

Abstract
Apolipoprotein L1 (APOL1) genetic variants are potent risk factors for glomerular disease, but one or more additional factors are required for expression of glomerular disease. Uncontrolled or poorly controlled HIV infection is the most potent susceptibility factor for APOL1 nephropathy that has been identified to date. APOL1 variants are associated with HIV-associated nephropathy (HIVAN), a podocyte disease, but not with HIV-immune complex disease, primarily a disease of the mesangium. The mechanism by which HIV brings out the latent glomerular disease risk remains to be defined. There are at least two classes of candidate mechanisms to explain the potent interaction between HIV-1 and APOL1. First, APOL1 variant proteins and HIV accessory proteins implicated in HIVAN may target the same or related intracellular pathways in podocytes. Recent data suggests roles for interleukin 1b and transcription factor EB (TFEB). Second, features of uncontrolled HIV-infection, including elevated circulating factors such as interferon, may drive APOL1 gene transcription or act upon podocytes in other ways. Deeper probing of APOL1-HIV interactions may yield insights that will aid understanding HIVAN, APOL1 nephropathy, and podocyte biology.
Keywords: Chronic kidney disease, interferon, Vpr, Nef
Introduction
APOL1 renal risk variants are strongly associated with kidney disease, and the strongest association that has been identified to date is with HIV-associated nephropathy. APOL1 risk variants are common, representing ~50% of alleles in West African populations and ~35% of alleles in African Americans; thus ~12% of African Americans have two risk alleles but only a small fraction, perhaps 15% or more will develop kidney disease during their lifetimes (Dummer et al. 2015). The strong association between HIV-1 infection and APOL1 nephropathy is important clinically wherever peoples of sub-Saharan descent live, and is important mechanistically in understand the pathophysiological mechanisms by which APOL1 variants damage the kidney.
APOL1 and hypertension-attributed chronic kidney disease, non-HIV FSGS and HIVAN
In 1984, early in the HIV epidemic, and years prior to widespread dissemination of combined anti-retroviral therapy (cART), the syndrome of HIVAN was noted to predominantly affect individuals of sub-Saharan Africa descent (Rao et al. 1984). A major advance was identification of two APOL1 genetic variants that explain much of this predilection (Genovese et al. 2010). These variants are termed G1 (a haplotype comprising two missense variants, S342G and I3484M) and G2 comprising a two amino acid deletion, 388NY.
Among the three common kidney diseases most strongly associated with APOL1 genetic variants, there is a hierarchy of increasing odds ratios (OR) for carriage of two variants (G1/G1, G1/G2 or G2/G2), with hypertension attributed chronic kidney disease (HA-CKD) < focal segmental glomerulosclerosis (FSGS) < HIVAN. In the AASK study, Lipkowitz and colleagues showed that for HA-CKD, which often correlates with arterionephrosclerosis on kidney biopsy (although kidney biopsies were not performed), the OR of having two risk alleles among was 2.6 (confidence interval 1.8, 3.6) for HA-CKD subjects compared to control subjects, and 19% of subjects had two risk alleles (Lipkowitz et al. 2013). Among the 35% of subject who had progressed to having a serum creatinine >3 mg/dL compared to control subjects, the OR was 4. This finding indicates that APOL1 risk variants are particularly associated with progressive kidney disease, a theme that recurs throughout the literature on APOL1 associations with chronic kidney disease.
The results of studies examining association of APOL1 and kidney disease in the setting of HIV infection are summarized in Table 1. In study from the USA, the odds ratio (OR) for non-HIV associated FSGS was 17 (95% confidence interval (CI): 11, 26) (Kopp et al. 2011); 72% of 217 subjects with FSGS had two risk alleles, compared to ~13% of individuals of the general population African American control subjects. Among 54 individuals with biopsy-proven HIVAN in the USA, the OR for carriage of two risk variants was 29 (95%CI: 13, 68) with 72% of HIVAN cases having two risk alleles and 8% of the hypernormal comparator group (subjects with HIV infection for >8 years, with normal serum creatinine and lacking proteinuria) having two risk APOL1 variants. While the absolute values for the OR differ between FSGS and HIVAN, the confidence intervals overlap. Further, for HIVAN, the comparator group has a lower allele frequency, as the authors were able to define a group with the exposure and without disease, in a way that was not possible to do for idiopathic FSGS (where the provocative factor is unknown) and this would tend to arithmetically increase the OR, as the HIV control group was predicted to be (and was) depleted in subjects with two APOL1 risk alleles. Thus, with regard to APOL1 genetic variants, the OR for FSGS and HIVAN are statistically similar in the USA. Atta and colleagues showed that among individuals with HIVAN, the presence of two risk alleles was not associated with differences in renal pathology or in renal survival, with median renal survival <12 months for those with and without 2 APOL1 risk alleles (Atta et al. 2012).
Table 1.
Prevalence of APOL1 risk alleles among HIV renal histologies.
| Disease | Country | N cases | APOL1 2 RA prevalence in reference population (HIV status) | APOL1 2 RA prevalence in disease | Odds ratio (CI) for kidney disease, when 2 APOL1 risk alleles are present | Reference |
|---|---|---|---|---|---|---|
| HIVAN | USA | 54 | 8% (HIV+) | 72% | 29 (14, 68) | (Kopp et al. 2011) |
| USA | 60 | ND | 62% | ND | (Atta et al. 2012) | |
| South Africa | 38 | 3.7% (HIV+) | 79% | 89 (18, 912) | (Kasembeli et al. 2015) | |
| HIVICK | USA | 31 | ND | 3% | ND | (Fine et al. 2012) |
| South Africa | 49 | 3.7% (HIV+) | 9% | 2.6 (NS) | (Kasembeli et al. 2015) | |
| HIV+ FSGS | USA | 35 | 8% (HIV+) | 63% | 20 (8, 45) | (Fine et al. 2012) |
| USA | 203-X | ND | X | 3 (1.5, 6) | (Atta et al. 2016) | |
| South Africa | 22 | 3.7% (HIV+) | 8% | 2.1 (NS) | (Kasembeli et al. 2015) | |
| HIV-negative FSGS | USA | 217 | 13% (HIV-) | 72% | 17 (11,26) | (Kopp et al. 2011) |
Shown are the results of studies that have examined the prevalence of APOL1 high-risk individuals (those carrying two APOL1 risk alleles) among HIV+ individuals with particular renal histologies. The rates of APOL1 two risk allele carriage for the control population are 13% in the general US African American population in the Kopp et al study, 4% in the general South African population in the Kasembeli et al study, and 8% among HIV+ African Americans lacking kidney disease in Kopp et al study. Odds ratio was calculated from data in Fine et al, using data from the HIV+ African American population, lacking kidney disease, taken from the Kopp et al study.
HIVAN, HIV nephropathy. HIVICK, HIV-associated immune complex kidney disease. ND, not determined. NS, not significant.
In a study from South Africa, comparing 38 individuals with HIVAN to 108 HIV-positive subjects with normal renal function, the OR for carrying two APOL1 risk alleles was 89 (95% CI: 18, 912) (Kasembeli et al. 2015). Due to the small sample size, the confidence intervals overlapped with those for HIVAN in the USA. The authors noted that this may be the highest OR yet reported for a genetic variant associated with a complex disease (i.e., one in which genetic and environmental factors combine to induce a phenotype). An important insight from this study is that a single copy APOL1 G1 risk allele (i.e., when present in a heterogyzous state with the G0 allele) is sufficient to increase the risk for HIVAN (Figure 1). This probably represents an effect of the G1 allele that is present in other populations, but that can only be demonstrated to reach statistical significance when the OR for the association of APOL1 variants with HIVAN is very high in a population, as is the case in South Africa.
Figure 1. Effect of APOL1 in NIH and South African cohorts.
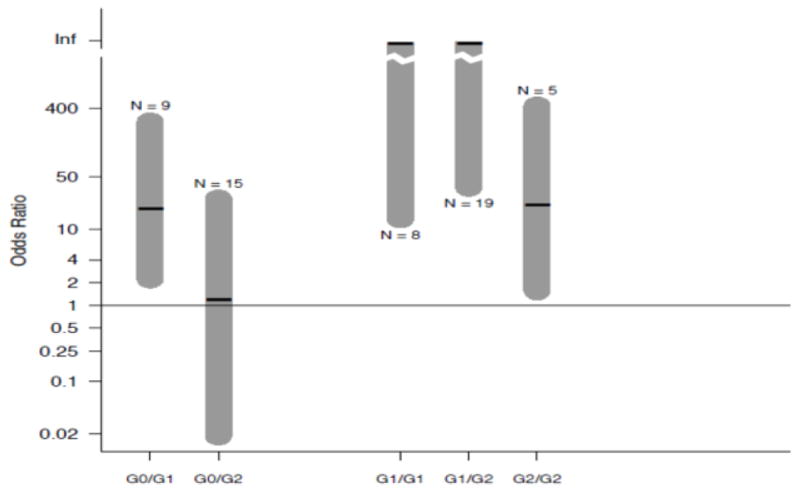
Shown are the odds ratios for APOL1 and HIVAN in South Africa. On the right are the associations with two APOL1 risk alleles. On the left are the associations with one risk APOL1 risk allele. Even with small sample size, an effect can be seen for a single copy of the G1 allele but not for the G2 allele, even though sample sizes were similar. Used with permission (Kasembeli et al. 2015).
Purswani and colleagues reported that for HIV+ youth, APOL1 high-risk status conferred an odd ratio for CKD of 4.4 (1.5, 11.2), but without kidney biopsy, it is not known how many of the subjects had HIVAN (Purswani et al. 2016).
The conclusion to be drawn from these studies is while there is a trend for the OR for carriage of two APOL1 risk variants to be higher in HIVAN than with FSGS, numerical differences have not been conclusively demonstrated due to modest sample sizes and features of study design particularly with regard to the characteristics of the control groups. Nevertheless, HIV is clearly a potent interactor with APOL1 risk alleles to induce glomerular injury.
APOL1 and other HIV-associated kidney diseases
APOL1 risk variants are also associated with FSGS that presents in the setting of HIV infection; contrasting factors associated with different renal disease are summarized in Table 2. Atta and colleagues showed that compared with HIV+ control subjects lacking both HIVAN and FSGS, the odds ratio for FSGS was 3.0 (95% CI 1.5, 5.8) (Atta et al. 2016). The distinction between collapsing FSGS (or collapsing glomerulopathy) and idiopathic FSGS is readily apparent in some cases and can be subtle in other cases. The former manifests podocyte hyperplasia, with an increase in cell number along the basement membrane, and/or the appearance of cells within Bowman space. These cells were previously believed to be dysregulated visceral epithelial cells (podocytes) (Barisoni et al. 1999). More recently have been proposed to be dysregulated parietal epithelial cells, possibly having progressed part of the way along the differentiation pathway to podocytes; evidence using fate-mapping mice support this theory (Shankland et al. 2014). Other characteristic features of HIVAN include microcystic tubular dilatation and an interstitial mononuclear cell infiltrate.
Table 2.
Demographic and clinical associations with HIVAN and HIVICK.
| HIVAN | HIVICK | |
|---|---|---|
| Black race | 10.3 (1.3, 80) | NS |
| HIV RNA >400 | 11.7 (1.2, 5.2) | 2.5 (1.2, 5.2) |
| Diabetes | NS | 2.8 (1.1, 6.8) |
| Hypertension | NS | 2.3 (1.2, 4.5) |
As demonstrated by Foy and colleagues, HIV-associated nephropathy (HIVAN) and HIV-associated immune complex disease (HIVICK) have distinct patterns of association with race and with the presence of other diseases (Foy et al. 2013).
By contrast, HIV-associated FSGS lacks prominent abnormalities in the podocyte, while manifesting segmental glomerular scars, and lacks microcystic tubular dilatation. The pathogenesis of HV-associated FSGS remains unclear. It has become more common following the widespread adoption of combined anti-retroviral therapy, which has been associated with HIV-positive individuals living longer and being at increased risk for obesity, metabolic syndrome, and metabolic diseases, including diabetes and atherosclerosis (Table 3). These individuals have systemic immune activation and a pro-coagulant state, both which may contribute to this suite of diseases and to premature aging. It is plausible that these processes predispose to FSGS, although the mechanistic pathway is largely unknown. In particular, they may predispose to adaptive FSGS, where the pathophysiology includes glomerular enlargement, likely on a hemodynamic basis, due to increased body size. It is also plausible that microvascular damage in the kidney, associated with metabolic disease and atherosclerosis, contributes to glomerular injury. The role of APOL1 variants in arterionephrosclerosis occurring in the setting of HIV infection deserves further investigation.
Table 3.
HIV histopathologies
| Without cART | With cART |
|---|---|
| HIVAN | Drug toxicities (tenofovir) |
| HIVICK | FSGS |
| Diabetic nephropathy | |
| Arterionephrosclerosis |
As shown, the most common renal histolopathologic findings have shifted as combined anti-retroviral therapies (cART) have come into wide-spread use.
HIV-associated immune complex kidney disease (HIVICK) may present with multiple immunoglobulin deposits in the glomerulus, or with IgA alone, and has lower predilection to progress to end-stage kidney disease (Foy et al. 2013). HIVICK is not associated with the APOL1 renal risk alleles (Fine et al. 2012; Kasembeli et al. 2015). Fine et al. noted that among 25 HIV+ subjects who underwent kidney biopsy and had zero APOL1 risk alleles, only 3 (12%) had FSGS while over 40% had HIVICK; the remainder had diabetic nephropathy or arterionephrosclerosis (Fine et al. 2012).
Mechanisms of APOL1 interaction with HIV: possible role of interferon
In a study of 333 African American men with HIV infection in the longitudinal Multicenter AIDS Cohort Study (MACS), Estrella and colleagues reported the rate of eGFR decline was faster in high-risk individuals (those with two APOL1 risk alleles) compared to low risk individuals, but only when viral load was not suppressed by anti-retroviral therapy (<400 copies/mL) (Estrella et al. 2015). Assuming that eGFR decline rate is a reasonable proxy for glomerular injury (proteinuria might be more sensitive), active viral replication may promote, or may even be essential for APOL1-mediated renal injury, or at least for collapsing glomerulopathy (HIVAN).
Plausibly, the link between viral replication, APOL1 genotype, and glomerular injury could be explained by either viral gene product(s) or by the host immune response to viral infection. There are no clinical studies that address interactions between viral gene products and APOL1, although there are basic science studies that explore this relationship, as discussed below; this would be fruitful area for future investigation.
There are several links between HIV infection and immune activation that may have relevance for understanding the role of APOL1. As an innate immune protein, APOL1 gene expression is driven by interferon, with potency γ > β > α interferon (Nichols et al. 2014). Interferon signaling activates STAT3, which occupies binding sites located within the APOL1 promoter. Acute and chronic HIV-1 infection is characterized by an intense pro-inflammatory state, including elevated levels of interferons in plasma and tissue (Doyle, Goujon, and Malim 2015). Among HIV+ African-American subjects, plasma levels of inflammatory markers interleukin-6, soluble tumor necrosis factor (TNF) receptors 1 and 2, chemokine (CC) motif ligand 2 (CC2), and beta2-microglobulin are higher among those with eGFR <60 ml/min/1.73m2 compared to those with higher eGFR. Among nephrotic subjects compared to others, plasma levels of CCL5 and interferon-α were also higher compared to those without kidney disease (Bruggeman et al. 2014). While kidney biopsies were not available in this study, these data do associate systemic markers of inflammation with kidney disease. There were no differences in cytokine levels among subjects with risk and non-risk APOL1 genotypes. Further, plasma APOL1 levels in HIV-positive subjects did not correlate with APOL1 genotype, CKD status, or plasma level of proinflammatory cytokines. It is remains possible that either brief or intermittent stimulation of interferon production predisposes to APOL1-induced glomerular injury (which would be difficult to detect even with a longitudinal study) and/or that plasma levels of APOL1 do not correlate closely with glomerular expression of APOL1.
Role of TLR4 signaling
There is emerging evidence that APOL1 renal risk variants enhance signaling downstream from toll-like receptor (TLR) 4 (Wakashin H and Kopp 2015). HIV infection is also associated with TLR4 signaling, suggesting synergy with APOL1 risk variants in promoting innate immune responses. HIV infection is associated with increased expression of TLR2 (but not TLR4) on freshly isolated monocytes and exposure to the HIV envelope protein gp120 further increased expression (Heggelund et al. 2004). A subsequent study confirmed these findings, and found that both TLR2 and TLR4 expression are increased in plasmacytoid dendritic cells obtained from HIV-positive subjects (Hernandez et al. 2012). Contrary findings, namely reduced TLR4 expression, have been reported for alveolar macrophages from HIV-positive subjects and in HIV-infected U1 monocytoid cells (Nicol et al. 2008), and thus the interplay between HIV infection and the TLR pathway is likely context dependent. The HIV regulatory protein Tat stimulates cytokine production in macrophages in a TLR4 dependent fashion (Ben Haij et al. 2015); whether it acts similarly in podocytes remains to be determined.
APOL1 as an HIV restriction factor
As noted above, APOL1 is an innate immune factor, known to target parasites, specifically Trypanosoma brucei. An intriguing recent observation by Popik and colleagues suggests that, in addition to its anti-parasite functions, that APOL1 acts as a viral restriction factor (Taylor, Khatua, and Popik 2014). In 293T human macrophages infected with HIV-1, overexpression of APOL1 reduced Gag p55 protein (core structural protein) and p24 protein (a capsid protein) by 90%. In human 937 monocytic cells stimulated by interferon-γ over-expression of APOL1 reduced production of infectious virus by 60–90%, depending on the particular HIV strain used.
APOL1 may exert these effects via two pathways, as shown by Popik and colleagues (Taylor, Khatua, and Popik 2014). First, APOL1 promotes nuclear translocation of transcription factor EB (TFEB), although the mechanism has not been defined. TFEB increases lysosomal biogenesis by increasing expression of genes encoding the lysosomal proteins LAMP1, LAMP2, and CD63, and thus increased endosomal activity. APOL1-mediated alterations in endolysosomal activity may be responsible for its anti-viral activity, as blockade of endolysosomal function suppressed the antiviral activity. Second, APOL1 reduced accumulation of the regulatory and accessory HIV proteins Nef, Vif, and Vpu (Tat and Vpr were not studied); these effects would be expected to disrupt the viral life cycle. In particular, Vif levels within APOL1-over-expessing cells were depleted by degradation in lysosomes and secretion from the cells in microvesicles, possibly exosomes. Finally, Popik and colleagues made a prediction that APOL1 might suppress mTOR activity, a hypothesis that is worth testing (Taylor, Khatua, and Popik 2014).
More recently, Mclaren and colleagues confirmed the anti-retroviral properties of APOL1 (McLaren et al. 2015). They started with a bioinformatics approach, seeking genes that are shared among at least some of 9 primate species and that showed evidence for evolutionary selection, i.e. an excess of synonymous over non-synonymous changing mutations. The underlying assumption of this analysis is that genes that have demonstrate more altered codons in a way that alters the encoded amino acid are likely undergoing these changes in response to selective pressure, rather than a result of a random process. The authors found that 841 (3.9%) of 21,389 primate genes showed strong evidence positive selection. They next identified 30 genes of this set that were up-regulated during HIV infection of (human) HEK393T cells or were known from previous work to interact with HIV proteins. They selected 27 genes not previously known to interact with HIV proteins, and found that following transient transfection, 16 gene products reduced HIV infection. Four of these gene products reduced retroviral infection by >50%: APOL1, APOL3, APOL6 and TNFRS10D. These data suggest that the APOL family members, in addition to having evolved to play an anti-parasite role, also have anti-viral roles and provide an important new perspective on this gene family. Interestingly, like APOL1, TNFRS10D expression is induced by interferon, as are APOL3 (or by the interferon-mimic poly-inosine-cytosine) (Uzureau et al. 2016) and APOL6 (Zhaorigetu et al. 2011).
APOL1 and the molecular and cellular mechanisms of HIVAN
Mclaren and colleagues also tested the ability of the HIV gene products to reduce HIV infection with a proviral construct that lacked a combination of three HIV proteins, the regulatory protein Nef and accessory proteins, viral protein R (Vpr) and viral protein U (Vpu). In the absence of these proteins, the inhibitory effect of 8 restriction factors including APOL1 and APOL6, was increased. The authors cautioned that since the restriction factors could efficiently inhibit even wild-type HIV constructs containing the full retroviral genome, the infection-enhancing of removing these HIV genes was modest. Nevertheless, these finding do suggest that there may be a functional interaction (not necessarily a molecular interaction) between APOL1 and one more of these HIV proteins. This finding is quite interesting, as data from transgenic mouse models have implicated two of these HIV gene products (from among the six HIV regulatory and accessory genes) as a cause of HIVAN: Nef (Simard et al. 2002) (Rahim et al. 2009; Zuo et al. 2006) and Vpr (Hiramatsu et al. 2007) (Zuo et al. 2006).
Singhal and colleagues have performed a series of informative experiments that illustrate how APOL1 variants exert toxicity, or enhanced toxicity, in cultured cells and provide insight into the pathogenesis of HIVAN. Previously they showed that an HIV construct lacking the gag and pol but encoding the six regulatory and accessory genes induces lysosomal injury in cultured human podocytes, with release of cathepsin L into the cytoplasm (Chandel et al. 2013). Recently, they studied human podocytes subjected to lentiviral infection to induce over-expression of each APOL1 genotype (Lan et al. 2014). They found that the risk variants increased lysosomal permeability in response to various injurious stimuli, the most potent was lentiviral delivery of the same HIV construct.
Most recently, Mikulak and colleagues studied the effect of APOL1 transfection on HIV infection of cultured podocytes (Mikulak et al. 2016). The APOL1 G0 variant reduced viral accumulation, while APOL1 risk variants increased viral accumulation. They found that IL-1b increased expression of the HIV receptor dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) in podocytes, and that HIV infection itself further increased Il-1 β expression, creating a positive-feedback loop enhancing infection. In APOL1-over-expressing cells, there was increased production of Il-1β, although there were no data on whether the cells with risk variants had further increased IL1- β production. They next examined the mechanisms of HIV accumulation within the cell. They found that the APOL1 risk variants, but not the common variant, increases mRNA levels of transcription factor EB (TFEB), the master regulator of lysosomal synthesis. They concluded APOL1 risk variants promote intracellular accumulation of HIV-1 through interfering with the endolysosomal function.
Conclusions
While HIVAN is becoming less frequent as cART has become widely available globally, other glomerular diseases associated with APOL1 are becoming more common in the HIV population. There are important lessons for glomerular disease to be learned from interactions among HIV proteins, the inflammatory consequences of HIV infection, and APOL1 variant driven gene expression.
Acknowledgments
This work was supported by the NIDDK Intramural Research Program, NIH, Bethesda, MD; by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research; and in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN26120080001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Conflict of interest statement and financial support for this work: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atta MG, Estrella MM, Kuperman M, Foy MC, Fine DM, Racusen LC, Lucas GM, Nelson GW, Warner AC, Winkler CA, Kopp JB. HIV-associated nephropathy patients with and without apolipoprotein L1 gene variants have similar clinical and pathological characteristics. Kidney international. 2012;82:338–43. doi: 10.1038/ki.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atta MG, Estrella MM, Skorecki KL, Kopp JB, Winkler CA, Wasser WG, Shemer R, Racusen LC, Kuperman M, Foy MC, Lucas GM, Fine DM. Association of APOL1 Genotype with Renal Histology among Black HIV-Positive Patients Undergoing Kidney Biopsy. Clin J Am Soc Nephrol. 2016;11:262–70. doi: 10.2215/CJN.07490715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barisoni L, Kriz W, Mundel P, D’Agati V. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 1999;10:51–61. doi: 10.1681/ASN.V10151. [DOI] [PubMed] [Google Scholar]
- Ben Haij N, Planes R, Leghmari K, Serrero M, Delobel P, Izopet J, BenMohamed L, Bahraoui E. HIV-1 Tat Protein Induces Production of Proinflammatory Cytokines by Human Dendritic Cells and Monocytes/Macrophages through Engagement of TLR4-MD2-CD14 Complex and Activation of NF-kappaB Pathway. PLoS One. 2015;10:e0129425. doi: 10.1371/journal.pone.0129425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggeman LA, O’Toole JF, Ross MD, Madhavan SM, Smurzynski M, Wu K, Bosch RJ, Gupta S, Pollak MR, Sedor JR, Kalayjian RC. Plasma apolipoprotein L1 levels do not correlate with CKD. J Am Soc Nephrol. 2014;25:634–44. doi: 10.1681/ASN.2013070700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel N, Sharma B, Husain M, Salhan D, Singh T, Rai P, Mathieson PW, Saleem MA, Malhotra A, Singhal PC. HIV compromises integrity of the podocyte actin cytoskeleton through downregulation of the vitamin D receptor. Am J Physiol Renal Physiol. 2013;304:F1347–57. doi: 10.1152/ajprenal.00717.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle T, Goujon C, Malim MH. HIV-1 and interferons: who’s interfering with whom? Nat Rev Microbiol. 2015;13:403–13. doi: 10.1038/nrmicro3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dummer PD, Limou S, Rosenberg AZ, Heymann J, Nelson G, Winkler CA, Kopp JB. APOL1 Kidney Disease Risk Variants: An Evolving Landscape. Semin Nephrol. 2015;35:222–36. doi: 10.1016/j.semnephrol.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrella MM, Li M, Tin A, Abraham AG, Shlipak MG, Penugonda S, Hussain SK, Palella FJ, Jr, Wolinsky SM, Martinson JJ, Parekh RS, Kao WH. The association between APOL1 risk alleles and longitudinal kidney function differs by HIV viral suppression status. Clin Infect Dis. 2015;60:646–52. doi: 10.1093/cid/ciu765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine DM, Wasser WG, Estrella MM, Atta MG, Kuperman M, Shemer R, Rajasekaran A, Tzur S, Racusen LC, Skorecki K. APOL1 risk variants predict histopathology and progression to ESRD in HIV-related kidney disease. J Am Soc Nephrol. 2012;23:343–50. doi: 10.1681/ASN.2011060562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy MC, Estrella MM, Lucas GM, Tahir F, Fine DM, Moore RD, Atta MG. Comparison of risk factors and outcomes in HIV immune complex kidney disease and HIV-associated nephropathy. Clin J Am Soc Nephrol. 2013;8:1524–32. doi: 10.2215/CJN.10991012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–5. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heggelund L, Muller F, Lien E, Yndestad A, Ueland T, Kristiansen KI, Espevik T, Aukrust P, Froland SS. Increased expression of toll-like receptor 2 on monocytes in HIV infection: possible roles in inflammation and viral replication. Clin Infect Dis. 2004;39:264–9. doi: 10.1086/421780. [DOI] [PubMed] [Google Scholar]
- Hernandez JC, Stevenson M, Latz E, Urcuqui-Inchima S. HIV type 1 infection up-regulates TLR2 and TLR4 expression and function in vivo and in vitro. AIDS Res Hum Retroviruses. 2012;28:1313–28. doi: 10.1089/aid.2011.0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiramatsu N, Hiromura K, Shigehara T, Kuroiwa T, Ideura H, Sakurai N, Takeuchi S, Tomioka M, Ikeuchi H, Kaneko Y, Ueki K, Kopp JB, Nojima Y. Angiotensin II type 1 receptor blockade inhibits the development and progression of HIV-associated nephropathy in a mouse model. J Am Soc Nephrol. 2007;18:515–27. doi: 10.1681/ASN.2006030217. [DOI] [PubMed] [Google Scholar]
- Kasembeli AN, Duarte R, Ramsay M, Mosiane P, Dickens C, Dix-Peek T, Limou S, Sezgin E, Nelson GW, Fogo AB, Goetsch S, Kopp JB, Winkler CA, Naicker S. APOL1 Risk Variants Are Strongly Associated with HIV-Associated Nephropathy in Black South Africans. J Am Soc Nephrol. 2015 doi: 10.1681/ASN.2014050469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, Friedman D, Briggs W, Dart R, Korbet S, Mokrzycki MH, Kimmel PL, Limou S, Ahuja TS, Berns JS, Fryc J, Simon EE, Smith MC, Trachtman H, Michel DM, Schelling JR, Vlahov D, Pollak M, Winkler CA. APOL1 Genetic Variants in Focal Segmental Glomerulosclerosis and HIV-Associated Nephropathy. Journal of the American Society of Nephrology: JASN. 2011;22:2129–37. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan X, Jhaveri A, Cheng K, Wen H, Saleem MA, Mathieson PW, Mikulak J, Aviram S, Malhotra A, Skorecki K, Singhal PC. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol Renal Physiol. 2014;307:F326–36. doi: 10.1152/ajprenal.00647.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkowitz MS, Freedman BI, Langefeld CD, Comeau ME, Bowden DW, Kao WH, Astor BC, Bottinger EP, Iyengar SK, Klotman PE, Freedman RG, Zhang W, Parekh RS, Choi MJ, Nelson GW, Winkler CA, Kopp JB. Apolipoprotein L1 gene variants associate with hypertension-attributed nephropathy and the rate of kidney function decline in African Americans. Kidney international. 2013;83:114–20. doi: 10.1038/ki.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren PJ, Gawanbacht A, Pyndiah N, Krapp C, Hotter D, Kluge SF, Gotz N, Heilmann J, Mack K, Sauter D, Thompson D, Perreaud J, Rausell A, Munoz M, Ciuffi A, Kirchhoff F, Telenti A. Identification of potential HIV restriction factors by combining evolutionary genomic signatures with functional analyses. Retrovirology. 2015;12:41. doi: 10.1186/s12977-015-0165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikulak J, Oriolo F, Portale F, Tentorio P, Lan X, Saleem MA, Skorecki K, Singhal PC, Mavilio D. Impact of APOL1 polymorphism and IL-1beta priming in the entry and persistence of HIV-1 in human podocytes. Retrovirology. 2016;13:63. doi: 10.1186/s12977-016-0296-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols B, Jog P, Lee JH, Blackler D, Wilmot M, D’Agati V, Markowitz G, Kopp JB, Alper SL, Pollak MR, Friedman DJ. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney international. 2014 doi: 10.1038/ki.2014.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol MQ, Mathys JM, Pereira A, Ollington K, Ieong MH, Skolnik PR. Human immunodeficiency virus infection alters tumor necrosis factor alpha production via Toll-like receptor-dependent pathways in alveolar macrophages and U1 cells. J Virol. 2008;82:7790–8. doi: 10.1128/JVI.00362-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purswani MU, Patel K, Winkler CA, Spector SA, Hazra R, Seage GR, 3rd, Mofenson L, Karalius B, Scott GB, Van Dyke RB, Kopp JB Hivaids Cohort Study Pediatric. Brief Report: APOL1 Renal Risk Variants Are Associated With Chronic Kidney Disease in Children and Youth With Perinatal HIV Infection. J Acquir Immune Defic Syndr. 2016;73:63–8. doi: 10.1097/QAI.0000000000001010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahim MM, Chrobak P, Hu C, Hanna Z, Jolicoeur P. Adult AIDS-like disease in a novel inducible human immunodeficiency virus type 1 Nef transgenic mouse model: CD4+ T-cell activation is Nef dependent and can occur in the absence of lymphophenia. J Virol. 2009;83:11830–46. doi: 10.1128/JVI.01466-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao TK, Filippone EJ, Nicastri AD, Landesman SH, Frank E, Chen CK, Friedman EA. Associated focal and segmental glomerulosclerosis in the acquired immunodeficiency syndrome. N Engl J Med. 1984;310:669–73. doi: 10.1056/NEJM198403153101101. [DOI] [PubMed] [Google Scholar]
- Shankland SJ, Smeets B, Pippin JW, Moeller MJ. The emergence of the glomerular parietal epithelial cell. Nat Rev Nephrol. 2014;10:158–73. doi: 10.1038/nrneph.2014.1. [DOI] [PubMed] [Google Scholar]
- Simard MC, Chrobak P, Kay DG, Hanna Z, Jothy S, Jolicoeur P. Expression of simian immunodeficiency virus nef in immune cells of transgenic mice leads to a severe AIDS-like disease. J Virol. 2002;76:3981–95. doi: 10.1128/JVI.76.8.3981-3995.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor HE, Khatua AK, Popik W. The innate immune factor apolipoprotein L1 restricts HIV-1 infection. J Virol. 2014;88:592–603. doi: 10.1128/JVI.02828-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzureau S, Coquerelle C, Vermeiren C, Uzureau P, Van Acker A, Pilotte L, Monteyne D, Acolty V, Vanhollebeke B, Van den Eynde B, Perez-Morga D, Moser M, Pays E. Apolipoproteins L control cell death triggered by TLR3/TRIF signaling in dendritic cells. Eur J Immunol. 2016;46:1854–66. doi: 10.1002/eji.201546252. [DOI] [PubMed] [Google Scholar]
- Wakashin H, Kopp JB. Apolipoprotein L1 has diverse RNA and protein isoforms and APOL1-B3 activates pro-inflammatory signaling. American Society of Nephrology annual meeting; San Diego, CA. 2015. [Google Scholar]
- Zhaorigetu S, Yang Z, Toma I, McCaffrey TA, Hu CA. Apolipoprotein L6, induced in atherosclerotic lesions, promotes apoptosis and blocks Beclin 1-dependent autophagy in atherosclerotic cells. The Journal of biological chemistry. 2011;286:27389–98. doi: 10.1074/jbc.M110.210245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Matsusaka T, Zhong J, Ma J, Ma LJ, Hanna Z, Jolicoeur P, Fogo AB, Ichikawa I. HIV-1 genes vpr and nef synergistically damage podocytes, leading to glomerulosclerosis. J Am Soc Nephrol. 2006;17:2832–43. doi: 10.1681/ASN.2005080878. [DOI] [PubMed] [Google Scholar]