.5 ± 7.0 | 297.1 ± 7.1 | 300.1 ± 10.1 | 303.75 ±0.40 |
Subscript digits denote the first insignificant figure. Errors are reported at the 95% C.L. (TWIMS: nmixtures = 24, npure = 23 (FRMYGG) or npure = 24 (all others); DTIMS: n = 24 (for each peptide))
Collision cross sections calculated for the AT-corrected deconvoluted IM profiles were statistically indistinguishable from the corresponding individually analyzed peptides (Table 2 and supporting information, Spreadsheet S2). Although results for neither raw nor AT-corrected deconvoluted IM data were statistically different from the results obtained for the individual peptides, average CCS values for the corrected-AT deconvolution results were closer to the average CCS values for the individually analyzed peptides. For example, the calculated CCS for individually measured [FRMYGG +2H]2+ was 301.5 ± 7.0 Å2, which is closer to the calculated CCS for the AT-corrected deconvoluted data of 300.1 ± 10.1 Å2 than to the estimated CCS value of 297.1 ± 7.1 Å2 for deconvoluted raw data (prior to AT-correction). These results demonstrate that AT-correction improves CCS calculations (both for data from Synapt G1 and G2-S instruments) and can be applied to data from complex mixtures containing more than two IM-overlapping components.
To further demonstrate the accuracy of CCS values calculated from AT-corrected deconvoluted IM profiles, we analyzed the same enkephalin peptides on a DTIM-MS and experimentally measured their individual CCS values. Results from DTIM-MS measurements are summarized in Table 2. As listed in Table 2, CCS values calculated from TW and DT type instruments are in close agreement, with <1.3% deviation from DT values. Moreover, CCS values derived from AT-corrected post-TWIM/CID MS deconvoluted data are not significantly different than CCS values derived for peptides that were individually analyzed using the DTIM-MS instrument (i.e., agree at the 95% confidence level; see Spreadsheet S2).
The number of significant figures in CCS values calculated using TWIM-MS (Tables 1 and 2) were limited by the number of significant figures (viz., accuracy) reported for calibrant CCS literature values. Therefore, TWIM-MS CCS values are reported to three significant figures whereas DTIM-MS CCS values (which are not limited by the calibrant CCS values) were reported to four significant figures. All experimental errors reported in this manuscript were calculated by propagating error through each step of CCS calculations. For DTIM-MS measurements, the primary error is in the recorded ATs and in the AT correction (i.e., to compensate for time outside of the IM portion of the instrument; see Spreadsheet S3). For TWIM-MS measurements, there are errors associated with calibrant and analyte ATs (and error in “shift factors” used for AT-correction), fitting parameters, and exponential calibration factors derived from calibrant ATs. TWIM-MS calibration errors were calculated using the linear least squares fit and errors (LINEST) function in Microsoft Excel (see Spreadsheets S1 and S2). Although error propagation calculations can be time consuming, they more accurately represent the precision of IM measurements and underscore the importance of developing a calibration independent TWIM-MS CCS equation. Nevertheless, our results suggest that CCS values for IM-unresolved isomers can be calculated using deconvolution of post-IM/CID MS data; moreover, statistical comparisons of CCS values calculated using deconvoluted TWIM-MS data and directly measured DTIM-MS data indicate that these values are indistinguishable.
In addition to the DT CCS values obtained for the hexapeptide isomers, theoretical CCS values were also determined for an ensemble of conformations resulting from molecular dynamics simulations. Good agreement is observed between experimentally measured (AT-corrected deconvoluted TWIM-MS) CCS values (vertical lines) and the conformational space plots (calculated using PSA with a preliminary parameter set for nitrogen) in Figure 5. Root-mean-squared deviation (RMSD) clustering representative conformations for each of the isomers that fall within the experimental range (i.e., experimentally determined CCS ±95% C.L.) are also shown in Figure 5.61 The structure of [RYGGFM + 2H]2+, with the two glycine amino acids in the center of the peptide chain (Figure 5a), is expected to have increased flexibility in the gas-phase and corresponds to the peptide ion with the lowest CCS among its four isomers in Table 2. The other three isomers have similar CCS values (as confirmed by their overlapping IM traces shown in Figure 4). The one internal glycine gives [RMFGYG + 2H]2+ (Figure 5b) some flexibility. he peptide ion, [RMFGYG + 2H]2+ (Figure 5b) with one internal glycine is predicted to have some flexibility and this ion possesses the next largest CCS of the series. On the other hand, the N-terminal glycines present in both [MFRYGG + 2H]2+ and [FRMYGG + 2H]2+ (Figure 5c and d) presumably restrict the flexibility of these ions in the gas-phase. Interaction of the positive arginine residue with the C-terminus carboxylate group in Figure 5d is expected to extend the structure by pushing out the tryptophan and phenylalanine benzene rings of [FRMYGG + 2H]2+ which we interpret to contribute to the largest CCS among the four isomers. Additional RMSD clustered representative conformations corresponding to the experimental range of the four isomers are provided in the supplemental materials (Figures S3–S6).
Figure 5.
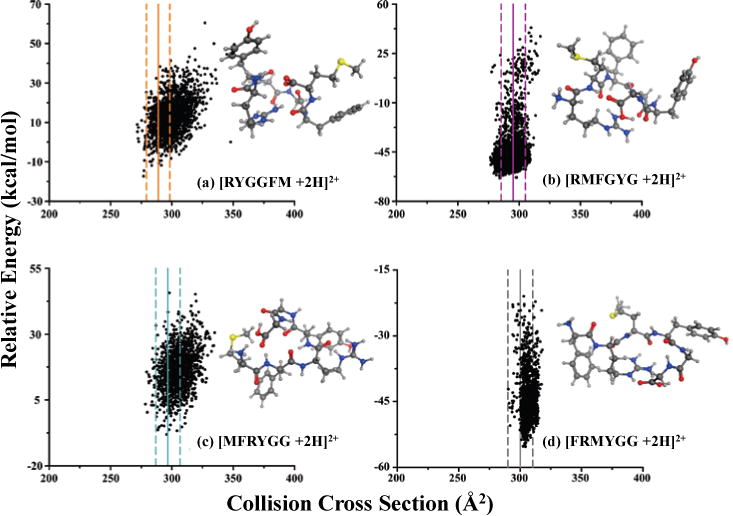
Computational results for calculating theoretical CCS values of the four enkephalin hexapeptides including (a) [RYGGFM + 2H]2+, (b) [RMFGYG + 2H]2+, (c) [MFRYGG + 2H]2+, and (d) [FRMYGG + 2H]2+. Solid vertical lines on each plot represent the AT-corrected deconvoluted TWIM-MS CCS values and the associated 95% C.L. are indicated with dashed lines; line colors match the corresponding deconvoluted IM profiles for hexapeptides in Figure 4. RMSD clustering representative conformations for each isomer are also included. Yellow, red, blue, gray, and white balls represent sulfur, oxygen, nitrogen, carbon, and hydrogen atoms, respectively.
4. CONCLUSION
We presented a method to calculate the CCS values of IM-unresolved isomers from TWIM-MS measurements using a modified version of the calibration protocol proposed by Ruotolo et al.11 To accurately measure CCS values for unresolved isomers, it was necessary to calculate IM AT “shift factors” which were used to correct the ATs of chemometrically deconvoluted IM profiles. Use of these shift factors improved the measured CCS values as compared to CCS values obtained from pure analytical standards analyzed on both TWIM-MS and DTIM-MS instruments. Moreover, the CCS values calculated using IM-deconvoluted data matched closely with theoretical conformational space scatter plots constructed using PSA.
Currently, CCS calibration for TWIM-MS yields measured CCS values with larger errors (e.g., ∼2%–3% of the mean CCS values, see Tables 1 and 2) than comparable DTIM-MS values (e.g., < 1% of the mean CCS values, see Table 2). Because of these large experimental errors (relative to DTIM-MS measurements), CCS values of some AT non-corrected deconvoluted data were not statistically different than individually measured values (see discussion of data presented in Tables 1 and 2 above). However, agreements between the individually measured CCS values and deconvoluted CCS values improved after applying AT correction (see Tables 1 and 2). As more precise calibration and instrumental techniques are developed, CCS measurement errors should (ideally) decrease. Similarly, as intra-instrument errors decrease, the use of AT-correction for post-IM acceleration will become more important for calculating more accurate CCS values.
In this report, we utilized TWIM-MS instruments for convolution experiments; however, the proposed time-corrected deconvolution technique is not limited to TWIM-MS and AT shift factors can be calculated for any IM-MS instrument capable of post-IM/CID. Moreover, the reported protocol could also be utilized in other MS techniques that exploit post-IM/CID33 to determine the presence of unresolved species and extract CCS information from subsequently deconvoluted spectra.
Supplementary Material
Acknowledgments
We thank Baylor University and National Science Foundation (NSF-IDBR, award number 1455668) for financial support. Support for Vanderbilt authors was provided by the National Institutes of Health (Grants 2R01GM092218, 5UH3TR000491-04, and 3UH3TR000491-0451), and the National Science Foundation (CHE-1229341). We also thank Matthew Brantley for assistance with calculating CID mass spectra matching scores and Brooke Brown for performing limited experimental replicates.
References
- 1.Cumeras R, Figueras E, Davis CE, Baumbach JI, Gracia I. Analyst. 2015;140:1376–1390. doi: 10.1039/c4an01100g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.May JC, McLean JA. Anal Chem. 2015;87:1422–1436. doi: 10.1021/ac504720m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruotolo BT, Giles K, Campuzano I, Sandercock AM, Bateman RH, Robinson CV. Science. 2005;310:1658–1661. doi: 10.1126/science.1120177. [DOI] [PubMed] [Google Scholar]
- 4.Scarff CA, Thalassinos K, Hilton GR, Scrivens JH. Rapid Communications in Mass Spectrometry. 2008;22:3297–3304. doi: 10.1002/rcm.3737. [DOI] [PubMed] [Google Scholar]
- 5.Wyttenbach T, Bowers MT. The Journal of Physical Chemistry B. 2011;115:12266–12275. doi: 10.1021/jp206867a. [DOI] [PubMed] [Google Scholar]
- 6.Bleiholder C, Dupuis NF, Wyttenbach T, Bowers MT. Nat Chem. 2011;3:172–177. doi: 10.1038/nchem.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhong Y, Hyung S-J, Ruotolo BT. Expert Review of Proteomics. 2012;9:47–58. doi: 10.1586/epr.11.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jurneczko E, Barran PE. Analyst. 2011;136:20–28. doi: 10.1039/c0an00373e. [DOI] [PubMed] [Google Scholar]
- 9.Chen S-H, Russell D. Journal of The American Society for Mass Spectrometry. 2015;26:1433–1443. doi: 10.1007/s13361-015-1191-1. [DOI] [PubMed] [Google Scholar]
- 10.Cumeras R, Figueras E, Davis C, Baumbach J, Gràcia I. Analyst. 2015;140:1391–1410. doi: 10.1039/c4an01101e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruotolo BT, Benesch JLP, Sandercock AM, Hyung S-J, Robinson CV. Nat Protocols. 2008;3:1139–1152. doi: 10.1038/nprot.2008.78. [DOI] [PubMed] [Google Scholar]
- 12.Rokushika S, Hatano H, Baim MA, Hill HH., Jr Anal Chem. 1985;57:1902–1907. [Google Scholar]
- 13.Siems WF, Wu C, Tarver EE, Hill HH, Jr, Larsen PR, McMinn DG. Anal Chem. 1994;66:4195–4201. [Google Scholar]
- 14.Shvartsburg AA, Smith RD. Anal Chem. 2008;80:9689–9699. doi: 10.1021/ac8016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.May JC, Dodds JN, Kurulugama RT, Stafford GC, Fjeldsted JC, McLean JA. Analyst. 2015;140:6824–6833. doi: 10.1039/c5an00923e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dwivedi P, Wu C, Matz LM, Clowers BH, Siems WF, Hill HH., Jr Anal Chem. 2006;78:8200–8206. doi: 10.1021/ac0608772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shvartsburg AA, Smith RD. Analytical Chemistry. 2011;83:9159–9166. doi: 10.1021/ac202386w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fasciotti M, Sanvido GB, Santos VG, Lalli PM, McCullagh M, de Sá GF, Daroda RJ, Peter MG, Eberlin MN. J Mass Spectrom. 2012;47:1643–1647. doi: 10.1002/jms.3089. [DOI] [PubMed] [Google Scholar]
- 19.Liang X, Wang X, Wang W, Zhou Q, Chen C, Peng L, Wen M, Qu T, Wang Z, Zhao K, Li J, Li H. Talanta. 2014;121:215–219. doi: 10.1016/j.talanta.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Purves RW, Ozog AR, Ambrose SJ, Prasad S, Belford M, Dunyach J-J. Journal of the American Society for Mass Spectrometry. 2014;25:1274–1284. doi: 10.1007/s13361-014-0878-z. [DOI] [PubMed] [Google Scholar]
- 21.May J, McLean J. Int J Ion Mobil Spec. 2013;16:85–94. doi: 10.1007/s12127-013-0123-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Clemmer DE. Analytical Chemistry. 1997;69:2504–2509. doi: 10.1021/ac9701344. [DOI] [PubMed] [Google Scholar]
- 23.Hoaglund-Hyzer CS, Li J, Clemmer DE. Anal Chem. 2000;72:2737–2740. doi: 10.1021/ac0000170. [DOI] [PubMed] [Google Scholar]
- 24.Clowers B, Dwivedi P, Steiner W, Hill H, Bendiak B. Journal of the American Society for Mass Spectrometry. 2005;16:660–669. doi: 10.1016/j.jasms.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 25.Clowers BH, Ibrahim YM, Prior DC, Danielson WF, Belov ME, Smith RD. Anal Chem. 2008;80:612–623. doi: 10.1021/ac701648p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zucker S, Lee S, Webber N, Valentine S, Reilly J, Clemmer D. Journal of The American Society for Mass Spectrometry. 2011;22:1477–1485. doi: 10.1007/s13361-011-0179-8. [DOI] [PubMed] [Google Scholar]
- 27.Hernandez DR, DeBord JD, Ridgeway ME, Kaplan DA, Park MA; Fernandez-Lima F. Analyst. 2014;139:1913–1921. doi: 10.1039/c3an02174b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giles K, Williams JP, Campuzano I. Rapid Commun Mass Spectrom. 2011;25:1559–1566. doi: 10.1002/rcm.5013. [DOI] [PubMed] [Google Scholar]
- 29.Xuan Y, Creese AJ, Horner JA, Cooper HJ. Rapid Communications in Mass Spectrometry. 2009;23:1963–1969. doi: 10.1002/rcm.4101. [DOI] [PubMed] [Google Scholar]
- 30.Creese AJ, Cooper HJ. Analytical Chemistry. 2012;84:2597–2601. doi: 10.1021/ac203321y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee S, Li Z, Valentine SJ, Zucker SM, Webber N, Reilly JP, Clemmer DE. International journal of mass spectrometry. 2012;309:154–160. doi: 10.1016/j.ijms.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu F, Lee S, Valentine S, Reilly J, Clemmer D. Journal of The American Society for Mass Spectrometry. 2012;23:2158–2166. doi: 10.1007/s13361-012-0491-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffmann W, Hofmann J, Pagel K. J Am Soc Mass Spectrom. 2014;25:471–479. doi: 10.1007/s13361-013-0780-0. [DOI] [PubMed] [Google Scholar]
- 34.Khakinejad M, Kondalaji S, Maleki H, Arndt J, Donohoe G, Valentine S. Journal of The American Society for Mass Spectrometry. 2014;25:2103–2115. doi: 10.1007/s13361-014-0990-0. [DOI] [PubMed] [Google Scholar]
- 35.Harvey D, Crispin M, Bonomelli C, Scrivens J. Journal of The American Society for Mass Spectrometry. 2015:1–14. doi: 10.1007/s13361-015-1163-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colby BN. Journal of the American Society for Mass Spectrometry. 1992;3:558–562. doi: 10.1016/1044-0305(92)85033-G. [DOI] [PubMed] [Google Scholar]
- 37.Zekavat B, Solouki T. J Am Soc Mass Spectrom. 2012;23:1873–1884. doi: 10.1007/s13361-012-0471-2. [DOI] [PubMed] [Google Scholar]
- 38.Windig W, Guilment J. Analytical Chemistry. 1991;63:1425–1432. [Google Scholar]
- 39.Brantley M, Zekavat B, Harper B, Mason R, Solouki T. J Am Soc Mass Spectrom. 2014;25:1810–1819. doi: 10.1007/s13361-014-0963-3. [DOI] [PubMed] [Google Scholar]
- 40.Meloun M, Čapek J, Mikšík P, Brereton RG. Analytica Chimica Acta. 2000;423:51–68. [Google Scholar]
- 41.Malinowski ER. Analytical Chemistry. 1977;49:612–617. [Google Scholar]
- 42.Zekavat B, Miladi M, Becker C, Munisamy S, Solouki T. Journal of The American Society for Mass Spectrometry. 2013;24:1355–1365. doi: 10.1007/s13361-013-0673-2. [DOI] [PubMed] [Google Scholar]
- 43.Zekavat B, Miladi M, Al-Fdeilat A, Somogyi A, Solouki T. Journal of The American Society for Mass Spectrometry. 2014;25:226–236. doi: 10.1007/s13361-013-0768-9. [DOI] [PubMed] [Google Scholar]
- 44.Harper B, Miladi M, Solouki T. Journal of The American Society for Mass Spectrometry. 2014;25:1716–1729. doi: 10.1007/s13361-014-0955-3. [DOI] [PubMed] [Google Scholar]
- 45.Bush MF, Campuzano IDG, Robinson CV. Anal Chem. 2012;84:7124–7130. doi: 10.1021/ac3014498. [DOI] [PubMed] [Google Scholar]
- 46.Pettit ME, Harper B, Brantley MR, Solouki T. Analyst. 2015;140:6886–6896. doi: 10.1039/c5an00940e. [DOI] [PubMed] [Google Scholar]
- 47.May JC, Goodwin CR, Lareau NM, Leaptrot KL, Morris CB, Kurulugama RT, Mordehai A, Klein C, Barry W, Darland E, Overney G, Imatani K, Stafford GC, Fjeldsted JC, McLean JA. Anal Chem. 2014;86:2107–2116. doi: 10.1021/ac4038448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.May JC, McLean JA. Proteomics. 2015 [Google Scholar]
- 49.Mason EA, Schamp HW., Jr Annals of Physics. 1958;4:233–270. [Google Scholar]
- 50.Case DA, Babin V, Berryman J, Betz RM, Cai Q, Cerutti DS, Cheatham TE, III, Darden TA, Duke RE, Gohlke H. 2014 [Google Scholar]
- 51.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA. Inc, Wallingford, CT. 2009;2:4. [Google Scholar]
- 52.Forsythe JG, Stow SM, Nefzger H, Kwiecien NW, May JC, McLean JA, Hercules DM. Anal Chem. 2014;86:4362–4370. doi: 10.1021/ac5001652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stow SM, Onifer TM, Forsythe JG, Nefzger H, Kwiecien NW, May JC, McLean JA, Hercules DM. Anal Chem. 2015;87:6288–6296. doi: 10.1021/acs.analchem.5b01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.von Helden G, Hsu MT, Gotts N, Bowers MT. The Journal of Physical Chemistry. 1993;97:8182–8192. [Google Scholar]
- 55.Mesleh MF, Hunter JM, Shvartsburg AA, Schatz GC, Jarrold MF. The Journal of Physical Chemistry. 1996;100:16082–16086. [Google Scholar]
- 56.Shvartsburg AA, Jarrold MF. Chemical physics letters. 1996;261:86–91. [Google Scholar]
- 57.Campuzano I, Bush MF, Robinson CV, Beaumont C, Richardson K, Kim H, Kim HI. Analytical Chemistry. 2012;84:1026–1033. doi: 10.1021/ac202625t. [DOI] [PubMed] [Google Scholar]
- 58.Bleiholder C, Wyttenbach T, Bowers MT. International Journal of Mass Spectrometry. 2011;308:1–10. [Google Scholar]
- 59.Bleiholder C, Contreras S, Do TD, Bowers MT. International Journal of Mass Spectrometry. 2013;345:89–96. [Google Scholar]
- 60.Anderson SE, Bleiholder C, Brocker ER, Stang PJ, Bowers MT. Int J Mass Spectrom. 2012;330-332:78–84. [Google Scholar]
- 61.Bleiholder C, Contreras S, Bowers MT. International Journal of Mass Spectrometry. 2013;354:275–280. [Google Scholar]
- 62.http://luschka.bic.ucsb.edu:8080/WebPSA/index.jsp.
- 63.Gidden J, Wyttenbach T, Jackson AT, Scrivens JH, Bowers MT. Journal of the American Chemical Society. 2000;122:4692–4699. [Google Scholar]
- 64.Forsythe JG, Petrov AS, Walker CA, Allen SJ, Pellissier JS, Bush MF, Hud NV, Fernandez FM. Analyst. 2015;140:6853–6861. doi: 10.1039/c5an00946d. [DOI] [PubMed] [Google Scholar]
- 65.Garmón-Lobato S, Abad-García B, Sánchez-Ilárduya MB, Romera-Fernández M, Berrueta LA, Gallo B, Vicente F. Anal Chim Acta. 2013;771:56–64. doi: 10.1016/j.aca.2013.01.065. [DOI] [PubMed] [Google Scholar]
- 66.Pringle SD, Giles K, Wildgoose JL, Williams JP, Slade SE, Thalassinos K, Bateman RH, Bowers MT, Scrivens JH. Int J Mass Spectrom. 2007;261:1–12. [Google Scholar]
- 67.Giles K, Pringle SD, Worthington KR, Little D, Wildgoose JL, Bateman RH. Rapid Communications in Mass Spectrometry. 2004;18:2401–2414. doi: 10.1002/rcm.1641. [DOI] [PubMed] [Google Scholar]
- 68.Radionova A, Filippov I, Derrick PJ. Mass Spectrometry Reviews. 2015 doi: 10.1002/mas.21470. n/a-n/a. [DOI] [PubMed] [Google Scholar]
- 69.Bush MF, Hall Z, Giles K, Hoyes J, Robinson CV, Ruotolo BT. Analytical chemistry. 2010;82:9557–9565. doi: 10.1021/ac1022953. [DOI] [PubMed] [Google Scholar]
- 70.Mäkinen M, Sillanpää M, Viitanen AK, Knap A, Mäkelä JM, Puton J. Talanta. 2011;84:116–121. doi: 10.1016/j.talanta.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 71.Mayer T, Borsdorf H. Analytical Chemistry. 2014;86:5069–5076. doi: 10.1021/ac5007393. [DOI] [PubMed] [Google Scholar]
- 72.Karpas Z, Berant Z. The Journal of Physical Chemistry. 1989;93:3021–3025. [Google Scholar]
- 73.Asbury GR, Hill HH. Analytical Chemistry. 2000;72:580–584. doi: 10.1021/ac9908952. [DOI] [PubMed] [Google Scholar]
- 74.Beegle LW, Kanik I, Matz L, Hill HH., Jr International Journal of Mass Spectrometry. 2002;216:257–268. [Google Scholar]
- 75.Srebalus Barnes CA, Hilderbrand AE, Valentine SJ, Clemmer DE. Analytical Chemistry. 2002;74:26–36. doi: 10.1021/ac0108562. [DOI] [PubMed] [Google Scholar]
- 76.Miladi M, Zekavat B, Munisamy SM, Solouki T. Int J Mass Spectrom. 2012;316-318:164–173. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.