

SUMMARY
Chromatin remodelers catalyze dynamic packaging of the genome by carrying out nucleosome assembly/disassembly, histone exchange and nucleosome repositioning. Remodeling results in evenly spaced nucleosomes, which requires probing both sides of the nucleosome, yet it is not understood how remodelers organize sliding activity to achieve this task. Here we show that the monomeric Chd1 remodeler shifts DNA back and forth by dynamically alternating between different segments of the nucleosome. During sliding, Chd1 generates unstable remodeling intermediates that spontaneously relax to a pre-remodeled position. We demonstrate that nucleosome sliding is tightly controlled by two regulatory domains: the DNA-binding domain, which interferes with sliding when its range is limited by a truncated linking segment, and the chromodomains, which play a key role in substrate discrimination. We propose that active interplay of the ATPase motor with the regulatory domains may promote dynamic nucleosome structures uniquely suited for histone exchange and chromatin reorganization during transcription.
TOC image
Chd1 is an ATP-driven chromatin remodeler that evenly repositions nucleosomes along DNA. Qiu et al. demonstrate that a single Chd1 molecule can dynamically shift nucleosomes back and forth. Such bidirectional activity is tightly controlled by two regulatory domains that guide nucleosome sliding and substrate selection.
INTRODUCTION
Chromatin remodelers are specialized ATP-dependent DNA translocases that can reposition, evict and replace histones within the nucleosome (Narlikar et al., 2013). The need for such activities arises from the compact organization of chromosomal DNA into nucleosomes that requires accessibility for essential genomic processes such as replication, transcription and DNA repair. Subsequent to such disruptive events, chromatin needs to be properly repackaged to maintain genomic integrity. Accomplishing these tasks requires multiple families of remodelers that are specialized for achieving particular remodeling outcomes. Each remodeler family can be identified by unique regulatory domains that determine substrate specificities and control action of a conserved helicase-like ATPase motor. The interplay of regulatory domains with the ATPase motor occurs in the context of temporally regulated epigenetic modifications critical for cellular differentiation, development and human diseases. As exemplified by the Chd1 chromatin remodeler, disruption of individual remodelers can have profound consequences such as loss of stem cell pluripotency or stimulation of cancer cell proliferation (Burkhardt et al., 2013; Gaspar-Maia et al., 2009; Zhao et al., 2017)
Action of the Chd1 remodeler is tightly coupled to transcription, as evidenced by direct interaction of Chd1 with several elongation factors (Kelley et al., 1999; Krogan et al., 2002; Simic et al., 2003), subunits of mediator and the spliceosome (Lin et al., 2011; Sims et al., 2007) and, in metazoans, the histone H3K4 methylation mark (Flanagan et al., 2005). Chd1 catalyzes both nucleosome assembly and array spacing (Fei et al., 2015; Gkikopoulos et al., 2011; Lusser et al., 2005), which are important in reestablishing the chromatin barrier after passage of RNA polymerase II (Smolle et al., 2012). Additionally, Chd1 has also been shown to facilitate or be required for exchange of histone H3 variants (Konev et al., 2007), a poorly understood process that requires significant structural reorganization of histone-DNA interactions.
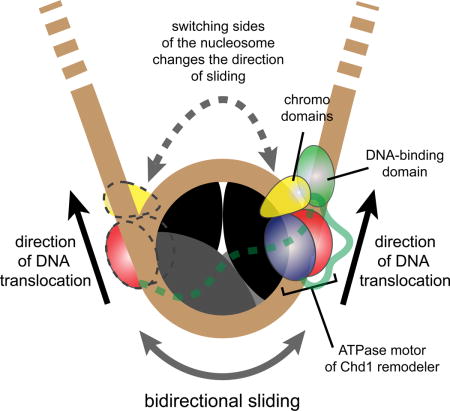
Chd1 possesses two prominent regulatory domains: a sequence non-specific DNA-binding domain (DBD) located C-terminal to the ATPase motor, and a pair of chromodomains immediately N-terminal to the ATPase motor. A crystal structure of the chromo-ATPase portion of Chd1 showed that the chromodomains can directly block a DNA-binding surface of the ATPase motor via an acidic helix (Hauk et al., 2010). This interaction appears autoinhibitory, as disruption of this interface increased ATPase stimulation by naked DNA and recovered sliding of nucleosomes lacking the H4 tail (Hauk et al., 2010). While these activities are consistent with the chromodomains serving as a selectivity filter, it has been unclear what natural nucleosome substrates may be blocked by chromodomain inhibition.
The DBD of Chd1 was initially found to have an important role in tethering the remodeler to nucleosome substrates. Deletion of the DBD severely impaired nucleosome sliding activity, yet substituting foreign binding domains restored robust sliding, indicating that the DBD is not mechanically required for nucleosome repositioning (McKnight et al., 2011; Nodelman and Bowman, 2013; Patel et al., 2013). More recently, the finding that the DBD communicates with the ATPase motor when bound to DNA flanking the nucleosome has suggested that the DBD also plays a regulatory role (Nodelman et al., 2017). Like SWI/SNF and ISWI remodelers, the ATPase motor of Chd1 translocates on DNA at superhelix location 2 (SHL2), an internal site ~20 bp from the nucleosome dyad (McKnight et al., 2011; Saha et al., 2005; Schwanbeck et al., 2004; Zofall et al., 2006). DNA translocation by these remodeler ATPases is believed to be unidirectional, and therefore relative to the SHL2 site where the ATPase motor is engaged, DNA flanking one side of the nucleosome shifts onto the core (entry side) while DNA on the other turn or gyre of DNA shifts further away from the nucleosome core (exit side). The ATPase motor and DBD of Chd1 can therefore be in two distinct organizations on the nucleosome: they can be either on opposite DNA gyres and spatially close together on the same “edge” of the nucleosome, or on the same DNA gyre and separated from each other across the face of the nucleosome (Nodelman et al., 2017). When on the same gyre and across the face of the nucleosome, the DBD can assist the ATPase motor via tethering, but would be too far to directly contact the ATPase motor. As shown with Chd1 fusion remodelers (McKnight et al., 2011; Patel et al., 2013), this separated organization is stimulating. In contrast, when the DBD and ATPase are on opposite DNA gyres and therefore physically close, these domains can communicate with each other. As suggested by faster nucleosome sliding away from Lac repressor and dampening of ATPase activity (Nodelman et al., 2017; Nodelman et al., 2016), this cross-gyre communication appears to interfere with nucleosome sliding. The dynamics by which Chd1 switches between active and inhibited states has not previously been examined.
In this study, we took advantage of a single molecule fluorescence approach to dissect the nucleosome sliding activity of Chd1. Our results reveal that Chd1 repositions nucleosomes in a stepwise manner, dependent on ATP hydrolysis. Surprisingly, we discovered that Chd1 shifts nucleosomal DNA back and forth as a monomer. ATP-dependent translocation of DNA was consistently followed by ATP-independent reversals, resulting in the DNA snapping back to a previous position. We believe this behavior reveals unstable remodeling intermediates, which provide potential checkpoints for regulatory elements. By mutational analysis, we investigated roles of both the chromodomains and DBD. We discovered that the chromodomains are responsible for blocking hexasome sliding, and therefore provide a critical safeguard against sliding incomplete nucleosomes. We also found that deletion of a linker segment between the ATPase motor and DBD, previously shown to have virtually no sliding activity (Nodelman and Bowman, 2013), yields dynamic but unstable movement of exit-side DNA, suggesting active inhibition due to the DBD. Taken together, our results reveal dynamic action and regulation of Chd1 that are likely central for assembling and evenly spacing nucleosomes throughout the genome.
RESULTS
Chd1 repositions the nucleosome in a stepwise manner
Following the single molecule design from a previous study (Deindl et al., 2013), we generated FRET-labeled nucleosomes to study remodeling by Chd1. Using the Widom 601 positioning sequence (Lowary and Widom, 1998), we prepared end-positioned nucleosomes (called 3N80), with the short 3 bp end labeled with Cy3, which is close enough to FRET with Cy5 on the H2A C-terminus (T120C) (Figure 1A) (Li and Widom, 2004). The 80 bp side of the DNA was biotinylated for immobilization to a NeutrAvidin-coated PEG surface (Ngo et al., 2015; Roy et al., 2008), and movement of the short end away from the histone core is reported by a decrease in FRET (Deindl et al., 2013). For single molecule detection, we added 100 pM of FRET labeled nucleosome to the surface, which yielded approximately 400 spatially separated FRET spots in one field of view (25 × 75 μm2).
Figure 1. Chd1 repositions the nucleosome in a stepwise manner.
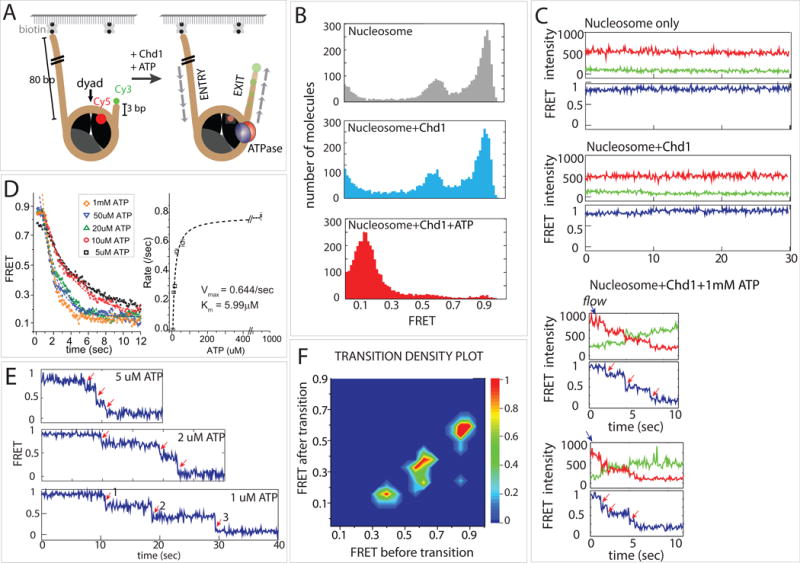
(A) Nucleosome-FRET construct was labeled with Cy3 on exit side DNA and Cy5 on histone H2A(T120C). Only the proximal Cy5 dye is shown.
(B) Histograms of FRET values before and after addition of Chd1. FRET histograms of nucleosomes alone displayed a bimodal distribution, correlating with nucleosomes having labeled H2A in a proximal (high FRET) or distal (mid-FRET) position (gray, top). Addition of Chd1 without nucleotide (light blue, middle) did not show significant differences from nucleosome alone. After addition of ATP (red, bottom), FRET histograms were dominated by a single, low FRET peak.
(C) Representative single molecule FRET traces for each condition in (B).
(D) Averaged FRET traces at varying ATP concentrations and the corresponding Michaelis-Menten fit.
(E) Single molecule traces at low ATP, which display stepwise decreases in FRET.
(F) Transition density plot showing three steps of discrete FRET transitions.
Chd1 (20 nM) and ATP (1 mM) were added in succession to FRET labeled nucleosomes immobilized on the PEGylated surface. Fifteen to twenty images were taken and the FRET values collected from 6000–8000 molecules were built into a FRET histogram. The major peak appears at high FRET of 0.9 as expected from the proximity between the Cy3 and Cy5 dyes (Figure 1B, top). A minor mid-FRET peak at 0.6 is likely due to the Cy5-labeled H2A at a distal position as seen in previous studies (Deindl et al., 2013; Levendosky et al., 2016). FRET peaks did not change upon addition of Chd1 protein alone without ATP (Figure 1B, middle). When ATP was added, all molecules shifted to low FRET (0.1) (Figure 1B, bottom), indicating that nucleosome repositioning by Chd1 was ATP dependent.
Consistent with the FRET histograms, the representative single molecule FRET traces show a steady high FRET signal for nucleosome alone and after addition of Chd1 without nucleotide (Figure 1C, top and middle). Immediately after addition of ATP (blue arrow), the high FRET transitioned to low FRET, indicating that Chd1 repositioned nucleosomes. A closer examination of single molecule traces revealed individual FRET steps, denoted by red arrows (Figure 1C, bottom). To reduce the stepping rate, repositioning activity was tested at varying ATP concentrations. The result was analyzed by collecting FRET values corresponding to the repositioning activity (0.9 to 0.1) from over 100 single molecule traces at different ATP concentrations and plotting the average FRET signal over time (Figure 1D, left). As expected, the rate of FRET decrease is the highest (~0.75/sec) at 1 mM ATP and substantially lower at 5–10 μM (~0.25/sec). Based on the calculated rate, we plotted the ATP dependent repositioning rate and fit to the Michaelis-Menten equation to determine Vmax and KM (Figure 1D, right). To improve the resolution of the stepwise FRET change, we performed the same measurement with low ATP concentrations (1–5 μM). Here, we observed three distinct steps of FRET values as indicated by red arrows (Figure 1E). We collected individual FRET values from over 100 traces and plotted as a transition density plot in which the x- and y-axis represent FRET values before and after a transition, respectively. This analysis shows that Chd1 takes discrete steps represented by FRET transition from 0.9 to 0.65, 0.65 to 0.4, and 0.4 to 0.1 in succession (Figure 1F). As this behavior is analogous to the stepping previously observed for ISWI remodelers (Deindl et al., 2013), these data suggest that Chd1 shifts DNA past the nucleosome core in bursts of multiple base pairs.
Monomeric Chd1 is sufficient for shifting nucleosomal DNA back and forth
After reaching a low FRET state (0.1), we noticed a prominent pattern of FRET fluctuation in a significant fraction of single molecule traces, suggesting movement of DNA in the opposite direction following the initial repositioning (Figure 2A). We interpret the regain in FRET as movement of the short DNA end back toward the nucleosome core. Given that nucleosomes possess 2-fold symmetry, one possible explanation for the back-and-forth motion could be alternating action of Chd1 on either side of the nucleosome. In these experiments, we flushed away excess Chd1 protein upon addition of ATP, yet it was possible that after initial exposure to Chd1, nucleosomes retained a Chd1 molecule on each side. An alternative possibility was that a single Chd1 molecule could also achieve bidirectional sliding, which would require that the ATPase motor hop to different nucleosome locations without dissociation of the remodeler. To determine whether a single Chd1 protein could stimulate bidirectional motion of nucleosomal DNA, we performed sliding reactions with freely diffusing nucleosomes and immobilized Chd1. Following a successful strategy from our previous studies (Hwang et al., 2014a; Hwang et al., 2014b; Qiu et al., 2013; Tippana et al., 2014), we tethered Chd1 to the surface using a FLAG:anti-FLAG interaction, which provided the advantage of observing activities of single Chd1 proteins. With this arrangement, the nucleosomes had the same FRET labeling scheme but were non-biotinylated, and therefore no fluorescence signals were detected until nucleosomes bound to surface-immobilized Chd1 (Figure 2B) The FRET histogram taken after adding FRET labeled nucleosome showed a FRET peak at around 0.8–0.9 (Figure 2C, top), similar to the original experiment shown in Figure 1B. These FRET signals obtained in the absence of ATP confirm that nucleosome binding to Chd1 does not rely on ATP. Upon addition of ATP, FRET decreased to low levels as before (Figure 2C, bottom). The nearly complete shift in FRET histogram signified that the majority of nucleosomes that engaged with the surface-bound Chd1 underwent active repositioning. As expected, the FRET signals remained high when nucleosomes bound Chd1 in the absence of ATP (Figure 2D, top). Remarkably, the single molecule FRET traces taken in ATP showed the same pattern of initial FRET decrease followed by periodic regain and loss of FRET (Figure 2D, bottom). This bidirectional movement of DNA relative to the histone core indicates that even as a monomer, Chd1 can reverse the direction of DNA translocation without dissociation.
Figure 2. Chd1 monomer is sufficient for repositioning single nucleosomes back and forth.
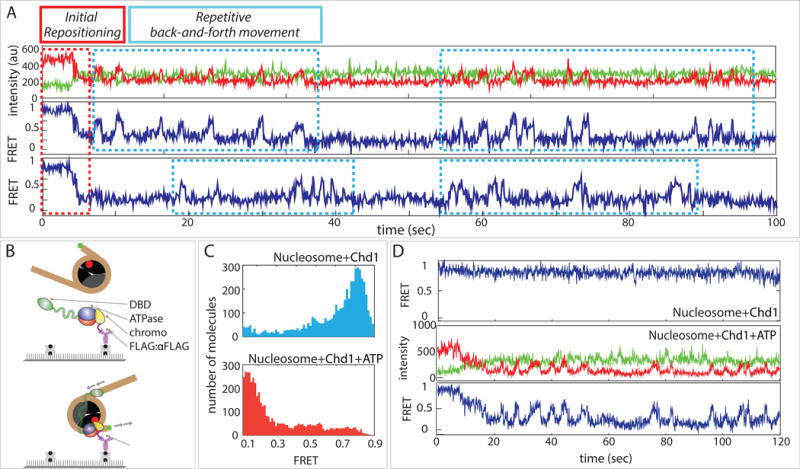
(A) Single molecule traces of biotin-tethered nucleosomes, displaying initial repositioning (decrease in FRET) followed by repetitive increases in FRET, signifying sliding in the opposite direction.
(B) Schematic of surface-immobilized Chd1 and FRET-labeled nucleosome applied to single molecule platform.
(C) FRET histograms of non-biotinylated nucleosomes with surface-immobilized Chd1 before and after ATP addition.
(D) Single molecule traces of FRET labeled nucleosomes bound to surface-immobilized Chd1 before (top) and after (middle, bottom) ATP addition.
Bidirectional sliding reveals unstable remodeling intermediates
After reaching a low FRET state (0.1), continuous FRET fluctuations were observed where the highest level achieved was typically ~0.7, which was lower than the initial FRET (~0.9). Such FRET fluctuations, which we interpret as a back-and-forth motion of DNA relative to the histone core, typically occurred many times in succession (Figure 2A). Analysis of 2000 traces over a 15-minute period showed that approximately 30% of molecules exhibited a fluctuating FRET pattern, indicating that the activity was continuous (Figure 3A).
Figure 3. Repetitive sliding reveals an unstable intermediate during remodeling.
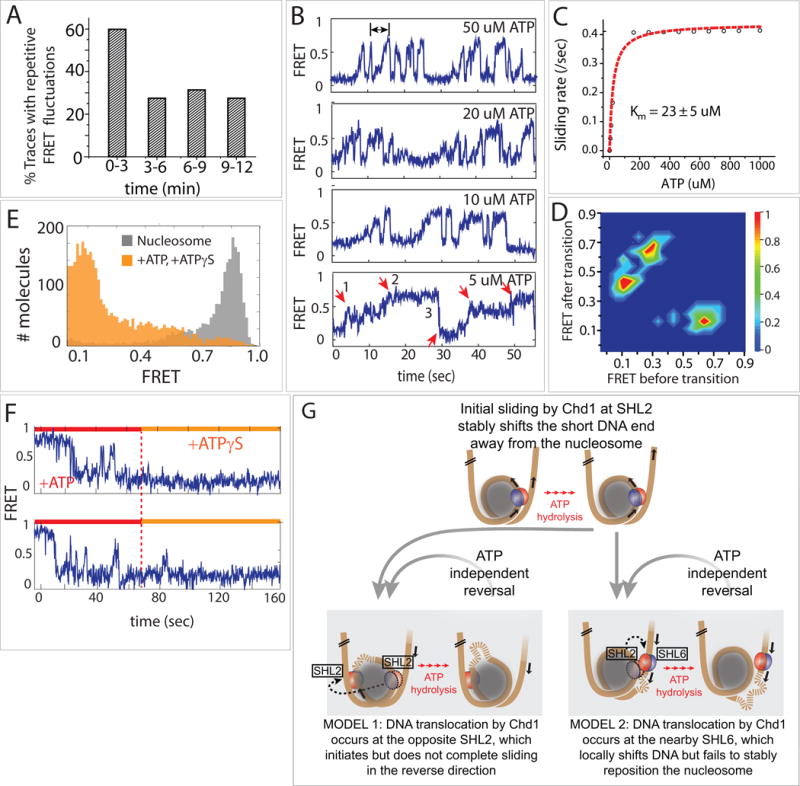
(A) Percent of traces (n=2000) showing repetitive FRET fluctuations over time.
(B) Single molecule FRET traces taken at various ATP concentrations.
(C) Michaelis-Menten fit of repetitive FRET fluctuations.
(D) Transition density plot showing distinct FRET states visited during the repetitive movement.
(E) FRET histogram before (gray) and after addition of ATP and ATPɣS (orange).
(F) Single molecule traces showing ATP and effect of subsequent ATPɣS addition.
(G) Two possible models to explain the bi-directional movement of nucleosomal DNA. In model 1, Chd1 changes the direction of DNA movement by engaging at the opposite SHL2, whereas in model 2, Chd1 instead engages around SHL6 located on the opposite gyre from the initial SHL2 site. In both cases, the shifted DNA is unstable without continuous ATP hydrolysis and snaps back to a remodeled state with the short DNA end away from the histone core.
The higher percentage of FRET fluctuation in the early phases of the remodeling reaction (0–3 min) may result from more nucleosomes being closer to the starting position, where FRET is most sensitive. As the nucleosomes are distributed along the DNA in subsequent times (3–12 min), most would be beyond the FRET detectable range.
Interestingly, we observed an asymmetric shape of the FRET fluctuations, where FRET increased gradually and decreased rapidly. This asymmetry was even more pronounced at low ATP concentrations (≤ 5 μM), which only slowed down the increase in FRET without affecting the sudden FRET drop (Figure 3B). This behavior suggests that the gradual FRET increase was ATP dependent while the abrupt FRET decrease was ATP independent. We calculated the frequency of this repetitive sliding by taking the inverse of time intervals corresponding to FRET increase, denoted by a double arrow (Figure 3B, top trace). The rates collected from over 1000 molecules under varying ATP concentrations were plotted against ATP concentration and fitted to the Michaelis-Menten equation, which yielded a KM of 23 μM ATP and Vmax of ~ 0.4/sec (Figure 3C). We analyzed the distributions of high and low FRET levels achieved during these fluctuations by taking FRET values before and after transitions from over 100 traces and plotting the values into a transition density plot (Figure 3D). Due to the way FRET transitions are plotted, the left top and right bottom triangles represent FRET stepping up and down, respectively. As shown, FRET steps up from 0.1 to 0.4 and 0.4 to 0.7 followed by stepping down from 0.7 to 0.1.
Although the gradual increase in FRET was consistent with Chd1 pulling the short DNA end back toward the histone core, the sudden ATP-independent drop suggested that the positioning of DNA was not stable. We hypothesized that when Chd1 switched the direction of DNA movement, correlating with increasing FRET, ATP hydrolysis was not only required for DNA movement but also for maintaining the DNA end closer to the nucleosome. To test this notion, we initiated sliding reactions with 1 mM ATP and then introduced 1 mM ATPɣS one minute later, while the nucleosomes were undergoing back and forth motion. With this subsequent addition of ATPɣS, which removed all residual ATP, the repetitive FRET fluctuations ceased. Importantly, all molecules transitioned to the low FRET value instead of stalling at different FRET states (Figure 3E, F), suggesting that the higher FRET states (0.7 FRET) were unstable intermediates. We therefore conclude that ATP hydrolysis was necessary to both achieve and maintain these higher FRET states. As described in the Discussion, this behavior suggests that Chd1 pulled DNA onto the nucleosome, perhaps forming a loop or an alternative structure, that was unable to be propagated around the histone core to allow for stable repositioning (Figure 3G).
The landscape for chromatin remodeling by Chd1 is impacted by strong nucleosome positioning sequences
Previous work has highlighted how Chd1 preferentially shifts mononucleosomes away from DNA ends (McKnight et al., 2011; Stockdale et al., 2006). Consistent with those findings, our smFRET experiments indicate that Chd1 can shift the histone octamer away from the short DNA end of 3N80 nucleosomes, and is unable to stably reposition the nucleosome back toward the short end. A basic question is therefore how Chd1 might be able to distinguish between the two sides of the nucleosome based on flanking DNA. Acting at SHL2, the remodeler ATPase motor pulls DNA onto the nucleosome from one side (entry DNA), which results in DNA being pushed out the other side (exit DNA). Initially, movement of 3N80 nucleosomes toward the 80 bp side means that this longer flanking DNA is the entry DNA and the shorter 3 bp side is the exit side. With the subsequent reversal in sliding direction, the shorter flanking DNA becomes the entry side.
One possible explanation for preferential sliding onto the 80 bp side was that shifting nucleosomes toward the short side was inefficient due to the limited length of flanking DNA. To explore this possibility, we tested sliding for a set of nucleosomes with increasing DNA lengths on the shorter side (Figure 4A). As expected from the farther initial placement of the Cy3 donor on the short flanking DNA (6–12 base pairs), the initial FRET values for these nucleosomes were progressively lower. Upon addition of Chd1 and ATP, FRET values initially decreased, indicating movement in the same direction, with the Cy3-DNA end as exit DNA. As previously observed for 3N80 nucleosomes, all nucleosomes displayed FRET fluctuations suggestive of sliding in the opposite direction, yet the majority of these traces failed to achieve higher FRET than the starting values (Figure 4B). These results suggest that despite the longer flanking DNA available for 6, 9, and 12 bp constructs, sliding stalled at approximately the same locations with respect to the Widom 601 positioning sequence (Figure 4C). This behavior suggests that for these nucleosomes, the DNA sequence and not length of flanking DNA was the major determinant for the locations where remodeling intermediates were unstable.
Figure 4. For the direction of nucleosome sliding by Chd1, DNA sequence can dominate over the length of flanking DNA.
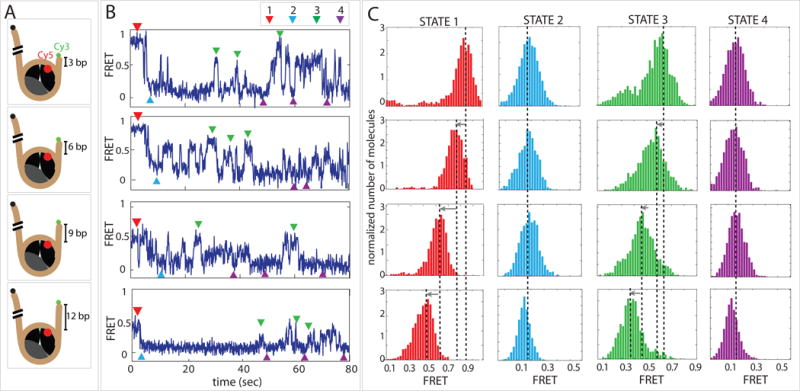
(A) Schematic diagrams of four FRET labeled nucleosome constructs with varying lengths of flanking DNA.
(B) Representative single molecule FRET traces obtained for each nucleosome construct.
(C) FRET histograms of four distinct states: starting nucleosome position (STATE 1, red), short DNA end shifted away from nucleosome (STATE 2, light blue), short DNA end pulled back toward nucleosome (STATE 3, green) and repetitive low FRET state where DNA is more distant from nucleosome (STATE 4, purple).
All of the nucleosomes described so far were generated using the Widom 601 positioning sequence, and one concern was that the apparent instability of remodeling intermediates may be particular to this DNA sequence. We therefore made two additional nucleosomes (4N80 and 80N4) based on the 603 positioning sequence, which was also generated by Widom (Lowary/Widom 1998) but shares only 28% sequence identity with 601 (Figure S1A). The 603 nucleosome showed similar smFRET patterns, shifting the short DNA end away from the nucleosome core, followed by periodic increases and decreases in FRET (Fig S1). As for 601, the FRET fluctuations were asymmetric, with slower increases in FRET compared to faster FRET decreases (Figure S1). These results support the conclusion that unstable remodeling intermediates are not unique for 601 nucleosomes. Since both 601 and 603 are strong positioning sequences, however, the high affinity for the histone core may be a major factor that destabilized remodeling intermediates in these experiments.
Limiting the range of the Chd1 DBD interferes with nucleosome sliding
The DBD of S. cerevisiae Chd1 is attached to the rest of the remodeler via an intrinsically disordered linker segment. We previously showed that while ≤29 residue deletions within the region spanning residues 961–1005 were well tolerated, removal of the entire 45 residue stretch abrogated sliding activity (Nodelman and Bowman, 2013). Despite virtually no nucleosome sliding activity, this variant lacking residues 961–1005, which we call Chd1-SL (for short linker), still displayed significant nucleosome-stimulated ATPase activity, suggesting that the remodeler initially engaged with nucleosomes but was unable to productively couple hydrolysis with sliding (Nodelman and Bowman, 2013). Based on the model for exit side inhibition, one explanation for the inability of Chd1-SL to slide nucleosomes could be from the persistent presence of the DBD on the exit side of the nucleosome, which is closer to SHL2 where the ATPase motor acts. Strikingly, when we tested nucleosome sliding activity of Chd1-SL by smFRET, we observed that the addition of ATP promoted an intermediate FRET state centered around 0.2 that was distinct from the fully repositioned state (0.1 FRET) described above (Figure 5A, B). Single molecule traces revealed that Chd1-SL and ATP induced dynamic DNA movement without stably attaining the lowest FRET state (Figure 5B). Overall, the FRET values shifted between 0.9 and 0.2 with intermittent excursions to other mid-FRET states, which were responsible for the two broad peaks observed in Figure 5A. When Chd1-SL was added to nucleosomes in the presence of AMP-PNP, remodeler-dependent FRET fluctuations were not observed (Supplementary Figure S2), suggesting that ATP hydrolysis was required for altering the position of nucleosomal DNA. These results demonstrate that Chd1-SL binds and can alter the DNA organization of nucleosomes in an ATP-dependent fashion, yet is somehow incapable of stably shifting DNA relative to the histone core. We speculate that this behavior reflects inhibitory action of the DBD on the nucleosome sliding process, amplified by the shortened linker. Whereas the normal linker allows the DBD to sample both entry and exit DNA, we surmise that the shortened linker restricts the DBD to exit DNA, where it interferes with nucleosome sliding even with limited DNA flanking the nucleosome.
Figure 5. Restricting the DBD to exit DNA results in unstable remodeling intermediates.
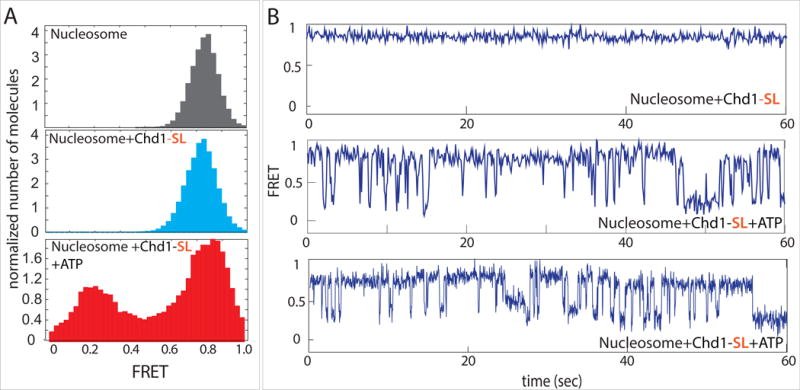
(A) FRET histograms taken for nucleosome alone (gray) and nucleosomes with Chd1-SL before (light blue) and after addition of ATP (red).
(B) Single molecule traces for nucleosome and Chd1-SL without (top) and with ATP (middle and bottom).
The N-terminal chromodomains of Chd1 guard against sliding hexasomes
Given the sensitivity of smFRET for detecting transient DNA movements, we decided to investigate the impact of Chd1 action on hexasomes. Compared to nucleosomes, hexasomes lack one histone H2A/H2B dimer, and we recently demonstrated that Chd1 was unable to robustly shift hexasomes toward the side lacking H2A/H2B (Levendosky et al., 2016). To monitor Chd1 activity on poor hexasome substrates, we produced end-positioned hexasomes with the side lacking the H2A/H2B dimer adjacent to the long flanking DNA, such that the Cy3-labeled DNA end was on the side with the remaining H2A/H2B dimer (Figure 6A). As expected, these hexasomes yielded a high FRET peak similar to nucleosomes (Figure 6B, top). With the absence of one H2A/H2B dimer, DNA wrapping of hexasomes appears to be weaker than that of nucleosomes, increasing the breadth of the histogram (Figure 1B). We confirmed that hexasomes engage with Chd1 in the absence of ATP by capturing FRET-labeled hexasomes with surface-immobilized Chd1 (Figure S3). Consistent with poor sliding of hexasomes, the FRET histogram in the presence of Chd1 and ATP showed only modest differences from hexasomes alone, with only a small increase in lower FRET populations (Figure 6B, bottom). Inspection of individual FRET traces, however, revealed large repetitive FRET fluctuations (Figure 6C). These dynamic FRET fluctuations were ATP-dependent, and the lower FRET states were visited only transiently, rapidly reverting to the initial high FRET state. These data are in accord with the bulk observations of poor hexasome sliding, yet reveal an unexpected activity of Chd1 toward hexasomes. Rather than having a mechanistic defect in hexasome sliding, we suspected that the inability to stably maintain shifted positions reflected a dominant regulatory process that prevented the normal progression of the remodeling cycle.
Figure 6. Chromodomains prevent Chd1 from repositioning a hexasome.
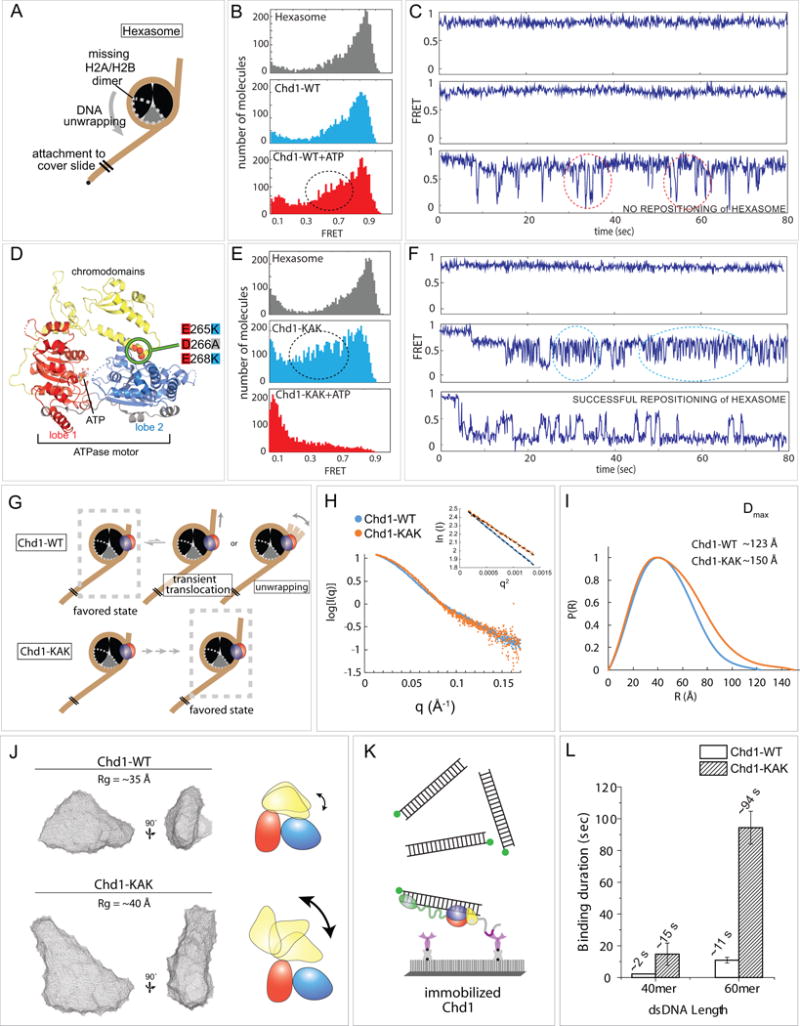
(A) Schematic diagram of hexasome conformation with the dotted gray outline indicating the location of the missing H2A-H2B dimer.
(B) FRET histograms of hexasome alone (gray) and hexasomes plus Chd1 before (light blue) and after ATP addition (red).
(C) Single molecule FRET traces corresponding with conditions in (B).
(D) Crystal structure of the chromodomain-ATPase portion of Chd1 (Hauk et al., 2010), highlighting the location of KAK mutation at the chromo-ATPase interface.
(E) FRET histograms of hexasome alone and hexasomes plus Chd1-KAK mutant, before and after ATP addition.
(F) Single molecule traces corresponding with conditions in (E).
(G) Interpretations of wildtype Chd1 and Chd1-KAK mutant activities on hexasomes.
(H) Small angle X-ray scattering (SAXS) profiles for Chd1-WT and Chd1-KAK proteins consisting of just the chromodomain and ATPase motor. Guinier plot analysis (inset) shows that samples were free from aggregation.
(I) P(R) distributions for SAXS data shown in (H).
(J) Ab initio bead models generated by DAMMIN. Cartoons on the right illustrate possible structural changes associated with the KAK mutation.
(K) Schematic of experiment in which Cy3 labeled dsDNA (40 or 60 bp) was added to surface immobilized Chd1-WT or Chd1-KAK proteins. Note that these constructs contain the DBD.
(L) Binding duration for dsDNA to both proteins (n=500 binding events).
In previous work, we found that disruption of the chromo-ATPase interface allowed naked DNA to activate the Chd1 ATPase similarly to nucleosomes, suggesting a loss in nucleosome-specific recognition (Hauk et al., 2010). Given the dynamic FRET profile of hexasomes, we wondered whether autoregulation by the chromodomains might contribute to the poor sliding of hexasomes. To investigate this possibility, we performed single molecule sliding experiments with a Chd1 variant containing three mutations at the chromo-ATPase interface (E265K/D266A/E268K), which we refer to as Chd1-KAK (Figure 6D). On nucleosomes, remodeling by Chd1-KAK closely resembled wild type Chd1, with characteristic ATP-dependent steps (Figure S4). On hexasome substrates, Chd1-KAK unexpectedly yielded rapid FRET fluctuations in an ATP-independent manner (Figure 6E,F middle panels). Previous work from many labs has shown that DNA sliding by remodelers requires ATP hydrolysis, and therefore we believe that the magnitude of FRET fluctuations we observed is most easily explained as dynamic DNA unwrapping from the Cy3 side of the hexasome. Interestingly, the Owen-Hughes group has reported that Chd1 can unwrap nucleosomes in the presence of AMP-PNP (Sundaramoorthy et al., 2017). In our experiments, Chd1-KAK stimulated FRET fluctuations in the absence of nucleotide, yet for wildtype Chd1, we failed to observe significant FRET fluctuations of hexasomes even with AMP-PNP (Figure S5). One possible explanation for the reported differences in 601 unwrapping for wildtype Chd1 could stem from the intrinsic asymmetry of the 601, which unwraps more readily from the one side than the other (Ngo, et al. 2015). Here we only monitored the more tightly wrapped TA-rich side of the nucleosomes, whereas Sundaramoorthy et al. followed the TA-poor side that more easily unwraps.
Unlike wildtype Chd1, upon addition of ATP, Chd1-KAK dramatically reduced FRET levels of hexasomes (Figure 6E, lower panel), suggestive of bona fide sliding. Individual smFRET traces showed a FRET decrease followed by repetitive FRET fluctuations, analogous to wild type Chd1 with nucleosomes (Figure 6E, F, lower panels). To confirm that hexasomes were repositioned by Chd1-KAK, we performed histone mapping experiments, which reveal histone locations on DNA before and after exposure to Chd1 and ATP. As shown in Figure S6, Chd1-KAK was capable of redistributing hexasomes whereas wildtype Chd1 was not. It is not clear whether the ATP-dependent FRET fluctuations for wildtype Chd1 represent DNA unwrapping or transient DNA translocations past the histone core (Figure 6G). Regardless of the effect, these experiments support that wild type Chd1 to cannot effectively reposition hexasomes. Thus, disruption of chromodomain autoinhibition in Chd1-KAK allowed Chd1 to reposition hexasomes, revealing that nucleosome specificity is achieved by the ability of the chromodomains to interrupt the remodeling reaction.
Disruption of the inhibitory chromodomain-ATPase interface results in a more extended conformation of Chd1 and more stable binding to naked DNA
What property of Chd1-KAK enables proficient repositioning of a hexasome? We hypothesized that the disruption of the chromo-ATPase interface with the KAK substitutions altered the ability of the chromodomains to stably pack against the ATPase motor and thereby reduce autoinhibition. To investigate this idea, we collected small angle X-ray scattering (SAXS) profiles for the chromo-ATPase portion of Chd1, equivalent to that used in the crystal structure (Figure 6D). Both wild type and KAK variants were well behaved, with low angle scattering demonstrating that the samples were free from aggregation (Figure 6H). Consistent with the KAK substitutions weakening interdomain interactions, Chd1-KAK possessed a significantly larger radius of gyration (Rg) of 39.8 ± 0.1 Å compared with an Rg for the wild type chromo-ATPase of 35.6 ± 0.1 Å. The Dmax of KAK mutant was also larger than wild type (150 versus 123 Å), indicating that disrupting this interaction between the chromodomains and ATPase motor results in a more extended conformation of the protein (Figure 6I). An extended conformation for KAK mutant was also supported by bead modeling, which suggested much looser domain-domain contacts in the absence of the wild type chromo-ATPase interface (Figure 6J). Interestingly, while bead models of the wild type chromo-ATPase showed a better fit to the crystal structure, the SAXS-derived models did not perfectly fit the shape of the chromo-ATPase crystal structure. This mismatch likely reflects dynamics of protein domains in solution that cannot be accurately represented with a static bead model. We believe that the SAXS models indicate inherent mobility of the chromodomains that are greatly exaggerated upon disruption of the chromo-ATPase interface.
One prediction of a more opened domain organization is that the ATPase motor of the KAK mutant should be able to engage more stably with DNA since the chromo-ATPase interface is disrupted. We previously reported that the KAK substitution enabled the isolated chromodomain-ATPase portion of Chd1 to weakly interact with DNA (Hauk et al., 2010); however, more recent experiments showed that the weak binding suggested by a gel shift was likely due to a contaminating factor, which could have reached significant levels relative to the 25 nM DNA probe given the high (110 μM) concentrations of Chd1 used. Detecting DNA binding by native gel shifts is therefore not sensitive enough to reveal DNA-binding interference by the chromodomains, since the isolated chromo-ATPase does not form a stable complex with naked DNA and inclusion of the DBD masks contributions from the chromo-ATPase. We therefore turned to single molecule observation to determine the extent that DNA binding was affected by disruption of the chromo-ATPase interface. Using Chd1 constructs containing the DBD, we tethered wildtype and KAK mutant proteins to the surface via a FLAG:anti-FLAG attachment. The protein molecules of Chd1 were seeded at single molecule density to which Cy3-labeled double-stranded (ds) DNA of 40 and 60 bp were added (Figure 6K). Nonspecific binding of DNA was minimal as almost no fluorescent spots appeared when 1 nM Cy3 dsDNA was added to surface without the protein. To assess the stability of wildtype Chd1 and Chd1-KAK for binding naked dsDNA, we measured the dwell time of the Cy3 signal, which signifies the duration that dsDNA bound to the surface-immobilized protein. As shown in Figure 6L, the KAK mutant had approximately seven- to eight-fold longer retention time with 40 and 60 bp dsDNA, respectively. In addition, the KAK mutant displayed a higher apparent affinity to dsDNA compared with wildtype Chd1 (Figure S7). This higher stability of the KAK mutant with naked DNA is consistent with a more opened organization of the chromodomains that provides the ATPase motor with greater access to DNA. Given the marked gain of the KAK mutant for binding DNA and sliding hexasomes, we propose that the chromodomains are poised to disrupt interactions between the ATPase motor and nucleosomal DNA, which in turn determines how productively the remodeler engages with its nucleosome substrates.
DISCUSSION
This work advances our understanding of the Chd1 chromatin remodeler, and puts forward several concepts that may also apply to other remodeling enzymes. We demonstrate that Chd1 shifts nucleosomes in predictable steps that likely encompass multiple base pairs. This behavior is consistent with the stepwise translocation observed for the related but distinct ISWI family of chromatin remodelers (Deindl et al., 2013). For ISWI, individual 1 bp steps were clustered in bursts of ~3–7 bp steps, with pauses delineating discrete units of translocation. Interestingly, discrete multi-base pair steps were not observed for the SWI/SNF-type remodeler RSC, which continuously shifted DNA using a 1–2 bp step size (Harada et al., 2016). The characteristic pauses that punctuate smFRET sliding patterns of Chd1 and ISWI remodelers are therefore not essential for nucleosome repositioning. We propose that these pauses provide opportunities for regulatory domains to influence the remodeling reaction. For Chd1, the N-terminal chromodomains and C-terminal DNA-binding domain (DBD) have both been shown to influence the remodeling reaction, and these domains may take advantage of unstable intermediate states to regulate nucleosome sliding.
Previous work with DNA gaps demonstrated that the ATPase motors of Chd1, ISWI, and SWI/SNF-type remodelers drive nucleosome repositioning by translocating on nucleosomal DNA at SHL2 (McKnight et al., 2011; Saha et al., 2005; Schwanbeck et al., 2004; Zofall et al., 2006). The ATP-dependent chromatin assembly and remodeling factor (ACF), an ISWI-type remodeler, ensures back-and-forth sliding by cooperatively binding to nucleosomes as dimers, with each remodeler ATPase poised at an SHL2 site (Racki et al., 2009). While the nucleosome can simultaneously accommodate two Chd1 proteins, one at each SHL2 site (Nodelman et al., 2017), here we make the unexpected discovery that back-and-forth movement of nucleosomal DNA can be achieved by a single Chd1 remodeler (Figure 2). After initially shifting the nucleosome away from the short end, however, Chd1 appears unable to stably shift nucleosomes in the reverse direction (Figure 3 and S1). The reason for this instability is unclear, but was observed with two different positioning sequences. While present evidence suggests that Chd1 shifts nucleosomes when at SHL2 (Nodelman et al., 2017), two recent studies have shown that remodeler ATPases can also engage with the outer gyre of DNA: the isolated SWI/SNF ATPase, in a cryoEM study, was shown to bind SHL6 as well as SHL2 (Liu et al., 2017), and yeast INO80 was shown to reposition nucleosomes by translocating on DNA around SHL5 (Brahma et al., 2017). For Chd1, reversal of DNA movement may therefore result from translocation of the ATPase at the SHL2 site on the opposite side of the nucleosome (Figure 3G, model 1), or by reorienting to SHL5 or SHL6 on the opposite gyre, which is close to the first SHL2 site (Figure 3G, model 2).
We speculate that the generation of unstable remodeling intermediates provides an important regulatory checkpoint. For both 601 and 603 nucleosomes, Chd1 appeared unable to stably shift nucleosomes back to the starting position, exhibiting a highly repetitive sliding behavior (Figure 3 and S1). While this repetitive sliding may have been exacerbated by the absence of a C-terminal Chd1 domain of unknown function (Mohanty et al., 2016) that was absent in our construct, we note that many helicases translocate on nucleic acids in a highly repetitive manner (Koh et al., 2014; Myong et al., 2007; Myong et al., 2009; Myong and Ha, 2010; Myong et al., 2005; Park et al., 2010; Qiu et al., 2013; Tippana et al., 2016). Analogous to keeping nucleic acids unwound, repetitive sliding by Chd1 may keep nucleosomes in alternative states. Chd1 is required for both histone replacement (Konev et al., 2007) and nucleosome assembly (Fei et al., 2015), processes that necessitate transient disruptions of histone-histone and histone-DNA contacts, and we speculate that such repetitive remodeling events may facilitate these dramatic and fundamental reorganizations of the nucleosome.
Although the present data is insufficient for determining whether Chd1 engages with SHL6 or only switches between both SHL2 sites, the ability of the ATPase motor to sample different segments of nucleosomal DNA likely arises from flexible tethering by the DBD. The ATPase motor and DBD of Chd1 are separated by a flexible protein segment (Nodelman and Bowman, 2013) that is long enough to allow the ATPase motor to bind to SHL2 while the DBD is bound to flanking DNA on either side of the nucleosome. We propose that the ATPase motor and DBD assist each other in nucleosome binding, keeping the remodeler close to its substrate. By being tethered to the nucleosome through one domain, the high effective concentration increases the likelihood that the other domain will reengage, and also offers the possibility to sample other locations on the nucleosome. In addition to changing the direction of sliding, changes in Chd1 domain organization is also expected to affect activity. Our experiments with the shortened linker between the DBD and ATPase motor (Chd1-SL) supports an inhibitory role of the DBD on exit DNA. A shortened linker would favor a close association of the DBD and ATPase motor on opposite DNA gyres. The Chd1-SL variant shows continuous FRET fluctuations, suggestive of DNA movement, yet was unable to attain a low-FRET state, indicative of stable nucleosome repositioning (Figure 5). These results suggest that when the DBD remains close to the ATPase motor, the nucleosome sliding reaction can be interrupted, making repositioning ineffective. The location of the ATPase motor therefore determines the direction of productive nucleosome sliding, whereas placement of the DBD relative to the ATPase motor modulates sliding activity.
We note that although our experiments described here were limited to mononucleosomes, the dynamic reengagement of the remodeler suggests an ability to diffuse along chromatin fibers. In a fiber, neighboring nucleosomes share the same segment of flanking DNA, and tethering by the DBD would be expected to allow the ATPase motor to also transfer to another nucleosome. In our experiments, we found a remarkably high retention of Chd1 on single nucleosomes over >20 min periods. Since monomers of Chd1 immobilized to the surface exhibited repetitive movement, the continuous Chd1 activity suggested a preference for retention on nucleosomes over dissociation into solution. These observations raise the possibility that Chd1 primarily migrates along chromatin fibers (Figure 7), potentially also switching between fibers with close nucleosomes packing. Such a behavior would likely allow individual Chd1 remodelers to processively reorganize nucleosome positioning at a local level.
Figure 7.
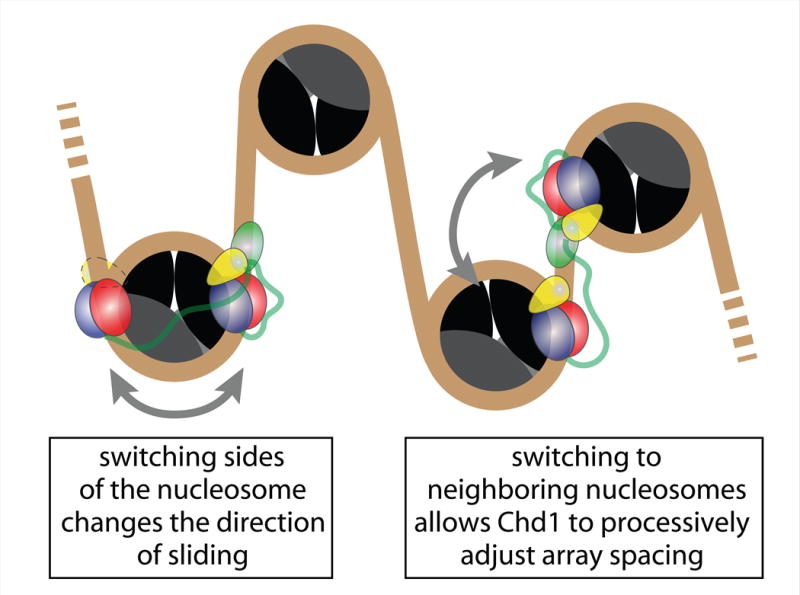
Proposed model of Chd1 generating back and forth motion to adjust nucleosomal spacing
STAR Methods section
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sua Myong (smyong@jhu.edu).
METHOD DETAILS
Preparation and Bulk Measurements of Nucleosomes and Chd1 Proteins
Histone purification
Histones were prepared essentially as previously described(Dyer et al., 2004). Xenopus laevis histones H2A, H2B, H3 and H4 were expressed in bacteria and FPLC purified. pET3a vectors containing expression constructs of each Xenopus laevis histone were transformed into Escherichia coli BL21(DE3) pLysS cells, grown at 37°C in 4 L of 2x TY media (containing ampicillin and chloramphenicol) and induced with IPTG at OD600=0.3–0.5. Cells were harvested at room temperature and resuspended to ~ 40mL in wash buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM EDTA pH 8, with 1 mM Benzamadine added fresh before use) and flash frozen in liquid nitrogen. The pellet was thawed, diluted to ~80 mL in wash buffer and sonicated. The insoluble fraction containing the histone-rich inclusion bodies was pelleted by centrifugation at 23,000 × g for 20 minutes at 4 °C. The supernatant was discarded and the pellet was thoroughly resuspended in 80 mL of wash buffer with 1% v/v Triton X-100 detergent followed by centrifugation for 10 minutes. The pellet was washed once more with wash buffer plus detergent and then two more times with wash buffer. After the last wash, the inclusion body pellet was spread in a thin layer on the inside of a 50 mL conical tube and stored at −20° C. To purify the histones from the incl usion bodies, the inclusion bodies were treated with 1 mL of DMSO and incubated at room temperature for 30 minutes and then the histones were unfolded with 40 mL of unfolding buffer (20 mM Tris-HCl pH 7.5, 7 M guanidine-HCl, 1 mM EDTA pH 8, 10 mM DTT, prepared fresh) and shaken at room temperature for one hour. The inclusion bodies were centrifuged at 23,000× g for 20 minutes at 18° C and the supernatant was passed over a desalting column (HiPrep 26/10; 17-5087-01) pre-equilibrated in desalting buffer (10 mM Tris-HCl pH 7.5, 7 M Urea, 1 mM EDTA, 100 mM NaCl, 5 mM β-Mercaptoethanol, prepared fresh and degassed) 20 mL at a time to remove the guanidine-HCl. Next, the histones were loaded onto a Q anion exchange column (HiPrep 16/10 Q FF, 17-5190-01) mounted on top of an S cation exchange column (HiPrep 16/10 SP FF, 17-5192-01) that were pre-equilibrated in ion exchange buffer A (10 mM Tris-HCl pH 7.5, 7 M Urea, 1 mM EDTA, 5 mM β-Mercaptoethanol, prepared fresh and degassed) with 10% ion exchange buffer B (ion exchange buffer A with 1 M NaCl). After loading, the tandem ion exchange columns were washed until the UV signal reached baseline. The Q column was then removed and the S column was washed further. A linear gradient from 10% to 60% ion exchange buffer B (100 mM to 600mM NaCl) over 30 column volumes was used to elute the histones from the S column. After evaluation by 18% SDS-PAGE, peak fractions were pooled and dialyzed in 3500 MWCO dialysis tubing against 4 L of 5 mM β-Mercaptoethanol with two more buffer changes with at least 3 hours between changes. The purified histones were lyophilized in 2 mg aliquots and stored at −20° C.
Refolding of histone dimer and histone tetramer
The histones were refolded into high salt buffer to form dimer (H2A/H2B) and tetramer ([H3/H4]2), which could later be combined in different ratios with DNA to form either nucleosomes or hexasomes. Each 2 mg histone aliquot was unfolded at room temperature in 1.5 mL unfolding buffer (20 mM Tris-HCl, pH 7.8, 6 M guanidine-HCl, 5 mM DTT, made fresh). After 1–2 hours, undissolved protein was removed by centrifugation at 16,100 × g for 10 minutes at room temperature. The unfolded histones were combined in equimolar ratios, adjusted to a final histone concentration of 1 mg/mL and placed in 3500 MWCO dialysis tubing. The histones were dialyzed into four changes of 500 mL refolding buffer (10 mM Tris, pH 7.8, 2 M NaCl, 1 mM EDTA, 5 mM β-mercaptoethanol added fresh) at least 3 hours apart.
For FRET experiments, residue 120 of H2A was mutated to cysteine and labeled with Cy5 maleimide. Histones to be labeled were unfolded with 1.5 mL labeling buffer (20 mM Tris, pH 7.0, 6 M guanidine-HCL, 5 mM EDTA) for each 2 mg aliquot. The cysteines were reduced by adding 4 μL of 500 mM TCEP and then the histones were incubated for 2 hours at room temperature. Each 2 mg aliquot of histone was labeled with 5 μM of Cy5 maleimide and incubated for 3 hours at room temperature in the dark. The labeling reaction was quenched with 80 mM β-Mercaptoethanol and the unreacted dye was removed by buffer exchanging with labeling buffer using an Amicon Ultra 3,500 MWCO concentrator. The labeled histone could then be combined with the other histones and dialyzed into refolding buffer.
Dimer and tetramer were purified individually by FPLC. The dimer or tetramer was concentrated in an Amicon Ultra 10,000 MWCO concentrator to ~ 1 mL and loaded onto a HiLoad 16/10 Superdex 75 size exclusion column pre-equilibrated in degassed refolding buffer. Peak fractions were analyzed by 18% SDS-PAGE and clean fractions with equal amounts of composite histones were pooled and concentrated. The dimer or tetramer was mixed 1:1 with freezing solution (10 mM Tris, pH 7.8, 2 M NaCl, 1 mM EDTA, 5 mM β-mercaptoethanol, 40% (v/v) glycerol), flash frozen in liquid nitrogen and stored at −80° C.
Preparation of Nucleosomal DNA
(See Supplementary Table S1 for a complete DNA sequence information)
DNA containing the Widom 601 or 603 nucleosome (Dechassa et al., 2010) positioning sequences were prepared by PCR in 5 or 10 mL reactions. DNA constructs were as follows (linker DNA lowercase and dyad bold/underlined):
601 (3N80), 5′cccTGGAGAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCCCCCGCGTTTTAACCGCCAAGGGGATTACTCCCTAGTCTCCAGGCACGTGTCAGATATATACATCCTGtgcatg tattgaacagcgaccttgccggtgccagtcggatagtgttccgagctcccactctagaggatccccgggtaccg;
603 (4N80), 5′tgccCAGTTCGCGCGCCCACCTACCGTGTGAAGTCGTCACTCGGGCTTCTAAGTACGCTTAGCGCACGGTAGAGCGCAATCCAAGGCTAACCACCGTGCATCGATGTTGAAAGGGGCCCTCCGTCCTTATTACTTCAAGTCCCTGGGGtacccg tttcgaggtcgactctagaggatcccgagagaatcccggtgccgaggccgctcaattggtcgtagacagctcta;
603 (80N4), 5′cacaggaaacagctatgaccatgattacgccaagcttcggaggacagtcctccgtgcaggtcgactctagaggatctgccCAGTTCGCGCGCCCACCTACCGTGTGAAGTCGTCACTCGGGCTTCTAAGTACGCTTAGCGCACGGTAGAGCGCAATCCAAGGCTAACCACCGTGCATCGATGTTGAAAGGGGCCCTCCGTCCTTATTACTTCAAGTCCCTGGGGtaca
Reactions contained 1× ThermoPol buffer, 2 mM MgSO4, 2 ng/μL pGEM 601 plasmid, 0.5 μM forward and reverse primers, 2 mM dNTP mixture, and Taq Polymerase. Primers containing fluorophores or biotin were ordered from IDT. The reaction was divided into 100 μL aliquots in thin-walled PCR tubes and placed in a thermocycler programed as follows: Step 1—95°C for 1 min, Step 2—95°C for 30 sec, Step 3—55°C for 30 sec, Step 4—72°C for 1 min, Step 5—go to Step 2 40 times, Step 6—72°C for 10 min. The individual aliquots were pooled and the PCR product was verified by 1.5% agarose electrophoresis with a 100 bp ladder.
Next, the target DNA was purified away from primers and incomplete products over a BioRad MiniPrep Cell apparatus. The PCR product was concentrated to ~ 50 μL in an Amicon Ultra 4 concentrator and sucrose was added to 8%. The PCR product was loaded on a 5.5 cm tall, 6% polyacrylamide (60:1 acrylamide:bisacrylamide) native MiniPrep Cell column and electrophoresed at 1 W using 0.5 × TBE running buffer and TE elution buffer (10 mM Tris pH 7.8, 1 mM EDTA pH 8). Elution fractions were collected at a rate of 3 minutes/fraction and analyzed on a 1.5% agarose gel. DNA usually eluted after about 2 hours. Clean peak fractions were pooled and concentrated before measuring the DNA concentration at A260–A310.
Nucleosome and Hexasome Reconstitution and Purification
The protocol for generating nucleosomes followed Dyer et al., 2004. Nucleosome and hexasome reconstitutions were assembled containing 5.95 μM purified DNA, 2 M NaCl, 10 mM Tris pH 7.8, 1 mM EDTA pH 8, 1 mM DTT, 6 μM [H3/H4]2 tetramer, and 12 μM H2A/H2B dimer (to make nucleosomes) or 7.2 μM H2A/H2B (to make hexasomes). The components were loaded into small 6–8,000 MWCO dialysis chambers and placed in 400 mL of cold, high-salt reconstitution buffer (RB high: 10 mM Tris-HCl pH 7.8, 2 M KCl, 1 mM EDTA pH 8, 1 mM DTT added fresh). The reconstitution buffer was stirred at 4° C in the dark while 2 L of a low salt buffer (RB low: same as RB high but containing 250 mM KCl) was exchanged with RB high at a rate of 1.5 mL/minute. After all of the RB low was been exchanged, the reconstitution was transferred to 400 mL of TED buffer (10mM Tris-HCl pH 7.8, 1 mM EDTA pH8, and 1 mM DTT added fresh) to dialyze for at least 3 more hours. The reconstitutions were concentrated to ~ 50 μL and brought to 8% sucrose in preparation for purification and stored on ice at 4° C.
Nucleosome or hexasome reconstitutions were purified using a BioRad MiniPrep Cell. The column was 7 cm tall, 7% polyacrylamide (60:1 acrylamide:bisacrylamide). The samples were electrophoresed at 1 W using 0.5 × TBE running buffer and TED elution buffer. Hexasomes usually eluted in 4.5 to 5 hours and nucleosomes eluted in 5 to 6 hours. Samples of eluted fractions were mixed 1:1 with 12% sucrose loading buffer and evaluated using 7% polyacrylamide (60:1 acylamide:bisacrylamide) native minigels, with a 100-fold dilution of the loaded reconstitutions providing a marker. Pure fractions were pooled and concentrated to at least 2 μM as measured by the DNA concentration. Pure nucleosome or hexasome was brought to 20% glycerol and 0.1 mg/mL BSA then flash frozen in liquid nitrogen and stored at −80° C.
Histone Mapping
The position of the histone octamer can be determined to near bp resolution using histone mapping as previously described (Kassabov and Bartholomew, 2004). Nucleosomes or hexasomes containing a single cysteine mapping site introduced at H2B (S53C) and fluorescently labeled DNA were buffer exchanged into TG buffer to remove DTT. The mapping site was labeled with 200–400 nM of the photactivatable crosslinker 4-azidophenacyl bromide (APB) at room temperature in the dark for 2.5 hours then quenched with DTT. Mapping reactions (50 μL) were assembled with 150 nM nucleosome or hexasome and 50 nM Chd1 in 1× slide buffer (20 mM Tris-HCl (pH 7.8), 50 mM KCl, 5 mM MgCl2, 5% sucrose (w/v), 0.1 mg/mL BSA, 1 mM DTT). Sliding reactions were initiated with the addition of 2 mM ATP and quenched at timepoints by mixing with 100 μL of quench buffer (20 mM Tris-HCl (pH 7.8), 50 mM KCl, 5% sucrose (w/v), 0.1 mg/mL BSA, 5 mM DTT, 5 mM EDTA, 150 ng/mL salmon sperm DNA) and chilled on ice. Quenched reactions were UV irradiated for 15 seconds to induce APB crosslinking to DNA. Irradiated samples were mixed with 150 μL of 20 mM Tris-HCl pH 8, 0.2% SDS, 50 mM NaCl and heated to 70°C for 20 minutes. Next, 300 μL of 5:1 Phenol:Chloroform was added, followed by vortexing and centrifugation at 16,100 × g for 2 minutes. About 250 μL of the top layer containing un-crosslinked DNA was removed. The sample was washed by adding 280 μL of 1M Tris-HCl pH 8 and 1% SDS and then vortexing, centrifuging, and removing 280 μL from the top layer. This wash step was repeated three more times. DNA was precipitated by addition of 33 μL NaAcetate pH 5.2, 1.5 μL salmon sperm DNA and 750 μL of 100% EtOH. Samples were mixed and stored on ice at 4°C overnight. Precipitated DNA was pelleted by centrifugation at 16,100 × g for 30 minutes at 4° C and the supernatant was discarded. The pellet was washed with 750 μL of 75% EtOH two times then allowed to dry. The DNA pellet was resuspended by adding 100 μL Ammonium Acetate, 2% SDS, 1mM EDTA pH 8, and vortexing for 1 minute. The DNA was cleaved at the crosslinking site through the addition of 5 μL NaOH and heating at 90° C for 40 minutes. The samples were neutralized with the addition of 105 μL of 20 mM Tris-HCl and 6 μL of 2 μM HCl and vortexed. The cleaved DNA was precipitated with the addition of 1 μL 2M MgCl2 and 480 μL 100% EtOH, and incubated at −20° C overnight. The precipitated DNA was pelleted, washed, and dried as before. The dry pellet was resuspended in 4 μL of deionized formamide loading buffer (89 mM Tris-borate pH 8, 5 mM EDTA pH 8, 95% (v/v) formamide, 0.2% (w/v) Orange G Dye). For reference, a sequencing ladder was prepared using USB-Affymetrix Thermo Sequenase Dye Primer Manual Cycle Sequencing Kit (cat# 79260) with the labeled primer used to generate the nucleosomal DNA. Sequencing reactions were mixed 1:1 with formamide loading buffer and heated to 70° C for 2 minutes. The mapping samples were heated at 95° C for ~30 se conds before loading on an 8% polyacrylamide (19:1 acrylamide:bisacrylamide) 7.7 M urea sequencing gel alongside the sequencing ladder. The gel was run for 1.25 hr at 65 W using 1× TBE running buffer and visualized on a Typhoon 9410 variable mode imager (GE Healthcare).
Chd1 protein purification
A truncated construct of Saccharomyces cerevisiae Chd1 (residues 118–1274), here referred to simply as Chd1, was purified as previously described (Nodelman et al., 2017; Patel et al., 2011). All Chd1 expression constructs contained an N-terminal 6 X His tag followed by a Prescission Protease cut site (LEVLFQ/GP). The N-termini of FLAG tag constructs were as follows (precisssion cleavage site bolded, and FLAG tags underlined; final D in sequence is residue 175): MSYYHHHHHHLESTSLYKKAGSAAAPFTGSLEVLFQGPQSTVKIPTRFSNRQNKTVNYNIDYSDDDLLESEDDYKDDDDKGSEEALSDLLESEDDYKD DDDKGSEEALSEENVHEASANPQPED. FLAG tagged constructs also lacked any with endogenous cysteines and contained two introduced cysteines at positions Q255C and K632C, which showed remodeling activity comparable to wild type Chd1. The Chd1 KAK mutant contained amino acid changes E265K, D266A, and E268K. Chd1-SL additionally contained an internal deletion of residues 961–1005 (Nodelman and Bowman, 2013)
Each expression construct, in a pDEST17 vector, was transformed into chemically competent Escherichia coli BL21(DE3) Trigger RIL cells for expression, plated on LB agar plates containing ampicillin (to retain pDEST17) and chloramphenicol (to retain the Trigger RIL plasmid). Overnight cultures started from single colonies were used to inoculate eight 1 L cultures of TB media containing ampicillin and chloramphenicol in baffled Fernbach flasks, and grown at 37° C. Onc e the expression cultures reached OD600 = 0.2–0.4, the temperature of the incubator was reduced to 18° C. At OD600 = 0.6–0.8, Chd1 expression was induced by the addition of 0.3 mM IPTG and the cultures were incubated for an additional 18 hours. Cells were harvested by centrifugation at 4000 × g for 10 minutes at 4° C. Cell pellets were flash frozen in liquid nitrogen and stored at −80° C.
The Chd1 proteins were FPLC purified. Cell pellets were thawed in room temperature water bath and immediately placed on ice. Pellets were resuspended in HisBind Buffer A (20mM Tris-HCl pH 7.8, 500 mM NaCl, 10 mM imidazole, 10% (v/v) glycerol, 0.2 μm filtered and degassed) to a volume of 100–150 mL. To lyse the cells, the cell slurry was brought to 0.1 M PMSF, 5 mM β-mercaptoethanol, 2.5 mM MgCl2, 0.5 mM CaCl2, 1 mg/mL lysozyme and 10 mg/mL DNase I and incubated on ice for 30 minutes. The cells were then sonicated and the lysate was centrifuged at 45,000 × g. The supernatant was loaded on 3 tandem HisTrap 5 mL Ni columns pre-equilibrated in HisBind Buffer A, and then washed extensively. When the UV signal reached baseline, the bound protein was eluted with a 505 mM imidazole bump by adding 50% HisBind Buffer B (HisBind Buffer A with 1 M imidazole). Peak fractions were evaluated by 12% SDS-PAGE and pooled. The protein was diluted 5 fold with TGzero (20 mM Tris-HCl pH 7.8, 10% (v/v) glycerol, 0.2 μm filtered and degassed) to bring the NaCl concentration to 100 mM for ion exchange chromatography. The protein was loaded on a cation exchange column (HiTrap SP FF) pre-equilibrated with TG0 with 10% TG1000 (TG0 with 1 M NaCl). After washing, the protein was eluted with a buffer gradient from 10% TG1000 to 50% TG1000 over 150 mL. Peak fractions were analyzed by SDS-PAGE and pooled. Protein usually eluted near 280 mM NaCl. The salt concentration was estimated from the peak and the protein was diluted to 200 mM NaCl by adding TG0. One mg of Prescission Protease was added to the protein and allowed to digest on ice at 4° C overnight. The digested protein was brought to 500 mM NaCl and 10% imidazole before loading onto a HisTrap column to remove the His tag and undigested protein. The flow through from the HisTrap column was concentrated to ~1 mL in an Amicon Ultra 15 10,000 MWCO and loaded onto a HiLoad 16/10 Superdex 75 size exclusion column pre-equilibrated in TG300D (20 mM Tris-HCl pH 7.8, 300mM NaCl, 10% (v/v) glycerol, 1 mM DTT added fresh, 0.2 μm filtered and degassed). Fractions were analyzed by 12% SDS-PAGE, pooled and concentrated. Aliquots were flash frozen in liquid nitrogen and stored at –80 °C.
SEC-SAXS (Size-Exclusion Chromatography-Small Angle X-ray Scattering)
SAXS data were collected at the Advanced Photon Source (BioCAT), beam line 18ID. The method of incorporating size-exclusion chromatography in-line with the equilibrium SAXS was carried out as described before (Mathew et al., 2004). To eliminate scattering from aggregates that could potentially make the data difficult to interpret, an in-line Superdex-200 10/300 gel-filtration column was used to purify the protein sample immediately upstream of the data collection chamber. Data acquisition was performed at a wavelength of 1.033 Å. Using a sample to detector distance of 3.5m, we were able to access a q range of ~0.006 Å to ~0.3 Å. One-second exposures were acquired through the entire duration of the SEC elution with a periodicity of 2 seconds. We were therefore able to use the exposures flanking the elution peak as buffer, which were averaged and subtracted from the exposures corresponding to the sample elution. Guinier approximation and pair distance distribution (P(r)) using PRIMUS (Konarev et al., 2003) were performed on buffer subtracted I(q) vs q curves corresponding to the peak of the elution profile to obtain the radiation of gyration (Rg) and the maximum dimension (Dmax) of the molecule. Ab initio bead models were calculated using DAMMIF, DAMAVER and DAMFILT (Franke et al., 2009). Bead models from 10–20 DAMMIF processes were averaged using DAMAVER.
Single Molecule Measurements of Nucleosomes and Chd1 Proteins
Nucleosomes
Preparation of nucleosomes with octamer or hexasome histones are described in the previous subsection. Each nucleosome consists of the 601 DNA sequence wrapped around the octamer histone, which have Cy5 labeled H2A histones. One side of the nucleosome has 80 bps of dsDNA with a biotinylated end and the other side has 0 or 3 bps of dsDNA and is labeled with Cy3 at the end.
Double-stranded DNA (dsDNA) Preparation
Complementary strands of oligonucleotides of random sequences were purchased from Integrated DNA technologies (Coralville, IA). Oligos with end-labeled Cy3 dye are ordered pre-labeled. dsDNA substrates were prepared by mixing the appropriate labeled and unlabeled oligonucleotides in a 1:1 molar ratio (to avoid excess of single strands) at 10 μM in DNA annealing buffer (10mM MgCl2, 10mM Tris-HCl (pH 8.0)). Double-stranded oligonucleotide mixtures were incubated at 95°C for 2 minutes followed by slow cooling to room temperature (at a rate of 2 degrees per minute) to complete the annealing reaction.
Nucleosome dilution buffer
50mM NaCl, 10mM Tris‐HCl (pH 8.0) and 5mM MgCl2.
Chd1 Proteins
Preparations and bulk measurements of the full length (Chd1-WT) and mutant Chd1 yeast proteins (Chd1-KAK, Chd1-120B-d234) were described in the previous sub-section. 2–20nM of proteins are used in each experiments as specified.
Reaction Buffer and Condition
Buffer consisting of 20 mM HEPES (pH 7.5), 5 mM MgCl2, 0.1 mM EDTA, 50 mM KCl, 1 mM DTT, 5% sucrose, 0.02% Nonidet P‐40 and 0.1 mg/mL BSA, was used with an oxygen scavenging system containing 1% v/v dextrose, 1 mg/ml glucose oxidase, 0.03 mg/ml catalase (Joo and Ha, 2008), and 2-mercaptoethanol (1% v/v), all items were purchased from Sigma-Aldrich (St. Louis, MO).
The measurements were performed at room temperature (21°C ± 1°C). ATP or non-hydrolyzing ATP analogues (ATPgS/AMP-PNP) was used in all experiments at a concentration of 1mM, unless otherwise specified.
Single‐Molecule Fluorescence Assay
We used home-built total internal reflection fluorescence microscope for single-molecule fluorescence assays. We excited the nucleosome samples containing Cy3 (donor) and Cy5 (acceptor) dyes with a solid-state 532 nm laser (75mW, Coherent CUBE) to measure the FRET signal. The emission signals were separated by using a dichroic mirror (cutoff: 630 nm) and detected by an EMCCD camera (iXon DU‐ 897ECS0‐#BV; Andor Technology). We applied FRET labeled nucleosome molecules (see above) to polyethylene glycol (PEG)-coated quartz surface via biotin-neutravidin linkage. The camera was controlled using homemade C++ program. Single‐molecule traces were extracted from the recorded video file by IDL software.
Slide Surface Preparation
In all cases of single molecule experiment, passivated slides were prepared ahead of time. Briefly, both the quartz slides and coverslips were washed with methanol and acetone, etched by sonication in 1 M KOH for 30 minutes, flamed for 30 seconds, treated with aminosilane for 20 minutes, and coated with a mixture of 98% mPEG (m-PEG-5000, Laysan Bio, Inc.) and 2% biotin PEG (biotin-PEG-5000, Laysan Bio, Inc). The PEG-coated quartz slides are assembled into multiple-channeled imaging chamber. NeutrAvidin is added as described in (Qiu and Myong, 2016). This allows proteins or oligonucleotide to bind to the surface via the biotin-NeutrAvidin linkage.
Chd1 remodeling immobilized nucleosomes
Chd1‐WT, Chd1-KAK, or Chd1‐120B d234 was mixed at 20nM with reaction buffer and added to a flow chamber that had 50–100pM nucleosomes specifically immobilized on the slide chamber surface. Excess proteins are washed away with a reaction buffer containing only ATP to observe the repositioning process in real-time. ATP concentrations ranging from 1uM to 1mM is used.
Tethered Chd1 remodeling nucleosomes
The full-length Chd1 and the KAK mutant proteins has a FLAG tag which can bind to biotinylated anti-FLAG antibody and subsequently tethered to the slide chamber surface. Anti-FLAG tag antibody (Biotin-M2) is obtained through Sigma-Aldrich (St. Louis, MO).
For nucleosome repositioning experiments with tethered proteins, biotinylated anti-FLAG antibody (1:200 dilution) was flow into a slide chamber with the neutravidin surface, then incubated for 5 minutes at room temperature. 2nM of Chd1-WT or Chd1-KAK in nucleosome dilution buffer were then added to the flow chamber and incubated for 1 minutes at room temperature. Then, 50–100pM of non-biotinylated nucleosomes in reaction buffer was added for the confirmation of protein-nucleosome binding (shown as corresponding Cy3 and Cy5 spots on screen), and finally, ATP in reaction buffer was added to the flow chamber to initiate the reaction.
Tethered Chd1 binding to dsDNA
Same full-length Chd1 and KAK mutant proteins with FLAG tag are used for dsDNA binding experiments.
For dsDNA binding experiments, slide chamber surface was prepared as in the case of the tethered protein, with anti-FLAG antibody and neutravidin. 10nM of Chd1-WT or Chd1-KAK were added to the slide chamber surface. Non-biotinylated dsDNA singly labeled with Cy3 ranging from 40 bps to 60 bps were added in concentrations ranging from 50pM to 2nM to observe protein-DNA binding affinity. Bound dsDNA is detected as single Cy3 spots on screen. 1mM ATP or ATPgS is then added (with imaging buffer) to observe any unbinding of dsDNA. (See Supplementary Table S1 for a complete DNA sequence information)
QUANTIFICATION AND STATISTICAL ANALYSIS
Single molecule traces were analyzed customized Matlab functions. FRET efficiency values were calculated as a ratio between acceptor intensity and total intensity.
For dwell time analyses
Peak-to-peak dwell time (δt) for each ATP concentration was collected from multiple FRET traces (>80) using Matlab and fitted to exponential curves using Origin (OriginLab Corporation, Northampton, MA) to obtain the rate (1/δt) for ATP concentrations ranging from 1uM to 1mM. These rates can then be fitted to the Michaelis-Menten equation using the Origin software in order to find Km, the concentration of ATP required for the reaction to reach half of the maximum rate.
For initial rate analyses
Initial FRET drop traces for nucleosome repositioning under various ATP concentrations were collected manually from individual FRET traces using Matlab. These traces are compiled to create average FRET-time traces and fitted using Origin to obtain rate k.
For dsDNA binding analysis
Setting the maximum Cy3 spot density to 600 (>600 = 100%), the number of Cy3 spots at various dsDNA concentration is plotted for each dsDNA-protein combination. These values can then be fitted into a Michaelis-Menten-like curve (Equation (2) above) to obtain binding affinity of dsDNA to different Chd1 protein.
DATA AND SOFTWARE AVAILABILITY
Single Molecule FRET data acquisition and analysis package can all be obtained freely from the website (https://cplc.illinois.edu/software/).
IDL (http://www.exelisvis.co.uk/ProductsServices/IDL.aspx) and Matlab (https://www.mathworks.com/) software with academic or individual licenses can be obtained from their respective software companies.
OriginLab (http://www.originlab.com/) software with academic or individual licenses can be obtained from the software company.
Supplementary Material
HIGHLIGHTS.
Monomeric Chd1 exhibits dynamic shifting of nucleosomal DNA back and forth
Bidirectional sliding by Chd1 entails unstable remodeling intermediates
Limiting the range of the Chd1 DBD interferes with nucleosome sliding
N-terminal chromodomains of Chd1 guard against sliding hexasomes
Acknowledgments
Ilana Nodelman prepared the 603 nucleosomes (4N80) and Chd1-SL protein used in this work. The authors thank members of Myong, Bowman lab and Taekjip Ha lab for helpful discussions. This work was supported by the Human Frontier Science Program (RGP0007/2012), American Cancer Society RSG-12-066-01-DMC, NIH 1DP2GM105453, National Science Foundation and Physics Frontiers Center Program (0822613) through the Center for the Physics of Living Cells to P.Q. and S.M., NIH P41-GM103622 to S. C. and NIH R01-GM084192 to G.D.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Y.Q. performed all single molecule experiments and analyzed data; R.F.L. generated nucleosome and Chd1 reagents, performed histone mapping experiments, and analyzed data; S.C. collected and analyzed SAXS data; A.P. produced nucleosome and Chd1 reagents; G.D.B and S.M. conceived of the project, supervised experiments, analyzed data, and wrote the paper with input from all authors.
References
- Brahma S, Udugama MI, Kim J, Hada A, Bhardwaj SK, Hailu SG, Lee TH, Bartholomew B. INO80 exchanges H2A.Z for H2A by translocating on DNA proximal to histone dimers. Nature communications. 2017;8:15616. doi: 10.1038/ncomms15616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt L, Fuchs S, Krohn A, Masser S, Mader M, Kluth M, Bachmann F, Huland H, Steuber T, Graefen M, et al. CHD1 is a 5q21 tumor suppressor required for ERG rearrangement in prostate cancer. Cancer Res. 2013;73:2795–2805. doi: 10.1158/0008-5472.CAN-12-1342. [DOI] [PubMed] [Google Scholar]
- Dechassa ML, Sabri A, Pondugula S, Kassabov SR, Chatterjee N, Kladde MP, Bartholomew B. SWI/SNF has intrinsic nucleosome disassembly activity that is dependent on adjacent nucleosomes. Mol Cell. 2010;38:590–602. doi: 10.1016/j.molcel.2010.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deindl S, Hwang WL, Hota SK, Blosser TR, Prasad P, Bartholomew B, Zhuang X. ISWI remodelers slide nucleosomes with coordinated multi-base-pair entry steps and single-base-pair exit steps. Cell. 2013;152:442–452. doi: 10.1016/j.cell.2012.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, Luger K. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 2004;375:23–44. doi: 10.1016/s0076-6879(03)75002-2. [DOI] [PubMed] [Google Scholar]
- Fei J, Torigoe SE, Brown CR, Khuong MT, Kassavetis GA, Boeger H, Kadonaga JT. The prenucleosome, a stable conformational isomer of the nucleosome. Genes Dev. 2015;29:2563–2575. doi: 10.1101/gad.272633.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim Y, Minor W, Rastinejad F, Khorasanizadeh S. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- Gaspar-Maia A, Alajem A, Polesso F, Sridharan R, Mason MJ, Heidersbach A, Ramalho-Santos J, McManus MT, Plath K, Meshorer E, et al. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature. 2009;460:863–868. doi: 10.1038/nature08212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkikopoulos T, Schofield P, Singh V, Pinskaya M, Mellor J, Smolle M, Workman JL, Barton GJ, Owen-Hughes T. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science. 2011;333:1758–1760. doi: 10.1126/science.1206097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada BT, Hwang WL, Deindl S, Chatterjee N, Bartholomew B, Zhuang X. Stepwise nucleosome translocation by RSC remodeling complexes. eLife. 2016;5 doi: 10.7554/eLife.10051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauk G, McKnight JN, Nodelman IM, Bowman GD. The chromodomains of the Chd1 chromatin remodeler regulate DNA access to the ATPase motor. Mol Cell. 2010;39:711–723. doi: 10.1016/j.molcel.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H, Kreig A, Calvert J, Lormand J, Kwon Y, Daley JM, Sung P, Opresko PL, Myong S. Telomeric overhang length determines structural dynamics and accessibility to telomerase and ALT-associated proteins. Structure. 2014a;22:842–853. doi: 10.1016/j.str.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H, Opresko P, Myong S. Single-molecule real-time detection of telomerase extension activity. Sci Rep. 2014b;4 doi: 10.1038/srep06391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo C, Ha T. Single-molecule FRET with total internal reflection microscopy. In: Selvin PR, Ha T, editors. Single-molecule techniques: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 2008. pp. 3–35. [DOI] [PubMed] [Google Scholar]
- Kassabov SR, Bartholomew B. Site-directed histone-DNA contact mapping for analysis of nucleosome dynamics. Methods Enzymol. 2004;375:193–210. doi: 10.1016/s0076-6879(03)75013-7. [DOI] [PubMed] [Google Scholar]
- Kelley DE, Stokes DG, Perry RP. CHD1 interacts with SSRP1 and depends on both its chromodomain and its ATPase/helicase-like domain for proper association with chromatin. Chromosoma. 1999;108:10–25. doi: 10.1007/s004120050347. [DOI] [PubMed] [Google Scholar]
- Koh HR, Xing L, Kleiman L, Myong S. Repetitive RNA unwinding by RNA helicase A facilitates RNA annealing. Nucleic Acids Res. 2014;42:8556–8564. doi: 10.1093/nar/gku523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konev AY, Tribus M, Park SY, Podhraski V, Lim CY, Emelyanov AV, Vershilova E, Pirrotta V, Kadonaga JT, Lusser A, et al. CHD1 motor protein is required for deposition of histone variant H3.3 into chromatin in vivo. Science. 2007;317:1087–1090. doi: 10.1126/science.1145339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Kim M, Ahn SH, Zhong G, Kobor MS, Cagney G, Emili A, Shilatifard A, Buratowski S, Greenblatt JF. RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Mol Cell Biol. 2002;22:6979–6992. doi: 10.1128/MCB.22.20.6979-6992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levendosky RF, Sabantsev A, Deindl S, Bowman GD. The Chd1 chromatin remodeler shifts hexasomes unidirectionally. eLife. 2016;5 doi: 10.7554/eLife.21356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Widom J. Nucleosomes facilitate their own invasion. Nat Struct Mol Biol. 2004;11:763–769. doi: 10.1038/nsmb801. [DOI] [PubMed] [Google Scholar]
- Lin JJ, Lehmann LW, Bonora G, Sridharan R, Vashisht AA, Tran N, Plath K, Wohlschlegel JA, Carey M. Mediator coordinates PIC assembly with recruitment of CHD1. Genes Dev. 2011;25:2198–2209. doi: 10.1101/gad.17554711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li M, Xia X, Li X, Chen Z. Mechanism of chromatin remodelling revealed by the Snf2-nucleosome structure. Nature. 2017;544:440–445. doi: 10.1038/nature22036. [DOI] [PubMed] [Google Scholar]
- Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- Lusser A, Urwin DL, Kadonaga JT. Distinct activities of CHD1 and ACF in ATP-dependent chromatin assembly. Nat Struct Mol Biol. 2005;12:160–166. doi: 10.1038/nsmb884. [DOI] [PubMed] [Google Scholar]
- Mathew E, Mirza A, Menhart N. Liquid-chromatography-coupled SAXS for accurate sizing of aggregating proteins. J Synchrotron Radiat. 2004;11:314–318. doi: 10.1107/S0909049504014086. [DOI] [PubMed] [Google Scholar]
- McKnight JN, Jenkins KR, Nodelman IM, Escobar T, Bowman GD. Extranucleosomal DNA binding directs nucleosome sliding by Chd1. Mol Cell Biol. 2011;31:4746–4759. doi: 10.1128/MCB.05735-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty B, Helder S, Silva AP, Mackay JP, Ryan DP. The Chromatin Remodelling Protein CHD1 Contains a Previously Unrecognised C-Terminal Helical Domain. J Mol Biol. 2016;428:4298–4314. doi: 10.1016/j.jmb.2016.08.028. [DOI] [PubMed] [Google Scholar]
- Myong S, Bruno MM, Pyle AM, Ha T. Spring-loaded mechanism of DNA unwinding by hepatitis C virus NS3 helicase. Science. 2007;317:513–516. doi: 10.1126/science.1144130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myong S, Cui S, Cornish PV, Kirchhofer A, Gack MU, Jung JU, Hopfner KP, Ha T. Cytosolic viral sensor RIG-I is a 5′-triphosphate-dependent translocase on double-stranded RNA. Science. 2009;323:1070–1074. doi: 10.1126/science.1168352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myong S, Ha T. Stepwise translocation of nucleic acid motors. Curr Opin Struct Biol. 2010;20:121–127. doi: 10.1016/j.sbi.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myong S, Rasnik I, Joo C, Lohman TM, Ha T. Repetitive shuttling of a motor protein on DNA. Nature. 2005;437:1321–1325. doi: 10.1038/nature04049. [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell. 2013;154:490–503. doi: 10.1016/j.cell.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo TT, Zhang Q, Zhou R, Yodh JG, Ha T. Asymmetric unwrapping of nucleosomes under tension directed by DNA local flexibility. Cell. 2015;160:1135–1144. doi: 10.1016/j.cell.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodelman IM, Bleichert F, Patel A, Ren R, Horvath KC, Berger JM, Bowman GD. Interdomain Communication of the Chd1 Chromatin Remodeler across the DNA Gyres of the Nucleosome. Mol Cell. 2017;65:447–459 e446. doi: 10.1016/j.molcel.2016.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodelman IM, Bowman GD. Nucleosome sliding by Chd1 does not require rigid coupling between DNA-binding and ATPase domains. EMBO Rep. 2013;14:1098–1103. doi: 10.1038/embor.2013.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodelman IM, Horvath KC, Levendosky RF, Winger J, Ren R, Patel A, Li M, Wang MD, Roberts E, Bowman GD. The Chd1 chromatin remodeler can sense both entry and exit sides of the nucleosome. Nucleic Acids Res. 2016;44:7580–7591. doi: 10.1093/nar/gkw406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Myong S, Niedziela-Majka A, Lee KS, Yu J, Lohman TM, Ha T. PcrA helicase dismantles RecA filaments by reeling in DNA in uniform steps. Cell. 2010;142:544–555. doi: 10.1016/j.cell.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Chakravarthy S, Morrone S, Nodelman IM, McKnight JN, Bowman GD. Decoupling nucleosome recognition from DNA binding dramatically alters the properties of the Chd1 chromatin remodeler. Nucleic Acids Res. 2013;41:1637–1648. doi: 10.1093/nar/gks1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, McKnight JN, Genzor P, Bowman GD. Identification of residues in chromodomain helicase DNA-binding protein 1 (Chd1) required for coupling ATP hydrolysis to nucleosome sliding. J Biol Chem. 2011;286:43984–43993. doi: 10.1074/jbc.M111.282970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y, Antony E, Doganay S, Koh HR, Lohman TM, Myong S. Srs2 prevents Rad51 filament formation by repetitive motion on DNA. Nature communications. 2013;4:2281. doi: 10.1038/ncomms3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y, Myong S. Single-Molecule Imaging With One Color Fluorescence. Methods in enzymology. 2016;581:33–51. doi: 10.1016/bs.mie.2016.08.011. [DOI] [PubMed] [Google Scholar]
- Racki LR, Yang JG, Naber N, Partensky PD, Acevedo A, Purcell TJ, Cooke R, Cheng Y, Narlikar GJ. The chromatin remodeller ACF acts as a dimeric motor to space nucleosomes. Nature. 2009;462:1016–1021. doi: 10.1038/nature08621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Hohng S, Ha T. A practical guide to single-molecule FRET. Nature methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A, Wittmeyer J, Cairns BR. Chromatin remodeling through directional DNA translocation from an internal nucleosomal site. Nat Struct Mol Biol. 2005;12:747–755. doi: 10.1038/nsmb973. [DOI] [PubMed] [Google Scholar]
- Schwanbeck R, Xiao H, Wu C. Spatial contacts and nucleosome step movements induced by the NURF chromatin remodeling complex. J Biol Chem. 2004;279:39933–39941. doi: 10.1074/jbc.M406060200. [DOI] [PubMed] [Google Scholar]
- Simic R, Lindstrom DL, Tran HG, Roinick KL, Costa PJ, Johnson AD, Hartzog GA, Arndt KM. Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. EMBO J. 2003;22:1846–1856. doi: 10.1093/emboj/cdg179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ, 3rd, Millhouse S, Chen CF, Lewis BA, Erdjument-Bromage H, Tempst P, Manley JL, Reinberg D. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol Cell. 2007;28:665–676. doi: 10.1016/j.molcel.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolle M, Venkatesh S, Gogol MM, Li H, Zhang Y, Florens L, Washburn MP, Workman JL. Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat Struct Mol Biol. 2012;19:884–892. doi: 10.1038/nsmb.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockdale C, Flaus A, Ferreira H, Owen-Hughes T. Analysis of nucleosome repositioning by yeast ISWI and Chd1 chromatin remodeling complexes. J Biol Chem. 2006;281:16279–16288. doi: 10.1074/jbc.M600682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaramoorthy R, Hughes AL, Singh V, Wiechens N, Ryan DP, El-Mkami H, Petoukhov M, Svergun DI, Treutlein B, Quack S, et al. Structural reorganization of the chromatin remodeling enzyme Chd1 upon engagement with nucleosomes. eLife. 2017;6 doi: 10.7554/eLife.22510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tippana R, Hwang H, Opresko PL, Bohr VA, Myong S. Single-molecule imaging reveals a common mechanism shared by G-quadruplex-resolving helicases. Proc Natl Acad Sci U S A. 2016;113:8448–8453. doi: 10.1073/pnas.1603724113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tippana R, Xiao W, Myong S. G-quadruplex conformation and dynamics are determined by loop length and sequence. Nucleic Acids Res. 2014;42:8106–8114. doi: 10.1093/nar/gku464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Lu X, Wang G, Lan Z, Liao W, Li J, Liang X, Chen JR, Shah S, Shang X, et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature. 2017;542:484–488. doi: 10.1038/nature21357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Persinger J, Kassabov SR, Bartholomew B. Chromatin remodeling by ISW2 and SWI/SNF requires DNA translocation inside the nucleosome. Nat Struct Mol Biol. 2006;13:339–346. doi: 10.1038/nsmb1071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.