

Abstract
Bone marrow (BM) transplantation has been used to study the cellular basis of genetic control of autoimmune diseases, but conclusions remain elusive due to the contradictory findings in different animal models. In the current study, we found that BM cells from myocarditis-susceptible A.SW mice can render irradiated, myocarditis-resistant B10.S recipient mice susceptible to myosin-induced myocarditis, indicating that hematopoietic cells express the genetic differences controlling susceptibility to autoimmune myocarditis. We then sought to differentiate the role of lymphoid vs nonlymphoid components of BM in the pathogenesis of myocarditis by comparing mixed chimeras receiving BM from A.SW wild-type or RAG−/− mice mixed with BM from B10.S wild-type mice. This experiment clearly demonstrated that T and B lymphocytes were indispensable for transferring the susceptible phenotype to disease-resistant recipients. Our findings significantly narrow the cellular expression of genetic polymorphisms controlling the EAM phenotype.
The pathogenesis of autoimmune diseases is generally attributed to effector functions of the adaptive immune system, but the humoral and cellular components of innate immune responses also play a substantial role in directing the later pathogenic adaptive immune responses (1, 2). In the past, bone marrow (BM)3 transplantation has been used to discriminate the contribution of hematopoietic vs nonhematopoietic cell lineages to disease susceptibility. Development of some autoimmune diseases, including spontaneous diabetes or lupus nephritis (3–5), originates from defects that reside within BM-derived stem cells of NOD or New Zealand White mice, respectively. Nonhematopoietic factors, however, have also been shown to control disease susceptibility as evidenced in experimental allergic encephalomyelitis (6, 7) and systemic lupus erythematosus in α-mannosidase II-deficient mice (1). Furthermore, in an animal model of experimental Lyme arthritis, resistance to disease induction was conserved by either hematopoietic or nonhematopoietic cellular compartments (8). These contradictory findings highlight the distinct pathogenic mechanisms in different autoimmune diseases.
Autoimmune myocarditis is an organ-specific autoimmune disease characterized by damage of the myocardium by infiltrating inflammatory cells. It often develops after an acute viral myocarditis in both humans (9) and rodents (10). Experimental autoimmune myocarditis (EAM) induced by cardiac myosin (CM) in susceptible mouse strains provides a virus-free model to study the immunopathogenic mechanisms of the disease. Like most autoimmune diseases, MHC haplotype is an important genetic factor for EAM susceptibility, but its effects are overshadowed by non-MHC traits (11, 12). MHC molecules are expressed prominently on hematopoietic cells, but varying levels of MHC class I molecules are also found on nonhematopoietic cells. Meanwhile, several non-H-2 Eam loci have been identified (13), but the cellular expression of these genetic polymorphisms remains unclear. For example, passive transfer of autoimmune myocardits by myosin-specific Ab depends upon target organ polymorphism (14). In addition, mutation or deletion in genes encoding cardiac Ags (15, 16) was found in patients with dilated cardiomyopathy, suggesting genes expressed in the target organ may contribute to disease susceptibility or severity. Our aim in the current study is to localize the cellular mediators that harbor the major genetic differences between myocarditis-susceptible and myocarditis-resistant mouse strains, and to further discriminate the roles of different cell lineages in genetic susceptibility to autoimmune myocarditis.
Materials and Methods
Mice and Ags
A.SW and B10.S (both H-2s haplotype) were purchased from The Jackson Laboratory, and have been bred and maintained in the conventional housing facilities at The Johns Hopkins University (Baltimore, MD). A.SW RAG−/− were generated by backcrossing A/J RAG−/− mice, a gift from Dr. D. Farris (University of Oklahoma, Oklahoma City, OK), to the A.SW background. Mouse CM was purified from frozen Swiss Webster white mouse hearts (Pel-Freez Biologicals) following procedures previously described (11).
Generation and characterization of BM chimeras
A.SW or B10.S recipients were lethally irradiated (~850 rad) and 12–24 h later, reconstituted with 2–5 × 106 donor BM cells, depleted of mature T and B cells. Animals were allowed to reconstitute for 12 wk before EAM induction. To assess the degree of chimerism, spleen cells (5 × 105/mouse) were stained with strain-specific markers of anti-Ly9.1 (FITC) or anti-Ly6.2 (clone 3A7), which recognize lymphocytes from A.SW or B10.S origin, respectively, in combination with cell surface markers, CD4, CD8, and CD19. Anti-Ly6.2 is a gift from Dr. K. L. Rock (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA) (17), and all other Abs were purchased from eBioscience.
Induction and assessment of EAM
EAM was elicited by immunization with mouse CM as previously described (18). At day 21, the hearts were collected, fixed in 10% formalin, and sent to Histoseve for embedding, sectioning, and H&E staining. The degree of EAM was scored based on the percentage of the area of infiltrated myocardium in the most affected section as follows: 0 = no infiltration, 1 = <10%, 2 = 10–30%, 3 = 30–50%, 4 = 50–80%, and 5 = >80%. The presence of myosin-specific IgG in immune sera was assessed by ELISA.
Statistical analysis
Differences in EAM severity and OD readings of Ab ELISA between groups were assessed for statistical significance by the nonparametric Mann-Whitney U test or t test in GraphPad Prism 4 software (http://www.graphpad.com).
Results
BM from susceptible mice impart EAM susceptibility to normally resistant recipients
To determine whether the genetic control of EAM was mainly mediated by cells of hematopoietic or nonhematopoietic origin, we generated BM chimeric mice from myocarditis-susceptible A.SW mice and myocarditis-resistant B10.S mice (both having the same H-2s haplotype). Regardless of whether BM transfer was performed with syngeneic or allogeneic donors, there was little evidence of graft-vs-host disease. Animals displaying any sign of graft-vs-host disease, such as weight loss or skin lesions, were excluded from the study. Twelve weeks following BM transplantation, chimeric mice were tested for EAM susceptibility by immunization with CM. Twenty-one days later, hearts were collected and histologically examined for the presence of myocardial lesions. At the same time, spleen cells were harvested and stained with Ly9.1 or Ly6A.2, which recognize lymphocytes of A.SW or B10.S origin, respectively, to test the repopulation of chimeric mice by donor BM. As shown in Fig. 1B, in B10.S mice receiving BM from A.SW donor (A→B) or conversely transplanted A.SW mice (B→A), 80% of spleen cells were derived from donor BM, whereas 20% were repopulated from residual host BM that survived after irradiation. In mice receiving autologous BM (A→A or B→B), 100% of spleen cells were either A- or B-derived, respectively. When myocarditis was examined, we found that BM-derived hematopoietic cells from A.SW were fully capable of transferring disease susceptibility to normally resistant B10.S mice (Figs. 1A and 2). Approximately 68% of B10.S mice receiving BM from A.SW donor (A→B mice) developed EAM with mean score of 1.22 ± 1.14, and both incidence and severity were comparable to control A→A mice. Interestingly, in the converse experiment, A.SW mice receiving BM from B10.S mice (B → A mice) developed slightly, but significantly, more EAM vs B → B controls (EAM with mean score of 0.70 ± 0.88 vs 0.21 ± 0.46, p = 0.013). This slight increase in EAM induction in B → A mice could be due to the hematopoietic cells originated from remaining A.SW BM after lethal irradiation (Fig. 1B) or minor contributions from nonhematopoietic cells.
FIGURE 1.
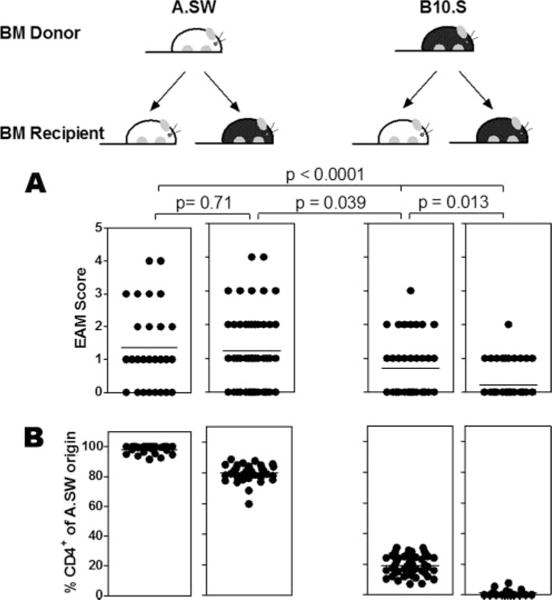
Phenotype of susceptibility to EAM is transferable through BM. After 12 wk of reconstitution, BM chimeric mice were subjected to EAM induction (A) and tested for repopulation from donor BM (B). A, The EAM score was assigned based on a grading scale describing the severity of myocarditis as shown in Fig. 2. Data are presented as individual values with mean (horizontal line). The differences between groups were analyzed by nonparametric Mann-Whitney U test in GraphPad Prism. B, Percentage of chimerism of spleen cells in each experimental group is expressed as individual values with mean (horizontal line).
FIGURE 2.
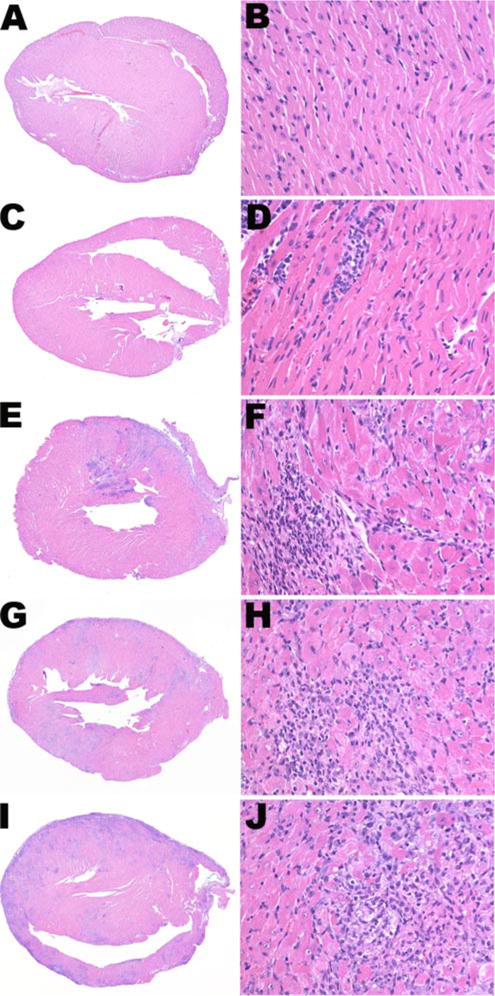
Representative histological appearance of EAM severity score. BM chimeric mice were immunized with CM in CFA at day 0 and day 7, with additional dose of pertussis toxin at day 0, to promote EAM induction. Twenty-one days later, hearts were collected for histological examination. A and B, Normal heart, EAM score = 0. C and D, Minimal myocarditis with <10% of myocardium involved, EAM = 1. E and F, Moderate myocarditis with 10–30% of myocardium involved, EAM = 2. G and H, Severe myocarditis with 30–50% of myocardium involved, EAM = 3. I and J, Extensive myocarditis with >50% of myocardium involved, EAM = 4. Magnification is ×5 (left column) and ×100 (right column).
The susceptibility to EAM is mainly determined by BM-derived lymphoid compartments
The finding that A.SW BM could transfer disease susceptibility to the normally resistant B10.S mice provides conclusive evidence that the predominant EAM susceptibility genes are expressed in BM-derived cells. To determine whether the lymphoid or nonlymphoid compartment within BM-derived cells express these susceptibility genes, irradiated B10.S were transplanted with 50/50 BM mixture of A.SW RAG−/− and B10.S wild-type (WT) mice (A−/−/B → B). As controls, irradiated B10.S recipients were also transplanted with BM from the following: 1) WT B10.S (B→B), 2) WT A.SW (A → B), or 3) a 50/50 mixture of WT A.SW and B10.S (A/B→B). The purpose of cotransfer of B10.S WT BM with RAG−/− was to render irradiated recipients immunocompetent, i.e., having normal T and B cell pools. In this experiment, re-population of the chimeric mice by different cell types was examined in the spleen. Like in Fig. 1B, chimeric status in A→B or B→A mice was ~80:20 of donor to host in CD4+ T cells (Fig. 3). In A/B→B mice, approximately half of CD4+ spleen cells came from A.SW origin and the other half from B10.S origin. CD4+ population in A−/−/B→B mice represent similar profile as B→B mice (Fig. 3). In all groups, CD8+ T cell and CD19+ B cell in spleens express similar profiles as observed in CD4+ T cells (Fig. 3B).
FIGURE 3.
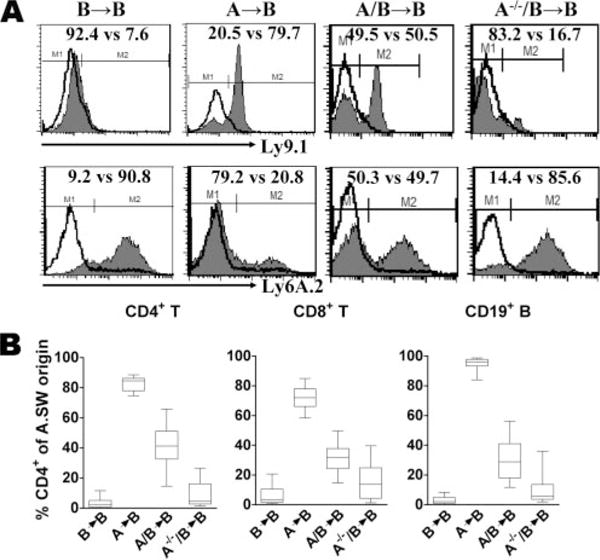
BM reconstitution of recipient mice transplanted with syngeneic or allogeneic donor BM. Recipient mice were lethally irradiated and injected i.v. with 2–5 × 106 BM cells from A.SW, B10.S, 50/50 mixture A.SW/B10.S, or 50/50 mixture A.SW RAG−/−/B10.S mice. After 12 wk, reconstitution of lymphocyte population in the spleen was analyzed by FACS. A, Surface expression of strain markers Ly9.1 (A.SW) and Ly6A.2 (B10.S) on CD4+ spleen cells from representative mouse of each experimental group is shown. The experimental population (gray shaded histogram) and isotype control (open histogram) are indicated. B, Histogram represents the mean percentage of chimerism of CD4+, CD8+, CD19+ lymphocyte population of individual mice in each experimental group.
These chimeras were also tested for their susceptibility to EAM. As shown in Fig. 4, B→B mice remained low responders, whereas A→B and A/B→B were almost equally susceptible to myocarditis. Approximately 80% of A→B and A/B→B mice developed myocarditis with mean EAM score of 1.65 ± 1.14 vs 1.45 ± 1.15, respectively. A−/−/B→B developed EAM with intermediate severity, but not significantly differ from B→B control and A/B→B, suggesting that disease susceptibility is determined by lymphocytes and possibly other hematopoietic cells such as NK cells and mast cells contributed by the RAG-deficient mice. In agreement with previous findings in the SCID mouse (19), RAG−/− mouse itself and B10.S transplanted with RAG−/− BM did not develop myocarditis following CM immunization (data not shown).
FIGURE 4.
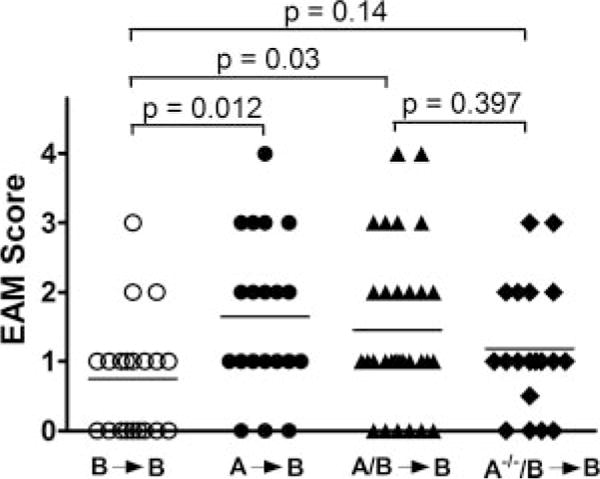
Phenotype of susceptibility to EAM is transferable through BM-derived lymphoid compartment. Similar to experiment in Fig. 3, chimeric mice were created by transferring BM from WT or RAG−/− A.SW alone or mixed with WT B10.S mice to irradiated WT B10.S recipients. EAM induction in these mice was tested after 12 wk of reconstitution, and represented as individual values with mean (horizontal line). EAM severity between groups was compared by nonparametric Mann-Whitney U test in GraphPad Prism.
Ab responses in BM chimeric mice
The production of CM-specific total IgG and its isotypes in these chimeric mice was measured by ELISA to determine their correlation with myocarditis phenotype. At total IgG level, a small difference was observed between A → B and control B → B mice (OD450 nm = 2.97 ± 0.57 vs 2.35 ± 0.63, p < 0.05), but not in A/B → B and A−/−/B → B mice. There was also no difference observed in WT A.SW and B10.S mice following EAM induction (data not shown). Interestingly, IgG1 levels were significantly higher in EAM-susceptible A → B and A/B → B (OD450 nm = 2.96 ± 0.77 and 2.58 ± 0.90, respectively, p <0.01), but not in resistant A−/−/B → B mice (OD450 nm = 2.30 ± 0.72), in comparison to B → B mice (OD 450 nm = 1.56 ± 1.14). IgG2a and IgG2c isotypes, encoded by different Ig H chain alleles, are expressed in A.SW and B10.S mice exclusively (20). Therefore, it is not surprising that a large amount of IgG2a was detected in B10.S recipients reconstituted with B cells of A.SW origin (OD450 nm = 2.12 ± 0.96 in A → B mice and OD450 nm = 1.02 ± 0.89 in A/B → B mice). IgG2c, although at a much lower amount with OD readings ranging from 0.17 to 0.21, was only detected in B10.S recipients reconstituted with B cells of B10.S origin (B → B, A → B, and A/B → B). Among the IgG isotypes, IgG1 and IgG2b were predominant Abs detected at large amounts, suggesting that there may be a Th2 component of EAM in the A strain mice (21).
Discussion
Clinical studies have reported that BM cells can transfer disease-causing autoreactive clones from donors to patients (22–24). This approach was used by us and other investigators to examine the cellular basis of autoimmune diseases in animal models. In accordance with previous findings in diabetes and lupus (3–5, 25), genetic susceptibility of myocarditis was shown to be controlled by donor-derived hematopoietic cells, not by the host background. Different findings were observed in other autoimmune diseases in which disease susceptibility or resistance was either mediated by nonhematopoietic factors (6, 7) or by both hematopoietic and nonhematopoietic factors (8). To further localize the EAM-controlling factors to lymphoid vs nonlymphoid compartment, mixed BM chimeras were created using BM mixture of RAG−/− A.SW and B10.S mice in the current study. Our results demonstrate that both lymphoid and nonlymphoid components of hematopoietic cells were the restriction elements for EAM susceptibility. These cell lineages may harbor polymorphic genes that are required for EAM pathogenesis but differ between strains.
BM transplantation created chimeric mouse consisting of donor BM-derived components on the host genetic background, which provides an ideal system to differentiate the role of hematopoietic vs nonhematopoietic cells. Good reconstitution status, i.e., 80:20 chimerism of donor to host, was achieved in these mice after 12 wk (Fig. 1). In all experiments, sham control B → B mice developed minimal myocarditis compared with their WT counterparts, indicating the damage to the heart was caused by autoimmune responses following CM immunization, but not transplantation-induced immunologic dysregulation. The degree of EAM susceptibility was positively correlated with the percentage of chimerism achieved, as A → A mice (100% of A strain) developed EAM with highest EAM incidence and severity, B → B mice (100% of B strain) have lowest disease and A/B → B mice (~50/50 for A to B strain) display intermediate phenotype. Coexistence of donor and host BM in mixed chimeras (A/B→B) has been suggested to facilitate the reciprocal induction of tolerance between donor and host, which minimize graft-vs-host and host-vs-graft disease (26, 27). We also showed that the autoimmune phenotype of A.SW recipients can be rescued by BM derived from resistant B10.S donor, as was previously shown in other models (5, 24, 28). These observations highlight the primary role of immune system (donor), but not the target organ (host), in the pathogenesis of autoimmune diseases. However, the contribution of the residual host BM to this process cannot be excluded because ~20% of lymphocytes were host-derived.
Hematopoietic cells of both innate and adaptive immune responses have been suggested to play role in the pathogenic process of EAM (2, 29). To study the contribution of lymphoid vs nonlymphoid cell lineages to the development of EAM separately, mixed chimeras receiving BM mixture of A.SW RAG−/− and B10.S were created. These animals had T and B cells of B10.S origin, whereas all other hematopoietic cells are mixtures of A.SW and B10.S contributions. Using this system, genetic susceptibility to EAM was mediated by T and B lymphocytes, and possibly other functional cell types in the RAG-deficient mice. The role of different lymphoid populations to EAM induction was first investigated by Smith and Allen (19) using SCID mice and was confirmed in this study in RAG-deficient mice (data not shown). EAM is believed to be mediated by CD4 T cells, as suggested by their predominance in the myocardial lesions, their capability to transfer disease following stimulation with CM, and the amelioration of EAM after their depletion with mAb in A/J mice (19, 29). Monoclonal Ab-mediated depletion of CD8 population reduced EAM severity but not incidence in A/J mice (19), whereas both disease incidence and severity were significantly enhanced in mice homozygous for CD8 mutation (CD8−/−) following immunization with CM (30). These paradoxical findings indicate that the CD8+ T cell may mediate both effector and suppressor functions in EAM via distinct pathways.
Other cells or cellular products including NK cells (2), mast cells (31), and complement (2) also influence disease susceptibility. Interestingly, a subset of TCRαβ+ CD4− CD8− double negative cells were found in lesions of autoimmune myocarditis (30), arthritis (32), and encephalomyelitis (33) developed in CD4-deficient mice, suggesting these cells may be capable of obviating the requirement for CD4+ T cells and subserve the effector function. B cells are generally considered not essential for EAM induction (34). The pathogenic mechanisms of the particular cell type may be altered in BM chimeras and their function will be investigated in future studies by creating chimeras using BM from mice deficient in CD4, CD8, or Btk (B cell) or depleting Ab.
Autoimmune myocarditis has been linked to both MHC haplotype and non-MHC genes, with non-MHC genes having strong effects (13). MHC molecules modulate adaptive immune responses, including autoimmune responses, via their unique role in Ag presentation. Yet, at least seven non-MHC loci have been reported to influence susceptibility to viral or immunization-induced autoimmune myocarditis in mouse (18, 35, 36). Linkage of these loci to apoptosis or tolerance induction was suggested; however, the mechanisms by which they modulate disease susceptibility remain to be investigated. In addition, polymorphisms in genes encoding cardiac Ags (15, 16) and CD45 (37) were found in patients with dilated cardiomyopathy, indicating potential association of these polymorphisms with myocarditis. The current findings significantly narrow our focus on identification of the predominant genetic polymorphisms responsible for EAM phenotype. Future studies will further subclassify the cells of interest by using genetically engineered animals that are deficient in particular subsets of T and B cells. This approach can also be used in other autoimmune disease models to dissect the cellular expression of a particular genetic trait.
Acknowledgments
We thank Monica V. Talor and Dongfeng Zheng for technical assistance.
Footnotes
This work is supported by Research Grants R01 HL-077611 and R01 HL-067290 from the National Institutes of Health.
Abbreviations used in this paper: BM, bone marrow; EAM, experimental autoimmune myocarditis; CM, cardiac myosin; WT, wild type.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Green RS, Stone EL, Tenno M, Lehtonen E, Farquhar MG, Marth JD. Mammalian N-glycan branching protects against innate immune self-recognition and inflammation in autoimmune disease pathogenesis. Immunity. 2007;27:308–320. doi: 10.1016/j.immuni.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 2.Kaya Z, Rose NR. Innate immunity in experimental autoimmune myocarditis. In: Zouali M, editor. Molecular Autoimmunity. Springer Science and Business Media, Inc.; New York: 2005. p. 1. [Google Scholar]
- 3.Ikehara S, Kawamura M, Takao F, Inaba M, Yasumizu R, Than S, Hisha H, Sugiura K, Koide Y, Yoshida TO, et al. Organ-specific and systemic autoimmune diseases originate from defects in hematopoietic stem cells. Proc Natl Acad Sci USA. 1990;87:8341–8344. doi: 10.1073/pnas.87.21.8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wicker LS, Miller BJ, Chai A, Terada M, Mullen Y. Expression of genetically determined diabetes and insulitis in the nonobese diabetic (NOD) mouse at the level of bone marrow-derived cells: transfer of diabetes and insulitis to nondiabetic (NOD X B10) F1 mice with bone marrow cells from NOD mice. J Exp Med. 1988;167:1801–1810. doi: 10.1084/jem.167.6.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.LaFace DM, Peck AB. Reciprocal allogeneic bone marrow transplantation between NOD mice and diabetes-nonsusceptible mice associated with transfer and prevention of autoimmune diabetes. Diabetes. 1989;38:894–901. doi: 10.2337/diab.38.7.894. [DOI] [PubMed] [Google Scholar]
- 6.Korngold R, Feldman A, Rorke LB, Lublin FD, Doherty PC. Acute experimental allergic encephalomyelitis in radiation bone marrow chimeras between high and low susceptible strains of mice. Immunogenetics. 1986;24:309–315. doi: 10.1007/BF00395536. [DOI] [PubMed] [Google Scholar]
- 7.Lublin FD, Knobler RL, Doherty PC, Korngold R. Relapsing experimental allergic encephalomyelitis in radiation bone marrow chimeras between high and low susceptible strains of mice. Clin Exp Immunol. 1986;66:491–496. [PMC free article] [PubMed] [Google Scholar]
- 8.Brown CR, Reiner SL. Bone-marrow chimeras reveal hemopoietic and nonhemopoietic control of resistance to experimental Lyme arthritis. J Immunol. 2000;165:1446–1452. doi: 10.4049/jimmunol.165.3.1446. [DOI] [PubMed] [Google Scholar]
- 9.Rose NR, Herskowitz A, Neumann DA, Neu N. Autoimmune myocarditis: a paradigm of post-infection autoimmune disease. Immunol Today. 1988;9:117–120. doi: 10.1016/0167-5699(88)91282-0. [DOI] [PubMed] [Google Scholar]
- 10.Rose NR, Wolfgram LJ, Herskowitz A, Beisel KW. Postinfectious autoimmunity: two distinct phases of coxsackievirus B3-induced myocarditis. Ann NY Acad Sci. 1986;475:146–156. doi: 10.1111/j.1749-6632.1986.tb20864.x. [DOI] [PubMed] [Google Scholar]
- 11.Neu N, Rose NR, Beisel KW, Herskowitz A, Gurri-Glass G, Craig SW. Cardiac myosin induces myocarditis in genetically predisposed mice. J Immunol. 1987;139:3630–3636. [PubMed] [Google Scholar]
- 12.Wolfgram LJ, Beisel KW, Herskowitz A, Rose NR. Variations in the susceptibility to coxsackievirus B3-induced myocarditis among different strains of mice. J Immunol. 1986;136:1846–1852. [PubMed] [Google Scholar]
- 13.Li HS, Ligons DL, Rose NR. Genetic complexity of autoimmune myocarditis. Autoimmun Rev. 2008;7:168–173. doi: 10.1016/j.autrev.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao L, Sindhwani R, Rojkind M, Factor S, Leinwand L, Diamond B. Antibody-mediated autoimmune myocarditis depends on genetically determined target organ sensitivity. J Exp Med. 1995;181:1123–1131. doi: 10.1084/jem.181.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 16.Olson TM, Michels VV, Thibodeau SN, Tai YS, Keating MT. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science. 1998;280:750–752. doi: 10.1126/science.280.5364.750. [DOI] [PubMed] [Google Scholar]
- 17.Rock KL, Yeh ET, Gramm CF, Haber SI, Reiser H, Benacerraf B. TAP, a novel T cell-activating protein involved in the stimulation of MHC-restricted T lymphocytes. J Exp Med. 1986;163:315–333. doi: 10.1084/jem.163.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guler ML, Ligons DL, Wang Y, Bianco M, Broman KW, Rose NR. Two autoimmune diabetes loci influencing T cell apoptosis control susceptibility to experimental autoimmune myocarditis. J Immunol. 2005;174:2167–2173. doi: 10.4049/jimmunol.174.4.2167. [DOI] [PubMed] [Google Scholar]
- 19.Smith SC, Allen PM. Myosin-induced acute myocarditis is a T cell-mediated disease. J Immunol. 1991;147:2141–2147. [PubMed] [Google Scholar]
- 20.Martin RM, Brady JL, Lew AM. The need for IgG2c specific antiserum when isotyping antibodies from C57BL/6 and NOD mice. J Immunol Methods. 1998;212:187–192. doi: 10.1016/s0022-1759(98)00015-5. [DOI] [PubMed] [Google Scholar]
- 21.Afanasyeva M, Wang Y, Kaya Z, Park S, Zilliox MJ, Schofield BH, Hill SL, Rose NR. Experimental autoimmune myocarditis in A/J mice is an interleukin-4-dependent disease with a Th2 phenotype. Am J Pathol. 2001;159:193–203. doi: 10.1016/S0002-9440(10)61685-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kishimoto Y, Yamamoto Y, Ito T, Matsumoto N, Ichiyoshi H, Katsurada T, Date M, Ohga S, Kitajima H, Ikehara S, Fukuhara S. Transfer of autoimmune thyroiditis and resolution of palmoplantar pustular psoriasis following allogeneic bone marrow transplantation. Bone Marrow Transplant. 1997;19:1041–1043. doi: 10.1038/sj.bmt.1700789. [DOI] [PubMed] [Google Scholar]
- 23.Neumeister P, Strunk D, Apfelbeck U, Sill H, Linkesch W. Adoptive transfer of vitiligo after allogeneic bone marrow transplantation for non-Hodgkin’s lymphoma. Lancet. 2000;355:1334–1335. doi: 10.1016/S0140-6736(00)02120-6. [DOI] [PubMed] [Google Scholar]
- 24.Sherer Y, Shoenfeld Y. Autoimmune diseases and autoimmunity post-bone marrow transplantation. Bone Marrow Transplant. 1998;22:873–881. doi: 10.1038/sj.bmt.1701437. [DOI] [PubMed] [Google Scholar]
- 25.Nakano K, Mordes JP, Handler ES, Greiner DL, Rossini AA. Role of host immune system in BB/Wor rat: predisposition to diabetes resides in bone marrow. Diabetes. 1988;37:520–525. doi: 10.2337/diab.37.5.520. [DOI] [PubMed] [Google Scholar]
- 26.Mathieu C, Casteels K, Bouillon R, Waer M. Protection against autoimmune diabetes in mixed bone marrow chimeras: mechanisms involved. J Immunol. 1997;158:1453–1457. [PubMed] [Google Scholar]
- 27.Good RA. Mixed chimerism and immunologic tolerance. N Engl J Med. 1993;328:801–802. doi: 10.1056/NEJM199303183281111. [DOI] [PubMed] [Google Scholar]
- 28.Adachi Y, Inaba M, Amoh Y, Yoshifusa H, Nakamura Y, Suzuka H, Akamatu S, Nakai S, Haruna H, Adachi M, et al. Effect of bone marrow transplantation on antiphospholipid antibody syndrome in murine lupus mice. Immunobiology. 1995;192:218–230. doi: 10.1016/S0171-2985(11)80099-9. [DOI] [PubMed] [Google Scholar]
- 29.Afanasyeva M, Georgakopoulos D, Rose NR. Autoimmune myocarditis: cellular mediators of cardiac dysfunction. Autoimmun Rev. 2004;3:476–486. doi: 10.1016/j.autrev.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 30.Penninger JM, Neu N, Timms E, Wallace VA, Koh DR, Kishihara K, Pummerer C, Mak TW. The induction of experimental autoimmune myocarditis in mice lacking CD4 or CD8 molecules. J Exp Med. 1993;178:1837–1842. doi: 10.1084/jem.178.5.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fairweather D, Frisancho-Kiss S, Gatewood S, Njoku D, Steele R, Barrett M, Rose NR. Mast cells and innate cytokines are associated with susceptibility to autoimmune heart disease following coxsackievirus B3 infection. Autoimmunity. 2004;37:131–145. doi: 10.1080/0891693042000196200. [DOI] [PubMed] [Google Scholar]
- 32.Tada Y, Ho A, Koh DR, Mak TW. Collagen-induced arthritis in CD4− or CD8-deficient mice: CD8 T cells play a role in initiation and regulate recovery phase of collagen-induced arthritis. J Immunol. 1996;156:4520–4526. [PubMed] [Google Scholar]
- 33.Koh DR, Ho A, Rahemtulla A, Penninger J, Mak TW. Experimental allergic encephalomyelitis (EAE) in mice lacking CD4 T cells. Eur J Immunol. 1994;24:2250–2253. doi: 10.1002/eji.1830240947. [DOI] [PubMed] [Google Scholar]
- 34.Malkiel S, Factor S, Diamond B. Autoimmune myocarditis does not require B cells for antigen presentation. J Immunol. 1999;163:5265–5268. [PubMed] [Google Scholar]
- 35.Aly M, Wiltshire S, Chahrour G, Osti JC, Vidal SM. Complex genetic control of host susceptibility to coxsackievirus B3-induced myocarditis. Genes Immun. 2007;8:193–204. doi: 10.1038/sj.gene.6364374. [DOI] [PubMed] [Google Scholar]
- 36.Kuan AP, Chamberlain W, Malkiel S, Lieu HD, Factor SM, Diamond B, Kotzin BL. Genetic control of autoimmune myocarditis mediated by myosin-specific antibodies. Immunogenetics. 1999;49:79–85. doi: 10.1007/s002510050466. [DOI] [PubMed] [Google Scholar]
- 37.Tchilian EZ, Gil J, Navarro ML, Fernandez-Cruz E, Chapel H, Misbah S, Ferry B, Renz H, Schwinzer R, Beverley PC. Unusual case presentations associated with the CD45 C77G polymorphism. Clin Exp Immunol. 2006;146:448–454. doi: 10.1111/j.1365-2249.2006.03230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]