

Abstract
Glucose oxidase has wide applications in the pharmaceutical, chemical, and food industries. Many recent studies have enhanced key properties of this enzyme using directed evolution, yet without being able to reveal why these mutations are actually beneficial. This work presents a synergistic combination of experimental and computational methods, indicating how mutations, even when distant from the active site, positively affect glucose oxidase catalysis. We have determined the crystal structures of glucose oxidase mutants containing molecular oxygen in the active site. The catalytically important His516 residue has been previously shown to be flexible in the wild-type enzyme. The molecular dynamics simulations performed in this work allow us to quantify this floppiness, revealing that His516 exists in two states: catalytic and noncatalytic. The relative populations of these two substates are almost identical in the wild-type enzyme, with His516 readily shuffling between them. In the glucose oxidase mutants, on the other hand, the mutations enrich the catalytic His516 conformation and reduce the flexibility of this residue, leading to an enhancement in their catalytic efficiency. This study stresses the benefit of active site preorganization with respect to enzyme conversion rates by reducing molecular reorientation needs. We further suggest that the computational approach based on Hamiltonian replica exchange molecular dynamics, used in this study, may be a general approach to screening in silico for improved enzyme variants involving flexible catalytic residues.
Keywords: molecular dynamics, Hamiltonian replica exchange, X-ray, enzyme floppiness, active-site preorganization, side-chain dynamics, anticorrelated motions
Introduction
Glucose oxidase (GOx) from Aspergillus niger is a β-d-glucose specific flavoprotein oxidase (EC 1.1.3.4) that efficiently catalyzes substrate conversion to d-glucono-δ-lactone. Due to its diverse potential applications in the fields of clinical, pharmaceutical, chemical and food industries, which reach far beyond the glucose biosensors typically used for blood sugar diagnostics, GOx has gained remarkable economic importance.1 In this respect, flavoprotein oxidases are generally attractive biocatalysts due to their high regio- and stereoselectivity and the ability to use molecular oxygen as an oxidizing agent.2 Furthermore, increasing the catalytic activity and stability of enzymes is a persisting necessity for many industrial applications.
The nondeterministic nature of evolution, both natural and directed, provides multiple uphill paths on the fitness landscape of an enzyme; most pathways, however, lead downhill.3,4 As some enzymes sacrifice their catalytic power for metabolic control or live under low evolutionary pressure, the location of a natural enzyme on the fitness landscape is not necessarily at the global optimum.5,6 Another constraint in enzyme evolution is diminishing returns: as an enzyme approaches its theoretical limit on the landscape, mutations keep having smaller additive benefits. The gain in one property often has a high cost for another (e.g., the apparent stability–activity tradeoff), and nature usually does not pay the price of complete catalytic optimization.7,8 GOx was postulated to be an “ideal enzyme” for biosensors because it fulfills three important criteria: high specificity, turnover, and stability.9 Although GOx is several orders of magnitude less efficient than the “perfect enzyme” triosephosphate isomerase, where the reaction is diffusion limited,5 GOx has a much higher rate constant than other oxidases, leading to its label “the ‘Ferrari’ of the oxidases”.10 The high efficiency and selectivity suggest that GOx is a highly-evolved enzyme that lies close to its catalytic limit. This is supported by several mutagenesis studies that managed to achieve only marginal improvements of the kinetic properties of GOx (e.g., up to around 5 times higher kcat or lower KM).11−15 However, despite the quite modest improvements in comparison to the wild-type enzyme (WT),15 their impact in relation to the multimillion dollar industry involving applications of GOx is still important.
In this work, we study several improved GOx mutants that were recently derived using a combination of directed evolution and ultrahigh-throughput screening.15 Since relatively unspectacular mutations, far from the active site, were responsible for the observed catalytic enhancement, we aimed to find out the underlying rationale for their improvement and studied them in more detail. GOx is described to operate by a ping-pong bi-bi mechanism, where the first step (reductive half-reaction) involves a concerted proton and hydride transfer from the anomeric carbon of glucose respectively to His516 and the N5 atom of the flavin adenine dinucleotide (FAD) cofactor (Figure 1a).16 Although not directly involved in the reaction mechanism, Glu412 and His559 are thought to act as a buffer for controlling the reactivity of the active site by maintaining the proper acidity (Figure 1b).16 The protonation state of His516 is crucial for the subsequent oxidative half-reaction,17 as it leads to increased oxygen binding and reactivity via stepwise single-electron transfers.10
Figure 1.

(a) In the reductive half-reaction, glucose binding is followed by concerted proton and hydride transfer from the C1 carbon of glucose to His516 and FAD, respectively. Electrons are then transferred, in the oxidative half-reaction, from reduced FAD to oxygen in two single-electron-transfer steps. (b) The active site of glucose oxidase from A. niger is buried in a pocket, and it is defined by Glu412, His516, His559, and FAD, which are shown as sticks, together with glucose, and colored by atom type (gray, C; blue, N; red, O; white, H; orange, P). The rest of the protein is shown in gray cartoon, and hydrogen bonds are indicated by dashed yellow lines.
The catalytic ability of an enzyme originates mostly from the stabilization of the transition state geometry for ligand conversion,18 where binding is based on shape and electrostatic complementarity.19 There are three main factors that can lead to suboptimal enzyme kinetics: (1) a slow chemical step, (2) dissociation of the initial enzyme–substrate encounter complexes before the advanced enzyme–substrate complex is being established, and (3) floppiness, that is, the coexistence of multiple closely related enzyme substates (e.g., multiple side chain rotamers), of which only some are productive.6,20 Floppiness increases the ratio of nonproductive to productive substates, leading to more futile enzyme–substrate encounters, which negatively affects enzymatic rates, an effect also observed by heating an enzyme.21 Therefore, a preorganized and rigid active site (i.e., a single substate) leads to the most efficient chemical step.6,22−26 The significance of productive and nonproductive substates in enzyme ensembles was recently shown, for example, for T4 lysozyme,27 cyclophilin A,28 and α-esterase 7.29 As a consequence of this active site preorganization, the relative residue rigidity has often been found to be higher for the catalytic than for the noncatalytic amino acids.22 Atomistic molecular dynamics (MD) simulations were shown to be a useful method for analyzing the floppiness of an enzyme and determining how mutations affect the enzyme substates.30−32 Thus, they nicely complement the analysis of static crystal structures by also describing the underlying dynamics of the protein.
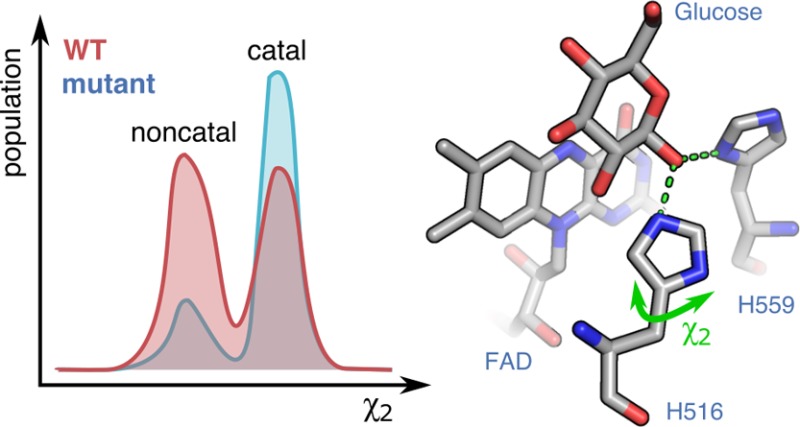
The aim of the current work is to understand the catalytic properties of GOx by studying several variants with improved catalytic activities and to create a basis for further improvement of the catalytic efficiency of this important enzyme. We report the very first crystal structures for GOx mutants, all bearing a molecule of oxygen in the active site. In addition to X-ray crystallography, we used MD simulations to corroborate our conclusions drawn from the crystal structures and to investigate the effects of the distant GOx mutations on the protein dynamics. Using Hamiltonian replica exchange MD, we further explored the conformational ensemble of the active site’s His516 that was previously reported as flexible in the wild-type enzyme.33,34 Our results provide structural and dynamic proofs that His516 is indeed flexible in the WT, where it can flip between catalytic and noncatalytic conformations, while in the most active mutant, A2, His516 is apparently locked in the catalytically active conformation.
Materials and Methods
Purification and Deglycosylation
The GOx mutants A2 and F9 (Table 1) were expressed in Pichia pastoris strain KM71H (Invitrogen) according to the manufacturer’s protocol. After 4 days of fermentation, the supernatant was concentrated to 20 mL on a Viva Flow 50 system (Sarotius) with a 50 kDa ultrafiltration membrane. The concentrate was dialyzed against 10 mM phosphate buffer (pH 6.0) overnight at 4 °C and loaded onto a 20 mL Fast Flow DEAE Sepharose column (GE Healthcare). The protein was purified using a linear gradient from 10 to 250 mM phosphate buffer (pH 6.0) over 12 column volumes. The GOx peaks were pooled together and concentrated to 1 mL using 10 kDa ultrafiltration columns. The enzyme solution was dialyzed against 50 mM sodium acetate buffer (pH 5.5) overnight at 4 °C.
Table 1. Mutations Present in the Simulated GOx Variants.
| GOx | mutations | ||||||
|---|---|---|---|---|---|---|---|
| P | T30V | I94V | A162T | ||||
| Pk | T30V | I94V | A162T | R537K | |||
| Pv | T30V | I94V | A162T | M556V | |||
| A2 | T30V | I94V | A162T | R537K | M556V | ||
| F9 | T30V | R37K | I94V | V106I | A162T | M556V | |
GOx deglycosylation was performed by incubating the protein solution with Endo H enzyme (30 U mg–1, NEB) for 20 h at 37 °C. The deglycosylated samples were loaded on a 120 mL Hi Load Superdex 75 gel filtration column using 10 mM sodium acetate buffer (pH 5.5) supplemented with 50 mM NaCl. The fractions with GOx activity were collected and concentrated to 25 mg mL–1 on a 10 kDa ultrafiltration column (Millipore).
GOx Crystallization
The concentrated solution was filtered through a 0.1 μm centrifugal filter (Millipore), and crystal growth conditions were screened initially using Hampton Screens I and II with the vapor diffusion sitting drop method on TAORAD crystallization plates. The first screening revealed that 1,4-dioxane is suitable to promote the crystallization of GOx, and optimal conditions were 100 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.0) in 40% 1,4-dioxane or 100 mM HEPES (pH 7.5) in 45% 1,4-dioxane. Crystals were picked from the droplets using cryo-loops and equilibrated stepwise for several seconds in the crystallization buffer containing increasing concentrations of polyethylene glycol (PEG400) or glycerol in order to cryoprotect the crystal before flash-freezing in a liquid nitrogen stream at −173 °C. The data sets were collected in house, using a Bruker FR591 rotating anode X-ray generator and a Mar345dtb detector.
The collected data were analyzed and processed using the software iMOSFLM, Pointless and scaled using SCALA, all belonging to the CCP4 suite.35 In order to solve structures of the F9 and A2 mutants, molecular replacement was carried out using the structure of the wild-type GOx from A. niger (PDB ID: 3QVP).36 All residues mutated in A2 or F9 were replaced by alanine residues in 3QVP for the model generation using Chainsaw. Molecular replacement was done using Molrep, and final refinement was carried out by iterative steps of modeling/refinement cycles with WinCoot37 and Refmac5.35 The Ramachandran plot analysis was performed with Rampage.38
Molecular Dynamics
Molecular dynamics simulations were performed for the WT (PDB ID: 1CF3),33P, A2 (PDB ID: 5NIT), and F9 (PDB ID: 5NIW) GOx variants. Two additional mutants were considered, Pk and Pv, to examine the effect of single mutations on P (Table 1). In the absence of crystal structures, starting coordinates of the P, Pk, and Pv mutants were obtained by reverting the corresponding residues in A2 GOx to a rotamer present in the wild-type enzyme. All structures were simulated in the oxidized form as holoenzyme (GOx + FAD) in complex with β-d-glucopyranose. The missing heavy atoms in the A2 and F9 structures were built using MODELER 9.14.39
Glucose was docked into the active site using AutoDock Vina.40 A binding mode positioned on the re face of FAD (Figure 1b) was chosen for further modeling, as this is the most reasonable mode according to the catalytic mechanism and as it resembles the previously proposed substrate position.33 In this orientation, the glucose H atom at the anomeric carbon C1 is directed toward the N5 atom of the isoalloxazine moiety of FAD, while the hydroxyl hydrogen from the same glucose carbon is oriented toward His516 and His559.
The β-d-glucopyranose and FAD topologies were created using ACPYPE41 and Antechamber.42 The glucose structure was optimized with Gaussian 0943 at the B3LYP/6-31G* level of theory, followed by the calculation of restrained electrostatic potential (RESP) charges at the HF/6-31G* level. The FAD charges were obtained from Todde et al.44
MD simulations were performed using the GROMACS 4.6.7 suite,45 with the Amber 99SB-ILDN force field46 and TIP3P explicit water.47 Hydrogen atoms were added, and the protonation states of all titratable residues were assigned on the basis of a PROPKA 3.1 analysis48 corresponding to a pH of 5.5, which is optimal for GOx activity. A disulfide bridge was defined between the Cys164 and Cys206 side chains. The protein was centered in a truncated octahedral box, at least 10 Å away from each of the box edges, and solvated with around 22000 water molecules. The net charge of the system was neutralized with sodium ions. The system was minimized in two stages: an initial minimization with steepest descent (maximum force of 500 kJ mol–1 nm–1), followed by a minimization with the conjugate gradient algorithm (maximum force of 100 kJ mol–1 nm–1).
Periodic boundary conditions were applied, and electrostatic interactions were treated using the particle mesh Ewald method.49 The cutoff distance for the short-range nonbonded interactions was 12 Å. An integration step of 2.0 fs was used, and bonds were constrained with the LINCS algorithm.50 The minimized system was gradually heated and equilibrated at 25 °C for 100 ps in the NVT ensemble with the protein and ligands restrained using a positional restraint force constant of 1000 kJ mol–1 nm–2. Following the equilibration under a constant volume, two stages of NpT equilibration were carried out. In the first phase, a 2 ns equilibration was performed with restraints on the protein and ligands. A second, 8 ns equilibration followed, with restrained protein backbone and FAD motion while glucose was free to move.
Production MD simulations were carried in the NpT ensemble for 100 ns (three independent simulations were performed for each GOx variant), collecting coordinates of the system every 20 ps. The modified Berendsen (v-rescale) thermostat51 and the Parrinello–Rahman barostat52 were employed. The production MD sampling time accumulated over all GOx variants amounted to 1.8 μs.
Hamiltonian Replica Exchange MD
The Hamiltonian replica exchange MD (HREX-MD) simulations were performed using GROMACS 4.6.7 in combination with the Plumed 2.1 plugin,53 as implemented by Bussi.54 The same conditions were applied as in the standard MD simulations. Four replicas were simulated for each GOx variant, where only the energy terms (i.e., the Hamiltonian) affecting His516 were scaled. The Hamiltonian scaling factors were exponentially distributed between 1.00 and 0.67 (exact scaling factors were 1.000, 0.874, 0.763, and 0.667), which corresponds to temperatures between 25 and 174 °C. The exchange of replicas was attempted every 4 ps during the 50 ns simulations. The exchange acceptance ratio was 30–70% in all HREX-MD simulations; only the Pv variant had an exchange rate of ∼15%. The sampling time accumulated over all HREX-MD simulations was 1.2 μs. Structures sampled for the HREX-MD simulation with the unperturbed Hamiltonian were used for the analysis.
Umbrella Sampling MD
The umbrella sampling (US-MD) simulations were performed using GROMACS 5.1.2 in combination with the Plumed 2.2 plugin. The same conditions were applied as in the previous MD simulations. The sampling was performed for the varying χ2 dihedral of His516 in the range of 25–235°, over 26 windows; exact χ2 restraining values are shown in Table S1 in the Supporting Information. The χ1 dihedral was restrained to 285° with a weak force of 50 kJ mol–1 rad–1 to keep this angle in the g– geometry. Each window was simulated for 50 ns. Dihedral angles were written every 0.5 ps, and the first 5 ns was discarded for the potential of mean force (PMF) calculations. The PMF was examined for the WT and A2 GOx, amounting to 2.6 μs US-MD sampling time. The PMF was calculated using the weighted histogram analysis method (WHAM),55 where the error was estimated using the blocking procedure.56 Briefly, each window from a US-MD simulation was split into 10 segments (i.e., blocks) of increasing length, ranging from 1 ns for the shortest block up to the full 45 ns per window for the longest block. The PMF was calculated for each block, and all PMFs were aligned to the final point at χ2 = 235°. The blocked standard error was calculated for each window using data from all 10 blocks.
Data Analysis
GROMACS tools, VMD 1.9.157 and MATLAB R2015b were used for the trajectory analysis. The dynamic cross-correlated motion analysis was performed in R 3.2.5 using the Bio3D package.58 The active site volumes were calculated with POVME.59 PyMOL60 and Chimera61 were used for figure rendering.
Results and Discussion
Crystallization and Structure Determination
In previous work,15 several GOx mutants with improved activity and stability were identified. The A2 mutant shows the highest catalytic activity, while the mutant F9 has the highest thermal stability. The first crystallization experiments were carried out using glycosylated GOx expressed in P. pastoris. Hampton Screens I and II were tested, but no promising crystallization conditions were found. Deglycosylation has been previously shown to be important for crystallization,62 as the process of crystallization demands highly uniform macromolecules. The glycosylation in P. pastoris is characterized by a uniform N-acetylglucosamine (NAG) core glycosylation with NAG-β(1,4)-NAG, followed by a heterogeneous glycosylation of a high mannose (β-d-mannose, BMA) content.63 In order to obtain uniform GOx molecules, the carbohydrate moieties were removed by enzymatic hydrolysis using glycosidases.
The crystallization experiments with the deglycosylated GOx were successful for the A2 and F9 GOx variants. The first screening using Hampton Screens I and II revealed that 1,4-dioxane is suitable to promote the crystallization of GOx. Further fine screening with different buffers, pHs, and 1,4-dioxane concentrations indicated that the crystallization works best using 100 mM HEPES (pH 7.0) in 40% 1,4-dioxane or 100 mM HEPES (pH 7.5) in 45% 1,4-dioxane. Regarding incompatibilities of classic crystallization plates, vapor diffusion crystallization was performed using TAORAD crystallization plates in their sitting drop configuration. The A2 and F9 GOx crystals grew in the form of long thick needles and showed an intense yellow color. The crystal growth took three to 5 days at room temperature and yielded crystals in the P3221 space group (Table 2). The cell content analysis gave a probability of 0.99 that one molecule is present per asymmetric unit with a water content of 57%.
Table 2. Data Collection and Refinement Statistics (Molecular Replacement)a.
| A2 (5NIT) | F9 (5NIW) | |
|---|---|---|
| Data Collection | ||
| space group | P3221 | P3221 |
| cell dimens | ||
| a, b, c (Å) | 128.7, 128.7, 77.7 | 128.1, 128.1, 77.7 |
| α, β, γ (deg) | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 |
| resolution range (Å) | 45.3–1.9 | 42.0–1.8 |
| Rmerge (%) | 13.6 (4.2/76) | 15.7 (3.8/82) |
| I/σ(I) | 12.6 (34.8/2.6) | 12.1 (29.8/2.8) |
| completeness (%) | 98.1 (99.8/88.1) | 98.3 (99.8/88.7) |
| redundancy | 8.2 (10.2/6.6) | 11.1 (11.1/10.4) |
| Refinement | ||
| resolution (Å) | 1.86 | 1.80 |
| no. of unique rflns | 61296 (2106/7940) | 67817 (2333/8856) |
| Rwork/Rfree (%) | 16.5/20.4 | 15.4/19.1 |
| no. of water molecules | 427 | 364 |
| B factors | ||
| protein | 19.6 | 24.2 |
| FAD | 15.6 | 19.4 |
| water | 28.9 | 33.3 |
| RMS deviations | ||
| bond lengths (Å) | 0.019 | 0.022 |
| bond angles (deg) | 1.94 | 2.20 |
| Ramachandran plot | ||
| favored region | 561 | 557 |
| allowed region | 18 | 17 |
| outlier region | 0 | 0 |
Values in parentheses are for the lowest- and highest-resolution shells.
The N-linked Asn glycosylation is related to specific motifs, i.e., Asn-X-Ser or Asn-X-Thr, where X can be any amino acid except Pro.64 Eight possible glycosylation sites (Asn43, Asn89, Asn161, Asn168, Asn258, Asn355, Asn388, and Asn473) are present in the GOx sequence. In the crystal structures, GOx was Asn-glycosylated at all sites, except Asn43. Interpretable electron density was observed for mutant A2 at four sites (89, 161, 355, and 388) and in mutant F9 at six sites (89, 161, 258, 355, 388, and 473), whereas the two additional sites showed a less pronounced electron density. In most cases, a single NAG moiety remained at each of these positions, while the deglycosylation removed the other initially present carbohydrates. At Asn89, the electron density indicates the presence of the core glycosylation, Asn89-NAG-NAG-BMA, which was not pruned by Endo H due to steric hindrance. Asn89 is located at the homodimeric interface of the GOx dimer, and BMA-rich glycosylation protrudes out of the dimer cleft. As the glycosylation is involved in the intermolecular interactions, it promotes the dimer state.
The cocrystallization approaches with d-glucose and d-glucal did not yield crystals. Soaking experiments were also unsuccessful and caused crystal degradation or did not yield visible ligand electron densities.
Protein Flexibility and Dynamics
To test the influence of the mutations on the protein dynamics, we performed MD simulations of several GOx variants. Figure S1 in the Supporting Information, which shows the root-mean-square deviation of the protein backbone motion, indicates that the WT and GOx mutants are stable during the 100 ns trajectories. A more detailed analysis of the enzyme dynamics reveals that the laboratory evolution of GOx led to a slight decrease in residue flexibility, especially in the active site region. This can be inferred from the associated standard deviations reflecting the change of the active site volumes (Table 3). Namely, the A2 mutant has a much smaller deviation than the WT enzyme, indicating a less flexible active site in A2. On the other hand, the residual root-mean-square fluctuations (Figure S2 in the Supporting Information) reveal a notable destabilization of the β-sheet D (residues 77–81, 93–97, 434–438, and 448–451) in the glucose binding domain of all mutants, which is caused by the I94V mutation lying at this β-sheet (Figure 2a). This mutation is present in all mutants, including the parent P. The F9 variant is much less flexible than the other tested mutants, which has a positive effect on its thermostability but makes the enzyme less efficient than P (Table 3). These results are in agreement with Fraser et al., who suggested that, although mutations should be directed toward a more rigid active site, second-shell residues should be flexible to ensure the efficiency of the numerous steps involved in catalysis.65
Table 3. Selected GOx Variants: Wild Type (WT), Parent (P), and Two Well-Performing Mutantsa.
| GOx | KM (mM)b | kcat (s–1)b | kcat/KM (mM–1 s–1)b | t1/2 (min)b | ⟨Vas⟩ (Å3) |
|---|---|---|---|---|---|
| WT | 28.26 ± 1.15 | 189.38 ± 8.94 | 6.7 | 10.50 ± 0.71 | 261.9 ± 103.6 |
| P | 14.98 ± 0.51 | 291.82 ± 10.10 | 19.5 | 9.00 ± 0.70 | 238.3 ± 77.8 |
| A2 | 18.54 ± 0.57 | 498.34 ± 15.12 | 26.9 | 11.74 ± 0.30 | 188.7 ± 65.2 |
| F9 | 19.76 ± 0.54 | 345.16 ± 14.79 | 17.5 | 15.75 ± 0.71 | 239.3 ± 73.0 |
Figure 2.

(a) A2 GOx crystal structure with glucose docked into the active site. The secondary structures involved in the anticorrelated motions are shown in yellow and pink, and the positions of mutations are designated by cyan spheres. His516, FAD, and glucose are shown by sticks and colored by atom type (see Figure 1 for the color code). (b) Dynamic cross-correlation maps (DCCMs) of the WT GOx (top half) and the A2 mutant (bottom half). The most discriminating regions are indicated by black rectangles: solid lines for anticorrelated and dashed lines for correlated motions. DCCMs for all GOx variants are given in Figure S3 in the Supporting Information. (c) Per-residue count of the correlated and anticorrelated motions (based on a cutoff of ±0.3) in the WT and A2 GOx. Correlated and anticorrelated motions are shown in red and blue, respectively. The positions of the mutations are represented with × and that of His516 with ★. The count plots for all variants are given in Figure S4 in the Supporting Information.
Another aspect of enzyme dynamics is the correlated nature of residue motion that facilitates many biochemical processes.66 Anticorrelated motions were previously related to enhanced catalysis in several enzymes.67,68 While we observe a general increase in both correlated and anticorrelated motions over the course of the GOx evolution (Figure 2b,c and Figures S3 and S4 in the Supporting Information), anticorrelated motions are particularly discriminating among the studied GOx mutants and the WT. Starting from the parent mutant P, the aforementioned I94V on β-sheet D, together with the T30V mutation located at helix H1, which is close to the phosphate groups of FAD, plays a very important role for anticorrelated motions in GOx. Namely, the motions of β-sheet D and α-helix H6 are anticorrelated to the motions of β-sheet C (glucose binding domain) and α-helices H1 (FAD binding domain) and H12, which extends to the active site’s His516 (Figure 2a). This kind of ordered motion is, to a varying degree, visible in all variants. The R537K mutation, although located on the surface, has a positive influence on the magnitude of both correlated and anticorrelated motions in the Pk and A2 variants (Figures S3 and S4). It further strengthens the anticorrelated motions already observed in the P mutant, while its effects on the correlated motions are especially high for the β-sheet C of the glucose binding domain (residues 211–213, 330–338, 347–353, 409–416, 420–427, and 484–489). From the Pv, A2, and F9 variants it can be seen that the M556V mutation, which is close to the active site, exhibits a positive effect on the anticorrelated and, even more, on the correlated motions of the same region as influenced by R537K (i.e., the β-sheet C; see Figures S3 and S4).
In the pentamutant A2, the mutations work together to considerably enhance both correlated and anticorrelated motions, as shown in Figure 2b,c. A principal component analysis of the fluctuations of the pairwise distances between the residues performing highly anticorrelated motions in A2 indicates that such motions contribute to the creation of a tighter active site in this variant (Figure S5 in the Supporting Information), which increases the probabilities of the contacts that directly stabilize the substrate in the proper position for the reaction to take place. This leads to the optimal orientation of the reactive atoms, which enhances catalysis and also lowers the KM value 1.5-fold. It should be noted that the nature of glucose binding by the WT and mutant GOx does not change, as the same residues are always involved (Figure S6 in the Supporting Information). However, this figure also shows that the mutations changed the priorities of certain residues in stabilizing the substrate in the active site.
His516 Conformational Ensemble in GOx Crystals
The structures of A2 (PDB ID: 5NIT) and F9 (PDB ID: 5NIW) are similar to those of the wild-type GOx. Major differences in these structures exist only at the active site. For the first time, we see an important electron density situated between His516 and FAD and interpret it as a molecule of oxygen (Figure 3). A water molecule, present in all A. niger GOx structures apart from 1GAL, bridges the Nε of His516 and the N5 atom of FAD (HOH1000 in A2) and is between 2.74 and 2.89 Å distant from His516.
Figure 3.
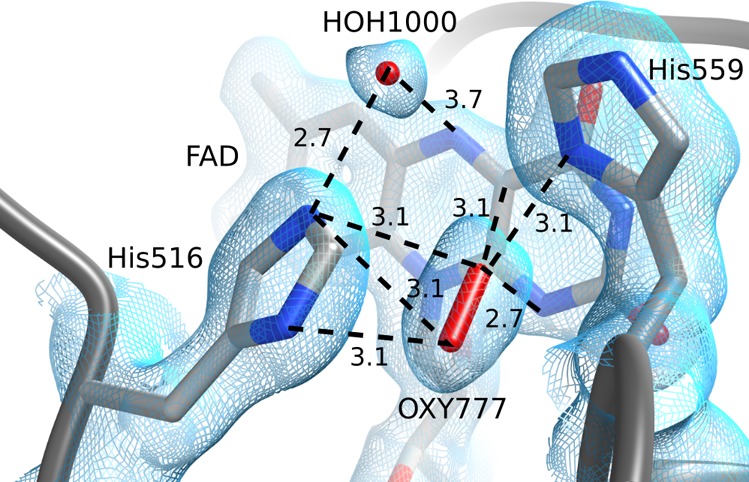
Structural view of the FAD re face of the A2 mutant, showing the catalytically important His516 and His559 residues. A water molecule (HOH1000) interacts with His516 and is oriented toward the N5 nitrogen of FAD. An oxygen molecule (OXY777) is well centered with respect to His516. The electron density is shown as a cyan mesh, and important distances (in Å) are indicated by black dashed lines.
In all of the structures from A. niger, the side chain of His516 populates the broadly defined (g–, Nt) rotamer with dihedral angles 240° < χ1< 360° and 150° < χ2 < 210° (Table 4). In the 1CF3 structure, where the His516 side chain deviates the most from the center of the (g–, Nt) rotamer population, the water molecule in the active site follows the His516 motion toward the tip of the oxygen molecule that is present in the A2 and F9 structures (Figure 4). Apart from movements within the (g–, Nt) rotamer observed in the wild-type A. niger structures, the (g–, Ng+) rotamer (240° < χ1< 360° and 30° < χ2< 90°) is structurally documented in GOx of Penicillium amagasakiense (PDB ID: 1GPE,33Table 4), where a water molecule is bridging His520 and His563, which are equivalent to His516 and His559, respectively, in GOx of A. niger. The (g–, Nt) rotamer of His516 is the geometry necessary for the proton transfer from glucose to occur and will be therefore denoted catalytic conformation. The same conformation of this conserved residue can be observed in many members of the superfamily, e.g., in aryl-alcohol oxidase,69 cholesterol oxidase,70 and cellobiose dehydrogenase,71 and in a recently crystallized glucose dehydrogenase (GDH) from Aspergillus flavus (35% sequence identity with A. niger GOx).72 GDH is oxygen-independent, yet it preserves the catalytic conformation in the unliganded state (PDB ID: 4YNT) and with gluconolactone (PDB ID: 4YNU) in the active site. QM/MM (quantum mechanics/molecular mechanics) calculations confirm that this conformation is the one present during catalysis in aryl-alcohol oxidase.73 In the (g–, Ng+) conformation, His516 has moved away from the substrate, making the active site geometrically and chemically unsuitable for the concerted proton and hydride transfer (Figure S7 in the Supporting Information). Thus, this conformation is called noncatalytic henceforth.
Table 4. Overview of A. niger and P. amagasakiense GOx and A. flavus GDH Crystal Structures Showing the Distribution of His516 Side Chain Dihedral Angles, the Number of Active Site Water Molecules in the PDB File, and the Distance between the Nε Atom of His516 and the Oxygen Atom of This Water Molecule.
| PDB ID | χ1 (deg) | χ2 (deg) | crystal water | HOH–His516 (Å) |
|---|---|---|---|---|
| 1GAL | 257 | 225 | a | |
| 1CF3 | 254 | 194 | 710 | 2.98 |
| 3QVP | 291 | 185 | 1094 | 2.77 |
| 3QVR | 295 | 195 | 1200 | 2.89 |
| 5NIT | 293 | 197 | 1000 | 2.75 |
| 5NIW | 288 | 199 | 1000 | 2.79 |
| 1GPE | 284 | 64 | 837 | 2.70b |
| 4YNT | 277 | 201 | 798 | 2.68c |
| 4YNU | 284 | 197 | d | d |
The absence of water might be due to the low resolution of the crystal structure.
Equivalent to His520 of P. amagasakiense GOx.
Equivalent to His505 of A. flavus GDH.
In 4YNU, the active site water is replaced by gluconolactone, whose O1 atom is positioned 2.79 Å from the His residue.
Figure 4.
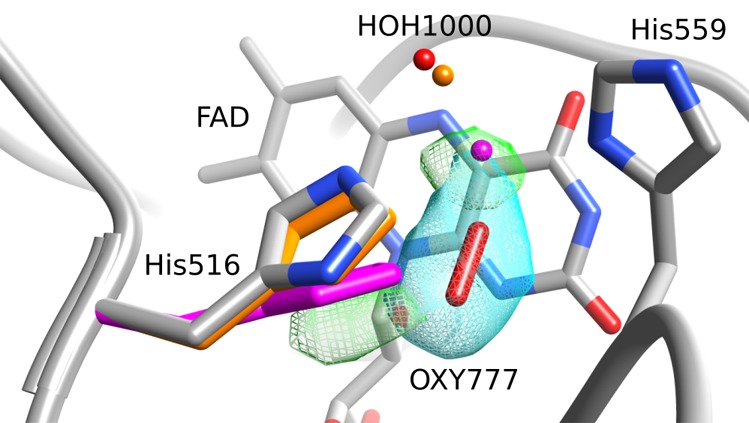
Structural view of the FAD re face of the F9 mutant, showing the electron density of the oxygen molecule (cyan mesh) as well as the remaining positive electron density (green). Aligned to the His516 side chain of F9 (colored by atom type), one can see the side chains of A. niger wild-type structures 3QVP (orange) and 1CF3 (magenta). Also shown are the oxygen atoms of the corresponding water molecules that are equivalent to HOH1000 in F9.
The conformation of His516 in the A2 and F9 structures is similar to those in A. niger GOx structures with PDB codes 3QVP and 3QVR. While the His516 conformation of 3QVR is almost identical with that in A2 and F9, that of 3QVP is slightly shifted toward the conformations found in the 1CF3 and 1GAL structures (Figure 4). The structures 3QVP and 3QVR resulted from an attempt to investigate the oxygen-binding site using chloride ions as oxygen substitutes,36 a method postulated to be an alternative to the approach using xenon for the identification of potential oxygen-binding sites. However, despite the similarity of conformations adopted by His516 to those in A2 and F9, no oxygen was reported in 3QVP and 3QVR. Instead, a water molecule was placed at the position occupied by the center of the oxygen molecule in A2 and F9.
In order to further investigate the active site, the electron densities of five wild-type GOx structures (1GAL, 1CF3, 3QVP, 3QVR, and 1GPE) were re-examined using structure factors from the PDB database. All structures show at least some positive and/or negative electron density near His516 (Figure S8 in the Supporting Information). The angular displacement of His516 from the conformation observed in the A2 mutant inversely follows the quality of the electron density around this residue and ends up with a partially missing electron density for the most deviating (g–, Nt) structures (1CF3 and 1GAL). This last observation was at the origin to indicate that the His516 side chain is flexible.33,34 A pH-induced conformational flexibility due to a different protonation state of His516 can be excluded, since crystals of the A2 and F9 mutants were grown at pH 7.0–7.5 and those of the 3QVP and 3QVR structures at pH 6.9 and 5.1, respectively, whereas crystals for the 1CF3 structure were obtained at an intermediate pH of 5.6. The 3QVR structure, with its His516 conformation closest to that observed in A2, shows only minor positive electron density on both sides of the water molecule, which was placed at the site of oxygen in A2, and may indicate the presence of oxygen already in this structure.
An interesting observation is the well-positioned oxygen in A2 and F9 with respect to the π-orbital system of His516 (Figure 3). As the oxygen reduction is spin forbidden by the triplet nature of molecular oxygen, a catalytic effect for the triplet–singlet transition might rely on the orbital coupling between oxygen and His516. Since the crystallographic electron density represents a mean observation of the conformational substates adopted by a protein and considering that the most active mutant A2 shows neither positive nor negative electron density around His516, its conformation can be seen as very well defined, corresponding to a pure catalytic conformer of His516. A more dynamic situation is observed for the F9 mutant, which mainly adopts the catalytic conformation as in A2. However, modest positive electron densities at the active site of F9 GOx indicate that the His516, with its water molecule, also samples small amounts of the displaced (g–, Nt) conformation observed in the 1CF3 structure (Figure 4).
His516 Conformational Dynamics from Simulations
In order to quantify the flexibility of the side chain of His516, we performed standard and enhanced MD simulations. Our initial MD simulations of A. niger wild-type GOx showed that χ1 is conserved to g– geometry (240–360°; Figure S9 in the Supporting Information). The χ2 dihedral samples two minima, Ng+ (30–90°) and Nt (150°–210°), neither of which corresponds to the most stable conformation in the His rotamer library. However, the noncatalytic (g–, Ng+) geometry is 4 times more probable in the backbone-independent rotamer library than the catalytic (g–, Nt) conformation.74 In WT, His516 is free to flip to a small cavity located in the vicinity of the active site. The M556V mutation in A2, which resides at the border of this cavity, significantly decreases the cavity size, making His516 sterically hindered (Figure 5). It is important to note that valine is the most common residue found at this position in the consensus of glucose oxidase sequences.15
Figure 5.
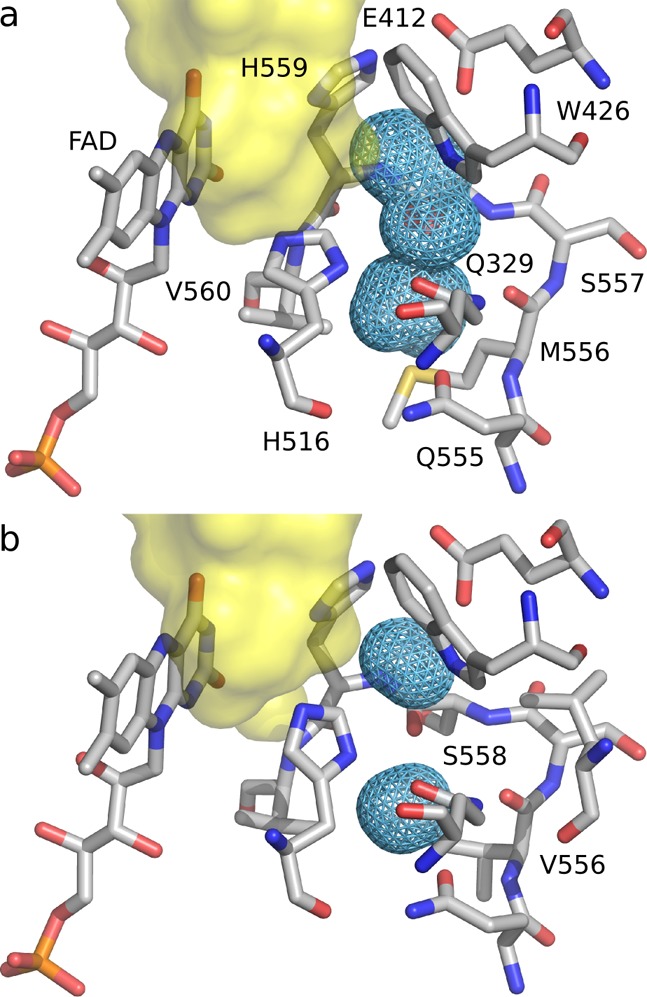
Cavity (light blue mesh) located in the vicinity of the active site (yellow surface) of (a) WT GOx and (b) A2 mutant. The M556V mutation returns this residue to its consensus sequence, which significantly decreases the cavity size preventing His516 flipping.
The subsequent Hamiltonian replica exchange MD simulations, performed to quantify the flexibility of the His516 side chain, revealed that the g– geometry is indeed dominant in all GOx variants. In WT, the catalytic (Nt) and noncatalytic (Ng+) conformations are quite equally distributed (Figure 6). The P mutant introduces a clear separation between the two conformations while simultaneously enriching the catalytic form, and R537K and M556V further reduce the noncatalytic geometry. The synergy of these effects conserves His516 mostly in the catalytic conformation in A2, increasing kcat 2.6 times and its efficiency 4-fold in comparison to that in WT. The F9 mutant bears the M556V mutation and, therefore, resembles the Pv variant, where the catalytic conformation is energetically more favorable. However, the absence of R537K and the addition of two other mutations (R37K/V106I) narrows the separation between the two conformations, making it more similar to WT. This indicates a lower energy barrier for the transition between the catalytic and noncatalytic states and, hence, slower catalysis than for the A2 mutant (increase in kcat 1.8 times and in efficiency 2.6 times in comparison to WT).
Figure 6.

His516 side chain dihedral angles (χ1 and χ2) distribution: (a) WT GOx, (b) parent mutant P, (c) Pk, (d) Pv, (e) A2, and (f) F9. The χ1 dihedral has a clear preference for g– geometry. The χ2 dihedral prefers either the catalytic Nt or noncatalytic Ng+ geometries in the different GOx variants. The normalized integrated distributions are shown as red curves on either side of the panels.
To properly quantify the energy barrier between the catalytic and noncatalytic His516 states, we performed umbrella sampling MD simulations of the WT and A2 variants in χ2 space. The global minimum of WT is at 60° (Ng+), and it is only 0.2 kcal mol–1 more stable than the minimum at 160° (Nt), which corresponds to an equilibrium mixture of 60:40 of noncatalytic to catalytic conformations at room temperature (Figure 7). Furthermore, having a rather high energy barrier of 2.9 kcal mol–1 for the Ng+ to Nt transition means that a significant amount of time is lost on making GOx conformationally fit for catalysis, indicating that WT GOx is not an optimal catalyst. In A2, on the other hand, the catalytic conformation is 30 times more probable, as it is 2.0 kcal mol–1 more stable than the noncatalytic form. Furthermore, the energy barrier for the conversion of the noncatalytic to the catalytic state is significantly lower (1.8 kcal mol–1) than for WT. Thus, the catalytic conformation can be achieved much more easily than for WT while the transformation back to the Ng+ conformation is slow due to the barrier of 3.8 kcal mol–1.
Figure 7.
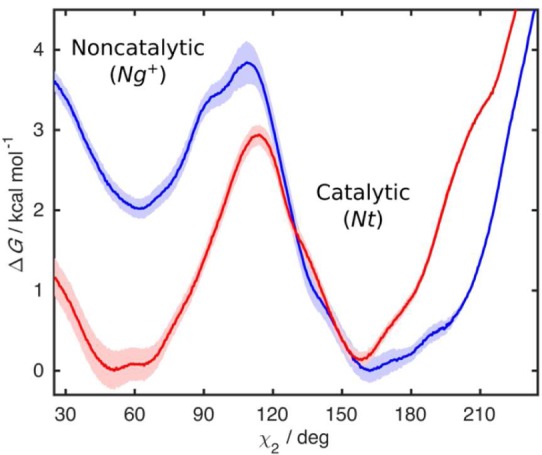
Free energy for the rotation around the χ2 dihedral angle of His516: (red) WT GOx; (blue) A2 mutant. The shaded areas around the free energy profiles represent the errors estimated using the blocking procedure.
The extensive US-MD simulations corroborate the relative energies of the minima determined from the HREX-MD simulations. Umbrella sampling, however, performs better in estimating barrier heights. On the other hand, HREX-MD is a very convenient and cost-effective technique and can thus represent an excellent screening method for identifying good enzyme designs that involve potentially flexible active site residues. Furthermore, it is fast enough to be used for guiding directed evolution experiments.
Conclusions
Glucose oxidase is an important industrial catalyst for which many mutations were proposed to enhance various properties. However, not much is known about the mode of action of these mutations. In order to fill this gap, we solved the first crystal structures of GOx mutants from A. niger and performed an extensive molecular dynamics investigation based on a total of 5.6 μs simulation time to correlate mutations with kinetic data. The crystal structures of the mutants A2 and F9 revealed molecular oxygen to be present at the active site and suggest that the side chain of His516, which is of utmost importance for the enzymatic reaction, is preorganized in the catalytic conformation and less flexible than in the wild-type GOx.
In the MD simulations, the most active mutant (A2) shows significant anticorrelated motions between secondary structure elements caused by the T30V and I94V mutations and both correlated and anticorrelated motions resulting from the R537K and M556V mutations. This long-range dynamic effect reduces the volume of the active site, which has a positive influence on catalytic efficiency. From all GOx variants studied here, A2 possesses the tightest and least floppy active site, where protein contacts stabilize the optimal geometry of glucose for its interconversion to gluconolactone. Our MD simulations thus confirm the observation from the crystal structures that His516 is flexible in the WT and more rigid in the mutants. Furthermore, we find that His516 can flip between the two substates, catalytic and noncatalytic. To study the relative populations of the two substates and barriers between them, we employed Hamiltonian replica exchange and umbrella sampling MD simulations. While both substates are equally populated in the WT enzyme, the most favorable conformation of His516 in the A2 mutant is the catalytic form. This results from the M556V mutation that reduces the size of a cavity in the vicinity of the active site and therefore restrains the movements of His516. As the turnover number of the discussed GOx variants is already very high (and probably very close to the theoretical limit), further design should be directed toward mutations that could provide higher binding affinities for glucose: mutations either in the first shells around the active site or at further positions (e.g., at the protein surface) that could positively modulate the correlated and anticorrelated motions.
From our study, we find that US-MD performs much better in estimating barrier heights, but both US-MD and HREX-MD are equally good for predicting positions and relative populations of the enzyme substates. Considering the relatively low computational cost and ease of use of HREX-MD simulations, we conclude that this method represents an attractive tool for in silico screening of enzyme variants involving flexible residues in the active sites.
Acknowledgments
D.P. was financially supported by the Jürgen Manchot Foundation. D.F. and K.H. were supported by the European Regional Development Fund (ERDF) and the European Union (“Die Europäische Kommission investiert in Ihre Zukunft”). B.S. thanks the “Strategischer Forschungsfonds” of the Heinrich-Heine University Düsseldorf (F2014/730-11) for financial support. S.C.L.K. thanks the Swedish Research Council (VR, Grant 2015-04928). The European Research Council provided financial support under the European Community’s Seventh Framework Programme (FP7/2007-2013)/ERC Grant Agreement 306474. S.C.L.K. is also a Wallenberg Academy Fellow. The authors gratefully acknowledge the computing time granted by the JARA-HPC Vergabegremium and VSR commission on the supercomputer JURECA (project ICS69) at Forschungszentrum Jülich.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.7b01575.
Umbrella sampling MD setup, additional figures showing electron densities for His516 of various wild-type and mutant GOx structures, and further MD simulation results (PDF)
Author Present Address
⊥ D.F.: Aquila Biolabs GmbH, Arnold-Sommerfeld-Ring 2, 52499 Baesweiler, Germany.
The authors declare no competing financial interest.
Supplementary Material
References
- Bankar S. B.; Bule M. V.; Singhal R. S.; Ananthanarayan L. Biotechnol. Adv. 2009, 27, 489–501. 10.1016/j.biotechadv.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Dijkman W. P.; de Gonzalo G.; Mattevi A.; Fraaije M. W. Appl. Microbiol. Biotechnol. 2013, 97, 5177–5188. 10.1007/s00253-013-4925-7. [DOI] [PubMed] [Google Scholar]
- Tracewell C. A.; Arnold F. H. Curr. Opin. Chem. Biol. 2009, 13, 3–9. 10.1016/j.cbpa.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms M. J.; Thornton J. W. Nat. Rev. Genet. 2013, 14, 559–571. 10.1038/nrg3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles J. R.; Albery W. J. Acc. Chem. Res. 1977, 10, 105–111. 10.1021/ar50112a001. [DOI] [Google Scholar]
- Bar-Even A.; Noor E.; Savir Y.; Liebermeister W.; Davidi D.; Tawfik D. S.; Milo R. Biochemistry 2011, 50, 4402–4410. 10.1021/bi2002289. [DOI] [PubMed] [Google Scholar]
- Arnold F. H.; Wintrode P. L.; Miyazaki K.; Gershenson A. Trends Biochem. Sci. 2001, 26, 100–106. 10.1016/S0968-0004(00)01755-2. [DOI] [PubMed] [Google Scholar]
- Tokuriki N.; Jackson C. J.; Afriat-Jurnou L.; Wyganowski K. T.; Tang R.; Tawfik D. S. Nat. Commun. 2012, 3, 1257. 10.1038/ncomms2246. [DOI] [PubMed] [Google Scholar]
- Wilson R.; Turner A. P. F. Biosens. Bioelectron. 1992, 7, 165–185. 10.1016/0956-5663(92)87013-F. [DOI] [Google Scholar]
- Mattevi A. Trends Biochem. Sci. 2006, 31, 276–283. 10.1016/j.tibs.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Zhu Z.; Momeu C.; Zakhartsev M.; Schwaneberg U. Biosens. Bioelectron. 2006, 21, 2046–2051. 10.1016/j.bios.2005.11.018. [DOI] [PubMed] [Google Scholar]
- Holland J. T.; Lau C.; Brozik S.; Atanassov P.; Banta S. J. Am. Chem. Soc. 2011, 133, 19262–19265. 10.1021/ja2071237. [DOI] [PubMed] [Google Scholar]
- Holland J. T.; Harper J. C.; Dolan P. L.; Manginell M. M.; Arango D. C.; Rawlings J. A.; Apblett C. A.; Brozik S. M. PLoS One 2012, 7, e37924. 10.1371/journal.pone.0037924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arango Gutierrez E.; Mundhada H.; Meier T.; Duefel H.; Bocola M.; Schwaneberg U. Biosens. Bioelectron. 2013, 50, 84–90. 10.1016/j.bios.2013.06.029. [DOI] [PubMed] [Google Scholar]
- Ostafe R.; Prodanovic R.; Nazor J.; Fischer R. Chem. Biol. 2014, 21, 414–421. 10.1016/j.chembiol.2014.01.010. [DOI] [PubMed] [Google Scholar]
- Leskovac V.; Trivić S.; Wohlfahrt G.; Kandrač J.; Peričin D. Int. J. Biochem. Cell Biol. 2005, 37, 731–750. 10.1016/j.biocel.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Roth J. P.; Klinman J. P. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 62–67. 10.1073/pnas.252644599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschlag D.; Natarajan A. Biochemistry 2013, 52, 2050–2067. 10.1021/bi4000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanoian P.; Liu C. T.; Hammes-Schiffer S.; Benkovic S. Acc. Chem. Res. 2015, 48, 482–489. 10.1021/ar500390e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P. K.; Doucet N.; Chennubhotla C.; Ramanathan A.; Narayanan C. Methods Enzymol. 2016, 578, 273–297. 10.1016/bs.mie.2016.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias M.; Wieczorek G.; Rosenne S.; Tawfik D. S. Trends Biochem. Sci. 2014, 39, 1–7. 10.1016/j.tibs.2013.11.001. [DOI] [PubMed] [Google Scholar]
- Smith A. J. T.; Müller R.; Toscano M. D.; Kast P.; Hellinga H. W.; Hilvert D.; Houk K. N. J. Am. Chem. Soc. 2008, 130, 15361–15373. 10.1021/ja803213p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A. Proc. Natl. Acad. Sci. U. S. A. 1978, 75, 5250–5254. 10.1073/pnas.75.11.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisliakov A. V.; Cao J.; Kamerlin S. C. L.; Warshel A. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 17359–17364. 10.1073/pnas.0909150106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A.; Sharma P. K.; Kato M.; Xiang Y.; Liu H.; Olsson M. H. M. Chem. Rev. 2006, 106, 3210–3235. 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- Roca M.; Oliva M.; Castillo R.; Moliner V.; Tuñón I. Chem. - Eur. J. 2010, 16, 11399–11411. 10.1002/chem.201000635. [DOI] [PubMed] [Google Scholar]
- Bouvignies G.; Vallurupalli P.; Hansen D. F.; Correia B. E.; Lange O.; Bah A.; Vernon R. M.; Dahlquist F. W.; Baker D.; Kay L. E. Nature 2011, 477, 111–114. 10.1038/nature10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keedy D. A.; Kenner L. R.; Warkentin M.; Woldeyes R. A.; Hopkins J. B.; Thompson M. C.; Brewster A. S.; Van Benschoten A. H.; Baxter E. L.; Uervirojnangkoorn M.; McPhillips S. E.; Song J.; Alonso-Mori R.; Holton J. M.; Weis W. I.; Brunger A. T.; Soltis S. M.; Lemke H.; Gonzalez A.; Sauter N. K.; Cohen A. E.; van den Bedem H.; Thorne R. E.; Fraser J. S. eLife 2015, 4, e07574. 10.7554/eLife.07574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabbitt P. D.; Correy G. J.; Meirelles T.; Fraser N. J.; Coote M. L.; Jackson C. J. Biochemistry 2016, 55, 1408–1417. 10.1021/acs.biochem.5b01322. [DOI] [PubMed] [Google Scholar]
- Osuna S.; Jiménez-Osés G.; Noey E. L.; Houk K. N. Acc. Chem. Res. 2015, 48, 1080–1089. 10.1021/ar500452q. [DOI] [PubMed] [Google Scholar]
- Ruscio J. Z.; Kohn J. E.; Ball K. A.; Head-Gordon T. J. Am. Chem. Soc. 2009, 131, 14111–14115. 10.1021/ja905396s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss G.; Çelebi-Ölçüm N.; Moretti R.; Baker D.; Houk K. N. Angew. Chem., Int. Ed. 2013, 52, 5700–5725. 10.1002/anie.201204077. [DOI] [PubMed] [Google Scholar]
- Wohlfahrt G.; Witt S.; Hendle J.; Schomburg D.; Kalisz H. M.; Hecht H. J. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999, 55, 969–977. 10.1107/S0907444999003431. [DOI] [PubMed] [Google Scholar]
- Hecht H. J.; Kalisz H. M.; Hendle J.; Schmid R. D.; Schomburg D. J. Mol. Biol. 1993, 229, 153–172. 10.1006/jmbi.1993.1015. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kommoju P.-R.; Chen Z.; Bruckner R. C.; Mathews F. S.; Jorns M. S. Biochemistry 2011, 50, 5521–5534. 10.1021/bi200388g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell S. C.; Davis I. W.; Arendall W. B.; de Bakker P. I. W.; Word J. M.; Prisant M. G.; Richardson J. S.; Richardson D. C. Proteins: Struct., Funct., Genet. 2003, 50, 437–450. 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Šali A.; Blundell T. L. J. Mol. Biol. 1993, 234, 779–815. 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. J. Comput. Chem. 2009, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa da Silva A. W.; Vranken W. F. BMC Res. Notes 2012, 5, 367. 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wang W.; Kollman P. A.; Case D. A. J. Mol. Graphics Modell. 2006, 25, 247–260. 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision A.02; Gaussian, Inc., Wallingford, CT, 2009.
- Todde G.; Hovmöller S.; Laaksonen A.; Mocci F. Proteins: Struct., Funct., Genet. 2014, 82, 2353–2363. 10.1002/prot.24596. [DOI] [PubMed] [Google Scholar]
- Pronk S.; Páll S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; van der Spoel D.; Hess B.; Lindahl E. Bioinformatics 2013, 29, 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Palmo K.; Maragakis P.; Klepeis J. L.; Dror R. O.; Shaw D. E. Proteins: Struct., Funct., Genet. 2010, 78, 1950–1958. 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. J. Chem. Phys. 1983, 79, 926. 10.1063/1.445869. [DOI] [Google Scholar]
- Søndergaard C. R.; Olsson M. H. M.; Rostkowski M.; Jensen J. H. J. Chem. Theory Comput. 2011, 7, 2284–2295. 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- Darden T.; York D.; Pedersen L. J. Chem. Phys. 1993, 98, 10089. 10.1063/1.464397. [DOI] [Google Scholar]
- Hess B. J. Chem. Theory Comput. 2008, 4, 116–122. 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. J. Chem. Phys. 2007, 126, 014101. 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. J. Appl. Phys. 1981, 52, 7182. 10.1063/1.328693. [DOI] [Google Scholar]
- Bonomi M.; Branduardi D.; Bussi G.; Camilloni C.; Provasi D.; Raiteri P.; Donadio D.; Marinelli F.; Pietrucci F.; Broglia R. A.; Parrinello M. Comput. Phys. Commun. 2009, 180, 1961–1972. 10.1016/j.cpc.2009.05.011. [DOI] [Google Scholar]
- Bussi G. Mol. Phys. 2014, 112, 379–384. 10.1080/00268976.2013.824126. [DOI] [Google Scholar]
- Grossfield A.WHAM: the weighted histogram analysis method, v. 2.0.9.
- Grossfield A.; Zuckerman D. M. Annu. Rep. Comput. Chem. 2009, 5, 23–48. 10.1016/S1574-1400(09)00502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Grant B. J.; Rodrigues A. P. C.; ElSawy K. M.; McCammon J. A.; Caves L. S. D. Bioinformatics 2006, 22, 2695–2696. 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- Durrant J. D.; de Oliveira C. A. F.; McCammon J. A. J. Mol. Graphics Modell. 2011, 29, 773–776. 10.1016/j.jmgm.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.6; Schrödinger LLC.
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Kalisz H. M.; Hecht H.-J.; Schomburg D.; Schmid R. D. J. Mol. Biol. 1990, 213, 207–209. 10.1016/S0022-2836(05)80179-2. [DOI] [PubMed] [Google Scholar]
- Bretthauer R. K.; Castellino F. J. Biotechnol. Appl. Biochem. 1999, 30, 193–200. 10.1111/j.1470-8744.1999.tb00770.x. [DOI] [PubMed] [Google Scholar]
- Mellquist J. L.; Kasturi L.; Spitalnik S. L.; Shakin-Eshleman S. H. Biochemistry 1998, 37, 6833–6837. 10.1021/bi972217k. [DOI] [PubMed] [Google Scholar]
- Bhabha G.; Biel J. T.; Fraser J. S. Acc. Chem. Res. 2015, 48, 423–430. 10.1021/ar5003158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick R. B.; Orellana L.; Esteban-Martín S.; Orozco M.; Salvatella X. Nat. Commun. 2014, 5, 4070. 10.1038/ncomms5070. [DOI] [PubMed] [Google Scholar]
- Ma H.; Szeler K.; Kamerlin S. C. L.; Widersten M. Chem. Sci. 2016, 7, 1415–1421. 10.1039/C5SC03666F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J.; Bruice T. C. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 13152–13156. 10.1073/pnas.0405502101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández I. S.; Ruíz-Dueñas F. J.; Santillana E.; Ferreira P.; Jesús Martínez M.; Martínez A. T.; Romero A. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 1196–1205. 10.1107/S0907444909035860. [DOI] [PubMed] [Google Scholar]
- Li J.; Vrielink A.; Brick P.; Blow D. M. Biochemistry 1993, 32, 11507–11515. 10.1021/bi00094a006. [DOI] [PubMed] [Google Scholar]
- Martin Hallberg B.; Henriksson G.; Pettersson G.; Divne C. J. Mol. Biol. 2002, 315, 421–434. 10.1006/jmbi.2001.5246. [DOI] [PubMed] [Google Scholar]
- Yoshida H.; Sakai G.; Mori K.; Kojima K.; Kamitori S.; Sode K. Sci. Rep. 2015, 5, 13498. 10.1038/srep13498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Ortega A.; Lucas F.; Ferreira P.; Medina M.; Guallar V.; Martínez A. T. Biochemistry 2012, 51, 6595–6608. 10.1021/bi300505z. [DOI] [PubMed] [Google Scholar]
- Scouras A. D.; Daggett V. Protein Sci. 2011, 20, 341–352. 10.1002/pro.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.