

Key Points
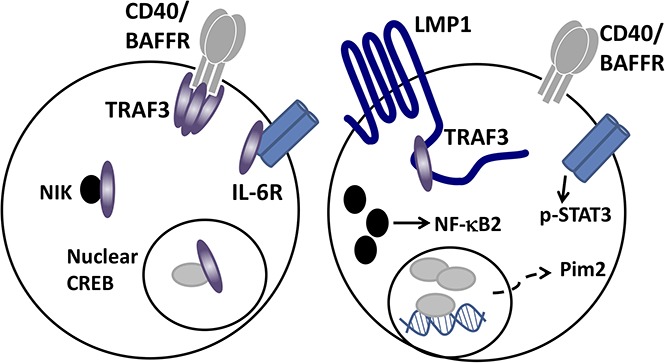
Expression of the Epstein-Barr virus–encoded oncoprotein LMP1 leads to sequestration of TRAF3 in B-lymphoma cells.
This sequestration inhibits TRAF3-negative regulation of prosurvival membrane, cytoplasmic, and nuclear signaling events in the B cell.
Abstract
Loss-of-function mutations in genes encoding the signaling protein tumor necrosis factor receptor–associated factor 3 (TRAF3) are commonly found in human B-cell malignancies, especially multiple myeloma and B-cell lymphoma (BCL). B-cell TRAF3 deficiency results in enhanced cell survival, elevated activation receptor signaling, and increased activity of certain transcriptional pathways regulating expression of prosurvival proteins. A recent analysis of TRAF3 protein staining of ∼300 human BCL tissue samples revealed that a higher proportion of samples expressing the oncogenic Epstein-Barr virus–encoded protein latent membrane protein 1 (LMP1) showed low/negative TRAF3 staining than predicted. LMP1, a dysregulated mimic of the CD40 receptor, binds TRAF3 more effectively than CD40. We hypothesized that LMP1 may sequester TRAF3, reducing its availability to inhibit prosurvival signaling pathways in the B cell. This hypothesis was addressed via 2 complementary approaches: (1) comparison of TRAF3-regulated activation and survival-related events with relative LMP1 expression in human BCL lines and (2) analysis of the impact upon such events in matched pairs of mouse BCL lines, both parental cells and subclones transfected with inducible LMP1, either wild-type LMP1 or a mutant LMP1 with defective TRAF3 binding. Results from both approaches showed that LMP1-expressing B cells display a phenotype highly similar to that of B cells lacking TRAF3 genes, indicating that LMP1 can render B cells functionally TRAF3 deficient without TRAF3 gene mutations, a finding of significant relevance to selecting pathway-targeted therapies for B-cell malignancies.
Visual Abstract
Introduction
Malignancies of B lymphocytes constitute the largest proportion of hematopoietic cell cancers, with B-cell lymphoma (BCL) representing the largest group.1 The human γ-herpesvirus Epstein-Barr virus (EBV), which infects >90% of humans, contributes to pathogenesis of Burkitt, Hodgkin, AIDS-associated, and posttransplant BCL.2 Additionally, a rarer form of EBV-associated diffuse large BCL (DLBCL) occurs in immunocompetent patients >50 years3; a similar type of DLBCL was recently reported in young patients.4
The EBV protein latent membrane protein 1 (LMP1) is expressed in EBV latency II and III programs, characteristic of Hodgkin, posttransplant, AIDS-associated,2 and DLBCL.4 LMP1 was implicated as oncogenic in the 1980s by its ability to transform cultured cells.5,6 Over the ensuing decade, it was revealed that B-cell LMP1 acts as a dysregulated mimic of CD40, inducing enhanced B-cell activation and survival via several pathways.7 Like CD40, the LMP1 cytoplasmic C terminus binds the tumor necrosis factor receptor–associated factor (TRAF) proteins, associating with TRAFs 1, 2, 3, 5, and 6; however, the 2 receptors use TRAFs in differential and sometimes contrasting ways.8,9 TRAF2 promotes CD40-mediated NF-κB activation in B cells, and TRAF1 amplifies this,10,11 but TRAFs 1 and 2 associate weakly with LMP1 and are dispensable for LMP1-mediated B-cell NF-κB activation.12 TRAF5 deficiency has only a modest impact upon CD40-mediated B-cell activation13 but causes major disruption in LMP1-mediated effects on B cells in vitro and in vivo in a mouse model.14 TRAF6 plays similar roles in activating B-cell signaling pathways downstream of CD40 and LMP1, but it binds a CD40 site distinct from the overlapping binding site for TRAFs 1, 2, 3 and 5, whereas TRAF6 binds to the shared TRAF-binding site of LMP1.15
The greatest contrast in TRAF utilization by CD40 vs LMP1 is for TRAF3. TRAF3 strongly inhibits both CD40 and B-cell–activating factor receptor (BAFFR) signals to B cells.12,16,17 However, TRAF3 is in contrast required for many LMP1-mediated activation events,12 as well as recruiting TRAF5.18 Interestingly, TRAF3 binds LMP1 with considerably greater avidity than CD40,12 corresponding to increased contact residues in LMP1-TRAF3 binding.19 It is also important to note that TRAF5’s association with LMP1 requires the binding of TRAF3,14 so the requirement for TRAF3 in various LMP1-mediated B-cell activation events may be a reflection of the necessary role of TRAF5 in promoting these events.
Although whole-mouse TRAF3 deficiency is neonatally lethal,20 conditional TRAF3 deletion in B cells (B-Traf3−/−) causes markedly enhanced homeostatic survival21,22; this is not observed in other TRAF3-deficient hematopoietic cell types.23-25 Older mice with B-cell TRAF3 deficiency develop BCLs.26 Enhanced B-cell survival results from multiple TRAF3-regulated, B-cell–specific mechanisms. These include constitutive noncanonical NF-κB2 activation, altered nuclear cyclic adenosine 5′-monophosphate response element–binding protein (CREB) –mediated transcription of genes encoding pro-survival proteins, increased interleukin-6 receptor (IL-6R) signaling and enhanced glucose metabolism.27-30
When the B-Traf3−/− mouse was first reported,21 inactivating mutations in the human TRAF3 gene were also noted, associated with multiple myeloma (MM).31,32 This has now been observed in multiple studies; such mutations are one of the top 11 seen in 66% of human MM.33 Loss-of-function TRAF3 mutations and/or changes in TRAF3 expression have also been associated with BCL.34-37
Because LMP1, a protein expressed constitutively in membrane rafts,38 binds TRAF3 with greater avidity than normal membrane receptors, we hypothesized that LMP1 sequesters TRAF3, preventing it from downregulating B-cell survival. Thus, LMP1 expression could result in a BCL-predisposing TRAF3-deficient phenotype, without mutation of TRAF3 genes, and this “functional” TRAF3 deficiency could contribute to the lymphomagenic properties of LMP1. The present report presents findings supporting this hypothesis and suggests that targeting TRAF3-regulated B-cell survival pathways may be useful in treating LMP1+ BCL.
Methods
Cell lines
The mouse BCL lines CH12.LX and M12.4.1 were previously described.39,40 The human BCL-derived lines Daudi (LMP1−), T5-1 (LMP1low), and SKW6.4 (LMP1high) were provided by ATCC (Manassas, VA). The Karpas 422 line (LMP1−) was obtained from George Weiner (University of Iowa, Iowa City, IA). BCL lines were cultured in RPMI 1640 supplemented with 10% fetal calf serum, 10 μM 2-ME, 2 mM L glutamine, and 100 μg/mL penicillin/streptomycin (BCM-10). Subclones of CH12.LX and M12.4.1 cells stably transfected with an inducible plasmid encoding wild-type (WT) LMP1 or the LMP1 mutant SUB2 (with 2 point mutations in the CTAR1 TRAF-binding site) under control of a Lac repressor were previously described.41 Transfected cell lines were maintained in BCM-10 containing 400 µg/mL geneticin (G418; Thermo Fisher Scientific, Grand Island, NY) and 200 µg/mL hygromycin B (Thermo Fisher Scientific).
Immunohistochemistry
Fixed and paraffin-embedded deidentified human DLBCL tissues were obtained from the University of Iowa Mayo Clinic Lymphoma Molecular Epidemiology Resource (MER). Tissue microarray staining was performed at the University of Iowa Pathology Research Laboratory. For TRAF3 staining, antigen retrieval was conducted in 10 mM Tris/HCl/1 mM EDTA buffer (pH 9.0). Primary rabbit monoclonal antibody (mAb) EP1730Y to TRAF3 (Abcam, Cambridge, MA) was used at a 1:50 dilution. Samples were stained with secondary horseradish peroxidase (HRP)-labeled goat anti-rabbit (Rb) immunoglobulin antibody (Dako/Agilent, Santa Clara, CA) for 30 minutes and then incubated for 5 minutes with diaminobenzene (Dako). Slides were rinsed with buffer, and Dako diaminobenzene enhancer applied for 3 minutes. Samples were rinsed in dH2O and counterstained for 1 minute with Harris hematoxylin. For LMP1 staining, antigen retrieval used 10 mM citrate (pH 6.0) for 15 minutes in a Decloaker (Biocare Medical) at 110°F. Samples were treated with 3% H2O2 for 8 minutes to quench, and S12 mAb to LMP1 was used at 1:25 dilution, with the Mouse Envision secondary antibody (Dako), and treatment as for TRAF3 staining. Stained tissue specimens were photographed using a Leica Aperio Ariol slide scanner in the University of Iowa Central Microscopy Facility, and staining was evaluated using Aperio ImageScope software. Images in Figure 1 were photographed using an Olympus BX-51 light microscope with a SPOT camera using SPOT Advanced image capture software (SPOT Imaging).
Figure 1.

TRAF3 and LMP1 protein expression in human DLBCL samples. Formalin-fixed, paraffin-embedded tissue samples were stained for TRAF3 protein as described in “Methods.” (A) The top row shows representative samples identified by whole-exome sequencing as having decreased copy number for the TRAF3 gene stained for TRAF3 protein (brown); the bottom row shows representative samples identified as having a normal complement of TRAF3 genes stained for TRAF3 protein. (B) A control sample of normal human tonsil tissue stained for TRAF3 protein. (C) A sample with low/negative TRAF3 protein (previously determined by TRAF3 staining as in A) (left) stained in parallel for LMP1 (brown). An LMP1-negative specimen from the same tissue slide is shown on the right.
Antibodies and reagents
Isopropyl β-D-1-thiogalactopyranoside (IPTG) was from Sigma Chemical (St. Louis, MO). Stimulatory antibodies used for CD40 or BAFFR immunoprecipitation were 1C10 (from F. Lund, University of Alabama, Birmingham, AL) and G28-5 (ATCC). Antibodies used for immunoblotting were Rb anti-TRAF3 and Rb anti-mouse CD40 (Santa Cruz Biotechnology, Dallas, TX), Rb anti-phospho STAT3 (Y705), Rb anti-NIK antibody (Cell Signaling Technology, Danvers, MA), and Rb anti-BAFFR (Abcam). The S12 anti-LMP1 mAb was the gift of F. Wang (Harvard University, Boston, MA); mouse anti-actin antibody was from Millipore (Billerica, MA). HRP-conjugated polyclonal goat anti-mouse and anti-Rb antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA). Recombinant cytokines mouse and human interleukin-6 (IL-6) and human BAFF were from PeproTech (Rocky Hill, NJ).
CD40 and BAFFR immunoprecipitation
CD40 was immunoprecipitated with 1C10 (mouse) and G28.5 (human) mAbs. Protein G paramagnetic beads (Invitrogen) were coated with anti-CD40 mAbs as described previously.42 Cells (20 × 106/sample) were resuspended in 500 µL serum-free medium and stimulated with 10 µL antibody-coated beads for 10 minutes at 37°C. Cells were pelleted by centrifugation and supernatant discarded. Cells were lysed with immunoprecipitation cocktail as described elsewhere.42 Beads were pelleted using a magnet and extensively washed. For human cell samples for blots, CD40 was deglycosylated by incubating beads with 250 U PNGase F (New England Biolabs, Ipswich, MA) according to the manufacturer’s protocol. 20 µL of 2× sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer was added to beads and heated for 10 minutes at 95°C. BAFFR was immunoprecipitated, and immunoprecipitations were processed as for CD40 immunoprecipitations. BAFFR was also deglycosylated for western blotting.
IL-6 stimulation
BCL cell lines and subclones stably expressing inducible WT LMP1 were incubated with IPTG for 4 hours and then stimulated with 20 ng/mL recombinant mouse IL-6 or 40 ng/mL recombinant human IL-6 for the indicated times. Cell lysates were prepared and western blotting performed as below.
Western blotting
Whole-cell lysates and immunoprecipitation sample proteins were resolved on SDS-PAGE gels and transferred to PVDF membranes (Millipore, Billerica, MA) as described.42 Blots were agitated in 5% milk in TBST (120 mM NaCl, 0.08% Tween 20, and 40 mM Tris) for 1 hour at room temperature, and incubated overnight at 4°C with primary antibodies. Blots were then washed in TBST, and incubated with HRP-conjugated secondary antibodies for 2 hours. Blots were developed using Supersignal West Pico (Thermo Scientific). Chemiluminescence was read with an LAS-4000 low-light camera and analyzed with MultiGauge software (Fujifilm Life Science, Edison, NJ,)
Immunofluorescent microscopy
Cells were fixed with ice-cold methanol and slides blocked for 30 minutes using rat serum and anti-CD16/32 antibody (eBioscience) and then stained with anti-CREB antibody.29 Fluorescent images were acquired using an LSM 710 confocal microscope (Zeiss) with 63× magnification and Zen2009 software (Zeiss). 50 to 60 cells per slide were examined. Images were analyzed with ImageJ software (NIH).
Statistics
Table 1 used the Fisher's exact test to analyze the difference in TRAF3 staining in relation to LMP1 staining. Figures 2, 3, 4, and 7 used the 2-tailed unpaired Student t test to study differences between the 2 means of different experimental groups. For multiple group comparisons, Figure 5 used one-way analysis of variance (ANOVA), and Sidak’s multiple comparison test was used for Figure 6 (2-way ANOVA and Bonferroni method). P ≤ .05 was considered significant. Prism software (GraphPad, La Jolla, CA) was used for calculations.
Table 1.
TRAF3 deficiency in human DLBCL samples
| Parameter | Total number of samples | TRAF3 deficient, n (%) |
|---|---|---|
| Exome sequencing (of 51 samples tested) | 51 | 6 (11.7) by exome sequencing |
| TRAF3 protein staining (277 samples tested) | 277 | 95 (34.3) by immunohistochemistry |
| LMP1 protein positive (of 277 tested) | 50 | 30 (60.0) by immunohistochemistry |
| LMP1 protein negative (of 277 tested) | 227 | 64 (28.1) by immunohistochemistry |
LMP1-positive and -negative samples differ significantly in their percentage of TRAF3high and TRAF3low staining, as determined by the Fisher's exact test (P < .0001).
Figure 2.

Effect of LMP1 expression upon TRAF3 availability to bind membrane CD40 or BAFFR in human BCL lines. (A-C) Western blots representative of 3 similar experiments. CD40 was immunoprecipitated from lysates of the indicated BCL-derived lines as described in “Methods.” Immunoprecipitates (IP) or whole-cell lysates (WCL) were subjected to SDS-PAGE and western blotting with antibodies specific for the indicated proteins. (D) DAUDI or SKW6.4 cells were untreated (left two lanes) or treated for 2 hours with 50 ng/mL recombinant human BAFF (rBAFF), after which BAFFR was immunoprecipitated from cell lysates. (E) Relative levels (mean ± standard error [SE]) of BAFFR-associated TRAF3 from 3 independent experiments. Statistical significance of differences was calculated as described in “Methods”; *P < .0008.
Figure 3.

Impact of LMP1 expression on levels of proviability kinases in human BCL cell lines. (A) Representative experiments in which whole-cell lysates of the indicated cell lines were subjected to SDS-PAGE and western blotted for NIK, PIM2, LMP1, and actin as described in “Methods.” (B) Relative PIM2 protein levels (presented as mean ± SE from 3 independent experiments as in A), with PIM2 levels in SKW6.4 cells set as 1. (C) Relative NIK protein levels (presented as mean values ± SE from 3 independent experiments as in A), with NIK levels in SKW6.4 cells set as 1. Statistical significance of differences was calculated as described in “Methods”; *P < .006.
Figure 4.

CD40-associated TRAF3 and cellular NIK levels in matched LMP1+vs LMP1−clones of mouse BCL lines. (A) The mouse BCL cell line CH12.LX and a subclone stably transfected with IPTG-inducible WT LMP1 were cultured overnight with IPTG as described in “Methods.” CD40 was immunoprecipitated from whole-cell lysates as in Figure 2, and samples subjected to SDS-PAGE were analyzed for CD40-associated TRAF3 by western blot. A representative of 3 similar blots is presented. (B) Relative CD40-associated TRAF3 levels (presented as mean values ± SE from 3 independent experiments as above), with CD40-associated TRAF3 levels in parent CH12.LX cells set at 1. *P < .0001. (C) Effect of LMP1 expression on relative cellular NIK levels. Whole-cell lysates of the indicated cell lines were blotted for NIK and actin as described in “Methods.”
Figure 7.

Relationship between LMP1 expression and levels of nuclear CREB in BCL cells. (A) Parent and LMP1-expressing clones of the M12.4.1 BCL cell line were stained with fluorescent anti-CREB antibody and analyzed by immunofluorescent confocal microscopy as described in “Methods.” Representative images from 6 fields per slide are shown and are also representative of 3 independent experiments with similar results. (B) Whole-cell lysates from the indicated cells were subjected to SDS-PAGE and western blot analysis of the indicated proteins; a representative blot of 3 similar experiments is shown. (C) Relative CREB protein levels (mean ± SE from 3 independent experiments as in panel B) were quantified, with levels in parent M12.4.1 cells set as 1. *P = .01 (Student t test; see “Methods”).
Figure 5.

LMP1 sequestration of TRAF3 requires the major LMP1 cytoplasmic domain TRAF-binding site. (A) A schematic of the CTAR1 cytoplasmic region of LMP1, indicating its 3 TRAF-binding motifs. The SUB2 LMP1 molecule has the indicated mutation in the first of these motifs. (B) CD40 immunoprecipitations (IP) as in prior figures, from whole-cell lysates (WCL) of parent CH12.LX cells or transfected subclones expressing WT or SUB2 LMP1 molecules, induced to express LMP1 as in Figure 4. A representative western blot of 3 similar experiments is shown. (C) Relative CD40-associated TRAF3 levels (presented as mean values ± SE from 3 independent experiments as above), with CD40-associated TRAF3 levels in parent CH12.LX cells set at 1. A 1-way ANOVA was used to analyze results for statistical significance, and the adjusted P value was calculated using Sidak’s multiple comparison test (*P < .0001, **P = .001).
Figure 6.

Influence of LMP1 expression on responsiveness to IL-6 signaling. (A) The indicated cell lines were stimulated with recombinant mouse IL-6 (as described in “Methods”), and whole-cell lysates were subjected to SDS-PAGE and western blot analysis for the indicated proteins. A representative blot of 3 similar experiments is shown. (B) Relative STAT3 phosphorylation following IL-6 stimulation was quantified in 3 independent experiments, and mean values ± SE were calculated. A 2-way ANOVA was used to examine the results for statistical significance, and the adjusted P value was calculated using the Bonferroni method (n = 2 independent experiments; *P = .043, **P = .031).
Results
Relationship between LMP1 and TRAF3 status of DLBCL
The presence of a genetic sequence or its transcripts does not always predict protein expression. Loss-of-function mutations in TRAF3 genes are associated with human B-cell malignancies, so to learn the status of TRAF3 protein expression in human BCL, we employed the Lymphoma MER DLBCL sample bank, accumulated by University of Iowa and Mayo Clinic. Whole-exome sequencing of 51 samples by Anne Novak (Mayo Clinic)43 revealed that 6 samples had copy-number loss of TRAF3. We then stained fixed tissue samples in this bank for TRAF3 protein. Representative staining of samples identified as having normal TRAF3 status or TRAF3 loss are presented in Figure 1A and show a strong correlation between staining intensity and copy number. Interestingly, 34.3% of samples from 277 DLBCLs displayed low/negative TRAF3 staining, compared with 11.8% with demonstrable TRAF3 genetic loss detected in the smaller sample. Additionally, 50 out of 277 DLBCL samples stained as LMP1+ (Figure 1C, left). Based upon the percentage of TRAF3-low/negative samples of the entire group, we expected that only ∼17 of the LMP1+ DLBCL samples would be TRAF3 low/negative (34.3% of 50 = 17.1). However, 30 of the LMP1+ DLBCL samples (60%) scored low/negative for TRAF3, perhaps reflecting the change in localization of cytoplasmic TRAF3. This staining pattern is shown in a representative sample in Figure 1C. LMP1 staining is notable for its primarily membrane localization in discrete aggregates. The overrepresentation of TRAF3low staining in the LMP1+ samples was highly significant by Fisher's exact test (P < .0001). Our results (summarized in Table 1) prompted us to test the hypothesis that LMP1 sequesters TRAF3, resulting in functional TRAF3 protein deficiency.
Impact of LMP1 expression upon TRAF3’s association with CD40/BAFFR
Our hypothesis predicts that the relative amount of TRAF3 available to CD40 will be reduced when B cells express LMP1. We tested this prediction using the LMP1high BCL line SKW6.4 in pairwise comparison with the LMP1− BCL lines DAUDI or KARPAS or the LMP1low line T5-1. CD40 was immunoprecipitated from whole-cell lysates of cell lines, followed by TRAF3 western blotting. In 3 independent comparisons, only a small amount of TRAF3 associated with CD40 in SKW6.4 cells, whereas much larger amounts bound CD40 in BCL lines with low or no LMP1, although total cellular amounts of TRAF3 were similar (Figure 2A-C).
TRAF3 also binds BAFFR and inhibits BAFFR signaling. We thus also examined the association between TRAF3 and BAFFR in human BCL lines that do or do not express LMP1. Figure 2D-E indicates that less TRAF3 associated with BAFFR in SKW6.4 cells compared with DAUDI; this difference was more pronounced following binding of recombinant BAFF, which stimulates TRAF3 degradation.44 Thus, LMP1 expression correlates with reduced TRAF3 association with CD40 and BAFFR, 2 membrane receptors for which TRAF3 is a negative regulator.
LMP1 expression and proviability kinases
In normal resting B cells, most NF-κB inducing kinase (NIK) is complexed in the cytoplasm with TRAF3. TRAF3 recruits a complex of TRAF2 and cellular inhibitor of apoptosis (cIAP), which leads to constitutive NIK polyubiquitination and degradation. NIK promotes activation of the noncanonical NF-κB2 pathway, so TRAF3 restrains this activation. TRAF3 recruitment to CD40 or BAFFR, together with TRAF2-cIAP, leads instead to TRAF3 polyubiquitination and proteasomal degradation, releasing NIK to activate NF-κB.45-47 In contrast, association with LMP1 causes no TRAF3 degradation,48 possibly because TRAF2 binds LMP1 weakly compared with CD40.12,49 However, LMP1 signaling induces NIK activation,50 so this must use a mechanism other than promoting TRAF3 degradation. Data in Figures 2 and 3 predict that levels of LMP1 expression sufficient to decrease cytoplasmic TRAF3 allow increased cellular NIK. Figure 3A,C supports this prediction; significantly more NIK is seen in SKW6.4 than in T5-1 BCL cells.
We recently found that TRAF3−/− primary B cells have elevated levels of the prosurvival kinase Pim2.51 Pim2 is under consideration as a therapeutic target in BCL and MM.52,53 Figure 3A-B shows that high LMP1 expression is also associated with enhanced Pim2 expression.
Effect of induced LMP1 expression upon B-cell TRAF3-CD40 association
Figures 2 and 3 provide data addressing our hypothesis in human BCL lines, providing the advantage of models relevant to human BCL pathogenesis. It is difficult to obtain the number of B cells required for biochemical analyses from fresh human tumor specimens. Cultured tumor cells, while providing a valuable alternative, have many mutations that can impact any feature examined, and there are thus often many differences between different cell lines. This limited our ability to obtain correlative data with human BCL lines. To test our hypothesis more mechanistically, we turned to a model of inducible LMP1 expression in mouse BCL lines,54 which provides important advantages. The only EBV-produced protein expressed in these cells is LMP1. We have specific matched pairs consisting of a parental cell line and subclones that stably and inducibly express LMP1, minimizing cell line differences, and also have subclones expressing LMP1 mutants useful for testing cause and effect.
We first examined the relationship between LMP1 expression and TRAF3 availability. Figure 4A-B shows that induced LMP1expression resulted in almost complete inhibition of TRAF3-CD40 binding, although total CD40 and TRAF3 amounts were not reduced compared with parent cells. Consistent with this finding and with results in human BCL lines (Figure 3), the LMP1-expressing subclone showed substantially increased NIK levels (Figure 4C).
Requirement for LMP1 TRAF binding for effects on TRAF3-binding receptors
TRAFs 1, 2, 3, 5, and 6 share common binding sites in the LMP1 C terminus, referred to as the C-terminal activating region (CTAR).55 The LMP1 CTAR has 3 PxQxT/S motifs that potentially bind TRAFs. One is in CTAR1, with 2 additional motifs in intermediate and CTAR2 regions (Figure 5A). There is direct interaction between TRAF3 and CTAR1, as well as an indirect interaction with CTAR2.41,56 TRAF3-CTAR1 binding is crucial for B-cell activation.56 Our hypothesis thus predicts that LMP1-mediated TRAF3 sequestration requires the TRAF-binding CTAR1 motif.
To test this prediction, we employed SUB2, an LMP1 molecule with 2 point mutations in the CTAR1 TRAF-binding motif (Figure 5A). This mutant has an ∼80% reduction in LMP1-TRAF3 binding compared with WT LMP1.56 In B cells expressing SUB2, the relative amount of CD40-bound TRAF3, although lower than in LMP1− B cells, was considerably higher than the near-complete absence of a CD40-TRAF3 association in B cells expressing WT LMP1 (Figure 5B). Quantitative analysis in Figure 5C shows that TRAF3-CD40 binding in cells expressing SUB2 increases approximately fivefold compared with TRAF3-CD40 binding in cells expressing WT LMP1. These data show that the ability of LMP1 to sequester TRAF3 is largely dependent on direct TRAF3 binding to the LMP1 CTAR1.
Impact of LMP1 expression upon the B-cell IL-6 response
IL-6 induces B-cell differentiation and survival; enhanced IL-6 production and/or responsiveness is linked to pathogenesis of B-cell malignancies.57 EBV transformation of B cells is also associated with increased IL-6 production,58 and LMP1 expression can endow both primary and transformed B cells with enhanced IL-6 production.54,59 Our recent studies revealed that TRAF3-deficient primary mouse and human B cells have enhanced IL-6 responsiveness, showing increased STAT3 phosphorylation.28 We discovered that TRAF3 normally recruits the protein tyrosine phosphatase nonreceptor 22 (PTPN22) to the IL-6R, inhibiting STAT3 phosphorylation; in the absence of PTPN22 recruitment, STAT3 phosphorylation is heightened.28 Because LMP1 expression in primary B cells of systemic lupus erythematosus–prone mouse strains also enhances IL-6 production,60 we examined whether LMP1+ B cells are also more responsive to IL-6 signaling. LMP1+ CH12.LX B cells showed enhanced pSTAT3 (but normal total STAT3) both in the basal state and following IL-6 stimulation compared with parent CH12.LX cells (Figure 6). Thus, LMP1-expressing B cells also mimic this malignancy-relevant property of TRAF3-deficient B cells.
Nuclear CREB and B-cell LMP1
While investigating prosurvival signaling pathways enhanced in TRAF3-deficient B cells, we found that a portion of cellular TRAF3 resides in the nucleus, where it binds the transcription factor CREB, recruits TRAF2, and promotes TRAF2-mediated CREB ubiquitination and degradation. Drugs that target the CREB/CREB-binding protein complex in turn decrease the abnormally enhanced survival of TRAF3-deficient B cells. In B cells lacking TRAF3, however, nuclear CREB is increased, supporting prolonged survival.29
To determine whether LMP1-mediated sequestration of TRAF3 also impacts regulatory functions of nuclear TRAF3, we examined the mouse BCL line M12.4.1 and subclones that express WT LMP1.54 Confocal fluorescence microscopy showed increased nuclear CREB in LMP1+ subclones compared with parent B cells (Figure 7A). Complementary western blots of total cellular CREB also showed increased total CREB protein in LMP1+ B cells (Figure 7B); there is approximately double the total CREB protein in LMP1+ B cells (Figure 7C). Together, our results indicate that the ability of LMP1 to sequester cellular TRAF3 renders a B cell functionally TRAF3 deficient.
Discussion
This study was designed to examine one possible mechanism by which B cells could be rendered deficient in availability of the tumor suppressor protein TRAF3, without requiring mutation or loss of TRAF3 genes. This is an important distinction, because the concept of delivering cancer treatments targeted to specific aberrant pathways of an individual tumor has generated great excitement in the oncology community. Most efforts to identify such tumor features focus upon gene mutations.34,61-63 One challenge to this approach is determining which of the many mutations typically found are changes driving tumor growth, survival, and/or metastasis and should be prioritized for targeted therapies.64,65 It is also increasingly appreciated that nongenetic changes in tumors have a significant impact on pathogenesis. In addition to epigenetic alterations, abnormal cellular localization and expression of specific proteins can make important contributions.66-68 The manner of protein alteration investigated here, sequestration by a viral tumor-associated protein, is a new mechanism to consider in examining pathogenic changes found in malignancies.
Our findings on the relative expression of TRAF3 protein in human BCLs (Figure 1A; Table 1) motivated us to examine the potential mechanism of protein sequestration as a means by which on oncogenic receptor promotes tumorigenesis. LMP1 has long been implicated in EBV-associated BCL.69-71 LMP1 delivers survival and activation signals via TRAFs, with TRAFs 3, 5, and 6 being the most important for LMP1 signaling to B cells (reviewed in Stunz and Bishop55). The requirement of LMP1 for association with TRAF3 to activate B cells12 may be partly indirect. LMP1 is also dependent upon TRAF5 to mediate B-cell activation,14 and TRAF5 requires TRAF3 for recruitment to the LMP1 CTAR.18 This study reveals that the LMP1-TRAF3 association can also impact B-cell biology by sequestering TRAF3, thus inhibiting its negative regulatory functions. The LMP1 staining shown in Figure 1, revealing the mainly membrane localization and aggregation of LMP1, is consistent with this hypothesis.
TRAF3 plays multiple important roles in restraining B-cell signaling. Together, these result in markedly enhanced survival and activation of TRAF3-deficient B cells. Germline Traf3 deletion is lethal in mice,20 so it is unsurprising that only one case of human single-allele germline deficiency in TRAF3, with only preliminary characterization of the myeloid cell compartment, has been reported.72 However, following revelation of the dramatic phenotype of B-Traf3−/− mice,21 multiple studies document TRAF3 mutations or reduced copy number in human B-cell malignancies, including MM,31-33 BCL,34,37,73-75 Waldenstrom macroglobulinemia,76 and B-cell leukemia.74 The location of the human TRAF3 gene on chromosome 14q32 likely enhances the probability that such mutations (most of which are large deletions and truncations) result from gene translocations frequently seen in B-cell tumors.74
A striking aspect of TRAF3-deficient B cells is their greatly enhanced survival capacity in the absence of increased proliferation.21 We discovered in recent years that normal functions of B-cell TRAF3 at the plasma membrane, in the cytoplasm, and in the nucleus all contribute to this enhanced survival when TRAF3 is deficient. TRAF3 was initially characterized in B cells as an inhibitor of CD40 signaling16 and later found to also inhibit BAFFR signaling17; this role is likely due to competition with TRAFs 2 and 6 in heterocomplexes.77,78 TRAF3 restrains the size of the plasma cell compartment (relevant to TRAF3 mutations in MM) by associating with and inhibiting the function of IL-6R via recruitment of PTPN22.28 It will be of future interest to determine whether and how TRAF3 regulates additional B-cell membrane receptors. We show here that expression of LMP1, which binds TRAF3 more avidly than CD40,12 reduces the relative amount of cellular TRAF3 available to bind CD40 and BAFFR in both mouse and human B cells, and this requires the TRAF-binding site of LMP1. We also demonstrate that LMP1 expression, like B-cell genetic TRAF3 deficiency, results in enhanced IL-6R signaling.
In the cytoplasm, TRAF3 binds to the kinase NIK, recruiting the TRAF2-cIAP complex to polyubiquitinate NIK and enhance its proteasomal degradation.27 In this manner, TRAF3 homeostatically restrains NF-κB2 activation. Consistent with this role, constitutive activation of NF-κB2 is seen in all TRAF3-deficient cell types examined to date, including B and T lymphocytes and myeloid cells.21,23-25 The present report shows that LMP1 expression, like genetic TRAF3 deficiency, results in increased cytoplasmic NIK.
It was previously concluded that this role of TRAF3 is both necessary and sufficient to explain the elevated survival of TRAF3-deficient B cells.79 This particular TRAF3 function is not B-cell specific, but only TRAF3-deficient B cells show a hypersurvival phenotype. For this reason, we sought additional prosurvival pathways regulated by TRAF3 specifically in B cells. This search revealed a new NIK-dependent B-cell function for TRAF3: enhanced glucose metabolism. This metabolic reprogramming involves enhanced expression of the glucose transporter Glut1 and the glycolytic enzyme hexokinase 2.30 Tumor cells exhibit increased glucose metabolism downstream of various alterations, so we could not easily test whether LMP1 expression also induces this property in BCL line models, but it is an intriguing possibility.
Our search revealed that a major pathway altered by TRAF3 deficiency specifically in B cells is transcription mediated by CREB.29 In addition to promoting expression of prosurvival proteins such as Mcl-1,29 CREB can enhance Glut1 expression,80 so this pathway may also contribute to altered glucose utilization in TRAF3-deficient B cells. TRAF3 binds nuclear CREB, recruiting TRAF2-cIAP to polyubiquitinate and promote CREB degradation.29 Similarly, LMP1 expression was shown here to enhance nuclear CREB levels.
This study demonstrated that the oncogenic protein LMP1, through its strong binding of TRAF3, reduced the availability of TRAF3 as a regulator of other membrane receptors, as well as cytoplasmic and nuclear proteins promoting B-cell survival. LMP1 expression can thus render a B cell functionally TRAF3 deficient and promote its abnormal survival. This reveals a newly appreciated mechanism by which alterations in localization and availability of TRAF3 protein can impact B-cell biology, in addition to the already known impact of loss-of-function mutations in TRAF3. This may not be the only way in which a B cell can be made functionally TRAF3 deficient. It will be interesting to determine whether receptor-mediated degradation of TRAF3 by elevated or chronic CD40 and BAFFR signaling also produces functional TRAF3 deficiency. Abnormal CD4081-85 and BAFFR78,86,87 signaling are both linked to pathogenesis of B-cell malignancies. Our findings indicate that many more B-cell malignancies may have abnormal TRAF3-regulated survival pathways than just tumors that have genetic mutations in TRAF3, and exploring alterations in the proteome of human tumors may also be critical to effectively employ individualized targeted therapies.
Acknowledgments
The authors are grateful to our University of Iowa colleagues, including Carol Holman for advice on histology and Mariah Leidinger and Allyn Lambertz (Pathology Research Laboratory) for performing staining of tissue microarrays. Anne Novak (Mayo Clinic, Rochester, MN) performed the whole-exome sequencing studies cited in “Results”; the authors appreciate her generosity in sharing these data with us.
This work was supported by the National Institutes of Health (NIH), National Cancer Institute (NCI) (grants CA099997 and CA97274), US Department of Veterans Affairs merit review I01BX001702, and a US Department of Veterans Affairs Senior Research Career Scientist Award (G.A.B.). The authors acknowledge use of the University of Iowa Central Microscopy Facility and an NCI P30 award to the Holden Comprehensive Cancer Center (grant CA086862), which provides partial support to this facility. This work used instruments in the University of Iowa Central Microscopy Research Facilities, including an Ariol Slide scanning microscope and micro-bright-field stereology software and a Zeiss LSM710 confocal microscope, purchased with funding from NIH Shared Instrumentation Grants 1S10OD014165 and 1S10RR025439, respectively. P.B.-P. received support from the Children’s Miracle Network. N.M. received support from NIH, National Institute of Allergy and Infectious Diseases T32 award AI007485 and National Institute of General Medical Sciences award GM007337. This material is based upon work supported in part by facilities and equipment provided by the US Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development.
Authorship
Contribution: P.B.-P. participated in the design, performance, analysis, and interpretation of experiments, the preparation of figures, and writing the manuscript; L.L.S. participated in conceptualization, performance, analysis, and interpretation of experiments and the preparation of figures; N.M., A.L.W., and B.S.H. participated in the performance, analysis, and interpretation of experiments and the preparation of figures; G.A.B. performed primary conceptualization and participated in experimental design, analysis, and interpretation of experiments and writing the manuscript; and all authors contributed to editing the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Gail A. Bishop, 2296 Carver Biomedical Research Building, 285 Newton Rd, Iowa City, IA 52242; e-mail: gail-bishop@uiowa.edu.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29. [DOI] [PubMed] [Google Scholar]
- 2.Küppers R. B cells under influence: transformation of B cells by Epstein-Barr virus. Nat Rev Immunol. 2003;3(10):801-812. [DOI] [PubMed] [Google Scholar]
- 3.Gibson SE, Hsi ED. EBV+ BCL of the elderly at a US tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40(9):653-661. [DOI] [PubMed] [Google Scholar]
- 4.Nicolae A, Pittaluga S, Abdullah S, et al. . EBV-positive large B-cell lymphomas in young patients: a nodal lymphoma with evidence for a tolerogenic immune environment. Blood. 2015;126(7):863-872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang D, Liebowitz D, Kieff E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell. 1985;43(3 pt 2):831-840. [DOI] [PubMed] [Google Scholar]
- 6.Hammerschmidt W, Sugden B. Genetic analysis of immortalizing functions of Epstein-Barr virus in human B lymphocytes. Nature. 1989;340(6232):393-397. [DOI] [PubMed] [Google Scholar]
- 7.Bishop GA, Hostager BS. Signaling by CD40 and its mimics in B cell activation. Immunol Res. 2001;24(2):97-109. [DOI] [PubMed] [Google Scholar]
- 8.Graham JP, Arcipowski KM, Bishop GA. Differential B lymphocyte regulation by CD40 and its viral mimic, LMP1. Immunol Rev. 2010;237(1):226-248. [DOI] [PubMed] [Google Scholar]
- 9.Hildebrand JM, Yi Z, Buchta CM, Poovassery JS, Stunz LL, Bishop GA. Roles of TRAF3 and TRAF5 in immune cell function. Immunol Rev. 2011;244(1):55-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hostager BS, Haxhinasto SA, Rowland SL, Bishop GA. Tumor necrosis factor receptor-associated factor 2 (TRAF2)-deficient B lymphocytes reveal novel roles for TRAF2 in CD40 signaling. J Biol Chem. 2003;278(46):45382-45390. [DOI] [PubMed] [Google Scholar]
- 11.Xie P, Hostager BS, Munroe ME, Moore CR, Bishop GA. Cooperation between TRAFs 1 and 2 in CD40 signaling. J Immunol. 2006;176(9):5388-5400. [DOI] [PubMed] [Google Scholar]
- 12.Xie P, Hostager BS, Bishop GA. Requirement for TRAF3 in signaling by LMP1 but not CD40 in B lymphocytes. J Exp Med. 2004;199(5):661-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakano H, Sakon S, Koseki H, et al. . Targeted disruption of Traf5 gene causes defects in CD40- and CD27-mediated lymphocyte activation. Proc Natl Acad Sci USA. 1999;96(17):9803-9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kraus ZJ, Nakano H, Bishop GA. TRAF5 is a critical mediator of in vitro signals and in vivo functions of LMP1, the viral oncogenic mimic of CD40. Proc Natl Acad Sci USA. 2009;106(40):17140-17145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arcipowski KM, Bishop GA. TRAF6 as a critical regulator of B lymphocyte functions. Curr Trends Immunol. 2013;14(3):45-52. [Google Scholar]
- 16.Hostager BS, Bishop GA. Cutting Edge: Contrasting roles of TRAF2 and TRAF3 in CD40-mediated B lymphocyte activation. J Immunol. 1999;162(11):6307-6311. [PubMed] [Google Scholar]
- 17.Xu L-G, Shu H-B. TRAF3 is associated with BAFF-R and negatively regulates BAFF-R-mediated NF-kB activation and IL-10 production. J Immunol. 2002;169(12):6883-6889. [DOI] [PubMed] [Google Scholar]
- 18.Arcipowski KM, Stunz LL, Graham JP, Kraus ZJ, Vanden Bush TJ, Bishop GA. Molecular mechanisms of TRAF6 utilization by the oncogenic viral mimic of CD40, LMP1. J Biol Chem. 2011;286(12):9948-9955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu S, Xie P, Welsh K, et al. . LMP1 protein from EBV is a structural decoy in B lymphocytes for binding to TRAF3. J Biol Chem. 2005;280(39):33620-33626. [DOI] [PubMed] [Google Scholar]
- 20.Xu Y, Cheng G, Baltimore D. Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity. 1996;5(5):407-415. [DOI] [PubMed] [Google Scholar]
- 21.Xie P, Stunz LL, Larison KD, Yang B, Bishop GA. TRAF3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity. 2007;27(2):253-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity. 2008;28(3):391-401. [DOI] [PubMed] [Google Scholar]
- 23.Xie P, Kraus ZJ, Stunz LL, Liu Y, Bishop GA. TRAF3 is required for T cell-mediated immunity and TCR/CD28 signaling. J Immunol. 2011;186(1):143-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie P, Poovassery JS, Stunz LL, et al. . Enhanced TLR responses of TRAF3-deficient B lymphocytes. J Leukoc Biol. 2011;90(6):1149-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lalani AI, Moore CR, Luo C, et al. . Myeloid cell TRAF3 regulates immune responses and inhibits inflammation and tumor development in mice. J Immunol. 2015;194(1):334-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore CR, Liu Y, Shao C, Covey LR, Morse HC III, Xie P. Specific deletion of TRAF3 in B lymphocytes leads to B-lymphoma development in mice. Leukemia. 2012;26(5):1122-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao G, Zhang M, Harhaj EW, Sun S-C. Regulation of NIK by TRAF3-induced degradation. J Biol Chem. 2004;279(25):26243-26250. [DOI] [PubMed] [Google Scholar]
- 28.Lin WW, Yi Z, Stunz LL, Maine CJ, Sherman LA, Bishop GA. The adaptor protein TRAF3 inhibits IL-6 receptor signaling in B cells to limit plasma cell development. Sci Signal 2015;8(392):81-88. [DOI] [PMC free article] [PubMed]
- 29.Mambetsariev N, Lin WW, Stunz LL, Hanson BM, Hildebrand JM, Bishop GA. Nuclear TRAF3 is a negative regulator of CREB in B cells. Proc Natl Acad Sci USA. 2016;113(4):1032-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mambetsariev N, Lin WW, Wallis AM, Stunz LL, Bishop GA. TRAF3 deficiency promotes metabolic reprogramming in B cells. Nature Sci Reports. 2016;6(Oct 18):35349. [DOI] [PMC free article] [PubMed]
- 31.Annunziata CM, Davis RE, Demchenko Y, et al. . Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12(2):115-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keats JJ, Fonseca R, Chesi M, et al. . Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12(2):131-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.San Miguel JF. Introduction to a series of reviews on multiple myeloma. Blood. 2015;125(20):3039-3040. [DOI] [PubMed] [Google Scholar]
- 34.Vaqué JP, Martínez N, Batlle-López A, et al. . B-cell lymphoma mutations: improving diagnostics and enabling targeted therapies. Haematologica. 2014;99(2):222-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ortega-Molina A, Boss IW, Canela A, et al. . The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015;21(10):1199-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang B, Calado DP, Wang Z, et al. . An oncogenic role for alternative NF-κB signaling in DLBCL revealed upon deregulated BCL6 expression. Cell Reports. 2015;11(5):715-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bushell KR, Kim Y, Chan FC, et al. . Genetic inactivation of TRAF3 in canine and human B-cell lymphoma. Blood. 2015;125(6):999-1005. [DOI] [PubMed] [Google Scholar]
- 38.Kaykas A, Worringer K, Sugden B. CD40 and LMP-1 both signal from lipid rafts but LMP-1 assembles a distinct, more efficient signaling complex. EMBO J. 2001;20(11):2641-2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bishop GA, Haughton G. Induced differentiation of a transformed clone of Ly-1+ B cells by clonal T cells and antigen. Proc Natl Acad Sci USA. 1986;83(19):7410-7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hamano T, Kim KJ, Leiserson WM, Asofsky R. Establishment of B cell hybridomas with B cell surface antigens. J Immunol. 1982;129(4):1403-1406. [PubMed] [Google Scholar]
- 41.Busch LK, Bishop GA. Multiple carboxyl-terminal regions of the EBV oncoprotein, LMP1, cooperatively regulate signaling to B lymphocytes via TRAF-dependent and TRAF-independent mechanisms. J Immunol. 2001;167(10):5805-5813. [DOI] [PubMed] [Google Scholar]
- 42.Rowland SR, Tremblay ML, Ellison JM, Stunz LL, Bishop GA, Hostager BS. A novel mechanism for TRAF6-dependent CD40 signaling. J Immunol. 2007;179(7):4645-4653. [DOI] [PubMed] [Google Scholar]
- 43.Novak AJ, Asmann YW, Maurer MJ, et al. . Whole-exome analysis reveals novel somatic genomic alterations associated with outcome in immunochemotherapy-treated DLBCL. Blood Cancer J 2015;5(8):e346. [DOI] [PMC free article] [PubMed]
- 44.Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol. 2009;9(7):491-502. [DOI] [PubMed] [Google Scholar]
- 45.Sun SC. The noncanonical NF-κB pathway. Immunol Rev. 2012;246(1):125-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hostager BS, Bishop GA. CD40-mediated activation of the NF-κB2 pathway. Front Immunol 2013;4: 371-374. [DOI] [PMC free article] [PubMed]
- 47.Lin WW, Hostager BS, Bishop GA. TRAF3, ubiquitination, and B-lymphocyte regulation. Immunol Rev. 2015;266(1):46-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown KD, Hostager BS, Bishop GA Differential signaling and TRAF degradation by CD40 and the EBV oncoprotein LMP1. I 2001;193(8):943-954. [DOI] [PMC free article] [PubMed]
- 49.Graham JP, Moore CR, Bishop GA. Roles of the TRAF2/3 binding site in differential B cell signaling by CD40 and its viral oncogenic mimic, LMP1. J Immunol. 2009;183(5):2966-2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sylla BS, Hung SC, Davidson DM, et al. . EBV-transforming protein LMP1 activates transcription factor NF-κB through a pathway that includes NIK and the IκB kinases IKKα and IKKβ. Proc Natl Acad Sci USA. 1998;95(17):10106-10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mambetsariev N. Mechanisms of TRAF3-Mediated Regulation of B Cell Survival [dissertation]. Iowa City, IA; The University of Iowa; 2016. [Google Scholar]
- 52.Gómez-Abad C, Pisonero H, Blanco-Aparicio C, et al. . PIM2 inhibition as a rational therapeutic approach in B-cell lymphoma. Blood. 2011;118(20):5517-5527. [DOI] [PubMed] [Google Scholar]
- 53.Neri P, Bahlis NJ. Pinning down myeloma with Pim2 inhibitors! Blood. 2013;122(9):1534-1536. [DOI] [PubMed] [Google Scholar]
- 54.Busch LK, Bishop GA The EBV transforming protein, LMP1, mimics and cooperates with CD40 signaling in B lymphocytes. 1999;162(5):2555-2561. [PubMed]
- 55.Stunz LL, Bishop GA. LMP1 and the B lymphocyte: a complex relationship. CRC Crit Rev Immunol. 2014;34(3):177-198. [DOI] [PubMed] [Google Scholar]
- 56.Xie P, Bishop GA Roles of TRAF3 in signaling to B lymphocytes by CTAR regions 1 and 2 of the EBV-encoded oncoprotein LMP1. J Immunol. 2004;173(9):5546-5555. [DOI] [PubMed]
- 57.Hilbert DM, Kopf M, Mock BA, Köhler G, Rudikoff S IL-6 is essential for in vivo development of B lineage neoplasms. J Immunol. 1995;182(1):243-248. [DOI] [PMC free article] [PubMed]
- 58.Yokoi T, Miyawaki T, Yachie A, Kato K, Kasahara Y, Taniguchi N. Epstein-Barr virus-immortalized B cells produce IL-6 as an autocrine growth factor. Immunology. 1990;70(1):100-105. [PMC free article] [PubMed] [Google Scholar]
- 59.Stunz LL, Busch LK, Munroe ME, et al. . Expression of the cytoplasmic tail of LMP1 in mice induces hyperactivation of B lymphocytes and disordered lymphoid architecture. Immunity. 2004;21(2):255-266. [DOI] [PubMed] [Google Scholar]
- 60.Peters AL, Stunz LL, Meyerholz DK, Mohan C, Bishop GA. LMP1, the EBV encoded oncogenic mimic of CD40, accelerates autoimmunity in B6.Sle mice. J Immunol. 2010;185(7):4053-4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dancey JE, Bedard PL, Onetto N, Hudson TJ. The genetic basis for cancer treatment decisions. Cell. 2012;148(3):409-420. [DOI] [PubMed] [Google Scholar]
- 62.Pon JR, Marra MA. Clinical impact of molecular features in DLBCL and FL. Blood. 2016;127(2):181-186. [DOI] [PubMed] [Google Scholar]
- 63.Lim MS, Elenitoba-Johson KSJ. Precision medicine for DLBCL. Clin Cancer Res. 2016;22(12):2829-2831. [DOI] [PubMed] [Google Scholar]
- 64.Rashid NU, Sperling AS, Bolli N, et al. . Differential and limited expression of mutant alleles in multiple myeloma. Blood. 2014;124(20):3110-3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nielsen JS, Sedgwick CG, Shahid A, et al. . Toward personalized lymphoma immunotherapy: Identification of common driver mutations recognized by patient CD8+ T cells. Clin Cancer Res. 2016;22(9):2226-2236. [DOI] [PubMed] [Google Scholar]
- 66.Cai SF, Chen C-W, Armstrong SA. Drugging chromatin in cancer: recent advances and novel approaches. Mol Cell. 2015;60(4):561-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bianchi G, Richardson PG, Anderson KC. Promising therapies in multiple myeloma. Blood. 2015;126(3):300-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Culjkovic-Kraljacic B, Fernando TM, Marullo R, et al. . Combinatorial targeting of nuclear export and translation of RNA inhibits aggressive B-cell lymphomas. Blood. 2016;127(7):858-868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Farrell PJ, Cludts I, Stühler A. Epstein-Barr virus genes and cancer cells. Biomed Pharmacother. 1997;51(6-7):258-267. [DOI] [PubMed] [Google Scholar]
- 70.Oudejans JJ, Jiwa NM, Meijer CJLM. Epstein-Barr virus in Hodgkin’s disease: more than just an innocent bystander. J Pathol. 1997;181(4):353-356. [DOI] [PubMed] [Google Scholar]
- 71.Bishop GA, Busch LK. Molecular mechanisms of B-lymphocyte transformation by Epstein-Barr virus. Microbes Infect. 2002;4(8):853-857. [DOI] [PubMed] [Google Scholar]
- 72.Pérez de Diego R, Sancho-Shimizu V, Lorenzo L, et al. . Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity. 2010;33(3):400-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Compagno M, Lim WK, Grunn A, et al. . Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459(7247):717-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nagel I, Bug S, Tönnies H, et al. . Biallelic inactivation of TRAF3 in a subset of B-cell lymphomas with interstitial del(14)(q24.1q32.33). Leukemia. 2009;23(11):2153-2155. [DOI] [PubMed] [Google Scholar]
- 75.Otto C, Giefing M, Massow A, et al. . Genetic lesions of the TRAF3 and MAP3K14 genes in classical Hodgkin lymphoma. Br J Haematol. 2012;157(6):702-708. [DOI] [PubMed] [Google Scholar]
- 76.Braggio E, Keats JJ, Leleu X, et al. . Identification of copy number abnormalities and inactivating mutations in two negative regulators of nuclear factor-kappaB signaling pathways in Waldenstrom’s macroglobulinemia. Cancer Res. 2009;69(8):3579-3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Haxhinasto SA, Hostager BS, Bishop GA. Cutting Edge: Molecular mechanisms of synergy between CD40 and the BCR: role for TRAF2 in receptor interaction. J Immunol. 2002;169(3):1145-1149. [DOI] [PubMed] [Google Scholar]
- 78.Hildebrand JM, Luo Z, Manske MK, et al. . A BAFF-R mutation associated with Non-Hodgkin’s lymphoma exhibits altered TRAF association and reveals new insights into proximal BAFF-R signaling. J Exp Med. 2010;207(12):2569-2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Häcker H, Tseng P-H, Karin M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol. 2011;11(7):457-468. [DOI] [PubMed] [Google Scholar]
- 80.Kim MO, Lee YJ, Park JH, Ryu JM, Yun SP, Han HJ. PKA and cAMP stimulate proliferation of mouse embryonic stem cells by elevating GLUT1 expression mediated by the NF-κB and CREB/CBP signaling pathways. Biochim Biophys Acta. 2012;1820(10):1636-1646. [DOI] [PubMed] [Google Scholar]
- 81.Gruss H-J, Hirschstein D, Wright B, et al. . Expression and function of CD40 on Hodgkin and Reed-Sternberg cells and the possible relevance for Hodgkin’s disease. Blood. 1994;84(7):2305-2314. [PubMed] [Google Scholar]
- 82.Urashima M, Chauhan D, Uchiyama H, Freeman GJ, Anderson KC. CD40 ligand triggered interleukin-6 secretion in multiple myeloma. Blood. 1995;85(7):1903-1912. [PubMed] [Google Scholar]
- 83.Planken EV, Dijkstra NH, Bakkus M, Willemze R, Kluin-Nelemans JC. Proliferation of precursor B-lineage ACLL by activating the CD40 antigen. Br J Haematol. 1996;95(2):319-326. [DOI] [PubMed] [Google Scholar]
- 84.Greiner A, Knörr C, Qin Y, et al. . Low-grade B cell lymphomas of MALT require CD40-mediated signaling and Th2-type cytokines for in vitro growth and differentiation. Am J Pathol. 1997;150(5):1583-1593. [PMC free article] [PubMed] [Google Scholar]
- 85.Ghia P, Boussiotis VA, Schultze JL, et al. . Unbalanced expression of bcl-2 family proteins in follicular lymphoma: contribution of CD40 signaling in promoting survival. Blood. 1998;91(1):244-251. [PubMed] [Google Scholar]
- 86.Novak AJ, Darce JR, Arendt BK, et al. . Expression of BCMA, TACI, and BAFF-R in multiple myeloma: a mechanism for growth and survival. Blood. 2004;103(2):689-694. [DOI] [PubMed] [Google Scholar]
- 87.Pham LV, Fu L, Tamayo AT, et al. . Constitutive BR3 receptor signaling in diffuse LBCL stabilizes NIK while activating both canonical and alternative NF-κB pathways. Blood. 2011;117(1):200-210. [DOI] [PMC free article] [PubMed] [Google Scholar]