

Abstract
Substantial attention has recently been devoted to G protein-biased agonism of the μ-opioid receptor (MOR) as an ideal new mechanism for the design of analgesics devoid of serious side effects. However, designing opioids with appropriate efficacy and bias is challenging because it requires an understanding of the ligand binding process and of the allosteric modulation of the receptor. Here, we investigated these phenomena for TRV-130, a G protein-biased MOR small-molecule agonist that has been shown to exert analgesia with less respiratory depression and constipation than morphine and that is currently being evaluated in human clinical trials for acute pain management. Specifically, we carried out multimicrosecond, all-atom molecular dynamics (MD) simulations of the binding of this ligand to the activated MOR crystal structure. Analysis of >50 μs of these MD simulations provides insights into the energetically preferred binding pathway of TRV-130 and its stable pose at the orthosteric binding site of MOR. Information transfer from the TRV-130 binding pocket to the intracellular region of the receptor was also analyzed, and was compared to a similar analysis carried out on the receptor bound to the classical unbiased agonist morphine. Taken together, these studies lead to a series of testable hypotheses of ligand–receptor interactions that are expected to inform the structure-based design of improved opioid analgesics.
Graphical abstract
Morphine and its derivatives are among the most effective analgesics in clinical use, but their efficacy is accompanied by serious adverse effects, including respiratory depression, constipation, nausea, vomiting, and dependence. Addiction to opioid analgesics, which is often the first step toward heroin addiction,1 is one of the most severe forms of drug abuse and represents a significant public health concern worldwide.2 These serious problems have been the driving force behind continued efforts to develop effective therapeutic tools for pain management.
The antinociceptive action of morphine is initiated by the activation of the μ-opioid receptor (MOR)-mediated G protein signaling pathway, as demonstrated by the suppression of the drug’s analgesic efficacy in MOR knockout mice.3 On the other hand, β-arrestin recruitment by the MOR appears to contribute to some of the unwanted effects of classical opioids. For instance, studies in β-arrestin2 knockout mice have shown a significant reduction in the level of the respiratory depression and constipation induced by morphine, while analgesia was enhanced.4–6 The ability of G protein-coupled receptor (GPCR) ligands to activate one signaling pathway or another has been termed “functional selectivity”, “collateral efficacy”, or “biased agonism”7–12 in the literature. The current, prevailing paradigm is that MOR ligands that primarily activate the G protein pathway while exhibiting limited arrestin recruitment (i.e., “G protein-biased” MOR agonists) may constitute more effective therapeutics as they seem to provide effective analgesia with reduced adverse effects.13 Notably, the weaker desensitization of the receptor due to its reduced level of arrestin-mediated internalization leads to a potentially limited tolerance liability for G protein-biased MOR agonists.
There are currently only a few published examples of G protein-biased MOR small-molecule agonists with a demonstrated improved pharmacological profile in vivo. For instance, the MOR-selective ligand herkinorin from the atypical salvinorin A diterpenoid scaffold, which is fully efficacious as a MOR agonist at the G protein pathway and does not promote the recruitment of β-arrestin2 or induce receptor internalization,14 has been reported to produce peripheral antinociception with decreased tolerance liability in rats.15 More recently, structure-based optimization of a novel chemical scaffold identified by virtual screening led to the discovery of PZM21, another selective, atypical MOR agonist that preferentially activates the Gi signaling pathway over β-arrestin2.16 In vivo studies demonstrated that this compound is an efficacious analgesic that does not exhibit respiratory depression or morphine-like reinforcing activity at equianalgesic doses in mice. Another G protein-biased MOR small molecule that has been reported to provide potent analgesia with less respiratory depression and constipation than morphine17,18 is TRV-130 (also known as oliceride), which also features an atypical chemical scaffold. In particular, this compound is the only one that is currently being evaluated in human clinical trials for acute pain management.19–21 Other opioid ligands with promising therapeutic advantages are those that display dual characteristics of μ agonism/δ antagonism in vitro. One of these compounds is UMB 425,22 which has recently been demonstrated to exhibit reduced tolerance liability in vivo.
Although it is still unclear whether the potential therapeutic advantages of G protein-biased MOR ligands are due to their lower overall efficacy when compared to that of morphine (i.e., their partial agonism) or rather to an actual G protein bias,23 understanding how they bind to the receptor and the conformational ensemble they stabilize is expected to contribute critical information to the design of improved therapeutics. This is not a trivial undertaking because the chemical scaffolds in question differ significantly from the canonical morphinan structure for which structural information is available, and the approximate scoring functions of automated docking strategies are often unable to discriminate between alternative predicted binding poses in the only crystallographic structures that are currently available for the MOR.24,25
Here we report, for the first time, how TRV-130 binds and stabilizes an activated conformational state of MOR using long-scale unbiased molecular dynamics simulations. We also discuss the results of a rigorous analysis of the information transfer from the TRV-130 binding pocket to the intracellular region of the receptor and compare it to a similar analysis carried out on the classical unbiased agonist morphine bound to MOR.
MATERIALS AND METHODS
System Setup and Simulations of Ligand Binding from the Bulk Solution
A pre-equilibrated system, containing the active, nanobody-bound MOR crystal structure from Protein Data Bank (PDB) entry 5C1M,25 with the N-terminus truncated at residue S64, and embedded in a hydrated 8.0 nm × 8.0 nm 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)/10% cholesterol bilayer, was used as a starting point for all ligand binding simulations. The nanobody was kept in these simulations to prevent deactivation of the receptor. Ten TRV-130 molecules (corresponding to an effective concentration of ~44 mM) were placed in the extracellular bulk solution, at least 1.0 nm from the receptor. The system was solvated with TIP3P water molecules, and ions were added to neutralize the overall charge and reach a physiological NaCl concentration. Proteins and lipids were described using the Charmm36 force field,26–28 while the ligand was parametrized using the Charmm General Force Field (CGenFF) via the Paramchem Web site.29 Ligand parameters were verified and, when necessary, optimized, following the published protocols.29 All simulations were carried out either with Gromacs 5.0.630 or on the massively parallel supercomputer Anton.31 First, the system was minimized and the solvent equilibrated with restraints on the lipids, the proteins, and the ligands using Gromacs. Specifically, this step consisted of running 1 ns in the NVT ensemble (at 300 K, with the V-rescale thermostat32), followed by 1 ns in the NPT ensemble (1 atm, with the Parinello–Rahman barostat33). Nonbonded interactions were cut off at 1.2 nm using the Verlet scheme and a force switch at 1.0 nm for the van der Waals modifier. Restraints were progressively released from the lipids first and then from the proteins within a total of 3 ns, followed by a final 20 ns equilibration without any restraint.
To generate different starting configurations, eight different simulations with randomly set initial seeds for the V-rescale thermostat were run for an additional 40 ns each, starting from the last frame of the equilibration run, and prior to submitting the production runs.
Anton simulations were run in the NPT ensemble, using the Nose–Hoover34 thermostat with a reference temperature of 300 K and the MTTK barostat35 with a reference pressure of 1 bar. Integration was performed with the RESPA integrator with a 2 fs time step and a 6 fs time step for the long-range electrostatics. Van der Waals and short-range electrostatics were cut off at 1.25 nm, and the long-range electrostatics were calculated using the Gaussian split Ewald method,36 using a 64 × 64 × 64 grid with σ = 0.28 nm and σs = 0.16 nm. The force field used was the same as that described above, with the exception of the cholesterol, for which the original Charmm36 model was used, a necessity to be able to run Charmm efficiently on Anton. Eight production simulations were run with lengths between 1 and 8.4 μs each (see set 1 in Table S1 for details). Simulations in which all ligands were bound to the membrane after 1 μs were not continued. Twenty-five additional simulations (set 2 in Table S1) with a variable length of 150 ns to 1 μs were carried out using starting configurations extracted from the eight initial simulations, using randomized velocities and after a 5 ns equilibration (see Table S1 for details). Overall, ~44 μs of simulations were harvested.
System Setup and Simulations of the Ligand–Receptor Complex
To analyze the effect of bound ligands at the orthosteric binding pocket on the dynamics of the receptor, we carried out independent simulations of the ligand-free, morphine-bound MOR, and TRV-130-bound MOR (see Table S1). Unlike the ligand binding simulations, which were carried out in the presence of the nanobody, these simulations were run without the nanobody. For the morphine-bound MOR simulations, a pre-equilibrated system of the activated MOR structure was used as a starting point to dock morphine in the binding site using the position and orientation of the morphinan ligand in the active crystal structure (PDB entry 5C1M) as a template. The system was solvated and equilibrated following the same protocol described above. For the MOR–TRV-130 complex simulations, the last frame of a simulation with TRV-130 in the binding site was used as the initial coordinates. With the exception of the bound TRV-130, all other ligand molecules were removed, as well as the nanobody. Water molecules and ions were added as in the other simulations reported herein, and the same equilibration protocol was applied as described above. Additional simulations of the ligand-free receptor without the nanobody were carried out using the same simulation protocol. All simulations were run with Gromacs 5 in the NPT ensemble using the Charmm36 force field, the V-rescale temperature coupling, and the Parrinello–Rahman barostat. The integration time step was increased to 4 fs using hydrogen mass repartitioning. Van der Waals and Coulombic interactions were treated as in the simulations described above. Three independent simulations were run for each system, i.e., TRV-130-bound, morphine-bound, and ligand-free receptors, for a total of 3 μs for each system (see Table S1).
Simulation Analyses
The interactions of each TRV-130 molecule with the receptor, nanobody (because of periodic boundary conditions), or the membrane were encoded in binary fingerprints. To discriminate between the different ligand–receptor interactions, the chemical structure of TRV-130 was divided into four fragments, i.e., the methoxy-thiophene moiety, the pyridine, the 6-oxaspiro[4.5]decan-9-yl, and the amine moiety (see Figure 1a). Binary fingerprints were defined on the basis of the minimal proximity (with a cutoff of 0.4 nm) to each residue of the protein, or the nanobody, as well as the headgroups of the lipids. These fingerprints were used to calculate pose distances based on the Tanimoto dissimilarity coefficient and clustered on the basis of these distances using a density-based spatial clustering of applications with noise (DBSCAN) algorithm.37 This clustering method has an advantage in that, unlike k-means clustering, it does not require to specify the number of expected clusters. Instead, two parameters must be chosen: the minimum number of points around a putative core point and the distance cutoff (ε). Points in the proximity of core points are clustered together, as well as additional points that may be reached from one of the core points. Data points that could not be associated with any cluster are treated as noise.
Figure 1.
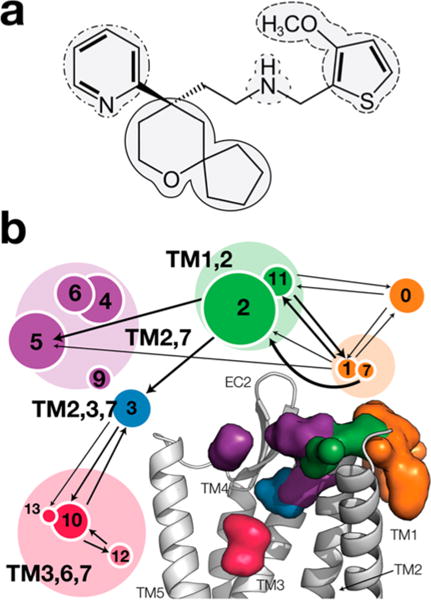
Chemical structure of TRV-130 and spatial distribution of its center of mass along the binding pathway. (a) Selection of the moieties used to define the interaction fingerprints employed in the analysis. The methoxy-thiophene, the pyridine, the spiro-fused tetrahydopyran-cyclopentane (6-oxaspiro[4.5]decan-9-yl moiety), and the amine moieties are delineated by dashed, dotted–dashed, solid, and dotted lines, respectively. (b) Clusters of the spatial distribution of the center of mass of TRV-130 along the binding pathway are represented by circles with areas proportional to their populations and grouped on the basis of their structural similarity. Specifically, the region in which the ligand is in contact with the membrane is shown as an orange surface, and the vestibule region is colored purple. Metastable states further inside the receptor are colored blue and green, and the orthosteric binding site is colored red. The arrows indicate transitions between clusters that were observed with higher probability during the binding simulations.
Thirteen clusters of sampled ligand conformations were derived from this clustering on the basis of ligand–receptor interaction fingerprints. The spatial distribution of the center of mass of TRV-130 along its binding pathway is shown in Figure 1 alongside the most frequently (more than five times) observed transitions among the 13 identified clusters. Illustrations of the type of ligand–receptor interactions formed with high probability by TRV-130 in the most populated clusters at each location with the largest spatial distribution of the center of mass of TRV-130, i.e., clusters 2, 3, 5, and 10, are reported in Figures S1–S4, respectively. Additional details of these interactions are reported in Table S2.
Markov State Model of TRV-130 Binding
To characterize the TRV-130 binding process, we calculated transition matrices for microstates obtained with k-means clustering (N = 100) with a maximum likelihood estimation using the pyEmma software,38 and a lag time of 50 ns.
Allosteric Signaling Analysis
Analysis of the dynamic signatures of the receptor was performed on three sets of unbiased simulations of ligand-free, TRV-130-bound, and morphine-bound receptors. The entropy of each degree of freedom, xiα, where i is the atom index and α a Cartesian direction, was estimated starting from a multivariate distribution of the joint probability distribution with covariance C estimated from the simulations as
Following the N-body Information Theory (NbIT) protocol described in the literature,39 the coordinates of the phenyl rings of phenylalanine and tyrosine side chains and of the α-carboxylic acid group of deprotonated glutamic and aspartic acid residues were symmetrized before calculating the correlation matrix. The covariance matrix for all the heavy atoms in the system was calculated using the Carma software.40 The configurational entropy of a set of degrees of freedom X41 is given by the expression:
where pX is the probability distribution of the coordinates in set X. In the following equations, we will make use of the definition of the entropy of a set of degrees of freedom conditional on another set as H(X|Y) = H(X∪Y) − H(Y). To identify coupled dynamics between clusters of residues {Xi}, we calculated the mutual information:
MI(X1,X2) measures the information shared by two sets of residues. To identify allosteric transmission of information, the co-information between groups of residues was calculated as follows: CI({Xi},X′) = MI({Xi}) − MI({Xi}|X′), where the conditional mutual information is defined as
For instance, for three groups of residues X1, X2, and X3, the three-body co-information MI(X1,X2;X3) measures the influence of X3 on the amount of information shared by X1 and X2. Thus, this measure can be used to derive a first estimate of information pathways between the set of residues encompassing the ligand binding pocket (i.e., the “transmitter”) and residues close to the intracellular region of the protein (i.e., the “receiver”). The contribution of a specific residue to an information measure M can be obtained by recalculating that measure after removing the contributing residue from the sets used in the original calculation, and conditioning M on the residue degrees of freedom:
where the backslash indicates a set difference. With this definition, the coinformation CI(X,Y;x) is equal to χMI(X,Y)(x)-MI(X,Y), so that it reflects the contribution of residue x to the mutual information between X and Y.
Residues within the binding pocket might have specific roles in stabilizing the bound state and transmitting the information to other regions of the protein. To single out the role of each degree of freedom in the allosteric coupling, we calculated the contribution of each residue to the (n-body) mutual information (“total correlation” in ref 39), defined as
To define the set of “transmitting” residues, we used the common receptor residues found within 5 Å of each ligand in the initial conformations of the simulations of TRV-130-bound and morphine-bound MOR. These residues were D1473.32, Y1483.33, M1513.36, V2365.42, W2936.48, I2966.51, H2976.52, V3006.55, W3187.35, I3227.39, and Y3267.43. The ligand’s atomic coordinates were also included in these sets. For the “receiver”, we considered the residues that are within 5 Å of the nanobody in the activelike MOR crystal structure.25 These residues were K100(IC1), T101(IC1), R1653.50, A1683.53, V1693.54, P172-(IC2), V173(IC2), A175(IC2), L176(IC2), D177(IC2), R179-(IC2), T180(IC2), P181(IC2), M2555.61, R2585.64, L2595.65, V2625.68, R263(IC3), A264(IC3), M265(IC3), S266(IC3), E2706.25, K2716.26, N2746.29, L2756.30, I2786.33, D3407.57, E341(H8), N342(H8), and R345(H8).
Hydration Sites
Spatial locations of stable water molecules within the helical bundle (hereafter “hydration sites”) were identified by calculating (with the volmap tool in VMD) the density of water molecules on a three-dimensional grid with a mesh of 0.5 Å, averaging it over 0.75 Å, and selecting points with a density at least twice the bulk water value. The point with the highest density was identified as a hydration site. After the removal of all grid points within 2 Å of this site, the next maximal density value was identified, repeating the operation until the whole set of high-density points was exhausted. Each hydration site was considered occupied in frames in which a water molecule was found within 1.0 Å of its center.
Interaction Networks
Interaction networks were generated from contact probabilities, averaged over all the simulation trajectories for a given ligand–receptor complex. Specifically, interactions of ligands or residues with hydration sites were taken into account when polar atoms of the ligand or of the residue side chains were within 4 Å of an occupied hydration site. Interactions between hydration sites were considered to be formed when two close (within 4 Å) hydration sites were simultaneously occupied by a water molecule. Nonpolar interactions were assumed to be formed when the centers of mass of the side chain atoms of two residues were closer than a fixed cutoff, fixed to 4 Å for pairs residues separated by fewer than four sites along the protein chain, and to 6 Å for all other pairs.
RESULTS
Binding Pathway of TRV-130 from the Bulk Solution to the MOR Orthosteric Binding Site
We aggregated and analyzed ~44 μs of simulations carried out in the presence of a high concentration of TRV-130 (see the chemical structure in Figure 1a) placed in the bulk solution (see Materials and Methods and Table S1).
A first set of simulations totaling ~39 μs showed ligand binding at different positions of the MOR. Only one binding event displayed the ligand at the MOR orthosteric binding site identified by crystallography, whereas at least one ligand was found interacting with the receptor in each of the remaining trajectories at different positions of the extracellular region of MOR, including what has been termed the “vestibule” region in the literature.42 To increase the degree of sampling of the conformational states along the binding pathway, we respawned some of the trajectories with the ligand at positions other than the orthosteric site and ran a second set of simulations (see Table S1) for a total of an additional ~5 μs. TRV-130 was ultimately found at the orthosteric site in eight of these simulations. Thus, we observed a total of nine binding events at the orthosteric site of the receptor during these simulations, although we cannot rule out the possibility that alternative binding modes may exist along the pathway.
A 75% average fraction of ligand molecules was calculated to be in contact with the membrane over the aggregated simulations. On the basis of a calculated average volume (VW) of 12700 × 0.029 nm3 ≈ 380 nm3 for the 12700 water molecules in the system setup (each with a specific volume of 0.029 nm3), and a calculated average volume of ~(137/2) × 0.60 × 3.5 nm3 ≈ 143 nm3 for the 137 POPC molecules in the two leaflets of the lipid bilayer (where 0.60 nm2 is the average area per lipid and 3.5 nm is the average membrane thickness), the effective TRV-130 concentration in the solvent was 1–75% × 10/(VWN0) ≈ 6.5 mM. We note that the partition coefficient implied by these concentrations is log PM/w = log(116/6.5) ≈ 1.8, where 116 mM is the effective ligand concentration in the membrane. Using a simple Poisson model, a kon ≈ (6.5 nM × 39 μs)−1 ≈ 0.3 × 108 (M min)−1 rate can be estimated from the simulations reported here. Notably, this calculated value is in general agreement with the experimental value of 15 × 108 (M min)−1.17
Because only a small fraction of the TRV-130 molecules interacted with the receptor during the total simulation time, we focused our analysis of the ligand binding pathway on the ligand trajectories in which the fraction of time the agonist spent in contact with the protein was >10%. Figure 1b shows the distribution of the center of mass of TRV-130 along the binding pathway. The sampled ligand conformations were grouped into 13 clusters using ligand–receptor interaction fingerprints based on the TRV-130 moieties indicated in Figure 1a. These 13 clusters are shown in Figure 1b alongside an illustration of the most frequently (more than five times) observed transitions between clusters. Specific ligand–receptor interactions formed with high probability by TRV-130 in the most populated clusters at each location with the largest spatial distribution of the center of mass of TRV-130 (i.e., clusters 2, 3, 5, and 10) are listed in Table S2. Panels a–d of Figure 2 show representative structures of clusters 2, 3, 5, and 10, respectively. Specifically, Figure 2a shows that initial contacts between TRV-130 and the receptor are formed at the extracellular side of TM1 and TM2, with the ligand also being partially in contact with the membrane and mostly exposed to the solvent. In the majority of poses in this cluster, the pyridine ring is enclosed in a small pocket lined by the side chains of A681.32, Y1282.64, and L1292.65 (see Figure 2a).
Figure 2.
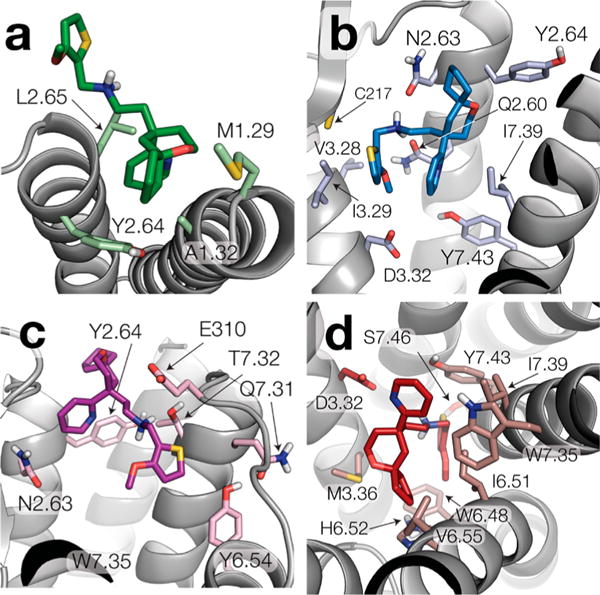
Representative structures of the clusters with the largest spatial distribution of the center of mass of TRV-130 at each MOR location. Specifically, panels a–d show ligand–receptor interactions of representative structures of clusters 2, 3, 5, and 10, respectively.
As found in previous studies of ligand binding to class A GPCRs, TRV-130 does not proceed directly to the orthosteric binding site after establishing the first contact with the receptor. Instead, it spends some time in the so-called vestibule, a region between the crystallographic orthosteric binding pocket and the extracellular side of the receptor (purple surface in Figure 1b), before it either unbinds or penetrates further inside the receptor (blue surface in Figure 1b), toward the orthosteric binding site (red surface in Figure 1b). While this multistep process is not new to small-molecule binding opioid receptors,42 we show, for the first time, that the process is regulated by two different ligand binding states in the vicinity of TM2, TM3, and TM7 that have different kinetic properties. Specifically, after its initial interactions with the receptor (Figure 2a), the ligand is found in either a metastable state that is likely to advance to the orthosteric binding site (blue surface and cluster 3 in Figure 1b) or another state characterized by alternative binding poses that are trapped in their position for a considerable amount of time and cannot proceed to the orthosteric site without unbinding first (purple surface and clusters 4–6 and 9 in Figure 1b).
In the representative structure of cluster 3 (Figure 2b and blue surface in Figure 1b), the 6-oxaspiro[4.5]decan-9-yl moiety is oriented toward TM2 and is located among Q1242.60, N1272.63, and Y1282.64. Polar interactions between the amine group of TRV-130 and N1272.63 or Q1242.60 appear to stabilize the ligand at this position. As shown in Figure 2b, the ligand’s methoxy-thiophen moiety is surrounded by hydrophobic residues located on TM3 (V1433.28 and I1443.29) and a cysteine in extracellular loop 2 (C217) while residues on TM7 (I3227.39 and Y3267.43) are close to the ligand’s pyridine ring.
Unlike conformations of cluster 3, representative TRV-130 poses of the larger cluster 5 (Figure 2c) are never seen to proceed to the orthosteric binding pocket. These poses either remain in the same position for the rest of the simulated time or unbind. Notably, at these positions, the ligand interacts mostly with residues on TM7 (Q3127.31, T3157.32, and W3187.35) and with residues on TM2 (N1272.63 and Y1282.64).
After visiting the state corresponding to cluster 3, TRV-130 proceeds toward the crystallographic orthosteric binding pocket, acquiring conformations that partially overlap with the cocrystallized ligand BU72 in the active MOR (Figure 2d and Figure S5). The 6-oxaspiro[4.5]decan-9-yl moiety of TRV-130 is oriented toward TM5-TM6 in these representative ligand poses of cluster 10, forming contact with M1513.36, V3006.55, I2966.51, and H2976.52. In contrast to the cocrystallized ligand BU72, which establishes a direct interaction with D1473.32, the interaction between TRV-130 and D1473.32 is water-mediated via two water molecules and involves the charged amine moiety of TRV-130 as well as the nitrogen of the pyridine ring (see Figure S5). As shown in Figure 2d, the pyridine ring of TRV-130 interacts with W3187.35 and I3227.39 but is also partially exposed to the solvent. The TRV-130 moiety that does not overlap with the cocrystallized ligand BU72 (see Figure S5) is the methoxy-thiophen moiety. As shown in Figure 2d, in the majority of TRV-130 representative poses of cluster 10, this moiety is oriented toward the center of the helical bundle, in the proximity of residues W2936.48, I2966.51, S3297.46, and Y3267.43 (see Table S2 for individual contact probabilities). However, a smaller population of ligand poses in this cluster have this moiety oriented toward the extracellular side of the receptor, forming interactions with residues Q1242.60, D1473.32, and I3227.39 (see Figure S6). Notably, the TRV-130 methoxy-thiophen moiety is also oriented toward the center of the helical bundle in the recently published docking pose of the ligand.16
Effect of Desolvation on Ligand Binding
Desolvation plays a crucial role in the binding of small molecules to proteins,43–45 including GPCRs.46 During the binding of TRV-130, several desolvation and resolvation events can be observed (see Figure 3) alongside the initial contact of the ligand with the receptor, as well as the binding to and unbinding from states along the binding pathway. In the vestibule, at the position that is closest to the orthosteric binding site (blue point in Figure 3), TRV-130 can be found to be less solvated (solvation of ~20) than at the orthosteric site (red point with a solvation of ~25 in Figure 3) or in the trapped states (purple point with a solvation of ~30 in Figure 3). Thus, transitions of the ligand to the orthosteric site or the extracellular side are accompanied by an increase in the number of water molecules around the small molecule.
Figure 3.
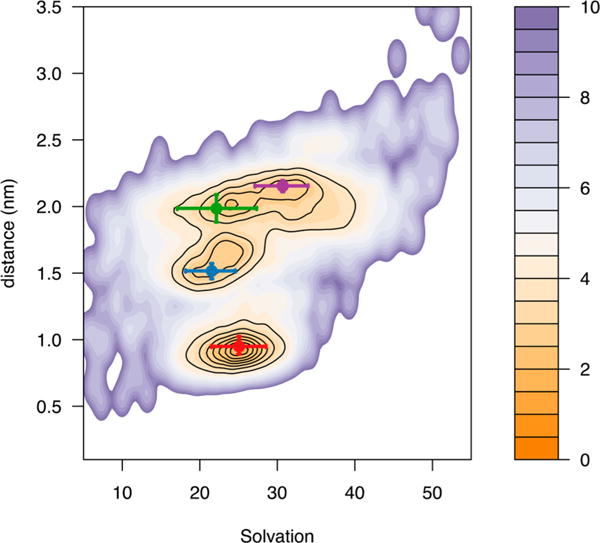
Ligand solvation upon binding. The mean values of the distance of the ligand from the center of mass of the receptor bundle as a function of ligand solvation are shown as green, blue, purple, and red points for clusters 2, 3, 5, and 10, respectively. Error bars indicate the 15 and 85% quantiles.
TRV-130 and Morphine Yield Different Signatures of Receptor Dynamics
To start elucidating how TRV-130 affects the conformational landscape and dynamic behavior of the bound receptor, we analyzed position correlations from three unbiased, microsecond-scale simulations of the TRV-130-bound MOR and compared them to those from independent simulations of the same receptor bound to the prototypical unbiased ligand morphine (see Table S1 for details of the simulations). Specifically, we defined a set of residues in contact with the ligands as the “transmitter” region and a set of residues at the intracellular end of MOR as the “receiver” region (see Materials and Methods for details) and applied information theory analysis to the receptor dynamics to investigate the communication between the two sets.39
The pairwise mutual information between these two sets of residues has positive values in the presence of either ligand (see the bold numbers in Table 1), confirming that the dynamics of the two regions is highly correlated. To investigate structural determinants of the allosteric coupling between the ligand binding region and the intracellular G protein/arrestin binding region of MOR, we first calculated the co-information among the transmitter region, the receiver region, and any other residue in the protein. When normalized by the mutual information (see Materials and Methods), co-information provides a measure of the impact of each residue on the mutual information between the transmitter and the receiver regions. Thus, residues with a large co-information value are either part of an allosteric channel or share a high level of mutual information with the channel itself. The top residues of MOR displaying the most significant contributions to co-information coupling in the presence of either TRV-130 or morphine are listed in Table 2 and illustrated as purple spheres in Figure 4.
Table 1.
Total Entropies (regular font) and Pairwise Mutual Information (bold font) between Residues within the Ligand Binding Pocket, the Receptor Intracellular Region, and the Sodium Binding Pocket As Derived from Analysis of the Simulations
| ligand binding pocket | receptor intracellular (IC) region | Na+ binding pocket | |
|---|---|---|---|
| ligand-free MOR | |||
| ligand binding pocket | −188 ± 66 | 17.1 ± 1.6 | 12.3 ± 1.0 |
| receptor IC region | – | 19.1 ± 12 | 9.43 ± 0.4 |
| Na+ binding pocket | – | – | −12.22 ± 6.45 |
| morphine-bound MOR | |||
| ligand binding pocket | −275 ± 5 | 16.5 ± 1.2 | 12.7 ± 1.3 |
| receptor IC region | – | −14.8 ± 2.4 | 9.4 ± 0.8 |
| Na+ binding pocket | – | – | −12.6 ± 1.9 |
| TRV-130-bound MOR | |||
| ligand binding pocket | −259 ± 7 | 17.3 ± 2.3 | 12.4 ± 0.7 |
| receptor IC region | – | −7.23 ± 3.51 | 11.0 ± 2.1 |
| Na+ binding pocket | – | – | −6.3 ± 1.4 |
Figure 4.
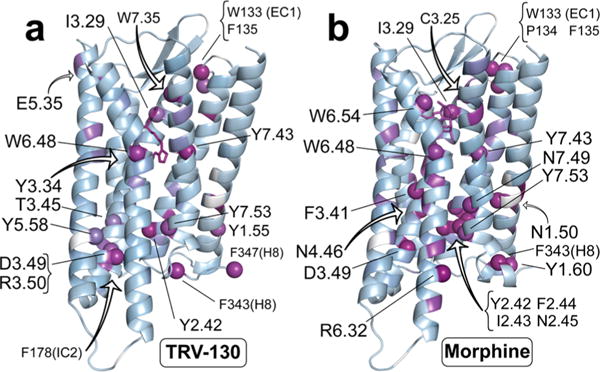
Most highly contributing residues to the allosteric coupling between the ligand binding pocket and the intracellular region. Specifically, residues involved in the allosteric coupling induced by TRV-130 are shown as purple spheres in panel a, whereas panel b shows residues involved in the allosteric coupling induced by morphine.
To add more structural context, we also calculated the interaction network formed by polar and nonpolar contacts among the receptor side chains, the ligand, and conserved hydration sites (displayed in Figures 5 and 6 for TRV-130 and morphine, respectively).
Figure 5.
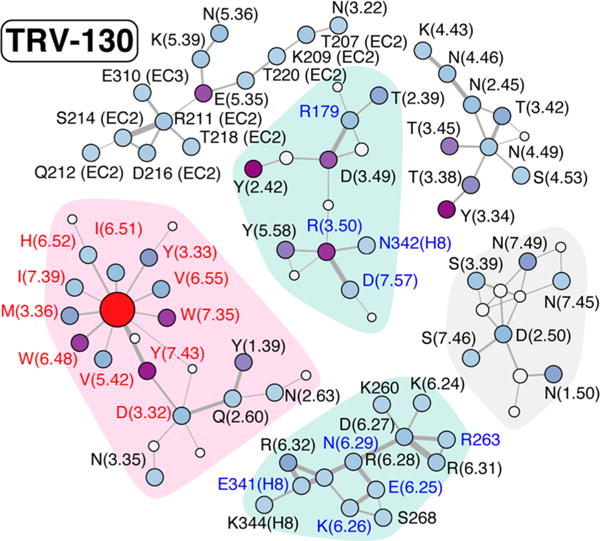
Interaction network connecting the side chains of residues in the TRV-130-bound MOR. Polar and nonpolar contacts are indicated by solid and dashed gray lines, respectively, with a thickness proportional to the interaction probability (>40%). Conserved hydration sites are denoted with gray circles, with the area being proportional to their occupancy. Residues defining the “transmitter” and the “receiver” sets are labeled in red and blue, respectively. The ligand is indicated by a red circle, while the other residues are colored according to their contribution to the co-information (increasing from light blue to purple). Only clusters of residues with five or more residues are displayed.
Figure 6.
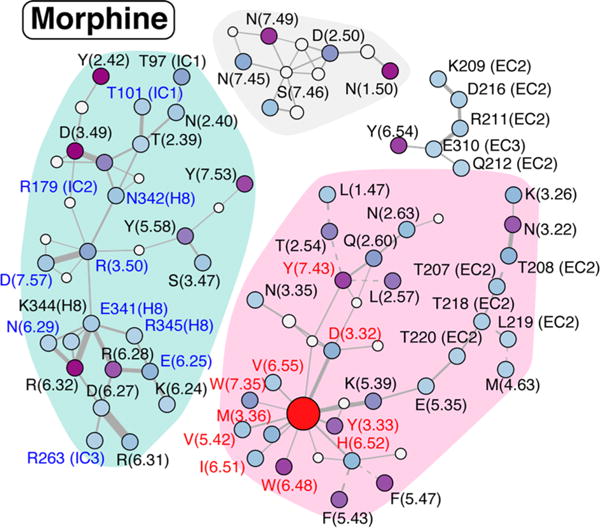
Interaction network connecting the side chains of residues in the morphine-bound MOR. See the legend of Figure 5 for details.
Striking differences in these interaction networks can be appreciated by comparing Figures 5 and 6. First, TM3 and TM6 residues in the intracellular region of the receptor cluster into two separate groups when TRV-130 is present at the orthosteric binding site (clusters with cyan shadowing in Figure 5). Notably, only the group with TM3 residues contains strong contributors to the co-information (specifically, Y1062.42, R1653.50, and D1643.49), while no strong coupling is observed for residues at the end of TM6. In contrast, morphine in the binding pocket allosterically regulates a substantial coupling to both the intracellular ends of TM3 and TM6. In the presence of morphine, intracellular residues interact through an extended network of polar and nonpolar interactions and form a single, strongly connected cluster, which is highlighted with a cyan background in Figure 6. As is evident from Figure 6, residues Y1062.42 and D1643.49 contribute to the co-information when morphine is at the orthosteric binding site as they did when TRV-130 was present. However, when morphine is bound to the receptor, these residues are connected to TM6 and TM7 residues R2776.32, R2736.28, and Y3367.53, which also strongly contribute to the co-information between transmitter and receiver regions.
The network of side chain interactions in the vicinity of the ligands is also strikingly different, with TRV-130 presenting a set of connected residues smaller than that of morphine. Residues Y1062.42, W133(EC1), Y3267.43, F343(H8), W2936.48, Y3367.53, F135(EC1), D1643.49, and I1443.29 are among those most strongly contributing to the allosteric coupling in the presence of either TRV-130 or morphine bound to the receptor, albeit with different strengths (see Table 2 for details). The observed role of residues such as W2936.48 and Y3267.43 was expected given the number of previous reports drawing attention to their importance in the allosteric process mediated by GPCRs (see, for instance, ref 47 and references therein).
Table 2.
Residues Contributing Most to the Mutual Information between the Ligand Binding Pocket and the Intracellular Region of the MORa
| TRV-130-bound MOR
|
morphine-bound MOR
|
||
|---|---|---|---|
| residue/ligand | contribution | residue/ligand | contribution |
| TRV-130 | 0.56 | D1643.49 | 0.75 |
| Y1062.42 | 0.44 | F1082.44 | 0.65 |
| W133(EC1) | 0.42 | Y1062.42 | 0.56 |
| Y1493.34 | 0.4 | I1072.43 | 0.55 |
| Y3267.43 | 0.37 | F343(H8) | 0.54 |
| F343(H8) | 0.36 | N1884.46 | 0.54 |
| F347(H8) | 0.35 | F135(EC1) | 0.53 |
| R1653.50 | 0.35 | R2776.32 | 0.52 |
| W2936.48 | 0.34 | F1563.41 | 0.50 |
| W3187.35 | 0.34 | W133(EC1) | 0.50 |
| Y911.55 | 0.34 | N861.50 | 0.49 |
| Y3367.53 | 0.33 | I1443.29 | 0.48 |
| F178(IC2) | 0.33 | Y961.60 | 0.48 |
| E2295.35 | 0.33 | N1092.45 | 0.48 |
| F135(EC1) | 0.33 | C1403.25 | 0.47 |
| D1643.49 | 0.32 | N3327.49 | 0.46 |
| I1443.29 | 0.31 | P134(EC1) | 0.46 |
| T1603.45 | 0.29 | morphine | 0.45 |
| Y2525.58 | 0.29 | Y2996.54 | 0.45 |
| W2936.48 | 0.45 | ||
| Y3267.43 | 0.45 | ||
| Y3367.53 | 0.44 | ||
Residues completely exposed to the lipid bilayer are not included in the table. Common residues in the two systems are highlighted in bold.
Our analysis also identifies W3187.35, R1653.50, Y1493.34, F347(H8), and Y911.55 as the most important players in the transmission of information for the TRV-130-bound receptor, while F1082.44, I1072.43, N1884.46, and R2776.32 are among those contributing only to the allosteric channel in the morphine-bound MOR (see Table 2). Notably, most of the residues in direct or water-mediated contact with the ligands do not contribute significantly to the co-information between the binding pocket and the intracellular region, including the highly conserved D1473.32.
To confirm the different functional roles of residues in the allosteric process, we calculated the contribution of individual residues in the binding pocket to the total correlation of the binding pocket (see Table 3). As reported in Table 3, the top residues that contribute to the total correlation are similar in the morphine-bound and TRV-130-bound receptors. In both systems, the ligand itself is the degree of freedom that maximally contributes to the total correlation of the binding pocket, along with residues H2976.52, W2936.48, I2966.51, and Y1483.33. Residues in the binding pocket that contribute to such a self-correlation act as “stabilizers” of the dynamics of the binding pocket (Table 3). On the other hand, residues of the binding pocket that maximally contribute to the coupling between the binding pocket and the intracellular region of the receptor function as “communicators” (Table 2). Notably, W2936.48 and Y3267.43 function as both stabilizers and communicators in both systems (compare Table 2 and Table 3). On the other hand, W3187.35, which is a stabilizer in the presence of either ligand, contributes only as a communicator between transmitter and receiver regions when TRV-130 is present.
Table 3.
Contribution of Individual Residues in the Ligand Binding Pocket to the Total Correlation of the Ligand Binding Pocket
| TRV-130-bound MOR
|
morphine-bound MOR
|
||
|---|---|---|---|
| residue | contribution | residue | contribution |
| TRV-130 | 0.68 | morphine | 0.73 |
| H2976.52 | 0.53 | W2936.48 | 0.66 |
| W2936.48 | 0.52 | H2976.52 | 0.59 |
| I2966.51 | 0.5 | Y1483.33 | 0.56 |
| Y1483.33 | 0.48 | I2966.51 | 0.54 |
| M1513.36 | 0.47 | W3187.35 | 0.51 |
| W3187.35 | 0.47 | D1473.32 | 0.5 |
| V3006.55 | 0.46 | Y3267.43 | 0.49 |
| D1473.32 | 0.44 | V3006.55 | 0.48 |
| I3227.39 | 0.43 | V2365.42 | 0.46 |
| V2365.42 | 0.43 | M1513.36 | 0.46 |
| Y3267.43 | 0.43 | I3227.39 | 0.46 |
Finally, the interaction network and co-information contribution from residues lining the vestibule are also appreciably different in the receptor simulated with the biased ligand TRV-130 or the unbiased agonist morphine bound at its orthosteric pocket. With TRV-130 bound to the receptor, a broad network of polar residues connects extracellular (EC) loops 2 and 3 to the extracellular ends of TM3 and TM5 (see the top left cluster in Figure 5). Among these residues, E2295.35 strongly contributes to transmitter–receiver coupling. On the other hand, when morphine is bound to the receptor, the interaction network between residues in the vestibule is less extended and contains fewer interactions. In this case, the main contributor to transmitter–receiver coupling is residue Y2996.54, at the extracellular end of TM6 (see Figure 6, top right cluster).
The Na+ Binding Site Presents Different Co-informa tion Patterns in the Presence of Morphine or TRV-130 at the Orthosteric Binding Site
Another interesting difference between the receptor dynamic signatures induced by the two simulated ligands concerns the different role of the residues lining the sodium binding site. Interestingly, these residues are part of separate clusters of the interaction networks derived from the simulations of the TRV-130–MOR or morphine– MOR complexes (gray backgrounds in Figures 5 and 6, respectively). While none of the residues in the gray cluster of Figure 5 contribute to the transmitter–receiver co-information derived from simulations of the TRV-130-bound receptor, N861.50 and N3327.49 strongly contribute when morphine is bound to the receptor (see Table 2). Notably, both N861.50 and N3327.49 are hydrogen-bonded through conserved hydration sites to D1142.50, which coordinates (together with N1503.35 and S1543.39) sodium in the ultra-high-resolution δ-opioid receptor crystal structure.48 The role of N3327.49 and Y3367.53, two residues of the conserved NPXXY motif of TM7, has also been extensively described in the literature in reference to the allosteric transmission of the signal from the exterior of the cell to its interior. Interestingly, mutation of N1503.35 to apolar side chains has been shown to enhance the constitutive activity in the β-arrestin pathway.48
To further clarify the role of the different residues in the sodium binding pocket in the modulation of allosteric coupling, we calculated the contribution of the residues that directly coordinate the sodium ion (i.e., D1142.50, N1503.35, and S1543.39) to the total correlation of the transmitter (i.e., ligand binding pocket) or receiver (i.e., intracellular side) regions on the receptor. The results reported in Table 4 show that the contribution of D1142.50 to the total correlation of the transmitter or receiver regions of MOR is slightly larger when morphine is bound to the receptor than when TRV-130 is bound. Notably, none of the three residues that coordinate directly the sodium ion contribute significantly to the co-information between the binding pocket and the intracellular side of the receptor (see Tables 2), notwithstanding their role in establishing correlations within the ligand binding pocket or the intracellular region of MOR.
Table 4.
Contributions of Residues in the Na+ Binding Pocket to the Total Correlation of the Ligand Binding Pocket and the IC Region of the Receptor
| TRV-130 | MOR | |
|---|---|---|
| ligand binding pocket | ||
| D1142.50 | 0.33 ± 0.01 | 0.38 ± 0.03 |
| N1503.35 | 0.39 ± 0.01 | 0.42 ± 0.03 |
| S1543.39 | 0.35 ± 0.02 | 0.35 ± 0.01 |
| IC region | ||
| D1142.50 | 0.23 ± 0.02 | 0.26 ± 0.002 |
| N1503.35 | 0.24 ± 0.01 | 0.26 ± 0.01 |
| S1543.39 | 0.26 ± 0.02 | 0.26 ± 0.01 |
DISCUSSION
We investigated the binding of TRV-130 to the activated crystal structure of MOR with long-scale, unbiased molecular dynamics simulations and analyzed the ligand’s induced allosteric communication between the accepted orthosteric binding pocket and the intracellular region of the receptor.
Our simulations offer unprecedented detail about the binding pathway of this G protein-biased agonist under current clinical evaluation for acute pain management, contributing testable hypotheses about its mechanism of molecular recognition, its mode of binding, and the role of different intermediate states within the so-called vestibule region of the receptor in modulating ligand binding kinetics. In particular, along with some metastable bound states along the binding pathway, we identified several bound states that trap the ligand into the vestibule region and that are not part of the ligand’s reactive binding pathway to the orthosteric pose. These states are hypothesized to modulate ligand binding kinetics, which was recently shown to play an important role in the profile of biased agonists.49 Notably, one of the residues that stabilize one of these intermediate states along the ligand’s reactive binding pathway, specifically the residue at position 2.63, is different among all opioid receptor subtypes. This residue is an asparagine in the MOR, a valine in the KOR, and a lysine in the DOR. Because the valine in the KOR is expected not to form the polar interaction that is seen between TRV-130 and N1272.63 in the MOR, whereas the lysine in the DOR would interfere with the ligand position in the pocket, it is tempting to speculate that N1272.63 may play an important role in determining the relative selectivity of TRV-130 for the MOR.17
After visiting the vestibule region, TRV-130 binds to the accepted orthosteric site of the MOR. At this site, the ligand adopts an energetically favorable pose that partially overlaps with the binding pose of the crystal ligand BU72, establishing direct and water-mediated interactions with conserved residues within the pocket. Interestingly, rigorous analysis of the receptor dynamics in the presence of the ligand shows that these direct and water-mediated ligand–receptor interactions are not those that contribute most significantly to information transfer (i.e., allosteric communication) between the binding pocket and the intracellular region of the receptor. Furthermore, by comparing simulations of the MOR bound to TRV-130 or the unbiased ligand morphine, we discovered that the interaction networks involved in allosteric communication are strikingly different depending on which ligand occupies the binding pocket. While a clear communication is seen between the TRV-130-bound binding pocket and the intracellular end of TM3, no strong coupling is observed with residues at the end of TM6. In contrast, in the presence of the unbiased morphine at the MOR orthosteric site, substantial coupling is observed between the binding pocket and the intracellular ends of both TM3 and TM6.
Differences in the dynamic signatures of the MOR in the presence of TRV-130 or morphine at the orthosteric binding site are seen not only at the level of residues in direct interaction with the ligand or the G protein but also at the accepted ion binding pocket and at the extracellular region of the receptor. For instance, in the TRV-130-bound MOR, residues in the EC2 and EC3 loops form a network of polar interactions that extend to the extracellular ends of TM3 and TM5, where some of the main contributors to the allosteric coupling between the binding pocket and intracellular region of the receptor reside. In contrast, a much less extended and connected interaction network involving extracellular residues is formed when morphine is bound to the receptor, with main contributors to the allosteric coupling found in this case at the extracellular end of TM6.
Altogether, information about the different interaction networks formed at the orthosteric binding pocket and different contributions to coupling between the ligand binding pocket and the intracellular side of the receptor provide new insights into the functional role of the residues involved that may be used in the rational design of drugs with tailored pharmacologic profiles. While some residues (see details in Results) strongly contribute to the stability of the intracellular region in the presence of either simulated ligand, others (e.g., W3187.35) act as communicators between the ligand binding pocket and intracellular regions only when TRV-130 is bound to the receptor. Among those residues most strongly contributing to the allosteric coupling in the presence of either TRV-130 or morphine bound to the receptor are Y1062.42, W133(EC1), Y3267.43, F343(H8), W2936.48, Y3367.53, F135(EC1), D1643.49, and I1443.29. While F1082.44, I1072.43, N1884.46, and R2776.32 are among the residues contributing to the allosteric channel in the morphine-bound MOR, our analysis suggests that W3187.35, R1653.50, Y1493.34, F347(H8), and Y911.55 are among the most important players in the transmission of information for the TRV-130-bound receptor. Experimental validation of these observations may suggest ways to fine-tune MOR signaling toward the desired therapeutic pathways and away from those mediating side effects.
Supplementary Material
Acknowledgments
The authors thank Dr. Michael LeVine for providing the preprocessing scripts and R functions used to calculate the quantities required by the n-body information theory. Computations were run on resources available through the Scientific Computing Facility at Mount Sinai, the Extreme Science and Engineering Discovery Environment under MCB080077, which is supported by National Science Foundation Grant ACI-1053575, and the Pittsburgh Supercomputing Center (PSC), which provided Anton computer time (under PSCA14006) through National Institutes of Health Grant R01GM116961. The Anton machine at PSC was generously made available by D. E. Shaw Research.
Funding
This work was supported by National Institutes of Health Grants DA026434, DA034049, and MH107053.
ABBREVIATIONS
- CGenFF
Charmm General Force Field
- DBSCAN
density-based spatial clustering of applications with noise
- DOR
δ-opioid receptor
- EC
extracellular
- KOR
κ-opioid receptor
- GPCR
G protein-coupled receptor
- MD
molecular dynamics
- MOR
μ-opioid receptor
- PDB
Protein Data Bank
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.6b00948.
Supplementary figures and tables and details of the simulation runs and their analyses (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Walwyn WM, Miotto KA, Evans CJ. Opioid pharmaceuticals and addiction: the issues, and research directions seeking solutions. Drug Alcohol Depend. 2010;108:156–165. doi: 10.1016/j.drugalcdep.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Results from the 2009 National Survey on Drug Use and Health: Vol I Summary of National Findings. Office of Applied Studies, National Survey on Drug Use and Health; Rockville, MD: 2010. [Google Scholar]
- 3.Kieffer BL. Opioids: first lessons from knockout mice. Trends Pharmacol Sci. 1999;20:19–26. doi: 10.1016/s0165-6147(98)01279-6. [DOI] [PubMed] [Google Scholar]
- 4.Raehal KM, Walker JK, Bohn LM. Morphine side effects in beta-arrestin 2 knockout mice. J Pharmacol Exp Ther. 2005;314:1195–1201. doi: 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- 5.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 6.Maguma HT, Dewey WL, Akbarali HI. Differences in the characteristics of tolerance to μ-opioid receptor agonists in the colon from wild type and β-arrestin2 knockout mice. Eur J Pharmacol. 2012;685:133–140. doi: 10.1016/j.ejphar.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kenakin T. New concepts in drug discovery: collateral efficacy and permissive antagonism. Nat Rev Drug Discovery. 2005;4:919–927. doi: 10.1038/nrd1875. [DOI] [PubMed] [Google Scholar]
- 8.Kenakin T. Collateral efficacy in drug discovery: taking advantage of the good (allosteric) nature of 7TM receptors. Trends Pharmacol Sci. 2007;28:407–415. doi: 10.1016/j.tips.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 9.Kenakin T. Biased agonism. F1000 Biol Rep. 2009;1:87. doi: 10.3410/B1-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mailman RB. GPCR functional selectivity has therapeutic impact. Trends Pharmacol Sci. 2007;28:390–396. doi: 10.1016/j.tips.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2006;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 12.Bohn LM. Selectivity for G protein or arrestin-mediated signaling. In: Neve K, editor. Functional Selectivity of G Protein-Coupled Receptor Ligands. Humana Press; Totowa, NJ: 2009. pp. 71–85. [Google Scholar]
- 13.Raehal KM, Bohn LM. Beta-arrestins: regulatory role and therapeutic potential in opioid and cannabinoid receptor-mediated analgesia. Handb Exp Pharmacol. 2014;219:427–443. doi: 10.1007/978-3-642-41199-1_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, Bohn LM. An opioid agonist that does not induce mu-opioid receptor–arrestin interactions or receptor internalization. Mol Pharmacol. 2006;71:549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamb K, Tidgewell K, Simpson DS, Bohn LM, Prisinzano TE. Antinociceptive effects of herkinorin, a MOP receptor agonist derived from salvinorin A in the formalin test in rats: new concepts in mu opioid receptor pharmacology: from a symposium on new concepts in mu-opioid pharmacology. Drug Alcohol Depend. 2012;121:181–188. doi: 10.1016/j.drugalcdep.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hübner H, Huang XP, Sassano MF, Giguère PM, Löber S, Da Duan Scherrer, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, Shoichet BK. Structure-based discovery of opioid analgesics with reduced side effects. Nature. 2016;537:185–190. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, Violin JD. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- 18.Chen XT, Pitis P, Liu G, Yuan C, Gotchev D, Cowan CL, Rominger DH, Koblish M, Dewire SM, Crombie AL, Violin JD, Yamashita DS. Structure-activity relationships and discovery of a G protein biased mu opioid receptor ligand, [(3-methoxythiophen-2-yl)methyl]({2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan-9-yl]ethyl})amine (TRV130), for the treatment of acute severe pain. J Med Chem. 2013;56:8019–8031. doi: 10.1021/jm4010829. [DOI] [PubMed] [Google Scholar]
- 19.Soergel DG, Subach RA, Sadler B, Connell J, Marion AS, Cowan CL, Violin JD, Lark MW. First clinical experience with TRV130: pharmacokinetics and pharmacodynamics in healthy volunteers. J Clin Pharmacol. 2014;54:351–357. doi: 10.1002/jcph.207. [DOI] [PubMed] [Google Scholar]
- 20.Viscusi ER, Webster L, Kuss M, Daniels S, Bolognese JA, Zuckerman S, Soergel DG, Subach RA, Cook E, Skobieranda F. A randomized, phase 2 study investigating TRV130, a biased ligand of the μ-opioid receptor, for the intravenous treatment of acute pain. Pain. 2016;157:264–272. doi: 10.1097/j.pain.0000000000000363. [DOI] [PubMed] [Google Scholar]
- 21.Soergel DG, Subach RA, Burnham N, Lark MW, James IE, Sadler BM, Skobieranda F, Violin JD, Webster LR. Biased agonism of the μ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain. 2014;155:1829–1835. doi: 10.1016/j.pain.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 22.Healy JR, Bezawada P, Shim J, Jones JW, Kane MA, MacKerell AD, Jr, Coop A, Matsumoto RR. Synthesis, modeling, and pharmacological evaluation of UMB 425, a mixed mu agonist/delta antagonist opioid analgesic with reduced tolerance liabilities. ACS Chem Neurosci. 2013;4:1256–1266. doi: 10.1021/cn4000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson GL, Kelly E, Christopoulos A, Canals M. Novel GPCR paradigms at the μ-opioid receptor. Br J Pharmacol. 2015;172:287–296. doi: 10.1111/bph.12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, Granier S, Gmeiner P, Husbands SM, Traynor JR, Weis WI, Steyaert J, Dror RO, Kobilka BK. Structural insights into mu-opioid receptor activation. Nature. 2015;524:315–321. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau TK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich B, Smith J, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 27.Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, MacKerell AD., Jr Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi1 and chi2 dihedral angles. J Chem Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD., Jr CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abraham M, Murtola T, Schulz R, Páll S, Smith J, Hess B, Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. [Google Scholar]
- 31.Shaw D, Deneroff M, Dror R, Kuskin J, Larson R, Salmon J, Young C, Batson B, Bowers K, Chao JC, Eastwood M, Gagliardo J, Grossman J, Ho C, Lerardi J, Kolossváry I, Klepeis J, Layman T, McLeavey C, Moraes M, Mueller R, Priest E, Shan Y, Spengler J, Theobald M, Towles B, Wang S. Anton, A Special-Purpose Machine for Molecular Dynamics Simulation. Commun ACM. 2008;51:91–97. [Google Scholar]
- 32.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 33.Parrinello M, Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys. 1981;52:7182. [Google Scholar]
- 34.Hoover WG. Canonical dynamics: Equilibrium phase-space distributions. Phys Rev A: At Mol Opt Phys. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 35.Martyna G, Klein M, Tuckerman M. Nosé-Hoover chains: the canonical ensemble via continuous dynamics. J Chem Phys. 1992;97:2635–2643. [Google Scholar]
- 36.Shan Y, Klepeis J, Eastwood M, Dror R, Shaw D. Gaussian split Ewald: A fast Ewald mesh method for molecular simulation. J Chem Phys. 2005;122:054101. doi: 10.1063/1.1839571. [DOI] [PubMed] [Google Scholar]
- 37.Sander J, Ester M, Kriegel HP, Xu X. Density-based clustering in spatial databases: the algorithm GDBSCAN and its applications. Data Mining and Knowledge Discovery. 1998;2:169–194. [Google Scholar]
- 38.Scherer MK, Trendelkamp-Schroer B, Paul F, Perez-Hernandez G, Hoffmann M, Plattner N, Wehmeyer C, Prinz JH, Noé F. PyEMMA 2: A software package for estimation, validation, and analysis of Markov models. J Chem Theory Comput. 2015;11:5525–5542. doi: 10.1021/acs.jctc.5b00743. [DOI] [PubMed] [Google Scholar]
- 39.LeVine MV, Weinstein H. NbIT–a new information theory-based analysis of allosteric mechanisms reveals residues that underlie function in the leucine transporter LeuT. PLoS Comput Biol. 2014;10:e1003603. doi: 10.1371/journal.pcbi.1003603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glykos NM. Carma: a molecular dynamics analysis program. J Comput Chem. 2006;27:1765–1768. doi: 10.1002/jcc.20482. [DOI] [PubMed] [Google Scholar]
- 41.Gokhale D, Ahmed N. Entropy Expressions and Their Estimators for Multivariate Distributions. IEEE Trans Inf Theory. 1989;35:688–692. [Google Scholar]
- 42.Granier S, Kobilka B. A new era of GPCR structural and chemical biology. Nat Chem Biol. 2012;8:670–673. doi: 10.1038/nchembio.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mondal J, Friesner RA, Berne BJ. Role of desolvation in thermodynamics and kinetics of ligand binding to a kinase. J Chem Theory Comput. 2014;10:5696–5705. doi: 10.1021/ct500584n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tiwary P, Mondal J, Morrone JA, Berne BJ. Role of water and steric constraints in the kinetics of cavity-ligand unbinding. Proc Natl Acad Sci U S A. 2015;112:12015–12019. doi: 10.1073/pnas.1516652112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Young T, Hua L, Huang X, Abel R, Friesner R, Berne BJ. Dewetting transitions in protein cavities. Proteins: Struct Funct Genet. 2010;78:1856–1869. doi: 10.1002/prot.22699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, Shaw DE. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc Natl Acad Sci U S A. 2011;108:13118–13123. doi: 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katritch V, Cherezov V, Stevens RC. Structure-function of the G-protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, Roth BL, Stevens RC. Molecular control of delta-opioid receptor signalling. Nature. 2014;506:191–196. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klein Herenbrink C, Sykes DA, Donthamsetti P, Canals M, Coudrat T, Shonberg J, Scammells PJ, Capuano B, Sexton PM, Charlton SJ, Javitch JA, Christopoulos A, Lane JR. The role of kinetic context in apparent biased agonism at GPCRs. Nat Commun. 2016;7:10842. doi: 10.1038/ncomms10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.