

Abstract
The microbiome’s involvement in health and disease, and the complexity of its composition and function, make it intriguing to consider human genetic factors that impact microbiome composition. Genes may influence health through their ability to promote a stable microbial community in the gut. Studies of heritability yield a consistent subset of microbes that are impacted by genes, but the use of genome-wide association studies (GWAS) to identify specific genetic variants associated with microbiota phenotypes has proven challenging. Processing microbiome datasets into traits to be modeled and reducing the burden of multiple testing are just some of the technical hurdles in microbiome GWAS. Studies to date are small by GWAS standards, making cross-study comparisons and validations particularly important in identifying authentic signals. Cross-study comparisons are hampered by differences in analytical approaches. Nevertheless, some consistent associations have emerged between populations, most notably between Bifidobacteria and the lactase non-persister genotype. These early successes open the way for the microbiome to be incorporated into studies that quantify interactions among genotype, environment, and the microbiome for predicting disease susceptibility.
Keywords: microbiome, GWAS, heritability, microbiota composition, human studies
Introduction
Microbiota coat the body’s surfaces. In the gut, microbial cells reach densities of 1012 per mL and in aggregate form a mass of up to a kilogram, constituting what amounts to an additional organ whose genome vastly expands the host’s in size and metabolic function. The host’s profound dependence on the microbiome for both establishment and maintenance of a normal phenotype is illustrated most vividly by comparisons of animals raised with and without a microbiome. Germfree animals, which are born, raised, and maintained aseptically, are devoid of microbial cells and therefore lack the many cues expected and necessary for their postnatal development and subsequent normal functioning. The abnormalities of germfree animals range across organ systems, from the immune to the cardiovascular system, and include basic functions such as lipid cycling, energy balance, and behavior (57). Many of the unusual phenotypes exhibited by germfree animals reverse upon colonization with a microbiome (27), yet others require microbial exposure at critical points in the animal’s development (1, 24, 52, 59). Given the clear importance of the microbiome for host function, a key goal is to understand the factors that determine colonization and abundance of commensal microbes. The host’s microbiome is acquired at birth and during life through contact with microbes in the environment, so unsurprisingly, environmental factors strongly influence its composition. The microbiome can contribute to fitness and illness (6, 22, 26, 71), therefore the host has a strong interest in shaping the microbiome in such a way to promote its own fitness.
As part of their partnership in the symbiosis, the microbiota perform functions beneficial to the host, from enhancing digestion to protection from invasion of pathogens. Natural selection acts on individual bacterial species to enhance their fitness, and to improve the function of the microbiota as a stable community. Selection pressure on the host itself can also result in selection of microbiota that perform functions beneficial to the host (39, 49). If members of the microbiota enhance host fitness (49), this could have the effect of ensuring the presence of host habitat for these microbiota over the longer term. Indeed, microbiota allow their hosts to exploit specific niches, for instance through detoxification of plant secondary compounds. As an example, goats can eat Leucaena when they harbor bacteria able to degrade 3,4-DHP (33). A mammal host may in turn exhibit behavioral or other traits to ensure the beneficial microbiota transfer to the next generation (49). Mechanisms for selecting, retaining and transferring key elements of the microbiome are likely to be genetically encoded by the host, and the discovery of these genetic factors will point to mechanisms underlying host-microbial symbioses.
One way to uncover potentially new host-microbe interactions is to search for genes with alleles that co-vary across a population with traits in the microbiome. It is likely that human alleles critical in maintaining essential microbial functions have gone to fixation. Indeed, the genetic underpinnings of human gut physiology and function that help maintain the microbial habitat may not present much variation that can be associated with differences in the microbiome across a population. However, microbiota and/or functions beneficial only in a specific context may show a signal of association with human genetic variation. For example, the strongest evidence of recent selection on the human genome is seen in geographically restricted areas that presented specific environmental challenges, such as high altitude, high pathogen load, and high toxicity, among others (54). It is likely that genetic evidence for selection on attributes of the microbiome may also be linked to specific challenges that humans have faced in recent evolution.
Indeed, one of the strongest signals of recent selection on humans consists of the genetic changes that enabled lactase persistence in adulthood and thereby the drinking of non-human milk. Remarkably, the most consistent signal to emerge from GWAS of the microbiome is related – it consists of an association between host genotype, milk consumption, and Bifidobacteria. In this instance, Bifidobacteria are more highly abundant in the gut microbiome of hosts who ingest milk post-weaning and that lack the lactase-persister genotype (see below for an expanded discussion of this association). Other examples of human genetic variation associating with variation in the microbiome are starting to emerge (Table 1 and Figure 1). Here, we review the recent findings from human-microbiome heritability and GWAS studies and the challenges emerging from the marriage of microbiome and human genetics.
Table 1.
Summary of human microbiome genome-wide association studies
| Reference | Body site | Sample size | Subject cohort | Subject population | Data type | Microbiome attributes examined | Number of variants |
Significance threshold |
|---|---|---|---|---|---|---|---|---|
| Blekhman et al. | 15 sites within the oral and nasal cavities, gastrointestinal tract, and on skin1 | 93 | Human Microbiome Project | United States | 16S | Beta-diversity (first 5 PCs), 615 taxa (genus to phylum) | 33,814 | Beta-diversity pathway-based analysis: P ≤ 10−6 Taxa: Genome- wide Q-value < 0.1 |
|
Main findings: Beta-diversity: enrichment of genes involved in leptin signaling in obesity, several other immunity- related pathways, and KEGG pathway primary bile acid biosynthesis. Taxa: 83 associations including HLA-DRA (Selenomonas in the throat), TLR1 (Lautropia in the tongue dorsum), LCT (Bifidobacterium in the GI tract). | ||||||||
| Davenport et al. | Stool | 127 | North American Hutterites | 16S | Alpha-diversity, 102 taxa (genus to phylum) 2 | 212,153 2 | Genome-wide Q-value < 0.2 |
|
|
Main findings: Significant associations with at least 8 bacterial taxa including Akkermansia with a variant near PLD1. Gene-set enrichment analysis identified enrichment of variants involved in olfactory receptor activity with 5 taxa. Stomach and intestines were identified as candidate tissues where host genetic variation may be acting to influence bacterial abundance of genus Faecalibacterium. | ||||||||
| Hua et al. | Lung | 147 | Environment And Genetics in Lung cancer Etiology | Italy | 16S | Beta diversity, alpha diversity, bacterial taxa (number not specified) | 383,263 | P < 5 × 10−8 |
|
Main findings: No significant associations after correcting for skewness and kurtosis of beta-diversity distributions. | ||||||||
| Goodrich et al. | Stool | 2,139 | TwinsUK | United Kingdom | 16S | Beta diversity, alpha diversity, 782 OTUs, 163 taxa (genus to phylum) | 1,300,091 | Genome-wide BH adjusted P- value < 0.1 |
|
Main findings: 92 significant associations with OTUs and taxa. Includes an association between the heritable taxon Unclassified Clostridiaceae with SLIT3 and Bifidobacterium with LCT. Beta diversity was associated with UHRF2. Association between Akkermansia and predicted tissue-specific expression of SIGLEC15 | ||||||||
| Bonder et al. | Stool | 1,514 Discovery: 984 Replication: 530 |
Discovery: LifeLines- Deep Replication: 500FG and MIBS-CO |
Netherlands | Meta- genomics | 219 microbial taxa, 636 MetaCyc pathways, 661 Gene Ontology (GO) |
8.1 million | Discovery: P < 5 × 10−5 Replication: P < 0.01 same direction Meta-analysis: P < 5 × 10−8 |
|
Main findings: Meta-analysis results: 9 loci associated to bacteria, 21 loci associated to MetaCyc and 12 loci associated to GO terms Strongest association with taxa: Blautia with SNPs near LINGO2 and Methanobacteriaceae with an extended long noncoding RNA (lncRNA). Strongest association with MetaCyc pathways: pathway involved in plant- derived steroid degradation with SORCS2 and SLIT3. Two GO terms were associated with SNPs near clusters of C-type lectin domain family 4 genes. GG genotype at SNP rs4988235 (near LCT gene) was associated with high Bifidobacterium. | ||||||||
| Turpin et al. | Stool | 1,561 Discovery: 1,098 Replication: 463 |
Genetic Environmental Microbial (GEM) Project | Discovery: Canada and United States Replication: Canada, United States, and Israel |
16S | Alpha diversity, 166 taxa (genus to phylum) | 3,727,707 | Genome-wide P < 5 × 10−8 Study-wise correcting for number of effective tests: P < 4.13 × 10−10 |
|
Main findings: 58 genome-wide significant associations with taxa, 6 of which achieved study-wise significance. 4 loci were replicated: Rikenellaceae with SNPs near UBR3, Faecalibacterium with CNTN6, Lachnospira with DMRTB1, and Eubacterium with SALL3. | ||||||||
| Wang et al. | Stool | Discovery: 1,812 Replication: 371 |
PopGen and FoCus Replication: FoCus obesity |
Germany | 16S | Beta diversity, 40 OTUs, 58 taxa (genus to phylum) | 6,344,846 | P < 5 × 10−8 |
|
Main findings: 42 loci associated with beta diversity including variants in the VDR gene. 21 of the 42 beta-diversity associated loci replicate. 54 significant associations with taxa. Unclassified Porphyromonadaceae was associated with SLC2A9. Several lncRNA including LINC01192 with Lactobacillales. | ||||||||
| Igartua et al. | Nasopharynx Nasal vestibule |
144 | North American Hutterites | 16S | nasal vestibule: 76 genera nasopharynx: 90 genera 2 | 148,653 2 | Genome-wide Q-value < 0.05 |
|
|
Main findings: 37 associations with genera, these genetic loci are enriched for genes in mucosal immunity pathways. Associations included Dermacoccus with a SNP near TINCR and an unclassified genus of family Micrococcaceae with PGLYRP4. | ||||||||
HMP specific sites tested: Attached keratinized gingiva (gums), Buccal mucosa (cheek), Hard palate, Palatine tonsils, Saliva, Subgingival plaque, Supragingival plaque, Throat, Tongue dorsum, Anterior nares (nostrils), Left and right antecubital fossa (inner elbow), Left and right retroauricular crease (behind the ear), and Stool.
Number reported is from the seasons combined analysis.
Figure 1. Heritability and genome-wide association studies of the human gut microbiome.
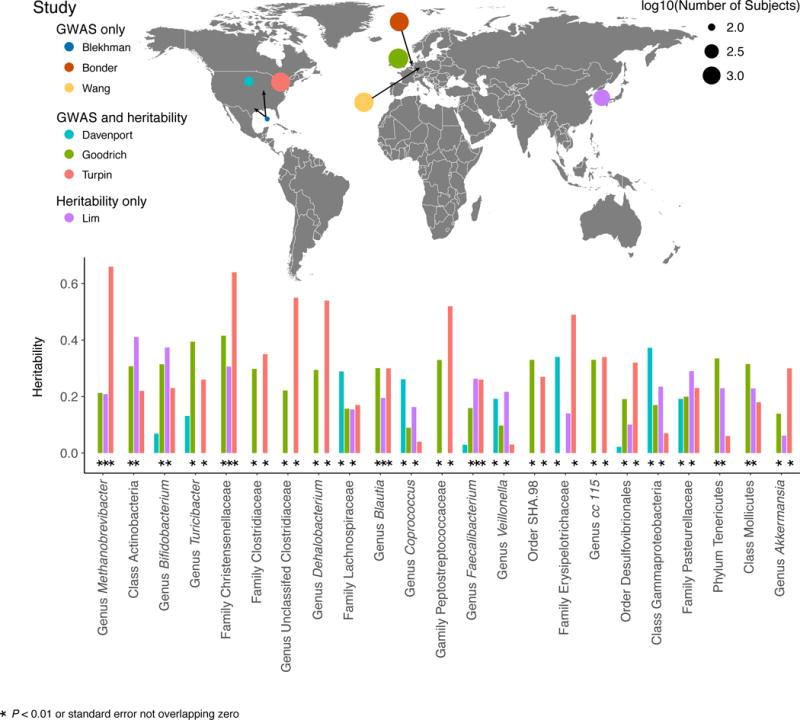
The top panel shows a world map indicating the locations and relative sample size of the currently published human gut microbiome heritability and GWA studies. Each colored circle represents a single study and the size of the circle indicates the study’s sample size. The bottom panel is a comparison of taxon heritability across studies. Only taxa found to have nominally significant heritability estimates (P < 0.05 or in the case of Davenport et al. ‘chip heritability’ estimates with a standard error not overlapping 0) in at least two of the four heritability studies are shown in the bar chart (bars are colored by study).
The microbiome as a complex trait in human genetics
The microbiome is a complex community of organisms and many attributes can be modeled in studies that examine the role of host genetics. Typically the microbiota are characterized either with 16S rRNA gene sequencing or through metagenome sequencing (20). These datasets allow the quantification of taxa or gene functions across samples and can also form the basis of various ecological metrics that characterize diversity in a sample or within a population. The microbiota in the human gut, for instance, consists of hundreds of operational taxonomic units (OTUs) per individual, with tens of thousands of OTUs represented across a population. These OTUs can be collapsed into higher taxonomic levels along their phylogeny (e.g., genus, family, order, etc.). In addition to considering specific taxa in the gut, host genetics may influence the total number and evenness of the taxa present (alpha-diversity). Meanwhile, genetic effects could become apparent when considering the extent of OTU sharing between individuals of varying relatedness (beta-diversity). Beyond taking a census approach to determine which microbes are present, shotgun sequencing can be used to characterize the functional metagenomic landscape of the microbiome. Microbial genes can be grouped into functional categories or pathways, and the abundances or presence/absence of those groups could be targets of modulation through host genetics. Any and all of these attributes can be characterized and modeled as a quantitative trait, for which heritability can be estimated and quantitative trait loci (QTLs) identified. Each of these microbiome attributes should be considered because it is unclear a priori how the host genome might influence the microbiome.
Heritable taxa of the human gut microbiome are increasingly validated across studies
The identification of heritable taxa from comparisons of related individuals preceded GWAS for two reasons: (i) to motivate GWAS, ensuring there was a genetic component determining microbiome composition, and (ii) to reduce the number of traits ascertained in a given GWAS by constraining it to the heritable list. The first unbiased search for heritable taxa among the human gut microbiota was conducted by Goodrich and colleagues, who studied genotyped twins from the TwinsUK registry. Stool samples were obtained from over 1,000 twin pairs (3,261 samples total) and the modeled data consisted of 16S rRNA gene sequences (18, 21). Genotyped twins allowed for both twin-based heritability analysis and GWA to identify host genes and metabolic pathways associated with these heritable taxa. Heritability analysis revealed that ~10% of the 945 taxa identified by 16S and shared by a minimum of 50% of the subjects had a heritability greater than 0.2 with 95% confidence intervals that did not overlap zero. Remarkably, of the 26 heritable taxa reported by Goodrich et al. (2014), 13 were nominally replicated in the Canadian Genetic Environmental Microbial (GEM) Project cohort (Figure 1; 270 related individuals from 123 families) (65). Six of the 26 could not be addressed by Turpin et al. for technical reasons, which implies that more than half of the heritable taxa that could be compared were heritable in a second population. Given that the human gut microbiome is variable across subjects and highly influenced by environmental factors such as diet, this congruence between studies argues strongly that specific, identifiable taxa are responsive to host genotype across populations and warrant a mechanistic follow-up.
For both the Goodrich 2014 and the Turpin 2016 studies, the list of heritable taxa that pass significance testing is a subset of the nominally heritable list. The nominally heritable list is generally assumed to be more likely to contain false positives and is usually not discussed in the main findings of the reports. However, an expansion of the population size and re-analysis of heritability in the UK twins showed that the list of nominally heritable taxa might be quite valuable. In the study of UK twins, the list of heritable taxa was generated twice, first with 416 twin pairs (21) and then again with an expanded set of 1,126 twin pairs (18). The tripling of the dataset revealed the following: (i) the list of taxa stayed constant, with minimal reshuffling of the heritability rankings, and (ii) the confidence intervals around the heritability estimates were reduced. This had the effect that taxa formerly excluded from the heritable list due to confidence intervals overlapping with zero were considered heritable through the expanded analysis. Furthermore, these observations underscore that the types of heritable taxa are not dependent on the specific set of individuals studied within the population. These results also demonstrate the expected increase in power to detect heritable microbes with larger sample sizes and the gain of confidence in the results from smaller sample sets. Small-scale analyses may therefore yield valuable insights into the heritability of taxa even when underpowered, and the nominally heritable taxa may be interesting to pursue further.
Most estimates of microbial abundance stem from 16S rRNA gene sequence data, which provides phylogenetic information but generally little functional information. Furthermore, many taxa are functionally redundant in the gut, and this is thought to contribute to the stability of the system (loss of a taxon does not lead to loss of function if the function is widely shared). So it is interesting that some taxa are indeed heritable, and this observation implies that (i) some attribute of the taxa is under selection, and (ii) that attribute is phylogenetically restricted.
Overall, heritability estimates of components of the gut microbiota are generally low compared to other heritable traits (53). Heritability estimates calculated for the UK twin fecal microbiome ranged from 0 to ~0.40 (18, 21). Heritability estimates have also been obtained using Korean twins and their families (Lim et al., range of 0–0.46), Canadian families consisting of mostly siblings (Turpin et al., range of 0-0.67), and the Hutterites (Davenport et al. seasons combined ‘chip-heritability’, range of 0-0.37). The low values of the heritability estimates may be linked to the fact that the data are derived from stool, which is a mix of mucosal and luminal contents. If a heritable microbe can be quantified in its original habitat (e.g., the mucosal surface), true heritability estimates may be higher. The low heritability should be considered not just a first pass but also a worst-case scenario, because more focused studies are bound to yield higher values.
Christensenellaceae
Goodrich et al. reported the most highly heritable taxon to be the family Christensenellaceae (21). Subsequently, the heritability of Christensenellaceae has been validated in Canadians of European descent (0.64) and in Koreans (0.31; Figure 1) (40, 65). Christensenellaceae is a family within Firmicutes that is relatively small (i.e., less branch length compared to a family such as the Ruminococcaceae), which might explain why the whole family is heritable. The heritability estimate for the whole family is driven by taxa that constitute branches of the phylogeny lacking cultured representatives at this time. Christensenella minuta, the first laboratory isolate, which leant its name to the family (47), has lower, non-significant heritability in the TwinsUK dataset (0.27) (21), but was however found to be heritable in the study of Turpin et al. (0.54) (65).
Goodrich et al. reported that the family Christensenellaceae constitutes the hub (i.e., most interconnected node) of a co-occurrence network consortium that includes the families Methanobacteriaceae, Dehalobacteriaceae, SHA-98, RF39 (Tenericutes) and ML615J-28 (Tenericutes), all of which are heritable. This consortium was also present in the data of Yatsunenko et al., derived from young adult twins from Missouri, USA (21, 72).
This Christensenellaceae consortium was, in addition to being heritable, enriched in lean versus obese individuals in the UK twins (21). It also positively correlated with alpha-diversity, which was higher in lean compared to obese subjects in the TwinsUK population. The association of Christensenellaceae with a lean phenotype was also observed in Missouri twins (64), the Dutch LifeLines- DEEP population (17), Koreans (40) and Japanese individuals (51). Christensenellaceae was subsequently linked to visceral fat phenotypes in the TwinsUK cohort (3), as well as with healthy levels of triglycerides in the Dutch LifeLines- DEEP cohort (17). Furthermore, the Christensenellaceae increased in relative abundance in stool of subjects consuming resistant starch, and correlated with levels of specific SCFAs in stool (66).
Taken together, these studies point to an interaction between the Christensenellaceae and its consortium members with diet, lipid metabolism and host adiposity. Goodrich and colleagues tested the causality of the association between a lean host phenotype and the relative abundance of the Christensenellaceae experimentally using fecal transplants into germfree mice. Amendment of an obese microbiome known to be extremely low in Christensenellaceae with live C. minuta cells protected germfree mouse recipients from the levels of adiposity gains observed in controls (i.e., same obese-derived microbiome with no-addition or with heat-killed C. minuta) (21). This finding linked the Christensenellaceae functionally to the lean phenotype and the underlying mechanisms are currently under investigation.
Methanogens
The co-occurrence of Christensenellaceae with methanogens as observed in UK twins was reported by Hansen et al. prior to the renaming of the family (25), and more recently in North Americans (66). Methanogens correlate with leanness in several studies (2, 38, 46, 56). Methanobrevibacter smithii (the dominant human gut methanogen) carriage was first shown to be heritable in Missouri twins (25). Corroborating this early finding, methanogen abundance was shown to be heritable in UK twins using both 16S rRNA data (Goodrich et al. 2016: 0.21) and metagenomic data (Xie et al. 2016: 0.38), as well as in Canadians of European descent (Turpin et al. 2016: 0.66) and in a cohort of Korean twins (Lim et al. 2016: 0.21). In a study of 1,514 individuals using fecal metagenomic data, Bonder et al. showed that methanogen abundance was associated with SNPs located within a long-noncoding RNA (5). Why methanogens are heritable and/or linked to this specific region of the genome remains unclear. Since methanogen abundances are typically correlated with other facets of the microbiome including specific taxa and alpha-diversity (see below), the association with the gene region may be driven by any of these co-occurring taxa, which complicates the task of understanding any mechanisms underlying the association.
Measures of richness
Alpha-diversity, expressed as various measures (e.g., number of observed OTUs, Shannon index, Faith’s Phylogenetic Diversity, etc.) (20), is heritable in at least three populations (10, 18, 65). Although moderate heritability has been observed for some alpha-diversity metrics, none of these studies reported significant associations with genetic variants (10, 18, 65). Alpha-diversity is commonly negatively associated with several chronic inflammatory diseases such as inflammatory bowel disease (IBD) and obesity (48). For example, alpha-diversity (assessed from 16S rRNA gene diversity analysis of fecal samples) has been observed to be lower in patients with Metabolic syndrome compared to controls (38, 40). The reasons why microbiomes exhibit lower alpha-diversity may differ between disease states.
One important factor that shapes the gut microbiome habitat and has been associated with alpha-diversity is gut transit time, which relates to stool consistency. The Bristol Stool Index (BSS) is often used as a proxy for colonic transit time because the two tend to be negatively associated (i.e., lower BSS indicative of longer transit time and harder stool). BSS has been negatively correlated with species richness and with the abundances of Methanobrevibacter and Akkermansia (i.e., these taxa and alpha-diversity are higher in hard stool) (67). In contrast, Tigchelaar et al. did not find a significant correlation between BSS and species richness, however, they did report that decreasing BSS score (i.e., harder stools) was significantly associated with Archaea (i.e., methanogens) and the bacterial families Christensenellaceae and Dehalobacteriaceae (all members of the heritable co-occurring consortium) (62). Roager et al. also reported species richness positively associated with colonic transit time (55). In addition, this group reported that three OTUs belonging to Christensenellaceae and one OTU classified as Methanobrevibacter positively associated with colonic transit time and with protein degradation products. In accord, in a study of Japanese subjects, the Christensenellaceae family negatively associated with bowel movement frequency (51). With longer transit time, microbiota have longer to work on substrates, liberate additional substrates (which can increase niche space and diversity), and potentially slower growing microbiota have the necessary gut retention time to reach measurable levels, all of which could lead to greater richness. Apart from associations between gut transit times and diseased states like cystic fibrosis and IBD, little work has been done on the genetics of gut transit time.
Identifying microbiome-host genotype associations
Attributes of the microbiome that are used as traits in GWAS (Table 1) include both (i) individual level measurements, such as alpha-diversity, relative abundances of specific taxa (OTUs, or taxa counts summarized at various levels of taxonomy), and functional pathways or gene ontology (GO) terms and (ii) cross-sample traits, such as beta-diversity metrics. Alpha-diversity and microbial or functional pathway abundances can simply be treated as individual quantitative traits where standard GWAS methods can be applied to each trait (7). The microbiome GWAS to date have used standard additive genetic modeling approaches (4, 10, 18, 30, 68), rank based correlations (5), or combination models, where common taxa are modeled as quantitative traits and rare taxa are modeled as binary traits (65).
Another avenue being explored for GWAS of microbiome attributes is the use of variable selection methods for high-dimensional data. Recently, Lynch et al. developed a pipeline called HOMINID, which uses a penalized regression method called Lasso (43). This pipeline performs a single regression for each genetic variant with all taxa as predictors. When HOMINID was applied to data from 93 participants in the Human Microbiome Project, six genetic variants remained significant following multiple testing correction. Application of this method to some of the recent microbiome GWAS that include thousands of individuals could be useful to identify more associations.
Association with beta-diversity is more complex because it is a measure of similarity or dissimilarity between two samples, resulting in a value for each pair of individuals. In the first GWAS of beta-diversity, Blekhman et al. performed principal coordinates analysis (PCoA) on the pairwise beta-diversity matrix and ran a GWAS for each of the first five principal coordinates (PCs) (4). This amounts to looking for human genetic variants that are associated with the majority of the microbiome variation in the dataset and allows for a reduction in the dimensionality of the data compared to testing the association of each taxon with all genetic variants. Recently, Wang et al. used the function ‘envfit’ in the ‘vegan’ R package to fit each genotype onto the main axes of the beta-diversity PCoA (by default the first two PCs) (68). In this method, genotype was treated as a categorical variable and SNPs associated with community composition are identified by determining if the centroids for the three genotypes (with respect to the main axes of the PCoA) are significantly different. Hua et al. developed a tool called microbiomeGWAS for associating beta-diversity with each genetic variant (28). MicrobiomeGWAS is based on the intuition that if a variant is associated with the microbiome, any two individuals with more alleles in common at a given locus (e.g., individuals have two alleles in common if both individuals are AA, and none in common if one is AA and the other is GG) will have more similar microbiota and therefore smaller beta-diversity distances.
Statistical challenges of microbiome GWAS
Treating the microbiome as a complex trait in genome-wide association studies is relatively new, and published studies have had small sample sizes (in the low thousands) by GWAS standards (hundreds of thousands). As such, it has been challenging for associations of specific alleles with microbiome traits to reach study-wide significance due to the burden of multiple testing in these small studies. Indeed, study-wide significance is a high bar when the number of tests is based on the total number of SNPs (hundreds of thousands to millions) combined with the total number of traits (typically in the high-hundreds to thousands).
There are a number of ways researchers reduce the number of tests performed in order to mitigate the large multiple testing burden. Some studies begin by focusing their analysis on metrics of overall community composition (alpha- and beta- diversity). Although this is an important initial step, its main limitation is that it does not provide information about which specific microbes are influenced by host genetics. Additionally, given the significant impact of environmental factors on the microbiome, any signal from an association with only a subset of the community will likely be drowned out when examining the community as a whole.
Of the thousands of OTUs inhabiting the gut, relatively few are shared among all or most individuals in a population (29). The presence of large numbers of rare taxa lead to the problem of zero-inflation when attempting to model all taxa in the gut (70). Most of the previously published studies reduced the number of tests by excluding taxa/functions with low abundance or prevalence, limiting the traits of interest to those that are more widely shared in the population. This strategy mitigates both the issues relating to modeling zero-inflated data, for which there would be low power to detect associations, and reduces the multiple testing burden. Even after this filtering, the number of traits that remain is typically in the hundreds, and performing an association for each taxon/function with each genetic variant results in a very large multiple testing burden. A strict Bonferroni correction for multiple testing would require studies to reach P values of 5 × 10−10 to 5 × 10−11.
In addition to filtering microbiome attributes, the number of tests can also be reduced by restricting which host genetic variants are examined. Constraining the SNPs tested to candidate gene sets is one strategy (19, 35). The drawback of this approach is that relevant genes not on the candidate gene list may be missed. Alternatively, others limit testing to SNPs only in genic regions (4). While this approach focuses on functional regions of the genome, it likely misses much of the signal, as human GWAS hits are often identified in intergenic regions thought to be regulatory in nature (12).
Replication cohorts can be used within a study to provide confidence in suggestive associations that do not pass a strict study-wide significance threshold in the discovery cohort (5, 65, 68). For instance, Bonder et al. used a three-step approach where all associations meeting a relaxed significance threshold (P < 5 × 10−5) in a discovery cohort were then examined in an independent cohort. If an association replicated in the independent cohort (same direction of association and a P < 0.01) it was tested in a meta-analysis using both the discovery and replication cohorts. The estimated study-wide FDR for associations that passed the meta-analysis significance (P = 5 × 10−8) was 12%.
Despite the challenges in modeling the microbiome with a true GWAS, there is immense value in reporting the results of an unbiased discovery approach. Results from separate studies can then be compared with each other, to identify taxa that are reproducibly associated with variants in the genome (9, 19, 68). This brings to light the critical importance of validating suggestive associations across studies until larger more powerful studies reduce the numbers of false positives.
Bifidobacterium and human lactase persistence
The most consistent signal to have emerged from human gut microbiome GWAS to date is the association between Bifidobacterium in the fecal microbiota and SNPs near the LCT gene on chromosome 2, first reported by Blekhman and colleagues in the Human Microbiome Project subjects (Blekhman et al., 2015). Remarkably, this association has since been replicated in twins from the United Kingdom (18), the North American Hutterites (18), a Dutch cohort (5), and individuals from northern Germany (68). The CC genotype of the SNP rs4988235 at this locus is associated with lactase non-persistence (13, 63) and elevated abundance of Bifidobacteria compared to the TT or TC genotypes. Bifidobacteria can utilize lactose, the milk sugar, as an energy source. These observations led Goodrich et al. to suggest that Bifidobacteria break down lactose and increase in abundance in hosts who are lactase non-persistent yet nevertheless consume lactose (19). This scenario implied a host genotype by diet interaction. This prediction was verified by Bonder et al. who observed the same association of the non-persister genotype with Bifidobacteria in 1,514 samples derived from 3 cohorts, and where the microbiome was characterized by shotgun metagenomics (5). Bonder et al. also had dietary information on the subjects, and observed the association only in those consuming milk. The exact nature of the association between Bifidobacterium, lactose in the diet and the non-persister genotype still requires experimental confirmation. More work is needed to decipher which species and strains of Bifidobacteria are implicated in this association. It is possible that the presence of the Bifidobacteria confers a degree of lactose tolerance to lactase-non-persisters.
To date, the lactase persistence genotype and Bifidobacteria association has been detected in persons of European descent only. Persons of African descent may exhibit independently acquired lactase persistence by their ancestors via a different genetic mechanism (63). However, it is intriguing to note that the phenotype (lactose tolerance or intolerance) predicted by the genotype is not always accurate (54), indicating that the microbiome may be mediating the phenotype. An association with Bifidobacteria may be expected in African populations as well, or it could be that the African equivalent occurs through lactase activity of a different gut microbe.
GWAS reveal tissues, pathways, and genes consistently associated with microbiome attributes
In addition to the replication of the Bifidobacterium - LCT association across multiple studies of the gut microbiome, a number of other broad trends emerge from cross-study comparisons of microbiome GWAS. First, select host tissues and pathways have been implicated across studies. Additionally, specific human genes repeatedly associate with the microbiome, although the corresponding taxa vary between studies. Finally, multiple lines of evidence point to human genetic influence on the abundance of distinct microbial pathways and functions.
Host tissues and pathways implicated
The most consistent finding between reports to date is that the regions containing variants associated with the microbiome are enriched for genes related to immunity. Through a pathway enrichment analysis, Blekhman et al. found enrichment for genes involved in the following immunity-related pathways: Leptin Signaling in Obesity, Melatonin Signaling, JAK/Stat Signaling, Chemokine Signaling, CXCR4 Signaling, and Role of Pattern Recognition Receptors in Recognition of Bacteria and Viruses (4). Genes associated with the microbiome of nasal, oral, and skin body sites drove most of the enrichment in these pathways. In a study of the nasal microbiota in a Hutterite population, Igartua et al. used the Ingenuity Pathway Analysis Knowledge Base to identify protein-protein interaction (PPI) networks from genes near nasal microbiome associated loci (30). Both of the significant PPI networks identified contain highly connected proteins (hubs in the PPI network) that play important roles in modulating mucosal immunity (including IgA, IgG, IL12/IL12RA, TCR and STAT5A/B).
Immune-related genes are also implicated in many of the gut microbiome GWAS. A targeted gene analysis in Bonder et al. revealed several significant associations of microbial and functional abundances with immune response genes (5). Their strongest signal in the targeted analysis was between the GO2000 term ‘cell–cell signaling’ and a SNP in the C11orf30–LRRC32 locus, which has been associated with multiple immune-related phenotypes. Other associations include genes implicated in IBD risk (CCL2, DAP2, IL23R), nucleotide-binding oligomerization domain genes NOD1 and NOD2, two CLEC loci, and two genetic variants in the MHC region. Additionally, the most significant association in Turpin et al. that was also validated in their replication cohort was between the abundance of the family Rikenellaceae and a locus containing the gene UBR3, which encodes for a protein involved in the protein ubiquitination pathway (65). The authors note that ubiquitination plays many crucial roles in the immune system.
In the gut microbiome studies, evidence suggests genetic variation may act in digestive tract tissues to affect microbiome composition. For instance, Wang et al. reported enrichment for genes expressed in the digestive tract (68). In the North American Hutterites, genetic variants associated with Faecalibacterium were enriched in DNase hypersensitivity sites (DHS) of intestine and stomach tissues (10). Goodrich et al. did not perform an enrichment analysis to identify candidate tissues, but when searching for an association between taxa and predicted gene expression across tissues, the only significant associations were with expression in the transverse colon (18).
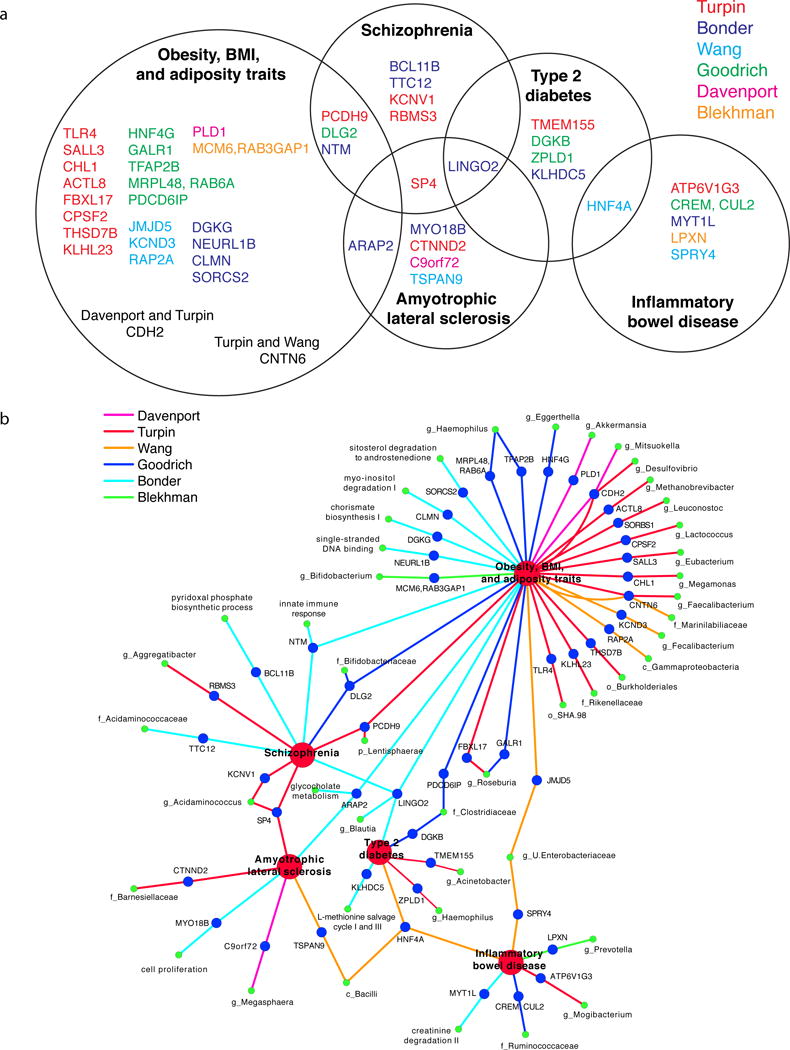
Many microbiome GWAS studies report genes associated with several of the same complex diseases. This includes IBD (4, 5, 65, 68), obesity (4, 5, 10, 65, 68) and type 2 diabetes (5, 65, 68), all of which are also associated with alterations in the gut microbiota (16, 37, 64). IBD risk genes are also repeatedly linked to gut microbiota composition in targeted association analyses (5, 15, 31, 35). Genetic variants near the genes PLD1 and LINGO2, which have been implicated in obesity GWAS (42, 50), associated with the abundance of Akkermansia (10) and Blautia (5) respectively. Both Akkermansia and Blautia have been linked to obesity related phenotypes (3, 14, 21). The overlap observed between genetic variants associated with both microbiome attributes and complex diseases motivates further investigation to better understand how human genetic variation impacts the microbiome in the context of these diseases.
Specific human genes and proteins implicated
The gene SLIT3 has been reported by three studies as having an association with some aspect of the microbiome. The most significant microbial pathway association observed by Bonder et al. was between SLIT3 and the sitosterol degradation to androstenedione pathway (involved in plant-derived steroid degradation) (5). Goodrich et al. also found an association with a variant in this gene and the abundance of unclassified Clostridiaceae (Heritability: 0.32) (18). The nasal microbiome GWAS performed by Igartua et al. identified a significant association between the abundance of Dermacoccus in the nasal vestibule and another variant in this gene (30). SLIT3 is a secreted protein expressed in several tissues including skin, stomach, small intestine, and colon (11). Hypermethylation at the SLIT3 5′ CpG island occurs in colorectal cancers (11). SLIT3 likely plays a role in inflammation: the expression of SLIT3 increases after LPS stimulation of mouse macrophages (60), and this gene has been associated with BMI in a GWAS for obesity (41).
Another emerging theme from the human microbiome GWAS is a link between host genetics, the microbiome and bile acids. One of the strongest signals of association with overall community composition reported by Wang et al. was with SNPs located in the gene that encodes for the vitamin D receptor (VDR). Further exploration into this association revealed that Parabacteroides was the taxon most highly associated with VDR and that Parabacteroides abundance was also significantly higher in VDR knockout mice compared to wild-type (68). VDR is a known receptor for secondary bile acids (45) and activation of VDR can inhibit bile acid synthesis (23). This led Wang et al. to profile serum bile acids: they reported significant correlations between the bile acid measurements, gut microbiome composition, and genetic variation at VDR as well as other loci. Interestingly, Blekhman et al. observed an enrichment of microbiome associated genes in the Primary Bile Acid Biosynthesis KEGG pathway (4). In addition, Xie et al. reported that the abundance of bile salt hydrolase genes was significantly heritable in UK Twins (Heritability = 0.29) (69), and Bonder et al. identified an association between the MetaCyc bacterial bile acid metabolism pathway and SNPs in the ARAP2 gene (5). All of these studies support an interaction between host genetics and the microbiome through regulation of bile acid metabolism.
One last example suggests a link for transit time (described above), a specific gene family and members of the gut microbiome. Jankipersadsing et al. conducted a GWAS using stool frequency as a trait in the LifeLines-Deep (LLD) population (n=1,546) and reported that the second strongest association is with the gene ALDH1A1 (32). The authors noted this gene’s role in xenobiotic metabolism. Goodrich et al. noted an association between another member of the aldehyde dehydrogenases gene family (ALDH1AL1) and SHA-98, a member of the heritable Christensenellaceae consortium that includes the methanogens, and noted this gene’s role in C1 metabolism. Whether and how these findings may be related remains to be clarified.
Microbial pathways implicated
To date, only Bonder et al. have used shotgun metagenomics to investigate the relationship between microbial pathways and host genetic variation genome-wide (5), preventing a cross-study comparison of microbial pathways that are associated with genetic variants. However, Xie et al. recently reported estimates of heritability for gut microbial pathways using 127 twin pairs from the TwinsUK cohort (69). As a result, it is possible to search for overlap between the significantly heritable pathways reported by Xie et al., and the pathways with a genetic association reported in Bonder et al. In addition to the bile acid metabolism example mentioned above, both studies suggested host genetics could have some influence on the abundance of microbial genes involved in riboflavin biosynthesis (Heritability = 0.51). Humans acquire riboflavin (vitamin B2) both through their diet and from riboflavin-producing gut microbes. The machinery required for riboflavin synthesis has been found in the genomes of most Bacteroidetes, Fusobacteria, and Proteobacteria examined, while a complete riboflavin operon was only present in about half of the Firmicutes and almost no Actinobacteria (44, 61). Riboflavin can be used as a redox mediator by Faecalibacterium prausnitzii to facilitate extracellular electron transfer, which consequently promotes its growth (34). Increased riboflavin metabolism has been observed in individuals with ulcerative colitis (36), while F. prausnitzii abundance is reduced in individuals with inflammatory bowel disease (58). Riboflavin biosynthesis was correlated with SNPs near the gene CLEC4A, which encodes a C-type lectin (5). Members of this gene family have a wide range of functions including important roles in inflammation and immunity (8). As more studies use metagenomics to investigate which microbial genes and functions are influenced by human genetics, comparisons across studies will be important to validate these initial findings.
Current difficulties when comparing across studies
Although common themes emerge when comparing GWAS results across studies providing insight into the genetic factors that influence the human microbiome, direct validation of specific associations is largely still lacking. Each study curates the uncovered associations and chooses to highlight only a subset, making comparisons across studies based on the findings mentioned in the text problematic. An association highlighted in one study may be present in another, but if it falls just under the significance threshold, it might not be reported. For example, the Bifidobacterium - LCT finding does not reach genome-wide significance in most studies, and it likely would not have been reported as a main finding without Blekhman et al. previously pointing it out. However, when specifically targeting the association between LCT and Bifidobacterium, most of the published studies observed this association. This is again related to the power issues in untargeted studies that make it difficult to differentiate between false positives and real signals.
There may be several more cases like the LCT example that are overlooked when comparing just the highlighted results of the reported findings. Additionally, any differences in the data analysis pipeline, for example in the filtering of less abundant and less prevalent taxa, makes comparisons across studies extremely difficult. To properly compare across studies requires all data to be analyzed in the same way, which implies a laborious re-processing of all datasets. Aspects of the analysis that are important to standardize include the method for OTU picking, the database and algorithm for taxonomy classification, cutoffs for taxa inclusion, the transformation method used on the microbiome data (if any), and the test for association (e.g., recent studies have used linear/logistic mixed models, negative binomial generalized linear models, log-normal generalized estimating equation models, and rank-based Spearman correlations). Only after this standardization can there be a more reliable comparison of associations across all studies for the same taxon × SNP pairs.
Prospectus
The microbiome is a complex trait. Initial forays into the identification of genes that co-vary with aspects of the microbiome are promising and have highlighted the role of immunity and diet in shaping the microbiome, although more direct comparisons between studies where all data are similarly processed are needed to improve the comparisons. Heritable taxa are so far remarkably consistent across studies, and many are health-associated. How the microbiome interacts with genotype to influence disease phenotype is an open frontier. Acquisition of SNP genotype information on common gut microbes based on deep metagenomic data will open opportunities to examine the evolutionary tuning of the microbiome to the human gut. Larger studies, across multiple populations, and in the context of disease susceptibility, should continue to shed light on human-microbiome interaction and co-evolution.
Figure 2.
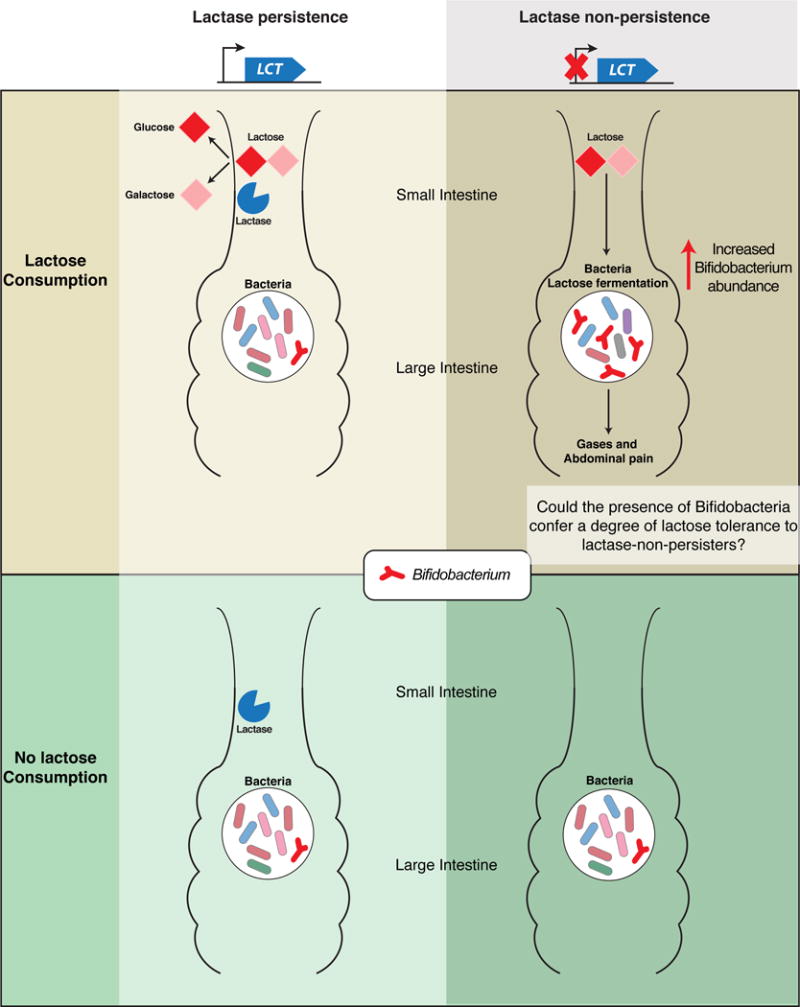
Figure 3.
Acknowledgments
Acknowledgements: We thank Angela Poole for comments on the manuscript. Support was provided by the Max Planck Society and by NIH grant R01 DK093595. ERD is funded by NIH F32 DK109595.
Literature Cited
- 1.An D, Oh SF, Olszak T, Neves JF, Avci FY, et al. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer t cells. Cell. 2014;156(1–2):123–33. doi: 10.1016/j.cell.2013.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armougom F, Henry M, Vialettes B, Raccah D, Raoult D. Monitoring bacterial community of human gut microbiota reveals an increase in lactobacillus in obese patients and methanogens in anorexic patients. PLoS One. 2009;4(9):e7125. doi: 10.1371/journal.pone.0007125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beaumont M, Goodrich JK, Jackson MA, Yet I, Davenport ER, et al. Heritable components of the human fecal microbiome are associated with visceral fat. Genome Biol. 2016;17(1):189. doi: 10.1186/s13059-016-1052-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blekhman R, Goodrich JK, Huang K, Sun Q, Bukowski R, et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015;16:191. doi: 10.1186/s13059-015-0759-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonder MJ, Kurilshikov A, Tigchelaar EF, Mujagic Z, Imhann F, et al. The effect of host genetics on the gut microbiome. Nat Genet. 2016;48(11):1407–12. doi: 10.1038/ng.3663. [DOI] [PubMed] [Google Scholar]
- 6.Budden KF, Gellatly SL, Wood DLA, Cooper MA, Morrison M, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol. 2017;15(1):55–63. doi: 10.1038/nrmicro.2016.142. [DOI] [PubMed] [Google Scholar]
- 7.Bush WS, Moore JH. Chapter 11: genome-wide association studies. PLoS Comput Biol. 2012;8(12):e1002822. doi: 10.1371/journal.pcbi.1002822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dambuza IM, Brown GD. C-type lectins in immunity: recent developments. Curr Opin Immunol. 2015;32:21–27. doi: 10.1016/j.coi.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davenport ER. Elucidating the role of the host genome in shaping microbiome composition. Gut Microbes. 2016:00–00. doi: 10.1080/19490976.2016.1155022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davenport ER, Cusanovich DA, Michelini K, Barreiro LB, Ober C, Gilad Y. Genome-wide association studies of the human gut microbiota. PLoS One. 2015;10(11):e0140301. doi: 10.1371/journal.pone.0140301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dickinson RE, Dallol A, Bieche I, Krex D, Morton D, et al. Epigenetic inactivation of slit3 and slit1 genes in human cancers. Br J Cancer. 2004;91(12):2071–78. doi: 10.1038/sj.bjc.6602222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards SL, Beesley J, French JD, Dunning AM. Beyond gwass: illuminating the dark road from association to function. Am J Hum Genet. 2013;93(5):779–97. doi: 10.1016/j.ajhg.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enattah NS, Sahi T, Savilahti E, Terwilliger JD, Peltonen L, Järvelä I. Identification of a variant associated with adult-type hypolactasia. Nat Genet. 2002;30(2):233–37. doi: 10.1038/ng826. [DOI] [PubMed] [Google Scholar]
- 14.Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, et al. Cross-talk between akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. 2013;110(22):9066–71. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17(1):179–84. doi: 10.1002/ibd.21339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104(34):13780–85. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu J, Bonder MJ, Cenit MC, Tigchelaar EF, Maatman A, et al. The gut microbiome contributes to a substantial proportion of the variation in blood lipids. Circ Res. 2015;117(9):817–24. doi: 10.1161/CIRCRESAHA.115.306807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodrich JK, Davenport ER, Beaumont M, Jackson MA, Knight R, et al. Genetic determinants of the gut microbiome in uk twins. Cell Host Microbe. 2016;19(5):731–43. doi: 10.1016/j.chom.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodrich JK, Davenport ER, Waters JL, Clark AG, Ley RE. Cross-species comparisons of host genetic associations with the microbiome. Science. 2016;352(6285):532–35. doi: 10.1126/science.aad9379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodrich JK, Di Rienzi SC, Poole AC, Koren O, Walters WA, et al. Conducting a microbiome study. Cell. 2014;158(2):250–62. doi: 10.1016/j.cell.2014.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, et al. Human genetics shape the gut microbiome. Cell. 2014;159(4):789–99. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hand TW, Vujkovic-Cvijin I, Ridaura VK, Belkaid Y. Linking the microbiota, chronic disease, and the immune system. Trends Endocrinol Metab. 2016;27(12):831–43. doi: 10.1016/j.tem.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han S, Chiang JYL. Mechanism of vitamin d receptor inhibition of cholesterol 7α-hydroxylase gene transcription in human hepatocytes. Drug Metab Dispos. 2009;37(3):469–78. doi: 10.1124/dmd.108.025155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen CHF, Nielsen DS, Kverka M, Zakostelska Z, Klimesova K, et al. Patterns of early gut colonization shape future immune responses of the host. PLoS One. 2012;7(3):e34043. doi: 10.1371/journal.pone.0034043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hansen EE, Lozupone CA, Rey FE, Wu M, Guruge JL, et al. Pan-genome of the dominant human gut-associated archaeon, methanobrevibacter smithii, studied in twins. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4599–4606. doi: 10.1073/pnas.1000071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. 2016;535(7610):75–84. doi: 10.1038/nature18848. [DOI] [PubMed] [Google Scholar]
- 27.Hooper LV. Bacterial contributions to mammalian gut development. Trends Microbiol. 2004;12(3):129–34. doi: 10.1016/j.tim.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 28.Hua X, Song L, Yu G, Goedert JJ, Abnet CC, et al. MicrobiomeGWAS: a tool for identifying host genetic variants associated with microbiome composition. Work Pap. 2015 doi: 10.3390/genes13071224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huse SM, Ye Y, Zhou Y, Fodor AA. A core human microbiome as viewed through 16s rrna sequence clusters. PLoS One. 2012;7(6):e34242. doi: 10.1371/journal.pone.0034242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Igartua C, Davenport ER, Gilad Y, Nicolae DL, Pinto J, Ober C. Host genetic variation in mucosal immunity pathways influences the upper airway microbiome. Microbiome. 2017;5(1):16. doi: 10.1186/s40168-016-0227-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imhann F, Vich Vila A, Bonder MJ, Fu J, Gevers D, et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut. 2016 doi: 10.1136/gutjnl-2016-312135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jankipersadsing SA, Hadizadeh F, Bonder MJ, Tigchelaar EF, Deelen P, et al. A gwas meta-analysis suggests roles for xenobiotic metabolism and ion channel activity in the biology of stool frequency. Gut. 2016 doi: 10.1136/gutjnl-2016-312398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones RJ, Megarrity RG. Successful transfer of dhp-degrading bacteria from hawaiian goats to australian ruminants to overcome the toxicity of leucaena. Aust Vet J. 1986;63(8):259–62. doi: 10.1111/j.1751-0813.1986.tb02990.x. [DOI] [PubMed] [Google Scholar]
- 34.Khan MT, Browne WR, van Dijl JM, Harmsen HJM. How can faecalibacterium prausnitzii employ riboflavin for extracellular electron transfer? Antioxid Redox Signal. 2012;17(10):1433–40. doi: 10.1089/ars.2012.4701. [DOI] [PubMed] [Google Scholar]
- 35.Knights D, Silverberg MS, Weersma RK, Gevers D, Dijkstra G, et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Med. 2014;6(12):107. doi: 10.1186/s13073-014-0107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146(6):1489–99. doi: 10.1053/j.gastro.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Larsen N, Vogensen FK, van den Berg FWJ, Nielsen DS, Andreasen AS, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5(2):e9085. doi: 10.1371/journal.pone.0009085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500(7464):541–46. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 39.Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124(4):837–48. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 40.Lim MY, You HJ, Yoon HS, Kwon B, Lee JY, et al. The effect of heritability and host genetics on the gut microbiota and metabolic syndrome. Gut. 2016 doi: 10.1136/gutjnl-2015-311326. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y-J, Liu X-G, Wang L, Dina C, Yan H, et al. Genome-wide association scans identified ctnnbl1 as a novel gene for obesity. Hum Mol Genet. 2008;17(12):1803–13. doi: 10.1093/hmg/ddn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518(7538):197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lynch J, Tang K, Sands J, Sands M, Tang E, et al. HOMINID: A framework for identifying associations between host genetic variation and microbiome composition. Work Pap. 2016 doi: 10.1093/gigascience/gix107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Magnúsdóttir S, Heinken A, Kutt L, Ravcheev DA, Bauer E, et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat Biotechnol. 2017;35(1):81–89. doi: 10.1038/nbt.3703. [DOI] [PubMed] [Google Scholar]
- 45.Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, et al. Vitamin d receptor as an intestinal bile acid sensor. Science. 2002;296(5571):1313–16. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 46.Million M, Maraninchi M, Henry M, Armougom F, Richet H, et al. Obesity-associated gut microbiota is enriched in lactobacillus reuteri and depleted in bifidobacterium animalis and methanobrevibacter smithii. Int J Obes. 2012;36(6):817–25. doi: 10.1038/ijo.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Morotomi M, Nagai F, Watanabe Y. Description of christensenella minuta gen. nov., sp. nov., isolated from human faeces, which forms a distinct branch in the order clostridiales, and proposal of christensenellaceae fam. nov. Int J Syst Evol Microbiol. 2012;62(1):144–49. doi: 10.1099/ijs.0.026989-0. [DOI] [PubMed] [Google Scholar]
- 48.Mosca A, Leclerc M, Hugot JP. Gut microbiota diversity and human diseases: should we reintroduce key predators in our ecosystem. Front Microbiol. 2016;7:455. doi: 10.3389/fmicb.2016.00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mueller UG, Sachs JL. Engineering microbiomes to improve plant and animal health. Trends Microbiol. 2015;23(10):606–17. doi: 10.1016/j.tim.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 50.Ng MCY, Hester JM, Wing MR, Li J, Xu J, et al. Genome-wide association of bmi in african americans. Obesity. 2012;20(3):622–27. doi: 10.1038/oby.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oki K, Toyama M, Banno T, Chonan O, Benno Y, Watanabe K. Comprehensive analysis of the fecal microbiota of healthy japanese adults reveals a new bacterial lineage associated with a phenotype characterized by a high frequency of bowel movements and a lean body type. BMC Microbiol. 2016;16(1):284. doi: 10.1186/s12866-016-0898-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olszak T, An D, Zeissig S, Vera MP, Richter J, et al. Microbial exposure during early life has persistent effects on natural killer t cell function. Science. 2012;336(6080):489–93. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Polderman TJC, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet. 2015;47(7):702–9. doi: 10.1038/ng.3285. [DOI] [PubMed] [Google Scholar]
- 54.Ranciaro A, Campbell MC, Hirbo JB, Ko W-Y, Froment A, et al. Genetic origins of lactase persistence and the spread of pastoralism in africa. Am J Hum Genet. 2014;94(4):496–510. doi: 10.1016/j.ajhg.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roager HM, Hansen LBS, Bahl MI, Frandsen HL, Carvalho V, et al. Colonic transit time is related to bacterial metabolism and mucosal turnover in the gut. Nat Microbiol. 2016;1(9):16093. doi: 10.1038/nmicrobiol.2016.93. [DOI] [PubMed] [Google Scholar]
- 56.Schwiertz A, Taras D, Schäfer K, Beijer S, Bos NA, et al. Microbiota and scfa in lean and overweight healthy subjects. Obesity. 2010;18(1):190–95. doi: 10.1038/oby.2009.167. [DOI] [PubMed] [Google Scholar]
- 57.Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol. 2007;19(2):59–69. doi: 10.1016/j.smim.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 58.Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, et al. Low counts of faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15(8):1183–89. doi: 10.1002/ibd.20903. [DOI] [PubMed] [Google Scholar]
- 59.Sudo N, Sawamura S, Tanaka K, Aiba Y, Kubo C, Koga Y. The requirement of intestinal bacterial flora for the development of an ige production system fully susceptible to oral tolerance induction. J Immunol. 1997;159(4):1739–45. [PubMed] [Google Scholar]
- 60.Tanno T, Fujiwara A, Sakaguchi K, Tanaka K, Takenaka S, Tsuyama S. Slit3 regulates cell motility through rac/cdc42 activation in lipopolysaccharide-stimulated macrophages. FEBS Lett. 2007;581(5):1022–26. doi: 10.1016/j.febslet.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 61.Thakur K, Tomar SK, De S. Lactic acid bacteria as a cell factory for riboflavin production. Microb Biotechnol. 2016;9(4):441–51. doi: 10.1111/1751-7915.12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tigchelaar EF, Bonder MJ, Jankipersadsing SA, Fu J, Wijmenga C, Zhernakova A. Gut microbiota composition associated with stool consistency. Gut. 2016;65(3):540–42. doi: 10.1136/gutjnl-2015-310328. [DOI] [PubMed] [Google Scholar]
- 63.Tishkoff SA, Reed FA, Ranciaro A, Voight BF, Babbitt CC, et al. Convergent adaptation of human lactase persistence in africa and europe. Nat Genet. 2007;39(1):31–40. doi: 10.1038/ng1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–84. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Turpin W, Espin-Garcia O, Xu W, Silverberg MS, Kevans D, et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat Genet. 2016;48(11):1413–17. doi: 10.1038/ng.3693. [DOI] [PubMed] [Google Scholar]
- 66.Upadhyaya B, McCormack L, Fardin-Kia AR, Juenemann R, Nichenametla S, et al. Impact of dietary resistant starch type 4 on human gut microbiota and immunometabolic functions. Sci Rep. 2016;6:28797. doi: 10.1038/srep28797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vandeputte D, Falony G, Vieira-Silva S, Tito RY, Joossens M, Raes J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut. 2016;65(1):57–62. doi: 10.1136/gutjnl-2015-309618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang J, Thingholm LB, Skiecevičienė J, Rausch P, Kummen M, et al. Genome-wide association analysis identifies variation in vitamin d receptor and other host factors influencing the gut microbiota. Nat Genet. 2016;48(11):1396–1406. doi: 10.1038/ng.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xie H, Guo R, Zhong H, Feng Q, Lan Z, et al. Shotgun metagenomics of 250 adult twins reveals genetic and environmental impacts on the gut microbiome. Cell Syst. 2016;3(6):572–84.e3. doi: 10.1016/j.cels.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu L, Paterson AD, Turpin W, Xu W. Assessment and selection of competing models for zero-inflated microbiome data. PLoS One. 2015;10(7):e0129606. doi: 10.1371/journal.pone.0129606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamamoto M, Matsumoto S. Gut microbiota and colorectal cancer. Genes Environ. 2016;38:11. doi: 10.1186/s41021-016-0038-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–27. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]