

Key Points
UNC13D gene transfer into HSCs corrects all clinical and biological signs of HLH in a mouse model.
Munc13-4 is expressed in mature CD8+ T cells allowing the correction of cytotoxic activity and consequently efficient viral restriction.
Abstract
Patients with mutations in the UNC13D gene (coding for Munc13-4 protein) suffer from familial hemophagocytic lymphohistiocytosis type 3 (FHL3), a life-threatening immune and hyperinflammatory disorder. The only curative treatment is allogeneic hematopoietic stem cell (HSC) transplantation, although the posttreatment survival rate is not satisfactory. Here, we demonstrate the curative potential of UNC13D gene correction of HSCs in a murine model of FHL3. We generated a self-inactivating lentiviral vector, used it to complement HSCs from Unc13d-deficient (Jinx) mice, and transplanted the cells back into the irradiated Jinx recipients. This procedure led to complete reconstitution of the immune system (ie, to wild-type levels). The recipients were then challenged with lymphocytic choriomeningitis virus to induce hemophagocytic lymphohistiocytosis (HLH)–like manifestations. All the clinical and biological signs of HLH were significantly reduced in mice having undergone HSC UNC13D gene correction than in nontreated animals. This beneficial effect was evidenced by the correction of blood cytopenia, body weight gain, normalization of the body temperature, decreased serum interferon-γ level, recovery of liver damage, and decreased viral load. These improvements can be explained by the restoration of the CD8+ T lymphocytes’ cytotoxic function (as demonstrated here in an in vitro degranulation assay). Overall, our results demonstrate the efficacy of HSC gene therapy in an FHL-like setting of immune dysregulation.
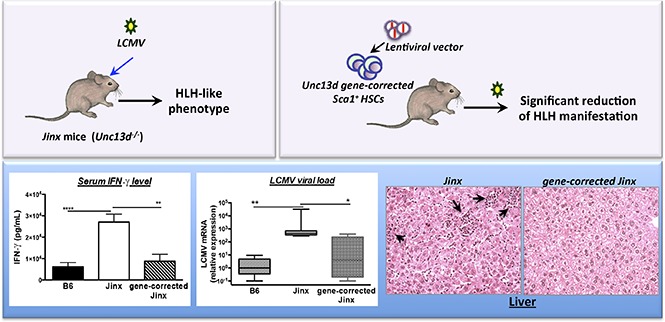
Visual Abstract
Introduction
Mutations in the UNC13D gene (coding for Munc13-4 protein, one of the components of the perforin-dependent cytotoxicity apparatus) cause familial hemophagocytic lymphohistiocytosis type 3 (FHL3). This condition accounts for up to 30% to 35% of all cases of FHLs.1 Munc13-4 helps to prime perforin-containing, cytotoxic granules before they fuse with the plasma membrane at the immunological synapse, and it’s defect results in defective cytotoxic granules exocytosis.2 Hemophagocytic lymphohistiocytosis (HLH) is a rare hyperinflammatory syndrome that is characterized by (1) the uncontrolled activation of macrophages and lymphocytes, and (2) a life-threatening cytokine storm, which together elicit hallmark features like nonremitting fever, pancytopenia, coagulopathy, hyperferritinemia, hemophagocytosis, and liver dysfunction.3 The short-term treatment of HLH is based on immunosuppression, whereas the only long-term, potentially curative treatment is allogeneic hematopoietic stem cell transplantation (HSCT).3
Our previous work on T cells from FHL3 patients demonstrated that lentiviral vector (LV)–derived expression of Munc13-4 protein was able to rescue in vitro and in vivo cytotoxic functions, highlighting T cells as a putative target for UNC13D gene correction and thus the curative treatment of HLH.4 Based on the outcomes of HSCT in FHL patients, T-cell chimerism of at least 10% to 15% is required for efficient remission of HLH.5,6 Hence, the T cells’ transduction efficiency is a key parameter in this strategy. We demonstrated that, by using a new lentiviral pseudotype (such as the H/F-envelope), high levels of T-cell transduction can be achieved.4 However, more preclinical studies are needed to further support the use of these new LVs in future clinical trials. In this context, the genetic correction of autologous hematopoietic stem cells (HSCs), which has provided convincing evidence of successful treatment of hematopoietic disorders,7,8 could be proposed for FHL3 patients. This strategy should be a more rapidly available therapeutic option than T-cell strategy with new LVs and would avoid the major complications associated with allogeneic HSCT.
Several mouse strains carrying defects in the lymphocyte granule-dependent cytotoxicity pathway have already been validated as models of human HLH.9-12 Of note, none of these mutant mouse strains develops HLH spontaneously, and they have to be infected with a viral trigger (ie, lymphocytic choriomeningitis virus [LCMV]) in order to develop HLH-like symptoms. Although the development of HLH in all these mouse strains is mainly because of the hyperactivation of T lymphocytes and high levels of interferon-γ (IFN-γ) secretion that subsequently induce macrophage activation, the severity of the clinical and immunological manifestations of HLH differ from one model to another. Hence, perforin- and Rab27-deficient mice develop the most severe HLH-like disease, whereas Munc13-4- and STX-11-deficient strains present a less severe phenotype.11,13 In the present study, we used a previously described Munc13-4-deficient strain (the Jinx mice) as a model of FHL3.14 After LCMV infection, Jinx mice develop nonfatal HLH characterized by a striking increase in CD8 T-cell activation, leukopenia, anemia, a sustained elevation of serum IFN-γ levels, and failure to control viral infection.14 Using this model, we showed that gene correction of murine Munc13-4-deficient HSCs (through a self-inactivating LV expressing a human UNC13D transgene) restored a functional immune system and effectively alleviated the HLH-like manifestations. Our encouraging preclinical results emphasize the feasibility of HSC gene therapy for FHL3, using an LV designed for clinical application in humans.
Materials and methods
Animal experiments and the induction of HLH via LCMV infection
C57BL/6J mice were purchased from Charles River Laboratories (Sulzfeld, Germany). Jinx mice harboring a homozygous mutation in Unc13d gene have been described elsewhere14 and were kindly provided by Genevieve de Saint Basile (Imagine Institute, Paris, France).
The WE strain of LCMV was kindly provided by Maries van de Broek and Rolf Zinkernagel (University of Zürich, Zürich, Switzerland). Control and gene-therapy-treated mice received a single intraperitoneal injection of 200 pfu of LCMV. Blood counts were checked after infection using an automated cell counter (Scil Vet abc Plus, ABX HORIBA). Serum levels of alanine transaminase and aspartate transaminase were determined using the VetTest Chemistry Analyzer (IDEXX Laboratories, Westbrook, ME). The serum levels of IFN-γ and other cytokines were determined using an enzyme-linked immunosorbent assay kit (eBioscience, San Diego, CA). We received the authorization to realize experiments using animals (mouse) from the French Ministry of National Education, Higher Education and Research under the reference number 05309.02.
Transduction and transplantation of Sca1+ cells
Bone marrow cells were harvested from femurs and tibias. Sca1+ progenitor cells were isolated using an anti-phycoerythrin (PE) Microbeads kit (Miltenyi Biotec, Bergisch Gladbach, Germany). The Sca1+ progenitors were prestimulated overnight in StemSpan serum-free expansion medium (SFEM) medium (StemCell Technologies, Grenoble, France) supplemented with 5% fetal bovine serum (for mouse B lymphoid, StemCell Technologies) and 1% gentamycin (GIBCO) with murine cytokines (100 ng/µL stem cell factor, 100 ng/µL Fms-like tyrosine kinase 3-ligand, 100 ng/µL thrombopoietin, 50 ng/µL interleukin-6 (IL-6), and 10 ng/µL IL-11 [all from Peprotech, Rocky Hill, NJ]), and then transduced at a multiplicity of infection of 100 with Munc13-4- or green fluorescent protein (GFP)–expressing LVs for 12 hours. Bulk transduced cells were harvested on the following day and then injected into the retro-orbital sinus of sublethally irradiated (7 Gy) Jinx mice between 6 and 8 weeks of age. Each animal received 2.5 × 105 bulk transduced Sca1+ cells.
The in vitro degranulation assay
Splenocytes from noninfected and infected mice were isolated and incubated in StemSpan SFEM medium (StemCell Technologies) supplemented with 5% fetal bovine serum (for mouse B lymphoid, StemCell Technologies), 1% gentamycin (GIBCO), 1% N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (GIBCO), 50 U/mL murine IL-2, and anti-CD107a-PE antibodies (clone: 1D4B; Sony Biotechnology, Champaign, IL) in absence or presence of 3 µg/mL coating anti-murine CD3 antibody (clone: 500A2; BD Bioscience, San Jose, CA). After a 3-hour incubation at 37°C, cells were stained with murine CD3-allophycocyanin (APC) and CD8-BV421 antibodies, and the viability dye 7-aminoactinomycin D. Flow cytometry analysis was performed using a MACSQuant analyzer (Miltenyi Biotec).
Antibodies and flow cytometry
Fluorochrome-conjugated anti-mouse CD45.1 APC-Cy7 (clone: A20), CD45.2 PC7 (clone: 104), CD3 APC-Cy7 (clone: 145-2C11), and CD19 PC7 (clone: 6D5) antibodies were from Sony Biotechnology. Anti-mouse anti NK1.1 V450 (clone: PK136) and CD11B APC (clone: M1/70) were from BD Biosciences. Flow cytometry analysis was performed using a MACSQuant analyzer (Miltenyi Biotec), and data were analyzed using Flow Jo version 10.0.7 (Tree Star, Ashland, OR).
Vector copy number (VCN) count and messenger RNA (mRNA) expression
Genomic DNA was isolated from splenocytes using a Wizard Genomic DNA Purification kit (Promega, Madison, WI). Real-time quantitative polymerase chain reactions (qPCRs) were performed with TaqMan probes design to detect a lentiviral sequence (Psi) and a sequence on murine genome (Titine). Serial dilutions of DNA plasmid containing the Psi and Titine sequences were used to plot a standard curve. Samples and serial dilutions were run in duplicate.
Total RNA was harvested from splenocytes with a RNeasy Micro Kit (Qiagen, Courtaboeuf, France) and then reverse-transcribed with a High Capacity complementary DNA (cDNA) Reverse Transcription Kit (Life Technologies, Carlsbad, CA). Diluted cDNA was used for real-time qPCR in TaqMan assays for the detection of codon-optimized Munc13-4. Levels of the Munc13-4 transcript were normalized against levels of endogenous glyceraldehyde-3-phosphate dehydrogenase transcripts. The PCRs were performed on a Viia7 Real-Time PCR System (ThermoFisher Scientific).
Quantification of viral titers
The LCMV load in the liver was assayed using qPCR, as described previously.11 Briefly, cDNA was isolated from tissue samples and analyzed with primers for LCMV (forward: 5′-TCTCATCCCAACCATTTGCA-3′; reverse: 5′-GGGAAATTTGACAGCACAACAA-3′) and murine β-actin (forward: 5′-CCAGCAGATGTGGATCAGCA-3′; reverse: 5′-CTTGCGGTGCACGATGG-3′), using SYBR Green PCR Master Mix (Applied Biosystems).
Statistics
Plots were generated using GraphPad Prism 6.0 (La Jolla, CA). Sahpiro-Wilk test was used to evaluate the normal distribution of the data. The significance was calculated using a Mann-Whitney U test for IL-6 and tumor necrosis factor α (TNF-α) levels, a Kruskal-Wallis multiple comparison for viral titer, and an unpaired Student t test for all other parameters (specified in figure legends). P <.05 was considered significant.
Results
UNC13D gene transfer in murine HSCs does not alter the development of the immune system
As described previously,4 we designed a self-inactivating LV carrying a codon-optimized human UNC13D or GFP cDNA under the control of a short elongation factor 1-α (EF1-α) promoter. In a series of independent experiments, Sca1+ bone marrow progenitors from Jinx CD45.2 donor mice were transduced ex vivo with a Munc13-4- or GFP-expressing LV and transplanted into sublethally irradiated Jinx recipients with a CD45.1/2 genetic background (referred to henceforth as the J/Munc [n = 12] and J/GFP [n = 8] groups, respectively) (Figure 1A). A group of mice (the J/B6 group, n = 11) having received Sca1+ cells from wild-type (WT) C57BL/6J mice served as a control group for the HSCT. Two other nonirradiated, nonmanipulated groups, WT (B6, n = 10) and Jinx mice (Jinx, n = 10), were used as controls in all experiments.
Figure 1.

Chimerism and immune system reconstitution in Jinx mice after transfer of the human UNC13D gene into murine HSCs. (A) A schematic representation of the transplantation and infection protocol in mice. Sublethally irradiated (7 Gy) Jinx 45.1/2 recipients were reconstituted with Sca1+ murine HSCs transduced with a Munc13-4- or GFP-expressing LV from Jinx 45.2 mice (the J/Munc and J/GFP groups, respectively) or nontransduced Sca1+ cells from B6 45.1 mice (the J/B6 group). Each mouse received total number of 2.5 × 105 Sca1+ cells. Mice 4 to 6 months posttransplantation were infected with LCMV (intraperitoneal injection, 200 pfu). Euthanasia time: 14 days after LCMV infection. (B) Flow cytometry analysis showing that more than 90% of the cells in the bone marrow and spleen were derived from the donor in all groups at euthanasia. (C) Flow cytometry analysis of various spleen cell subsets in the control groups (B6 and Jinx mice) and transplanted groups. Data are presented as the mean ± standard deviation percentage (first graph) and the absolute count (second graph) for each subset within the spleen. B6 (n = 10), Jinx (n = 10), J/GFP (n = 8), J/B6 (n = 11), and J/Munc (n = 12). *P < .05 using unpaired Student t test. (D) Flow cytometry analysis of Munc13-4/GFP (optimized human Munc13-4 fused to GFP) expression in Sca1+ cells, 48 hours after transduction (n = 3 mice).
Four to 6 months after transplantation, the percentage of donor cells exceeded 90% in the bone marrow and spleen. There were no significant differences between transplanted groups (Figure 1B). Immune reconstitution was analyzed by quantifying the percentage and the absolute number of the different immune subsets in the spleen. In the 3 transplanted groups, the percentages and the absolute counts of T cells (CD3+), B cells (CD19+), natural killer cells (NK1.1+), and myeloid cells (CD11B+) were similar to those measured in B6 mice (Figure 1C). To assess the transduction efficacy of our LVs, some mice received Sca1+ cells transduced with an LV expressing Munc13-4/GFP fusion protein; 40% of Sca1+ cells were found to express Munc13-4/GFP before infusion in mice (Figure 1D). Taken as a whole, these data demonstrate that our LVs efficiently transduced murine progenitor cells and did not alter the latter’s lineage-specific development.
The Munc13-4 transgene is stably expressed in peripheral immune cells
Four to 6 months after transplantation, provirus integration in the splenocytes resulted in a mean VCN per cell of 3.5 (1.3-5) in J/Munc mice and 4.5 (2.4-6) in J/GFP mice. Human UNC13D transgene mRNA was expressed in the recipients’ splenocytes in average 1.85 times higher compared with endogenous Unc13d mRNA in WT mice, but there was no correlation between the quantity of transgene mRNA and the VCN (r = 0.52) (Figure 2A). It should be noted that in experiments with a Munc13-4/GFP fusion–expressing LV, transgene expression was detected in >50% of bone marrow Sca1+ cells and in the CD3+, CD4+, CD8+, CD19+, CD11B+, and NK1.1+ cell subsets in the spleen (Figure 2B). Overall, these results confirm that HSC gene transfer was followed by vector integration and expression of the human transgene in peripheral immune cells.
Figure 2.

Human Munc13-4 transgene integration and expression. (A) The transgene VCN and mRNA expression were measured by qPCR in the J/Munc group (absolute and relative amounts, respectively), and the result is presented as a correlation graph (r = 0.52; P = .04). (B) Flow cytometry analysis showing expression of the Munc13-4/GFP fusion protein 4 to 6 months after transplantation in the bone marrow (BM) for Sca1+ cells and in the spleen for various cell subsets.
UNC13D gene transfer rescues LCMV-infected Jinx mice from HLH-like disease
WT and Jinx mice differ markedly in their response to LCMV infection. Whereas WT mice show robust resistance to the virus, Jinx mice display an intense immune response, T-cell activation, and then HLH-like biological and clinical manifestations.14 In most cases, elevated serum levels of IFN-γ at day 8 postinfection constitute the major biological manifestation in murine models of HLH.9,15 In order to study the effect of UNC13D gene correction of HSCs on the pathogenic events in Jinx mice, we challenged the animals with LCMV 4 to 6 months after transplantation, the time point for a full peripheral whole blood cell reconstitution from long-term HSCs. Age-matched B6 and Jinx controls were infected with LCMV. The serum IFN-γ levels in J/B6 mice and (more interestingly) in J/Munc recipients were similar to that measured in B6 mice and significantly lower than those in J and J/GFP recipients (Figure 3A). In murine models of HLH, the serum IFN-γ peak at day 8 postinfection is associated with elevated serum levels of other cytokines (especially IL-6 and TNF-α).16 In our experiment, the serum levels of IL-6 and TNF-α followed the same pattern as the IFN-γ levels (ie, no difference vs B6 mice challenged with LCMV, and lower levels than in the Jinx and J/GFP groups; Figure 3A).
Figure 3.

HLH-like manifestation after LCMV infection in different mouse groups. (A) Serum cytokine levels were assayed 8 days after LCMV infection. Levels were lower in the treated groups (J/B6 and J/Munc) compared with nontreated controls. (B) Body weight and temperature were measured every 2 days postinfection; the intergroup difference was significant at euthanasia (D14 postinfection). Body weight is presented as a percentage of the value on D0 (the day of infection). (C) At euthanasia, the WBC and RBC counts were measured. All data are presented as the mean ± standard error of the mean. Sahpiro-Wilk test was used to evaluate the normal distribution of the data. P values were calculated using a Mann-Whitney U test for IL-6 and TNF-α levels (which are not normally distributed) and an unpaired Student t test for all other parameters. *P < .05; **P < .01; ***P < .001; ****P < .0001. Hct, hematocrit.
We hypothesized that the low serum levels of IFN-γ and other cytokines in gene-corrected animals would be accompanied by attenuation of the severity of HLH-like disease. As expected (compared with B6 mice), Jinx mice developed characteristics clinical signs, such as body weight loss, a fall in body temperature, and pancytopenia including low white blood cell (WBC) and red blood cell (RBC) counts, and a low hematocrit, 14 days postinfection (the time at which the mice were euthanized in the present study based on previous published data on HLH manifestation in murine models9-11,14) (Figure 3B-C). It is noteworthy that GFP gene transfer (in the J/GFP group) had no beneficial effects on these biological signs. Remarkably, we found that treatment with gene-corrected HSCs in the J/Munc group lessened the fall in body temperature and the body weight loss to the levels seen in the J/B6 group (Figure 3B). Gene correction also rescued LCMV-infected Jinx mice from the development of cytopenia, as demonstrated by an increase in the WBC and RBC counts and the hematocrit (Figure 3C).
Hepatomegaly was also found to be attenuated in both the J/Munc and J/B6 groups, relative to the nontreated Jinx and J/GFP mice (Figure 4A). In accordance with this observation, plasma levels of the liver damage biomarkers aspartate transaminase, alanine transaminase, and lactate dehydrogenase were significantly lower in both treated groups and were thus similar to the values measured in control B6 mice (Figure 4B). In addition, liver sections evidenced prominent inflammatory foci in Jinx and J/GFP groups, barely detected in B6 and treated mice (Figure 4C).
Figure 4.

Hepatomegaly and liver damage. (A) Liver weight is presented as a percentage of body weight. (B) Serum levels of aspartate transaminase (ASAT), alanine transaminase (ALAT), and lactate dehydrogenase (LDH). Data are presented as the mean ± standard error of the mean. P values were calculated using an unpaired Student t test. (C) Hematoxylin and eosin staining of liver sections at day 14 postinfection. Inflammatory infiltrates are depicted with arrows (upper panel: 10× objective; lower panel: 25× objective). Representative sections are shown. *P < .05; **P < .01; ***P < .001.
Taken as a whole, these data demonstrate that the ex vivo correction of Munc13-4 deficiency in HSCs enabled Jinx recipients to control inflammation and immune dysfunction and to avoid organ damage after LCMV infection.
UNC13D gene transfer into HSCs restores cytotoxic function in CD8+ cytotoxic T lymphocytes (CTLs) in vitro and in vivo
To examine whether the reduction in HLH-like manifestations observed in J/Munc mice was correlated with the restoration of CTL activity, we evaluated the cells’ ex vivo degranulation capacity and in vivo cytotoxic activity. In the ex vivo degranulation assay, splenocytes from infected mice were incubated with murine anti-CD3 antibody. Unlike CD8+ splenocytes from Jinx or J/GFP mice, a significant proportion of splenic CD8+ T cells from J/Munc mice demonstrated evidence of degranulation as indicated by elevated cell surface expression of CD107a (which accompanies the release of cytotoxic granules) (Figure 5A).
Figure 5.

Functional and phenotypic characterization of CD8+CTLs. (A) To assess degranulation activity, splenic CD8+ T cells were stimulated with murine anti-CD3 (3 µg/mL). Surface expression of CD107a was determined by flow cytometry and presented as percentage of CD8+ population that was CD107a+. P values were calculated using an unpaired Student t test. (B) The viral titer in the liver was measured by a qPCR assay of amplified LCMV mRNA and was normalized against levels of endogenous murine β-actin. The data are presented in a box plot, and P values were determined using a Kruskal-Wallis test for multiple comparisons. Representative fluorescence-activated cell sorter plots (C) and a bar graph (D) showing the distribution of CD8+ T-cell subsets (mean ± standard deviation) at euthanasia (TN: CD62L+CD44−; TCM: CD62L+CD44+; TEM: CD44+CD62L−). *P < .1; **P < .01; ***P < .001; ****P < .0001.
The CTLs’ in vivo cytotoxic activity was indirectly assayed by measuring LCMV titers in the liver following infection. As expected, control B6 and J/B6 mice successfully restrain LCMV replication, but Jinx and J/GFP mice failed to do so and presented with high viral titers 14 days postinfection (Figure 5B). Interestingly, we observed that J/Munc mice presented with lower viral titers in the liver than control J/GFP mice did.
Phenotypic analysis of CD8+ splenocytes demonstrated that the majority of the latter cells had an effector memory T-cell phenotype (CD44highCD62L−) in all experimental groups (Figure 5C-D). This observation indicated that the expression of human Munc13-4 did not alter the CTLs’ final differentiation into the effector phenotype and confirmed that the failure to clear LCMV (as observed in Jinx and J/GFP mice) is because of a pronounced defect in degranulation rather than in antigen triggering or activation.
Overall, our results show that the improvement in clinical and biological parameters of HLH-like disease observed in gene-corrected J/Munc group was correlated with the restoration of the CTLs’ cytotoxic effector functions in vitro and in vivo.
Discussion
In the present study, we evaluated the efficacy of HSC gene therapy in a murine model of FHL3 (ie, a strain showing signs consistent with the human disease). The LV used in this study efficiently transduced murine Sca1+ progenitor cells and drove the Munc13-4 expression in differentiated cells. We found that UNC13D gene transfer into HSCs significantly reduced the manifestations of HLH induced by LCMV infection. The mechanism of HLH attenuation in gene corrected animals was because of the expression of Munc13-4 in CD8+ CTLs, which allowed the correction of the cells’ degranulation defect and the recovery of effective cytotoxic activity and efficient viral restriction.
Our findings are consistent with prior reports on HSC gene transfer in a perforin-deficient mouse model, demonstrating the efficacy of this strategy for restoring cytotoxic function in CTLs and correcting biological parameters (especially the WBC and RBC count and the serum IFN-γ level) in a HLH-like setting.17 One important aspect of the present study is that HSC gene correction with a Munc13-4-expressing LV not only corrected biological manifestation of HLH, but above all enabled effective viral clearance and recovery from liver damage. Unlike perforin (the expression of which is restricted to mature T and NK cells), Munc13-4 is ubiquitously expressed in human and mice,18 and so its vector-derived expression in HSCs may be less likely to induce undesirable side effects. Our results confirmed this hypothesis, as even the use of an LV with a short EF1-α ubiquitous promoter was not associated with an abnormal lineage-specific commitment of HSCs for up to 6 months after gene transfer. However, we are planning further preclinical studies under good laboratory practice conditions to assess the most important long-term adverse effect of gene transfer strategies (ie, off-target genotoxicity) in more detail.
We did not observe a correlation between the VCN and mRNA expression. It should be borne in mind that the LV construct used in this study encoded a human codon-optimized Munc13-4; hence, this human protein might not be optimally expressed in a murine background. In a clinical trial, one would expect to see optimized transgene expression and thus greater efficacy.
In previous work, we highlighted the potential of T-cell gene correction for FHL3 patients, given that the main defect in FHL3 is the absence of T-cell cytotoxicity.4 If approved by regulatory bodies, the use of gene-corrected autologous T cells would be a treatment option for FHL3 patients. However, it has recently been demonstrated that (1) NK cell cytotoxicity has a distinct immunoregulatory role in HLH disease,19 and (2) Munc13-4 has a critical function in platelets and neutrophils.20-23 By using an LV coding for a Munc13-4/GFP fusion protein, our present study demonstrated that gene transfer into murine HSCs resulted in transgene expression in all the hematopoietic lineages, including NK and myeloid cells. Thus, HSC gene correction (providing multilineage Munc13-4 expression) might be a valuable treatment option for FHL3 in patients. We aim to evaluate the efficacy of both strategies (T cell–based and HSC-based) in 2 independent phase 1/2 clinical trials. Slow T-cell reconstitution is regarded as being primarily responsible for poor infection control, the occurrence of graft-versus-host disease, and relapse after HSCT.24 Hence, at a later step and according to results achieved in the separate trials, patients could be transplanted with gene-corrected autologous HSCs as a curative long-term treatment and with gene-corrected autologous T cells.
It should be also noted that other proposed strategies like the administration of Janus kinase 1/2 (JAK1/2) inhibitors in HLH mouse models demonstrated an efficient remission from systemic inflammation but still could not restrict viral proliferation.16,25 JAK1/2 inhibitors might provide an additional benefit if administered prior to the infusion of gene-corrected HSCs (a definitive curative approach for HLH disease).
Overall, the present study set out the rationale for curative Munc13-4 gene therapy in FHL3. In the future, the use of this type of autologous cell-based strategy in a clinical trial might provide further tools for progressing the field of personalized medicine.
Acknowledgments
The authors thank Giséle Froment, Didier Nègre, and Caroline Costa from the lentivector production facility/SFR BioSciences Gerland–Lyon Sud (UMS3444/US8).
This study was funded by the French National Institutes of Health and Medical Research (INSERM), the Association Française contre les Myopathies (grant 17045), a European Research Council grant (ERC Regenerative Therapy, 269037), and a European Union FP7 grant (CELL-PID, 261387).
Authorship
Contribution: T.S. designed and conducted experiments, analyzed data, and wrote the manuscript; A.D. performed and analyzed experiments and read the manuscript; F.E.S. designed, performed, and analyzed experiments and read the manuscript; J.R. and H.S. performed experiments and read the manuscript; C.L.-P. discussed data and reviewed the manuscript for critical content; G.d.S.B. provided the Jinx strain and reviewed the manuscript for critical content; S.M. and F.M. provided vectors and read the manuscript; and M.C. and I.A.-S. designed and supervised the overall research and reviewed the manuscript for critical content.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliations for F.M. are University of Paris Descartes–Sorbonne Paris Cité, Imagine Institute, Paris, France, and Department of Life Sciences, University of Modena and Reggio Emilia, Modena, Italy.
Correspondence: Isabelle André-Schmutz, INSERM U1163, Imagine Institute, 24 Blvd de Montparnasse, 75015 Paris, France; e-mail: andre.schmutz@gmail.com.
References
- 1.de Saint Basile G, Ménasché G, Latour S. Inherited defects causing hemophagocytic lymphohistiocytic syndrome. Ann N Y Acad Sci. 2011;1246:64-76. [DOI] [PubMed] [Google Scholar]
- 2.Feldmann J, Callebaut I, Raposo G, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 2003;115(4):461-473. [DOI] [PubMed] [Google Scholar]
- 3.Bode SF, Lehmberg K, Maul-Pavicic A, et al. Recent advances in the diagnosis and treatment of hemophagocytic lymphohistiocytosis. Arthritis Res Ther. 2012;14(3):213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soheili T, Rivière J, Ricciardelli I, et al. Gene-corrected human Munc13-4-deficient CD8+ T cells can efficiently restrict EBV-driven lymphoproliferation in immunodeficient mice. Blood. 2016;128(24):2859-2862. [DOI] [PubMed] [Google Scholar]
- 5.Hartz B, Marsh R, Rao K, et al. The minimum required level of donor chimerism in hereditary hemophagocytic lymphohistiocytosis. Blood. 2016;127(25):3281-3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gholam C, Grigoriadou S, Gilmour KC, Gaspar HB. Familial haemophagocytic lymphohistiocytosis: advances in the genetic basis, diagnosis and management. Clin Exp Immunol. 2011;163(3):271-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Booth C, Gaspar HB, Thrasher AJ. Treating immunodeficiency through HSC gene therapy. Trends Mol Med. 2016;22(4):317-327. [DOI] [PubMed] [Google Scholar]
- 8.Touzot F, Hacein-Bey-Abina S, Fischer A, Cavazzana M. Gene therapy for inherited immunodeficiency. Expert Opin Biol Ther. 2014;14(6):789-798. [DOI] [PubMed] [Google Scholar]
- 9.Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104(3):735-743. [DOI] [PubMed] [Google Scholar]
- 10.Kögl T, Müller J, Jessen B, et al. Hemophagocytic lymphohistiocytosis in syntaxin-11-deficient mice: T-cell exhaustion limits fatal disease. Blood. 2013;121(4):604-613. [DOI] [PubMed] [Google Scholar]
- 11.Sepulveda FE, Debeurme F, Ménasché G, et al. Distinct severity of HLH in both human and murine mutants with complete loss of cytotoxic effector PRF1, RAB27A, and STX11. Blood. 2013;121(4):595-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pachlopnik Schmid J, Ho C-H, Diana J, et al. A Griscelli syndrome type 2 murine model of hemophagocytic lymphohistiocytosis (HLH). Eur J Immunol. 2008;38(11):3219-3225. [DOI] [PubMed] [Google Scholar]
- 13.Jessen B, Kögl T, Sepulveda FE, de Saint Basile G, Aichele P, Ehl S. Graded defects in cytotoxicity determine severity of hemophagocytic lymphohistiocytosis in humans and mice. Front Immunol. 2013;4:448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crozat K, Hoebe K, Ugolini S, et al. Jinx, an MCMV susceptibility phenotype caused by disruption of Unc13d: a mouse model of type 3 familial hemophagocytic lymphohistiocytosis [published correction appears in J Exp Med. 2008;205(3):737]. J Exp Med. 2007;204(4):853-863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pachlopnik Schmid J, Ho C-H, Chrétien F, et al. Neutralization of IFNgamma defeats haemophagocytosis in LCMV-infected perforin- and Rab27a-deficient mice. EMBO Mol Med. 2009;1(2):112-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maschalidi S, Sepulveda FE, Garrigue A, Fischer A, de Saint Basile G. Therapeutic effect of JAK1/2 blockade on the manifestations of hemophagocytic lymphohistiocytosis in mice. Blood. 2016;128(1):60-71. [DOI] [PubMed] [Google Scholar]
- 17.Carmo M, Risma KA, Arumugam P, et al. Perforin gene transfer into hematopoietic stem cells improves immune dysregulation in murine models of perforin deficiency. Mol Ther. 2015;23(4):737-745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koch H, Hofmann K, Brose N. Definition of Munc13-homology-domains and characterization of a novel ubiquitously expressed Munc13 isoform. Biochem J. 2000;349(1):247-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sepulveda FE, Maschalidi S, Vosshenrich CAJ, et al. A novel immunoregulatory role for NK-cell cytotoxicity in protection from HLH-like immunopathology in mice. Blood. 2015;125(9):1427-1434. [DOI] [PubMed] [Google Scholar]
- 20.Chicka MC, Ren Q, Richards D, et al. Role of Munc13-4 as a Ca2+-dependent tether during platelet secretion. Biochem J. 2016;473(5):627-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harper MT, van den Bosch MT, Hers I, Poole AW. Platelet dense granule secretion defects may obscure α-granule secretion mechanisms: evidence from Munc13-4-deficient platelets. Blood. 2015;125(19):3034-3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramadass M, Catz SD. Molecular mechanisms regulating secretory organelles and endosomes in neutrophils and their implications for inflammation. Immunol Rev. 2016;273(1):249-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monfregola J, Johnson JL, Meijler MM, Napolitano G, Catz SD. MUNC13-4 protein regulates the oxidative response and is essential for phagosomal maturation and bacterial killing in neutrophils. J Biol Chem. 2012;287(53):44603-44618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogonek J, Kralj Juric M, Ghimire S, et al. Immune reconstitution after allogeneic hematopoietic stem cell transplantation. Front Immunol. 2016;7:507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Das R, Guan P, Sprague L, et al. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood. 2016;127(13):1666-1675. [DOI] [PMC free article] [PubMed] [Google Scholar]