

Abstract
The rapid increase in the number and quality of fluorescent reporters and optogenetic actuators has yielded a powerful set of tools for recording and controlling cellular state and function. To achieve the full benefit of these tools requires improved optical systems with high light collection efficiency, high spatial and temporal resolution, and patterned optical stimulation, in a wide field of view (FOV). Here we describe our ‘Firefly’ microscope, which achieves these goals in a Ø6 mm FOV. The Firefly optical system is optimized for simultaneous photostimulation and fluorescence imaging in cultured cells. All but one of the optical elements are commercially available, yet the microscope achieves 10-fold higher light collection efficiency at its design magnification than the comparable commercially available microscope using the same objective. The Firefly microscope enables all-optical electrophysiology (‘Optopatch’) in cultured neurons with a throughput and information content unmatched by other neuronal phenotyping systems. This capability opens possibilities in disease modeling and phenotypic drug screening. We also demonstrate applications of the system to voltage and calcium recordings in human induced pluripotent stem cell derived cardiomyocytes.
OCIS codes: (180.2520) Fluorescence microscopy, (170.3880) Medical and biological imaging, (170.2655) Functional monitoring and imaging
1. Introduction
Recent advances in fluorescent reporters and optogenetic actuators have enhanced our ability to detect and control cellular function with light [1,2]. Novel small molecule [3] and protein-based fluorescent sensors enable monitoring of a wide variety of physiological parameters [2,4–6], including pH [7], calcium [8], transmembrane voltage [9], as well as second-messenger signaling molecules such as cyclic AMP [10] and protein activation, e.g. RAS [11].
Recently developed optogenetic actuators enable optical control over transmembrane voltage [12–14] as well as control over transcription [15], protein translocation [16], aggregation [17], dissociation [18], and binding and release [19,20]. By combining optogenetic actuators and reporters in the same cell, one can probe the input-output properties of individual cells with high throughput and information content. For instance, pairing of a channelrhodopsin actuator with a near-infrared fluorescent voltage indicator led to an ‘Optopatch’ construct for all-optical electrophysiology (Hochbaum et al. 2014). A recent review described the transformative potential of all-optical stimulation and readout of neural activity [21]. Patterned optogenetic stimulation paired with fluorescence imaging can also yield novel insights in many areas of developmental and cellular biology (see e.g [22].).
Here we introduce the Firefly microscope, optimized to use these optogenetic tools over a wide FOV, with high resolution in space and time. The system has applications in neuroscience, cardiac biology, disease modeling, and high-content screening. As an illustrative application, we consider the challenge of functional excitability measurements in cultured human induced pluripotent stem cell (hiPSC)-derived neurons, which are emerging as a powerful tool for disease modeling and drug discovery [23]. HiPSC-derived neurons can be highly heterogeneous in their morphology, gene expression profiles, firing patterns, and drug responses. One typically must record from > 100 cells to reveal statistically robust genetic or pharmacological effects. Neurons spike on the millisecond timescale, so it is necessary to have ~1 kHz frame rate and therefore high light collection efficiency. To fully characterize neuronal excitability typically requires 30 s of recording under a variety of stimulus patterns. This requirement for extended observation strongly disfavors tiling multiple fields of view with conventional high-magnification imaging. Tiling becomes prohibitively slow for more than a small number of measurements. To achieve the capacity to screen thousands of conditions, and hundreds of neurons per condition, requires recording from as many cells in parallel as possible.
Imaging over a very wide field at high frame-rate entails a partial loss of spatial resolution. In many applications this tradeoff is desirable. In Optopatch recordings, for example, one can stimulate and record whole-cell voltage dynamics without resolving the details of cellular sub-structure. Indeed, localizing the fluorescence from each cell on a small number of camera pixels minimizes the contribution of electronic read noise to the signal. In other cases, sub-cellular targeting of reporters can provide physiological information on cellular microcompartments (e.g. inside organelles) without the need for sub-cellular optical resolution. In these examples, imaging at low magnification increases the number of cells measured in parallel, without sacrificing the relevant biological information.
In other applications, simultaneous recording across a wide FOV is a fundamental necessity. For instance, to study network dynamics in cultured neurons, one must record simultaneously from cells separated by millimeter distances, corresponding to the length of axonal projections in vitro. To study action potential propagation in cultured cardiomyocytes, one must image a sufficient area with high time resolution to map propagation of wavefronts at ~20 cm/s (corresponding to 200 μm/ms) [24]. Finally to probe the acute response of any cell type to addition of a drug, one can typically only sample a single FOV per well. One should maximize the number of cells in the FOV to obtain the most robust results. The Firefly microscope meets the needs of wide-field optogenetic applications, with good light collection efficiency, sub-cellular spatial resolution for recording and stimulation, high temporal resolution, and many wavelengths available for combinatorial application of different reporters and actuators.
Much effort in the field has been devoted to large-area in vivo imaging of fluorescent sensors using, for example, light sheet microscopy [25,26] and two photon microscopy [27]. However, light sheet microscopy requires a 2-objective geometry that is not well suited to planar samples. Scanning confocal and two-photon microscopies have recently been extended to very wide FOV imaging in vivo [28–31]. However, point-scanning techniques face a fundamental tradeoff in spatial resolution, field of view, and temporal resolution. Pulsed lasers operating at 80 MHz can maximally probe 8x107 pixels/s. Typically one must average over several laser shots per pixel, leading to practical imaging rates of 1x107 pixels/s or lower. Modern scientific complementary metal-oxide semiconductor (sCMOS) cameras can record up to 4x108 pixels per second, offering the opportunity for simultaneously high spatial and temporal resolutions over a large FOV in a 1-photon fluorescence format. However, to benefit from these imaging capabilities requires careful attention to optics to maximize signal photons while minimizing sources of optical background and aberration. Existing low-magnification commercial microscopy systems lose too much light for many advanced applications, particularly in neuronal recording.
Here, we focus on in vitro imaging where flat samples with low scattering and absorption enable fast imaging with 1-photon fluorescence. We describe a microscope (‘Firefly’) built around an off-the-shelf low-magnification (2x) high numerical aperture (NA 0.5) objective. The Firefly microscope attains cellular resolution in a functional FOV of 6x6 mm at a frame rate of 100 Hz, well suited for calcium imaging, or in a truncated FOV of 0.6x6 mm at a frame rate of 1 kHz, suitable for voltage imaging in neurons. The high NA objective leads to efficient light collection, an essential attribute for high-speed imaging with high signal-to-noise ratio (SNR). The Firefly microscope also provides arbitrarily reconfigurable patterned light illumination for optogenetic stimulation, with 20 kHz update rate and 7 μm spatial resolution. In contrast to other ultrawidefield microscope systems, this microscope can be assembled for < $100,000 from mostly off-the-shelf components, yet attains 10x higher light collection efficiency than the commercially available microscope that uses the same objective lens.
We first introduce the layout of the Firefly microscope and characterize its optical performance. We then describe the capabilities for patterned optical stimulation. Next, we describe an unusual near-total internal reflection (TIR) illumination geometry to minimize background and heating. Finally, we demonstrate the microscope’s utility for all-optical electrophysiology in rat neurons, mouse dorsal root ganglion (DRG) neurons, human induced pluripotent stem cell (hiPSC)-derived motor neurons, and an extended cardiomyocyte syncytium. We anticipate that the ease and low cost of assembly, high optical quality, and modular design of the Firefly microscope system will give it broad application in functional biological imaging and screening.
2. Microscope design and optical path
The Firefly microscope has three main optical systems: (1) a high-NA, large FOV imaging path, (2) patterned illumination using a digital micromirror device (DMD), and (3) near-TIR illumination with a high-powered red laser coupled into the sample with a prism (Fig. 1).
Fig. 1.

Microscope optical diagram. Fluorescence from the sample passes through a pair of high NA, large field-of-view objectives to form an image at 2x magnification on an sCMOS camera. The camera records a Ø6 mm region with 3.25 μm spatial resolution and 10 ms temporal resolution. Illumination for optogenetic stimulation is patterned by reflection off a DMD, and focused onto a small mirror that couples the light into the objective en route to the sample. High-power red laser illumination passes through a custom fused silica prism to illuminate the sample at close to the angle for total internal reflection. This geometry maximizes illumination intensity at the sample while minimizing laser flux through on-axis optical components which would otherwise produce background autofluorescence. Six different color LEDs are combined and focused onto the sample from above for homogeneous illumination and high-speed modulation across much of the near-UV and visible spectrum. Omitted from the diagram is an iris in the LED system imaged onto the sample to control illumination size. Inset: design of the fused silica prism.
2.1 Imaging path
The Firefly microscope is designed around the objective, the most critical component in determining overall optical performance. The light gathering power per unit area of the sample is proportional to E = NA2, where NA is the numerical aperture of the objective. This parameter determines the signal level one can attain from single cells for a given illumination intensity, and also the amount of signal that can be acquired from a cell before photobleaching or phototoxicity set in. If the NA drops too low, then single-cell signals are lost.
The total quantity of light gathered across a sample (assuming homogeneous illumination intensity) is proportional to R = (FOV area ∙ NA2). This parameter measures the number of photons per frame detected on the camera, and thereby measures the total information acquired from the sample per unit time. The parameter R combines the signal per cell and the number of cells measured in a given sampling interval.
In most microscope objectives, there is a tradeoff between FOV size and NA (Table 1), so that one loses the ability to detect fast single-cell dynamics at low magnification. For example, in Olympus’ super apochromat objective series, there is a 71-fold loss in collection efficiency, E, between the 60x and 4x objectives, while there is a 227-fold increase in imaging area. If one is choosing between tiling high magnification, high NA FOVs vs. extending the exposure time on a low magnification, low NA FOV, the approach with the larger value of R will have the larger shot noise-limited SNR in a given measurement interval (assuming the same illumination intensity and same total FOV area in both scenarios).
Table 1. Comparison of commercially available objectives. In most microscope objectives there is a tradeoff between the numerical aperture (NA) and field of view (FOV). The total light collection is proportional to FOV area ∙ NA2. Here total light collection is measured relative to the 60x objective. The MVPLAPO 2 XC objective offers much higher light collection than the other objectives.
| Part number | Mag. | NA | Ø FOV (mm) | Relative light collection |
|---|---|---|---|---|
| UPLSAPO 60XO | 60x | 1.35 | 0.44 | 1.0 |
| UPLSAPO 20XO | 20x | 0.85 | 1.33 | 3.6 |
| UPLSAPO 10X2 | 10x | 0.4 | 2.65 | 3.2 |
| UPLSAPO 4X | 4x | 0.16 | 6.63 | 3.2 |
| MVPLAPO 2 XC | 2x | 0.5 | 17 | 204.8 |
For the Firefly microscope, we selected an objective that transcends the tradeoff between FOV and NA, which typically requires an increase in physical size of the objective. Several custom objective designs have been described along these lines [28–31], but we sought to use a commercially available objective to facilitate broad adoption of the microscope system. We used an Olympus MVPLAPO 2 XC, which is a 2x objective with a high NA of 0.5, a very large nominal FOV of 17 mm, a working distance of 2 cm and a back focal length of 5 cm (Fig. 1). This objective is far larger than standard microscope objectives: ~65 mm in diameter and ~120 mm tall.
We initially tested this objective in the Olympus stereomicroscope for which it was designed, the MVX10. Figure 2(a) shows a pair of isolated fluorescent beads at different “zoom” magnifications. The beads were illuminated from below (not through the zoom body), so the illumination intensity was constant at all zoom levels. Figure 2(b) shows the precipitous drop in total fluorescence collected from the beads at low magnifications, indicative of an optical loss in the imaging path. When the zoom body was set to 2x magnification, the optical throughput was only 10% of the throughput at the highest magnification. Thus at low magnification the zoom body discarded most of the light collected by the objective, losing the benefit of the high NA.
Fig. 2.

Light collection efficiency of the MVX10 zoom body as a function of zoom magnification. (a) Images of a pair of isolated fluorescent beads on the Olympus MVX microscope at different magnifications set by adjusting the zoom body. (b) Due to light losses in the zoom body, the total light collection efficiency, calculated by averaging the counts over the pair of beads, drops at low magnifications. The light collection efficiency at 2x magnification is only 10% of the efficiency at ≥8x magnification.
As an alternative to the MVX10 imaging path, we tested commercially available lenses to use for the “tube lens” to form the image on the camera. We found three lenses which had good light transmission and acceptable aberration across the field of view, each of which yielded a different total magnification. Two were objectives from the Olympus MVX stereomicroscope series: the MVPLAPO 1 X (NA = 0.25, back focal length = 10 cm) yielded a total magnification of 2x, and the MVPLAPO 0.63 X (NA = 0.15, back focal length = 16 cm) yielded a total magnification of 3.2x. The third was a Zeiss camera lens (135 mm f/2 Apo Sonnar T* ZF.2) which yielded a total magnification of 2.7x. In contrast to the MVX10 optical path, these lenses had sufficient aperture to transmit all the light collected by the objective, i.e. they achieved 10-fold higher light collection efficiency at low magnification. This increase in optical throughput was essential to achieving sufficiently high sensitivity to enable high-speed neural recordings.
Although the objective had a nominal FOV of 17 mm, the FOV in our microscope was set by the 13 mm width of the sCMOS image sensor, leading to a ~6 mm FOV at 2x magnification. In the infinity space between the objective back aperture and the tube lens, the fluorescence diverged in a cone with a half-angle of 3.5°, set by the rays from the edges of the FOV. To prevent the finite aperture of the tube lens from clipping this light, the tube lens was placed as close as possible (~5 cm) to the back of the objective. Large area fluorescence emission filters (Ø50 mm) were placed in an automated filter wheel (Edmund Optics, 84-889) between the objective and the tube lens.
Images were recorded on a Hamamatsu ORCA-Flash4.0 sCMOS camera with 6.5 μm pixels, which at 2x magnification resulted in a pixilation-limited resolution of 3.25 μm at the center of the FOV and a 6x6 mm FOV recordable at 100 Hz or a 0.6x6 mm FOV recordable at 1 kHz. The built-in microlens array immediately in front of the camera’s image sensor has an acceptance angle of ± 15°, which just exceeded the angular range of the incident light at 2x magnification. The sCMOS camera has a rolling shutter and thus a 1 frame-time delay between pixels in the center and top or bottom of the FOV at maximum frame rate. At the magnification of Firefly microscope, cell bodies span < 10 pixel rows and result in time shifts of < 100 μs across individual cells, which can typically be neglected in analysis. In the rare scenarios where this delay was a problem, we corrected the data in post-processing by time-shifting each row by an amount proportional to its distance from the image midline.
Figure 3 characterizes optical aberration and light collection efficiency using the Zeiss tube lens at 2.7x magnification. Figure 3(a) shows a 5 mm wide field of view of single 1 μm diameter TetraSpeck Microspheres (ThermoFisher #T-7282), fluorescent beads that absorb and emit across the visible spectrum. Figure 3(b) shows magnified views of the boxed regions in (a). Figure 3(c)-left axis shows the full width at half maximum (FWHM) diameter of individual beads as a function of distance from the center of the FOV. Across a 3 mm diameter region in the center of the FOV, the resolution was pixel-limited and a 1 μm bead mapped onto a single pixel. Near the edges of the FOV there was asymmetrical blurring (cyan box) from astigmatism (a difference in sagittal versus tangential focus). The focal plane curvature had different signs for rays focusing in the tangential and sagittal planes: as one moved away from the optic axis, the tangential focus dropped below the central focus (radius of curvature of −950 mm) while the sagittal focus rose above the central focus (radius of curvature of 220 mm). The light collection efficiency (Fig. 3(c)-right axis) was constant across a Ø3 mm FOV and dropped to around 70% at Ø6 mm. Despite these aberrations, in cellular samples it was straightforward to identify single fluorescent cells (see e.g. Figs. 5(a) & 5(b)), and to record spiking patterns of well-separated, individual neurons, all the way to Ø6 mm. We also quantified the small degree of chromatic aberration via the focal shift needed to bring beads of different color into focus (Fig. 3(d)). We did not detect any wavelength-dependent shifts in magnification or in image registration. The chromatic focal shift was readily corrected via an automated focus adjustment when switching color channels.
Fig. 3.

Optical performance of the Firefly microscope. (a) An image of 1 μm diameter beads, showing a 5 mm wide segment through the center of the field of view. The Zeiss tube lens provided 2.7x magnification. (b) Magnified view of sub-regions from the center of the FOV to the edge shown by colored boxes in (a). In the broad, central region the resolution was pixel limited, but astigmatism became detectable near the edges. (c) Left axis: FWHM diameter of single beads as a function of distance from image center, showing the degradation of resolution near the edges. Right axis: light collection efficiency of the optical system measured with a large, homogeneous sample illuminated with a distant source. (d) Chromatic focal shifts for beads of different colors. The color bands are defined as (central wavelength/pass band width): red (736/128 nm), orange (607/36 nm), and green (525/40). (e) Stripes of 2-pixels on/2-pixels off projected from the DMD onto a thin sample of dried dye, showing modulation at a 7 μm pitch. Diffraction orders off a 1 pixel-on/1-pixel off pattern missed the small mirror in the imaging path (Fig. 1) and were not resolved.
Fig. 5.

Wide-field excitability recordings in rat hippocampal neurons. (a) QuasAr fluorescence image of cultured rat hippocampal neurons expressing voltage actuator CheRiff and voltage reporter QuasAr. See Visualization 1 (10.1MB, avi) . Fluorescence recordings from the boxed cells are shown in (c). (b) White light trans-illumination image of the same FOV. (c) Voltage traces as recorded by fluorescence imaging of the selected neurons. The full FOV was stimulated simultaneously with successively more intense pulses of blue light to activate CheRiff. (d) An expanded view of the indicated spike train showing the details of the spiking waveform including after-hyperpolarizations.
2.2 Patterned illumination
The second optical subsystem of the Firefly microscope was for patterned optogenetic stimulation. In the context of Optopatch measurements, patterned blue light stimuli activated the channelrhodopsin CheRiff [9]. The patterned stimulation can be used, for example, to excite a single neuron while recording the response of its neighbors or to launch an action potential wave across a cardiomyocyte syncytium. Patterned optogenetic stimulation can also be used to activate genes in regions of developing embryos; or to initiate photochemical reactions in sub-regions of a sample. Our system had patterned illumination at 405 nm and 473 nm, but the optics were designed to be achromatic so that light of any visible wavelength could be patterned, for activation of a wide range of optogenetic and photochemical processes.
Patterned illumination was implemented with a DMD. The DMD (Digital light innovations, Discovery D4100 with DLP9500 chip and ALP 4.1 High-Speed control software) had a 0.95” 1080p micromirror array, a pixel size of 10.8 μm, and a triggerable binary image refresh rate of over 20 kHz. A 1 W, 473 nm laser (Dragon Lasers, MBL-N 473) and 300 mW, 405 nm laser (Lilly Electronics, LSR405NR) were combined and expanded to overfill the DMD. The laser light was directed onto the DMD from 24° off the array surface normal and orthogonal to the axis of micromirror rotation. The square micromirrors rotate to ± 12° along their diagonal, so the direct reflection off each micromirror propagated perpendicular to the array surface. Light was collected by a 0.63x MVX objective (160 mm back focal length) and directed toward the microscope.
The close proximity between the objective and tube lens precluded the typical epi-illumination configuration of having a dichroic mirror at 45° in the infinity space. Instead, a small 45° broadband mirror was affixed to the center of a large glass window and inserted into the infinity space (Fig. 1). The 4 mm mirror (Tower Optical, MPCH-4.0) obscured 5% of fluorescence emission, comparable to the losses one would anticipate from a dichroic mirror. The small mirror had the further merit of being compatible with illumination at any wavelength, while a dichroic mirror performing the same task would be wavelength specific.
The combination of the 0.63x MVX objective lens after the DMD and the 2x imaging objective led to a 3.2x demagnification of the DMD onto the sample. The small reflective aperture of the 45° mirror captured only a single diffraction order off the DMD, leading to an optical power efficiency from DMD to sample of 7%. In view of the inexpensive availability of high-power diode lasers, this loss of excitation power was not limiting. Sufficient illumination reached the sample for e.g. channelrhodopsin stimulation (50 mW/cm2 at 473 nm), fluorescence calcium imaging with GCaMP6F (200 mW/cm2 at 473 nm in cardiomyocytes), and photochemical reactions of aryl azide photocrosslinkers (1 W/cm2 at 405 nm).
The small reflective aperture of the coupling mirror also limited the DMD illumination to NA 0.013 at the sample, corresponding to a spatial resolution of 7 μm in the sample plane, or 2 DMD pixels (Fig. 3(e)). Using this optical system, it was possible to excite the sample with an arbitrarily reconfigurable light pattern with a temporal resolution of 50 µs and a spatial resolution of 7 µm. Although we only equipped the DMD with two light sources, other colors could be easily added by introducing additional dichroic beam combining mirrors upstream of the DMD.
The Firefly microscope was also equipped with LED sources mounted from above, to enable transmitted light and fluorescence imaging with additional colors (Thorlabs mounted LEDs). Transmitted white light images clearly resolved single cells (Fig. 5(b)). LED illumination for fluorescence imaging was collimated with aspheric lenses (Thorlabs ACL25416U-A), and the tails of the LED emission spectra were blocked with excitation filters. Light from multiple LEDs was combined with dichroic mirrors, enabling rapid, independent modulation for dynamic fluorescence measurements. The combined colors were imaged oversized onto a field aperture, which was in turn imaged onto the sample to yield a homogeneous spot encircling the FOV. We used LEDs at 375 nm, 455 nm, 485 nm, 530 nm, and 565 nm to complement the 405 nm, 470 nm, and 635 nm laser illumination wavelengths.
2.3 Near-TIR red illumination
A challenge of high-speed imaging is to illuminate the sample with sufficient intensity to compensate for the short exposure time. To illuminate a large FOV with high intensity implies a high total optical power. The required optical power becomes particularly large when the fractional fluorescence changes are small or the signals are intrinsically dim. For instance, QuasAr voltage reporters require high intensity red illumination (635 nm, ~100 W/cm2) to record neuronal voltage traces with good SNR at a 1 kHz frame rate. As the imaging area is increased from ~0.04 mm2 in a typical microscope using a 60x objective to ~4 mm2 here, the red excitation power must increase correspondingly, from ~40 mW to ~4 W. We used two DILAS 8 W lasers (M1B-638.3-8C-SS4.3-T3) combined with a polarizing beam splitter to provide the illumination.
The higher laser powers are not a concern from the perspective of phototoxicity, because the laser intensity—which determines photochemical reaction rates—is equal or lower in the wide-field than in the narrow-field instruments. However, with the high laser power, sample heating and autofluorescence from optical components such as the objective become major concerns, because these processes are proportional to total laser power. We found that passing the high-power red laser through the objective induced sufficient autofluorescence in the objective’s glass components to overwhelm the weak QuasAr fluorescence.
To avoid this high background, we directed the laser to the sample without passing through the objective (Fig. 1). A low-autofluorescence, fused silica prism coupled the light into the sample close to the critical angle just short of total internal reflection (TIR). This geometry compressed the optical beam so only a thin slice of the sample at the coverslip-aqueous interface was illuminated (Fig. 4(a)). As in light-sheet microscopy [26], the beam propagated nearly parallel to the image plane (Fig. 4(b)) and laterally exited the FOV, minimizing out-of-plane autofluorescence.
Fig. 4.
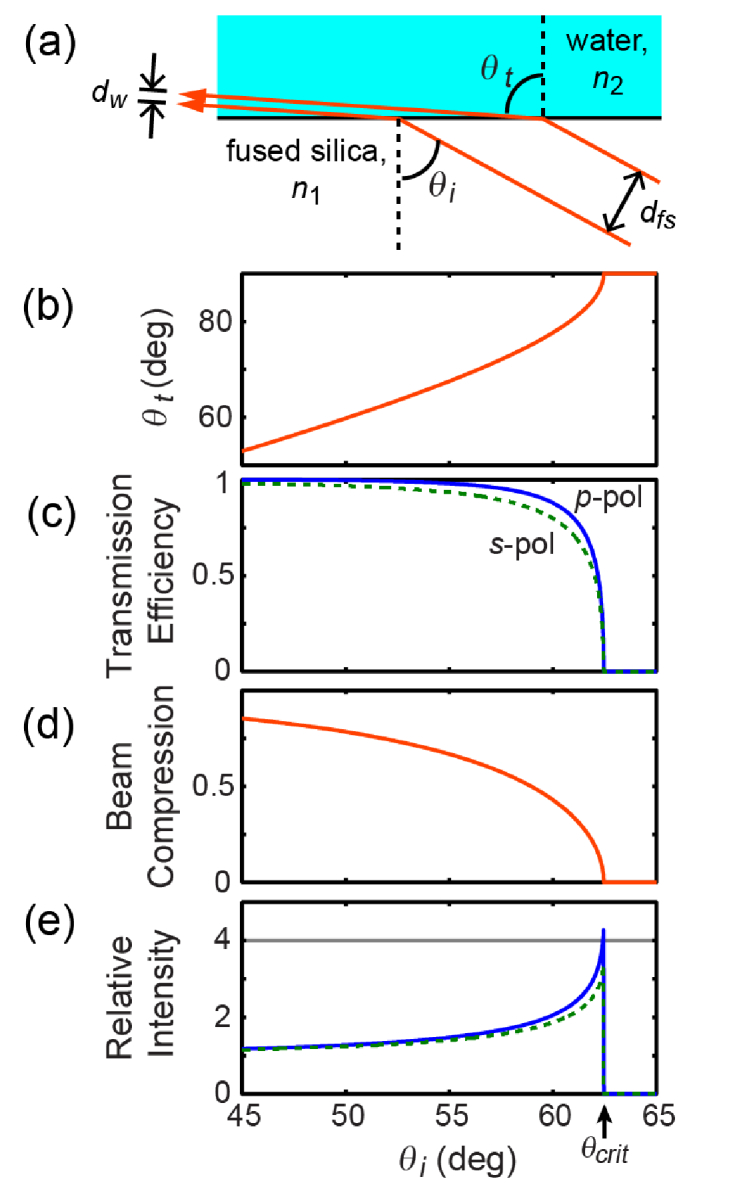
Near-TIR illumination. (a) Diagram of refraction and beam compression at the silica/water interface. (b) Angle of the transmitted beam near the critical angle as a function of the angle of the incident beam. (c) Transmission efficiency of the two light polarizations due to reflection loss. (d) The ray-optics beam compression due to refraction. (e) Net illumination intensity increases as the incidence angle approaches the critical angle because beam compression outweighs reflection losses.
Despite Fresnel reflection losses at the coverslip-aqueous interface, refractive beam compression increased the overall optical intensity at the surface. The Fresnel transmission coefficients at the glass-water interface [32] are given by (Fig. 4(c)):
| (1a) |
| (1b) |
Here the subscript p indicates p-polarization with the electric field in the plane of incidence and reflection and the subscript s indicates s-polarization where the E-field is perpendicular to this plane. θi and θt are the angles of incidence and transmission as shown in Fig. 4(a), related by Snell’s Law. Simple trigonometry shows that the beam width in the water (dw) is compressed relative to the width in the fused silica (dfs) by a factor η (Fig. 4(d)):
| (2) |
The relative light intensity as a function of angle, plotted in Fig. 4(e), is then:
| (3) |
At the transition to TIR, where θt = 90°, the expressions simplify to and. For fused silica (n1 = 1.46) and water (n2 = 1.33) the intensity enhancements are: Ip = 4.39 and Is = 3.64. This enhancement allowed us to use a lower overall laser power to achieve the same target illumination intensity, reducing sample heating and background autofluorescence from the substrate and imaging buffer.
To implement near-TIR illumination, the laser was coupled into the sample via a low-autofluorescence fused silica prism (custom from Mark Optics) with dimensions shown in Fig. 1. To couple light efficiently into the sample and to avoid TIR at the top prism surface, a drop of index-matching immersion oil was placed between the top of the fused silica prism and the bottom of the coverslip-bottomed dish containing the sample. This illumination geometry provided homogeneous near-TIR illumination over a readily defined FOV, while avoiding autofluorescence associated with passing the light through the objective.
We found that the immersion oil comprised a significant source of background autofluorescence, so we explored a variety of index-matching fluids. Silicone oil from Olympus (Z-81114) produced only 40% as much autofluorescence as the next-best oil tested. Cargille Type FF oil, ultrapure glycerol (ThermoFisher #15514-011), 68% sucrose in water, and Olympus Type F oil all produced similar levels of autofluorescence. For typical expression levels in rat hippocampal cells, QuasAr2 fluorescence was ~5x larger than oil autofluorescence so the oil choice had a minimal impact on measurement SNR. In that scenario, Cargille Type FF oil was chosen for its low price and convenient viscosity.
The laser was routed to the prism through a lens placed one focal length from the sample, and a mirror on a translation stage was mounted in the back focal plane of the lens (Fig. 1). Because the mirror was in the lens’s conjugate (Fourier) plane, a change in angle on the mirror re-positioned the laser in the sample, while movement of the translation stage carrying the mirror changed the incidence angle without changing the position.
As the laser incident angle approached the critical angle, the transmitted beam approached horizontal and the brightness of fluorescent cells increased according to Eq. (3). As the beam crossed into TIR, which occurred over a small range of incident angles because of angular content within the Gaussian beam, the transmitted beam vanished and the fluorescent cells became much dimmer because they were much thicker than the ~100 nm evanescent field penetration depth. To eliminate all extraneous excitation light, which added to out-of-focus autofluorescence and heating, the laser passed through a slit aperture that was imaged onto the sample (Fig. 1). The slit was adjusted to uniformly illuminate the FOV and no more.
In summary, the Firefly microscope design provided high speed, wide field of view recordings with excellent low-light performance, and had highly reconfigurable patterned illumination with multiple colors. We next show how this optical system can be used for wide-field all-optical electrophysiology and other applications.
3. Wide-field neural recording
The Firefly optical system was designed for wide-field all-optical electrophysiology recordings using the Optopatch genetic constructs [9], which comprise a blue light-activated nonselective cation channel, CheRiff, and a red light-excited near infrared fluorescent voltage reporter, QuasAr. By expressing both genes together, one can stimulate an action potential in an excitable cell such as a neuron or cardiomyocyte using blue light and simultaneously record the cell’s transmembrane voltage using red light. This combination provides a powerful tool for studying cellular electrophysiology.
Figure 5 shows voltage recording in cultured rat hippocampal neurons expressing the constructs CheRiff-mOrange2 and QuasAr-Citrine, both driven by the excitatory neuron-specific promoter CaMKII. Figure 5(a) shows the QuasAr fluorescence, with sub-cellular resolution across the full 4x0.5 mm FOV. A slightly defocused white light trans-illumination image of the same cells is shown in Fig. 5(b), demonstrating that most neurons visible in white light also express fluorescent protein. Synaptic blockers were added to the imaging medium to eliminate the complicating effects of synaptic transmission.
A movie of QuasAr fluorescence (Visualization 1 (10.1MB, avi) ) was recorded at 1 kHz (emission filter Semrock FF02-736/128) while the neurons were stimulated with successively more intense pulses of blue light, revealing the heterogeneous excitability of this population of cells. Figure 5(c) shows the fluorescence time traces from each cell boxed in Fig. 5(a). For each box, a raw time trace was calculated by averaging over all the pixels in the box, and the SNR was improved by weighting more strongly pixels whose fluorescence time trace was strongly correlated with the average time trace during spiking behavior, as described previously [33]. Figure 5(d) shows a magnified view of the time trace for one stimulus period of one cell, which shows high fractional fluorescence change, good SNR, and physiological action potential waveforms with clear after hyperpolarizations.
3.1 Minimizing substrate autofluorescence and sample heating
When performing neuronal imaging at high red laser illumination powers (~300 W/cm2), we initially detected significant background autofluorescence and sample heating even in the near-TIR geometry (Fig. 6). We originally tested glass-bottomed dishes from several suppliers (e.g. MatTek, In Vitro Scientific), but all showed a large autofluorescent background where the laser passed through the glass. In addition, there was clear evidence of sample heating as reported by increased electrical activity of temperature-sensing neurons from the mouse dorsal root ganglion (DRG) (Fig. 6(d)). A thermocouple placed into the imaging medium above the cells did not report a laser-induced temperature change, suggesting that the heating was localized to the cells or cellular substrate.
Fig. 6.

Reduction of autofluorescent background and sample heating with fused silica coverslips. (a, b) The red laser was coupled into a clean, cell-free coverslip-bottomed dish where the coverslip was made of (a) glass or (b) fused silica. Fluorescence images, collected with the QuasAr emission filter, are plotted on the same scale. (c) Histogram of background pixel counts from the illuminated region in each image showing 8x reduced background in fused silica. (d) Fluorescent voltage recordings from mouse sensory neurons (including temperature sensing neurons) plated on glass (bottom) or fused silica (top). Initially there were 2 seconds of recording of spontaneous activity, followed by three periods of CheRiff stimulation with successively stronger pulses of blue light. Below the example fluorescence recordings are raster plots showing a tick for each recorded action potential from each of the roughly 10 cells per FOV. As the intensity of the red excitation laser increased, which improves the SNR but can also heat the sample, action potential bursts were induced on glass but not the fused silica.
Suspecting the glass, we assembled tissue culture dishes (Willco Wells, KIT-3512) with fused silica coverslips (Esco Optics, R425000) cleaned with 2 M NaOH. In a dish containing imaging buffer but no cells, the fluorescent background was reduced by 8-fold relative to glass (Fig. 6(a)-(c)). Heating was also reduced, allowing us to image at higher red laser intensities or to image at temperatures closer to 37 °C without light-induced activation of DRG neurons. Figure 6(d) shows the different action potential firing patterns of DRG neurons transfected with Optopatch and plated on fused silica (top) or glass (bottom). In the first imaging epoch, spontaneous activity was recorded via QuasAr fluorescence without blue-light stimulation of CheRiff. At low red laser illumination powers (2-3 W), cells displayed minimal spontaneous activity. At higher powers (5-6 W), neurons grown on glass fired a train of action potentials after ~1 s of red laser illumination, indicative of laser-induced heating. Neurons grown on fused silica remained quiescent, suggesting that the lower autofluorescence of the fused silica correlated with lower optical absorption of the substrate, and hence lower heating.
An alternative to the fused silica substrate is the UV-compatible cyclic olefin copolymer (COC), which had significantly lower background autofluorescence and laser heating than glass. COC dishes are commercially available and can be tissue-culture treated for cell adhesion (e.g. Ibidi #81156). Although a fused silica or COC substrate is not strictly required for Optopatch recordings, it is preferred due to the lower background autofluorescence, improved SNR of voltage traces, and reduced laser-induced sample perturbation.
3.2 High-throughput recordings in hiPSC-derived neurons
An important application of all-optical electrophysiology is disease modeling and drug discovery in human neurons derived from induced pluripotent stem cells (iPSCs). These neurons can be derived from individual patients with known disease-causing mutations, offering the promise of connecting the genetic mutation to neuronal function and drug response. However, iPSC-derived neurons are extremely heterogeneous, so robust phenotyping requires measurements on ~100 neurons per condition. Manual patch clamp measurements at ~1 hr/cell are too slow for this task.
The high optical detection efficiency and low background autofluorescence of the Firefly microscope, combined with the minimization of laser-induced sample heating, permitted highly parallel, high-throughput Optopatch measurements on hiPSC-derived neurons. Figure 7 shows optical electrophysiology measurements acquired simultaneously on 85 human iPSC-derived motor neurons in a single FOV [23]. Figure 7(a) shows cell weight maps overlaid on the average image. First, the image was segmented into smaller regions with a watershed algorithm, and then pixel weight maps and time traces were extracted from each region using principal component analysis/independent component analysis (PCA/ICA). Example traces and the blue light induced CheRiff stimulation are shown in Fig. 7(b). Figure 7(c) shows a raster plot in which each row corresponds to one cell and each point represents one action potential, extracted from the fluorescence. Figure 7(d) shows the average firing rate for the population of cells during the stimulus protocol. The step stimuli probe firing rate adaptation under constant optogenetic drive. The pulse trains probe the maximum firing rate and activity-dependent channel inactivation, e.g. from state-dependent channel blockers. The shallow ramp stimulus probes rheobase and the onset of spiking, while the steep ramp stimulus probes depolarization block (where spiking ceases under strong drive).
Fig. 7.

High-throughput recordings from human iPSC motor neurons (adapted with permission from [23]). (a) Fluorescence image of the voltage sensor QuasAr in grayscale and the automatically identified single cells in color. (b) Example fluorescence recordings of voltage from three individual cells (top), and the blue light stimulus pattern (bottom). (c) Raster plot for one field of view showing the behavior of cells during initial (red) and repeat (teal) imaging. Each row is one neuron, and each point corresponds to one action potential. (d) Average firing rate of the population of neurons during the repeat imaging experiments. (e) The location of detected cell centroids from 4193 rat hippocampal neurons in 48 FOV’s in 16 wells, showing the homogeneous distribution of detected cells within the FOV. The SNR of each the detected spikes in each cell is indicated by color, showing uniform sensitivity across the FOV.
To demonstrate assay robustness, the FOV was measured twice under identical stimulus protocols; the population average shows excellent repeatability. To further demonstrate throughput and microscope performance, we recorded from 48 FOV’s in 16 wells and ran it through the automated analysis. Figure. 7(e) shows the location of the >4000 detected cells homogeneously distributed across the FOV, demonstrating that red and blue light illumination, fluorescence collection, and the segmentation algorithm perform uniformly across the imaging area. The microscope’s robust and high-throughput performance opens the prospect of disease modeling and drug discovery with the Firefly microscope.
3.3 Recording from cardiomyocytes
The wide FOV, high sensitivity, and high speed of the Firefly microscope also provide unique capabilities for functional mapping in human iPSC-derived cardiomyocytes (Visualization 2 (2.9MB, avi) ). Human induced pluripotent stem cell derived cardiomyocytes (hiPSC-CMs, CDI) were grown on a substrate patterned with different sized islands of fibronectin [24] (see protocols for details). In brief, a polyacrylamide gel, which cells cannot adhere to, was deposited in the dish. The acrylamide was chemically activated so it could covalently bond fibronectin, which was deposited by microcontact printing using a polydimethylsiloxane (PDMS) stamp. The stamp contained squares ranging in size from 50 µm to 1 mm. The hiPSC-CMs were transfected with CaViar [34], a construct comprising the QuasAr2 voltage indicator fused to the GCaMP6F calcium reporter [8], under control of the universal CMV promotor. The cells were plated using the vendor-specified protocol and infected with lentivirus after 4 days and imaged after 12 days in culture.
Figure 8(a) shows GCaMP6F fluorescence in a 6 x 6 mm FOV containing several hundred independently beating islands; Visualization 2 (2.9MB, avi) is the corresponding movie. The majority of islands beat spontaneously, showing robust action potentials, calcium transients, and mechanical contractions. Figure 8(b) shows fluorescent time traces reporting calcium in the islands boxed in (a). The ability to record from hundreds of independently beating islands and thousands of cells simultaneously is a powerful capability for high-throughput measurements, particularly in robustly characterizing drug response in many independently beating populations.
Fig. 8.

Wide-field recordings in human iPSC -derived cardiomyocytes. (a) A 6 x 6 mm image showing GCaMP6F fluorescence of several hundred islands of various sizes containing human iPSC-derived cardiomyocytes. See Visualization 2 (2.9MB, avi) . (b) Fluorescent time traces from islands shown in (a) showing spontaneous calcium spiking. (c) A composite fluorescence image of a continuous syncytium of cardiomyocytes expressing CheRiff-EGFP (green) and QuasAr2-mOrange2 (red). (d) Top: voltage reported by averaging QuasAr2 fluorescence over the entire FOV (black). Bottom: blue light stimulus to activate CheRiff (blue). (e) Fluorescence time traces reporting voltage recorded from the left and right of the FOV (purple and cyan rectangles in (c)). Initially, the spontaneous voltage wave propagated from left to right (purple upstroke precedes cyan) but after optogenetic stimulation on the right edge of the FOV (dark blue rectangle in (c) and trace in (e)) the propagation direction reversed (cyan upstroke precedes purple).
The Firefly microscope was also effective for recording electrical activity in spontaneously beating or optogenetically paced cardiomyocytes. Figure 8(c) shows a composite fluorescence image of cardiomyocytes co-infected with lentivirus for CheRiff-eGFP (green, GFP emission filter) and QuasAr2-mOrange2 (red, mOrange emission filter). The black trace in Fig. 8(d) shows membrane voltage as recorded by QuasAr2 fluorescence, calculated by averaging over the whole field of view before (first 5 pulses) and after full-field CheRiff stimulation with blue light (blue waveform). Upon onset of optogenetic pacing, the cardiomyocytes immediately synchronized to the optical stimulus.
The high time resolution imaging enabled mapping of electrical wave propagation in the syncytium. Figure 8(e) shows the average traces from the left edge (purple box in Fig. 8(c)) and right edge (cyan box in Fig. 8(c)) of the FOV. Initially, the spontaneous activity propagated from left to right at a velocity of 180 mm/sec: for the first two pulses in Fig. 8(e) the purple upstroke precedes the cyan upstroke. After using the DMD to selectively stimulate the right edge of the FOV (royal blue rectangle in (c) and trace in (e)), the voltage wave propagated from right to left (cyan upstroke preceded purple upstroke). To detect these rapidly propagating wavefronts required high-speed imaging over a wide FOV.
4. Discussion
The Firefly microscope described here combines high-speed, wide-field fluorescence imaging with high light collection efficiency and patterned illumination. It enables simultaneous optogenetic stimulation and voltage imaging of up to ~100 cultured neurons simultaneously. The system can also stimulate and track voltage wave propagation in syncytia of cardiomyocytes. With the rapidly expanding array of fluorescent sensors that report distinct facets of the cell state, and the growing toolkit of optical actuators for stimulating and otherwise manipulating cells, bidirectional optogenetic stimulation and enabling imaging systems promise to become ever more useful.
A key merit of the Firefly design is that the system is readily assembled from off-the-shelf components, yet achieves 10-fold greater light gathering efficiency than the comparable commercial microscope by replacing the commercial tube lens with a high-end camera lens (Fig. 2). The only custom optical element is the prism used for near-TIR illumination. For applications that do not require high-intensity planar illumination, this element can be omitted. While fully custom laser scanning systems can achieve higher spatial resolution and better optical sectioning for tissue imaging in e.g. the rodent brain, the simpler Firefly system provides an overall higher data-rate (pixels/s) for high-speed imaging of planar samples.
Here we have focused on applications of the Firefly microscope to optical electrophysiology. The experiments in cultured neurons are particularly relevant for stem cell-based disease modeling efforts, where one often wishes to record from large numbers of human stem cell-derived neurons or cardiomyocytes. In these cultures, cellular heterogeneity has bedeviled low-throughput manual patch clamp recordings, and the ability to record from hundreds of cells is essential for detecting subtle genetic or pharmacological effects.
A related application is to classifying functional responses in heterogeneous cultures comprising different cell sub-types. For example, cells can be classified based on (1) size and gross morphology, (2) a fluorescent tag expressed by a subtype selective promoter such as CaMKII for excitatory neurons [35] or Dlx for inhibitory neurons [36], or (3) fixing and immuno-labeling of cells after functional imaging, and registration of the immunocytochemistry images with functional results. It is desirable to record from many cells in a large FOV to sample the different sub-populations, particularly if some sub-types are rare. This strategy can be applied to Optopatch measurements or to other fluorescent reporters, which may be targeted to cellular compartments such as the mitochondria [37,38] or endoplasmic reticulum [39,40]. Sub-cellular information, encoded in genetically encoded trafficking sequences, can be extracted even with low-resolution imaging. With these slower sensors, Firefly’s full Ø6 mm FOV can be used to record from many thousands of individual cells in parallel.
The Firefly optical system enables many other applications beyond those discussed here. For instance, the FOV readily encompasses the entire body of a zebrafish embryo, enabling intra-vital Ca2+ imaging [41]. The high light collection efficiency could be used to enhance sensitivity in wide-field Ca2+ [42] and voltage [43] brain imaging applications. Patterned illumination at 405 nm wavelength can activate a variety of photochemical reactions, including activation of azobenzene-based crosslinkers and photoconversion of switchable fluorescent proteins. Patterned activation of a photochemical crosslinker has been used to selectively crosslink target cells to a dish, while non-target cells can then be washed away [44]. This ‘Photostick’ protocol facilitates pooled genetic screens in which cells with desired attributes can be optically selected from a heterogeneous mixture. These examples illustrate the diversity of applications that can be achieved with ultrawidefield, high-sensitivity imaging.
5. Conclusion
The Firefly microscope enables diverse measurements using fluorescent sensors and optogenetic actuators at low magnification and high throughput. This throughput is essential for disease modeling and high-content screening, and the large area enables measurements in neural network and extended electrical syncytia. The Firefly microscope will be a powerful tool for studying neuroscience and cardiac biology.
6. Protocols
6.1 Rat hippocampal culture
Primary rat cells were prepared from the hippocampus of E18 Sprague Dawley rats from BrainBits. Cells were shipped at 4 °C (never frozen), and dissociated following BrainBits protocols. They were plated at a density of 30k cells/cm2 onto glass-bottomed dishes (MatTek, P35G-1.5-10-C) pre-coated with poly-D-lysine (PDL) and laminin. At 7 days in vitro (DIV), cells were infected with lentivirus coding for QuasAr2-citrine and CheRiff-mOrange2. At 14 DIV, cells were imaged on the microscope. Immediately prior to imaging, the buffer was exchanged to Tyrode’s medium (containing, in mM: 125 NaCl, 2.5 KCl, 3 CaCl2, 1 MgCl2, 10 HEPES, 30 glucose, pH 7.3) with synaptic blockers (10 µM NBQX, 25 µM AP-V, 20 µM gabazine) to quench network activity.
6.2 Mouse DRG culture
Cells were dissected from the dorsal root ganglion of P7 mice pups (Taconic Biosciences). Cells were enzymatically digested and plated at a density of 12.5 k cells/cm2 onto glass- or fused silica-bottomed dishes pre-coated with PDL and laminin. At 3 DIV, cells were infected with lentivirus for QuasAr2 and CheRiff at a ratio of 5:1. Cells were cultured in a medium comprised of: 48.5 mL Neurobasal-A medium (Life Technologies; Cat# 10888-022), 1 mL B27 supplement (Life Technologies; Cat# 17504-044) and 0.5 mL GlutaMAX (Life Technologies; Cat# 61870-036), supplemented with 2 ng/mL of GDNF. At 15 DIV, cells were imaged after buffer exchange to Tyrode’s medium.
6.3 Lentivirus production
Lentivirus was prepared in HEK cells following established protocols [45]. In brief, low passage number HEK293T cells were plated onto gelatin-coated (Stemcell technologies, #07903) 15-cm dishes in DMEM + 10% FBS. When HEK cells reached 80% confluence, they were transfected in serum-free medium with polyethylenimine (PEI; Sigma 408727). Into 1.2 mL of DMEM with 14 μg of the vector plasmid, 9 μg of the 2nd generation packaging plasmid psPAX2 (Addgene #12260), and 4 μg of viral entry protein VSV-G plasmid pMD2.G (Addgene #12259) were added 36 μL of 1 mg/mL PEI, vortexed, incubated for 10 min, and added dropwise to the plate. After 4 hours, the medium was exchanged back to 16 mL DMEM10 to avoid PEI toxicity. The supernatant was harvested at 48 hours post transfection, centrifuged 5 min at 500 g to pellet cells and debris, and filtered through a 0.45 μm filter.
6.4 hiPSC cardiomyocyte culture
hiPS-CM (iCell cardiomyocytes), plating and maintenance medium were purchased from Cellular Dynamics International, plated and cultured according to manufacturer instructions. Cells were plated at a density of 50k/cm2 in glass-bottomed dishes with patterned fibronectin (prep described below); cells were plated initially in just the recessed region of the glass-bottomed dish. After 1 hour, a very gentle rinsing removed dead and un-adhered cells and the volume of Plating Medium was increased to 1 mL. 48 hours after plating, the plating medium was aspirated from the dish and replaced with the maintenance medium. Cells were infected with lentivirus on at 4 DIV, and imaged after 10 – 14 DIV. Prior to imaging, the medium was replaced with a low-autofluorescence imaging buffer (in mM was: 1.8 CaCl2, 2.5x10−4 Fe(NO3)3, 0.81 MgSO4, 5.3 KCl, 44 NaHCO3, 129 NaCl, 0.91 NaH2PO4, 1 sodium pyruvate, 10 D-( + )-galactose). Imaging on the microscope was performed at 37 °C and 5% CO2 using the microscope’s environmental chamber.
6.5 Cardiomyocyte patterning
Cell patterning was implemented by covalently binding fibronectin to a poly-acrylamide gel which had been bonded to the glass surface. The fibronectin was patterned using a polydimethylsiloxane (PDMS) stamp. In brief, glass-bottom dishes (In Vitro Scientific, D35-20-1.5-N). Dish surfaces were cleaned with 5 min. air plasma (SPI Plasma-Prep II). The glass was treated with silane solution comprising 0.5% vol/vol 3-(trimethoxysilyl)propyl methacrylate (Sigma 440159), 2% glacial acetic acid, and 97.5% anhydrous ethanol (200 μL). The dish was incubated for 30 min in an oxygen-free N2 atmosphere, rinsed with absolute ethanol, and baked at 65°C in a vacuum oven.
Next a soft polyacrylamide gel which resists cell adhesion was polymerized on the surface, where it covalently bound the methacrylate functionalized glass. The polyacrylamide was doped with NHS leaving groups for covalently binding fibronectin. As in Cretu et al. [46], the roughly 40 μm gel thickness was controlled by polymerizing between the activated glass and a siliconized, non-stick top coverslip (Hamilton Research, HR3-239). The polymerization solution was 8% W/V acrylamide (Sigma A4058), 0.2% W/V bis-acrylamide (Sigma M1533), 0.1% V/V TEMED (Sigma T7024), 1.2 mg/mL potassium persulfate (KPS; Sigma 216224), and 4.2 mg/mL acryl-NHS (Sigma A8060) in a 40 mM phosphate buffer at pH 7. Immediately after adding the TEMED and KPS, the solution was vortexed, 10 μL of the solution was pipetted onto the center of several glass-bottomed dishes, and the siliconized coverslip was placed on top. After polymerization was complete (a couple minutes), the top coverslip was pried off using a sewing needle, and the gel was rinsed.
To grow cells on islands of pre-determined size and shape, standard soft lithography techniques [47] were used to make patterned islands of fibronectin covalently bound to the polyacrylamide. In brief, patterns were printed on a Mylar transparency (CAD Art Services) and transferred to a layer of SU-8 3025 photoresist on a Si wafer via contact lithography. The SU-8 master was then used as a template for casting PDMS stamps. The PDMS stamp was coated with fibronectin (Yo Proteins #663, 0.05 mg/mL) for 30 min following by aspiration and air-dry for 10 min before printing the fibronectin pattern onto the NHS-functionalized acrylamide. The NHS covalently bound to the fibronectin, forming a stable cell-adherent pattern.
Acknowledgments
We thank Vaibhav Joshi for culturing neurons, and John Ferrante and Michael Murphy for performing neuronal recordings.
Funding
This work was supported by the Howard Hughes Medical Institute; NIH grant (1-R01-EB012498); ONR (N000141110-549); the Gordon and Betty Moore Foundation.
Disclosures
CAW: Harvard University (P), Q-State Biosciences (I,E). AEC: Harvard University (P), Q-State Biosciences (I,C).
References and links
- 1.Cohen A. E., “Optogenetics: Turning the Microscope on Its Head,” Biophys. J. 110(5), 997–1003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin M. Z., Schnitzer M. J., “Genetically encoded indicators of neuronal activity,” Nat. Neurosci. 19(9), 1142–1153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Terai T., Nagano T., “Small-molecule fluorophores and fluorescent probes for bioimaging,” Pflugers Arch. 465(3), 347–359 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Newman R. H., Fosbrink M. D., Zhang J., “Genetically encodable Fluorescent Biosensors for Tracking Signaling Dynamics in Living Cells,” Chem. Rev. 111(5), 3614–3666 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mehta S., Zhang J., “Reporting from the field: genetically encoded fluorescent reporters uncover signaling dynamics in living biological systems,” Annu. Rev. Biochem. 80, 375–401 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tantama M., Hung Y. P., Yellen G., Optogenetic Reporters: Fluorescent Protein-Based Genetically Encoded Indicators of Signaling and Metabolism in the Brain., 1st ed. (Elsevier B.V., 2012), Vol. 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miesenböck G., De Angelis D. A., Rothman J. E., “Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins,” Nature 394(6689), 192–195 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Chen T.-W., Wardill T. J., Sun Y., Pulver S. R., Renninger S. L., Baohan A., Schreiter E. R., Kerr R. A., Orger M. B., Jayaraman V., Looger L. L., Svoboda K., Kim D. S., “Ultrasensitive fluorescent proteins for imaging neuronal activity,” Nature 499(7458), 295–300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hochbaum D. R., Zhao Y., Farhi S. L., Klapoetke N., Werley C. A., Kapoor V., Zou P., Kralj J. M., Maclaurin D., Smedemark-Margulies N., Saulnier J. L., Boulting G. L., Straub C., Cho Y. K., Melkonian M., Wong G. K.-S., Harrison D. J., Murthy V. N., Sabatini B. L., Boyden E. S., Campbell R. E., Cohen A. E., “All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins,” Nat. Methods 11(8), 825–833 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DiPilato L. M., Zhang J., “The role of membrane microdomains in shaping β2-adrenergic receptor-mediated cAMP dynamics,” Mol. Biosyst. 5(8), 832–837 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Mochizuki N., Yamashita S., Kurokawa K., Ohba Y., Nagai T., Miyawaki A., Matsuda M., “Spatio-temporal images of growth-factor-induced activation of Ras and Rap1,” Nature 411(6841), 1065–1068 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Lin J. Y., “A user’s guide to channelrhodopsin variants: features, limitations and future developments,” Exp. Physiol. 96(1), 19–25 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klapoetke N. C., Murata Y., Kim S. S., Pulver S. R., Birdsey-Benson A., Cho Y. K., Morimoto T. K., Chuong A. S., Carpenter E. J., Tian Z., Wang J., Xie Y., Yan Z., Zhang Y., Chow B. Y., Surek B., Melkonian M., Jayaraman V., Constantine-Paton M., Wong G. K.-S., Boyden E. S., “Independent optical excitation of distinct neural populations,” Nat. Methods 11(3), 338–346 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider F., Grimm C., Hegemann P., “Biophysics of Channelrhodopsin,” Annu. Rev. Biophys. 44, 167–186 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Liu H., Gomez G., Lin S., Lin S., Lin C., “Optogenetic Control of Transcription in Zebrafish,” PLoS One 7(11), e50738 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yumerefendi H., Lerner A. M., Zimmerman S. P., Hahn K., Bear J. E., Strahl B. D., Kuhlman B., “Light-induced nuclear export reveals rapid dynamics of epigenetic modifications,” Nat. Chem. Biol. 12(6), 399–401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bugaj L. J., Choksi A. T., Mesuda C. K., Kane R. S., Schaffer D. V., “Optogenetic protein clustering and signaling activation in mammalian cells,” Nat. Methods 10(3), 249–252 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Wang H., Vilela M., Winkler A., Tarnawski M., Schlichting I., Yumerefendi H., Kuhlman B., Liu R., Danuser G., Hahn K. M., “LOVTRAP: An Optogenetic System for Photoinduced Protein Dissociation,” Nat. Methods 13(9), 755–758 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim B., Lin M. Z., “Optobiology: optical control of biological processes via protein engineering,” Biochem. Soc. Trans. 41(5), 1183–1188 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang K., Cui B., “Optogenetic control of intracellular signaling pathways,” Trends Biotechnol. 33(2), 92–100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emiliani V., Cohen A. E., Deisseroth K., Häusser M., “All-Optical Interrogation of Neural Circuits,” J. Neurosci. 35(41), 13917–13926 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson H. E., Goyal Y., Pannucci N. L., Schüpbach T., Shvartsman S. Y., Toettcher J. E., “The Spatiotemporal Limits of Developmental Erk Signaling,” Dev. Cell 40(2), 185–192 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hempel C. M., Werley C. A., Dempsey G. T., Gerber D. J., Ellen I., Biomap L. B., “Targeting neuronal function for CNS drug discovery,” Drug Discov. Today. Technol. 23, 17–25 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Werley C. A., Chien M.-P., Gaublomme J., Shekhar K., Butty V., Yi B. A., Kralj J. M., Bloxham W., Boyer L. A., Regev A., Cohen A. E., “Geometry-dependent functional changes in iPSC-derived cardiomyocytes probed by functional imaging and RNA sequencing,” PLoS One 12(3), e0172671 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahrens M. B., Orger M. B., Robson D. N., Li J. M., Keller P. J., “Whole-brain functional imaging at cellular resolution using light-sheet microscopy,” Nat. Methods 10(5), 413–420 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Huisken J., Swoger J., Del Bene F., Wittbrodt J., Stelzer E. H. K., “Live Embryos by Selective Plane Illumination Microscopy,” Science 305, 1007–1009 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Katona G., Szalay G., Maák P., Kaszás A., Veress M., Hillier D., Chiovini B., Vizi E. S., Roska B., Rózsa B., “Fast two-photon in vivo imaging with three-dimensional random-access scanning in large tissue volumes,” Nat. Methods 9(2), 201–208 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Stirman J. N., Smith I. T., Kudenov M. W., Smith S. L., “Wide field-of-view, multi-region, two-photon imaging of neuronal activity in the mammalian brain,” Nat. Biotechnol. 34(8), 857–862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai P. S., Mateo C., Field J. J., Schaffer C. B., Anderson M. E., Kleinfeld D., “Ultra-large field-of-view two-photon microscopy,” Opt. Express 23(11), 13833–13847 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McConnell G., Trägårdh J., Amor R., Dempster J., Reid E., Amos W. B., “A novel optical microscope for imaging large embryos and tissue volumes with sub-cellular resolution throughout,” eLife 5, e18659 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sofroniew N. J., Flickinger D., King J., Svoboda K., “A large field of view two-photon mesoscope with subcellular resolution for in vivo imaging,” eLife 5, 1–20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Born M., Wolf E., Principles of Optics, Seventh Ed (Cambridge University Press, 1999). [Google Scholar]
- 33.Kralj J. M., Douglass A. D., Hochbaum D. R., Maclaurin D., Cohen A. E., “Optical recording of action potentials in mammalian neurons using a microbial rhodopsin,” Nat. Methods 9(1), 90–95 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dempsey G. T., Chaudhary K. W., Atwater N., Nguyen C., Brown B. S., McNeish J. D., Cohen A. E., Kralj J. M., “Cardiotoxicity screening with simultaneous optogenetic pacing, voltage imaging and calcium imaging,” J. Pharmacol. Toxicol. Methods 81, 240–250 (2016). [DOI] [PubMed] [Google Scholar]
- 35.H. Hioki, H. Kameda, H. Nakamura, T. Okunomiya, K. Ohira, K. Nakamura, M. Kuroda, and T. Furuta, “Efficient gene transduction of neurons by lentivirus with enhanced neuron-specific promoters,” Gene Ther. 14, 872–882 (2007). [DOI] [PubMed]
- 36.Dimidschstein J., Chen Q., Tremblay R., Rogers S. L., Saldi G. A., Guo L., Xu Q., Liu R., Lu C., Chu J., Grimley J. S., Krostag A. R., Kaykas A., Avery M. C., Rashid M. S., Baek M., Jacob A. L., Smith G. B., Wilson D. E., Kosche G., Kruglikov I., Rusielewicz T., Kotak V. C., Mowery T. M., Anderson S. A., Callaway E. M., Dasen J. S., Fitzpatrick D., Fossati V., Long M. A., Noggle S., Reynolds J. H., Sanes D. H., Rudy B., Feng G., Fishell G., “A viral strategy for targeting and manipulating interneurons across vertebrate species,” Nat. Neurosci. 19(12), 1743–1749 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gutscher M., Pauleau A.-L., Marty L., Brach T., Wabnitz G. H. V., Samstag Y., Meyer A. J., Dick T. P., “Real-time imaging of the intracellular glutathione redox potential,” Nat. Methods 5(6), 553–559 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Terhzaz S., Southall T. D., Lilley K. S., Kean L., Allan A. K., Davies S. A., Dow J. A. T., “Differential gel electrophoresis and transgenic mitochondrial calcium reporters demonstrate spatiotemporal filtering in calcium control of mitochondria,” J. Biol. Chem. 281(27), 18849–18858 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Birk J., Meyer M., Aller I., Hansen H. G., Odermatt A., Dick T. P., Meyer A. J., Appenzeller-Herzog C., “Endoplasmic reticulum: reduced and oxidized glutathione revisited,” J. Cell Sci. 126(Pt 7), 1604–1617 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Henderson M. J., Baldwin H. A., Werley C. A., Boccardo S., Whitaker L. R., Yan X., Holt G. T., Schreiter E. R., Looger L. L., Cohen A. E., Kim D. S., Harvey B. K., “A low affinity GCaMP3 variant (GCaMPer) for imaging the endoplasmic reticulum calcium store,” PLoS One 10(10), e0139273 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hou J. H., Kralj J. M., Douglass A. D., Engert F., Cohen A. E., “Simultaneous mapping of membrane voltage and calcium in zebrafish heart in vivo reveals chamber-specific developmental transitions in ionic currents,” Front. Physiol. 5(AUG), 344 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim T. H., Zhang Y., Lecoq J., Jung J. C., Li J., Zeng H., Niell C. M., Schnitzer M. J., “Long-Term Optical Access to an Estimated One Million Neurons in the Live Mouse Cortex,” Cell Reports 17(12), 3385–3394 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petersen C. C. H., Sakmann B., “Functionally Independent Columns of Rat Somatosensory Barrel Cortex Revealed with Voltage-Sensitive Dye Imaging,” J. Neurosci. 21(21), 8435–8446 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chien M.-P., Werley C. A., Farhi S. L., Cohen A. E., “Photostick: a method for selective isolation of target cells from culture,” Chem. Sci. (Camb.) 6(3), 1701–1705 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geraerts M., Michiels M., Baekelandt V., Debyser Z., Gijsbers R., “Upscaling of lentiviral vector production by tangential flow filtration,” J. Gene Med. 7(10), 1299–1310 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Cretu A., Castagnino P., Assoian R., “Studying the effects of matrix stiffness on cellular function using acrylamide-based hydrogels,” J. Vis. Exp. 42, 7–11 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qin D., Xia Y., Whitesides G. M., “Soft lithography for micro- and nanoscale patterning,” Nat. Protoc. 5(3), 491–502 (2010). [DOI] [PubMed] [Google Scholar]