

Abstract
Background
Targets with multiple (prerequisite or allosteric) binding sites have an increasing importance in drug design. Experimental determination of atomic resolution structures of ligands weakly bound to multiple binding sites is often challenging. Blind docking has been widely used for fast mapping of the entire target surface for multiple binding sites. Reliability of blind docking is limited by approximations of hydration models, simplified handling of molecular flexibility, and imperfect search algorithms.
Results
To overcome such limitations, the present study introduces Wrap ‘n’ Shake (WnS), an atomic resolution method that systematically “wraps” the entire target into a monolayer of ligand molecules. Functional binding sites are extracted by a rapid molecular dynamics shaker. WnS is tested on biologically important systems such as mitogen-activated protein, tyrosine-protein kinases, key players of cellular signaling, and farnesyl pyrophosphate synthase, a target of antitumor agents.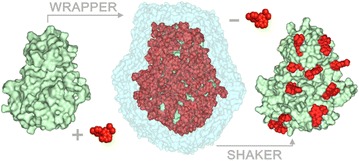
Electronic supplementary material
The online version of this article (10.1186/s13321-017-0255-6) contains supplementary material, which is available to authorized users.
Keywords: Peptide, Search, Pocket, Pharmacodynamics, Water, Interaction, Structure, Complex, Dissociation, Flexibility
Background
Molecular docking complements experimental structure determination and it has become a standard tool of drug discovery for the determination of protein–ligand complex structures [1]. The technique in practice is a compromise between computational cost and accuracy. Its high speed necessitates the use of severe approximations such as (i) restriction of the search space to the surroundings of the binding site, (ii) no or inadequate explicit hydration of the ligand-target interface, (iii) partial or complete neglect of target flexibility [2–5] during ligand binding, (iv) and non-deterministic search algorithms [1, 6] based on random number generation. Approximations i–iv seriously limit the applicability of docking methods for the following reasons. Restriction of the search to a primary binding site requires knowledge of its location and also neglects multiple sites such as allosteric ones [7, 8]. Water molecules often play a role in ligand binding [9–11] and ignoring interfacial water positions during docking may drive the ligands into pockets which are or should be filled with water molecules, resulting in incorrectly docked ligand poses [12]. Potential water release is also important during ligand binding especially through its entropic contributions [13, 14]. Neglecting or limiting the flexibility of target molecules is obviously incorrect at binding situations with induced fit [15]. Eventuality of random number generation in search engines such as Monte-Carlo or genetic algorithms [1, 5, 6] is a natural barrier of the reproducibility and reliability of the results.
The blind docking (BD) approach was introduced [16, 17] to extend the docking search to the entire target surface. In BD, previous knowledge and restriction of the search to a primary binding site are not necessary, and therefore, it can be used in search of multiple binding sites, as well. Indeed, BD has gained popularity [18–20] and has been used for finding allosteric [21–23], or multiple [24–28] binding sites. Thus, BD addresses the above first challenge and performs a global search instead of a focused one at an increased computational cost. However, approximations ii–iv cannot be remediated as simply as the first one. Promising approaches using explicit water molecules in the binding pocket [10] (approximation ii) and treating target flexibility (approximation iii) have been reported for focused docking [29]. However, such approaches have not been implemented in conjunction with solving the global search problem of BD on the entire target surface. Statistical evaluation of multiple docking trials has been shown to increase reproducibility of a BD search [17]; by using multiple randomized (approximation iv) initial ligand positions. Thus, it has become common to perform several docking trials with different initial positions in a BD search to ensure that the largest possible part of the target surface is scanned. However, even such a statistical evaluation cannot guarantee systematic and reproducible exploration of the entire target surface during BD.
Molecular dynamics (MD) simulations have an increasing impact on drug development [30–32]. A series of pioneering studies have reported the use of MD for tracking the ligand binding process [33–37], at atomic resolution. MD calculations also allow the use of explicit water molecules and flexible targets overcoming the above limitations from approximations ii and iii [38–40] potentially opening a new avenue for improvement of BD. MD simulations typically use random starting conformations for the ligands, likewise to BD. Generally, long MD calculation times are required for successful navigation of the ligand into the binding site such that the computational time necessary for accurate docking of a ligand may be prohibitive in practice. Pocket search methods were also developed, exploiting the above-mentioned advantages of MD [41]. A recent review [30] also concludes that “Improper preparation of the initial structure or insufficient equilibration of the initial structure(s) can impact the quality of the MD results”. The present study is aimed at overcoming the above uncertainties of present fast BD and molecular dynamics techniques, by combination of their advantages into a new strategy. Test applications are presented with successful identification of multiple binding sites on biologically important systems such as MAP and tyrosine-protein kinases, key players of cellular signaling as well as farnesyl pyrophosphate synthase, a target of antitumor agents.
Algorithm
Wrap ‘n’ Shake (WnS) is a new method composed of consecutive algorithms, the Wrapper and the Shaker (Fig. 1, Additional file 1: Supporting Movie 1) offering a systematic search for multiple binding sites and modes. WnS works in synergy with popular open source program packages AutoDock 4.2.3 [29] and GROMACS 5.0.2 [42].
Fig. 1.
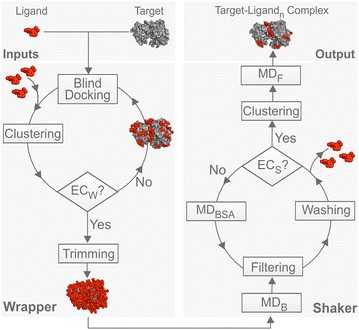
Wrap ‘n’ Shake flowchart featuring the main steps of the method. A quick overview is also presented in Additional file 1: Supporting Movie 1
Wrapper
Wrapper performs several fast BD cycles by AutoDock 4.2, and AutoGrid 4.2 [29] and systematically covers the entire surface of the target with a monolayer of ligand copies (Fig. 1). Each BD cycle is performed as described in Additional file 2: Table S1, and results in 100 docked ligand copies, which are ordered by their interaction energies with the target, and structurally clustered. To achieve a ligand monolayer, the ligand–ligand interactions are minimized through implementation of a weak repulsion between the docked ligand copies, and therefore blocking the formation of ligand aggregates (Additional file 2: Table S2). At the same time, target-ligand interactions are maximized (Additional file 2: Table S3) to ensure that the largest possible numbers of new ligand copies are placed on the surface in an actual BD cycle. The initial experiments (Additional file 2: Table S2) also showed that introduction of a weak repulsion is essential to avoid erroneous ligand geometries clashing with target atoms. Such unwanted clashes (Additional file 2: Table S2) were obtained if intermolecular electrostatic (ECoulomb) and van der Waals (ELJ, Eq. 1) interaction energy terms were simply switched off at the ligand atoms. Notably, calculation of total target-ligand intermolecular interaction energy (Einter) in AutoDock 4.2 is based on the scaled ECoulomb and ELJ terms of the Amber96 force field [43], and an estimate for de-solvation free energy changes (ΔGsol, Eq. 1). ELJ is the sum of Lennard-Jones potential energy values (V, Fig. 2) calculated for all target-ligand atom pairs.
| 1 |
Fig. 2.
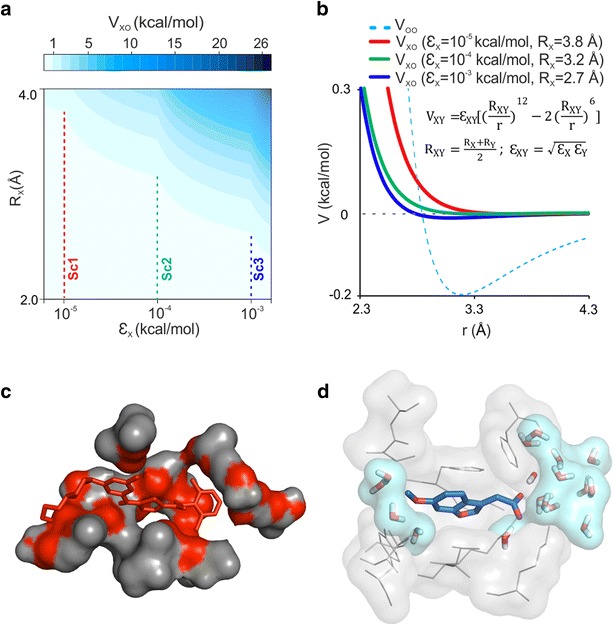
Systematic calibration of εX and RX. a A section of the VXO(r, εX, RX) LJ potential function at r = 2 Å. Scenarios Sc1-Sc3 are shown with the magnitude of the RX values corresponding to a short range repulsion of VXO ≈ 1 kcal/mol (dashed lines) b VXO LJ potential functions of scenarios Sc1-Sc3. VOO of an oxygen atom pair is also shown for comparison. c An example of excluded atoms X (red, Cycle 1, Rank 2, System 9). Docked ligand conformation, is presented with sticks and the binding pocket is shown as surface. d Ligand of System 2 (dark blue sticks) bound to its target farnesyl pyrophosphate synthase (grey lines and surface) after Shaker (MDF step). Explicit water molecules surrounding the ligand within 7 Å are shown with sticks and light blue surface
Finally, instead of the above-mentioned, oversimplified attempt of switching off all intermolecular terms of Einter we elaborated a new protocol which produced the desired ligand monolayer by introduction of an excluded atom type (X). In this protocol, all ligand copies docked in a cycle and their surrounding target atoms are excluded from the next cycle (red in Fig. 2c), and only the unbound target surface (grey) is used for a next BD cycle. The neighboring target atoms are selected by an interface tolerance of 3.5 Å, the maximal distance between a target heavy atom and the closest docked ligand heavy atom. The above exclusion of certain atoms during docking is physically achieved by modification of the non-bonding terms of Einter. For this, the new atom type X is assigned for excluded atoms (red in Fig. 2c) by a C program Wrp developed for this study. Wrp switches off ECoulomb by setting the partial charge of X to zero and also assigns new LJ parameters.
The new LJ parameters were fine-tuned for atom type X in order to produce the necessary weak repulsions described above. Briefly, the LJ parameters of X were calibrated considering the pairwise LJ potential between atom types X and Y (VXY) at three common atom types (Y=O, C and H). A systematic search of both equilibrium potential well-depth (εX, Fig. 2a) and inter-nuclear distance (RX) was conducted. Numerous docking runs were performed to evaluate the effect of the selected LJ parameters. A pre-defined value of r = 2 Å (ca. a covalent bond + 0.5 Å) was used as a minimal distance where short-range repulsion should act at a desired maximal value not exceeding a VXY≈1 kcal/mol. Three scenarios (Sc1-Sc3) were evaluated as shown in the r = 2 Å section of VXO (r, εX, RX) function (Fig. 2a) calculated for the XO atom type pair. Sc2 (green line, Fig. 2a, b) was identified as an optimal scenario with an εX = 10−4 kcal/mol and an RX of 3.2 Å (approximate distance between heavy atoms in an H-bond). In this case, available target surface is optimally used without large ligand-free zones in the monolayer. A short-range repulsion was achieved (green line in Fig. 2b) with a zero value beyond the repulsion zone. If RX was too large (Sc1, red in Fig. 2a, b) then the repulsion zone around the docked ligand copies would also increase with a VXO curve shifted to the right if compared to the green curve of Sc2 (Fig. 2b) resulting in large ligand-free zones, i.e. a non-optimal arrangement of the ligand copies in the monolayer. Importantly, the repulsion zone in the optimal VXO curve of Sc2 starts at lower distances (r) than in the VOO curve. VOO is shifted to the right of the red curve (Sc1), which would result in even larger ligand-free regions than Sc1. Thus, using only a repulsion term of VOO would have not been adequate for exclusions of atoms in wrapping. On the other hand, if RX was too small (Sc3, blue in Fig. 2a, b), then unwanted attractive effects such as aggregation between docked ligand copies would still happen similar to Trial 1, in Additional file 2: Table S2. Accordingly, in Sc3 the corresponding blue curve is shifted to the left from the green Sc2 curve (Fig. 2b). The same procedure was repeated for atom types Y = C and H and an average RX value of 3.6 Å was concluded (Additional file 2: Table S3) and used in Wrapper along with the above εX = 10−4 kcal/mol.
These calibrated LJ parameters of X allowed elimination of the above-mentioned unwanted interactions between the newly docked ligand copies and the already filled binding pockets (Fig. 2c). As the introduced repulsive potential acts on a short range, the ligands can still dock to other, unbound parts of the target surface. The new atom type and parameters also maximize target-ligand interactions adding the maximal number of ligand copies to the mono-layer during a BD cycle.
Wrapper cycles are terminated by either the drop of uncovered surface area of the target below one percent of its total (ligand-free, initial) surface area, or positive target-ligand interaction energy in every cluster representative (ECW in Fig. 1). As a last step, a trimming is performed to remove all ligand copies situated more than 3.5 Å from the target. Wrapper results in a target wrapped in N ligand copies (target-ligandN complex) provided as a single Protein Databank (PDB) file. Wrapper is implemented in a new open source package WnS as shell scripts and a C program Wrp available for download together with a User’s Manual at www.wnsdock.xyz.
Shaker
Shaker selects functional binding sites by removing non-specific, loosely bound ligand copies from the target surface. The target-ligandN complex is placed in a box filled with water and subjected to MD simulations in consecutive cycles. The cycles are performed until a 75% of the ligand copies are eliminated (Exit Criterion of Shaker, ECS Fig. 1). In each Shaker cycle, distance and energy metrics are calculated describing target-ligand interactions at each time step (frame) of a trajectory. The metrics include the closest distances between the target and the ligand as well as ELJ, calculated using Amber parameters. Based on these metrics, filtering (Additional file 2: Table S4) and subsequent removal of the corresponding ligand co-ordinates (Washing, Fig. 1) are applied to exclude ligand positions dissociated from their starting binding positions. The filtering involves two distance-based steps and two final steps based on ELJ.
Before the first cycle a 5-ns target backbone-restrained MD (MDB) is used to grossly shake off the weakly bound ligands. In cases where this initial MD is not enough to reach the required ECS (Additional file 2: Table S1 and Additional file 2: Table S7), multiple cycles with 20-ns simulated annealing (MDBSA) simulations are performed, using position restraints on the target backbone atoms. Depending on the molecular weight (MW, Table 1) of the ligands, SA was done, using two temperature protocols, up to 50 °C (MW ≤ 300) or 80 °C (MW ≥ 300). High temperature in SA accelerated the dissociation process as expected. After MDBSA cycles, a clustering and ranking step is performed, using the last frames of the remaining ligands. A refinement of 20-ns MD with full protein flexibility (MDF) is also performed on every target-ligand complex resulted after clustering (Additional file 2: Table S7 and Additional file 2: Table S8). The Shaker protocol (Additional file 2: Table S9) was formulated during multiple trials (Additional file 2: Tables S5 and S6) and results in a final solution structure of a target-ligandn complex, where n is the total number of final cluster representatives.
Table 1.
Test systems
| # | PDB IDa | Target | Ligand | MWb |
|---|---|---|---|---|
| 1 | 3ptb | bovine β-trypsin | benzamidine | 120 |
| 2 | 3n3 l | farnesyl pyrophosphate synthase | (6-methoxy-1-benzofuran-3-yl) acetic acid (MS0) | 206 |
| 3a | 3hvc | mitogen-activated protein kinase | 4-[3-(4-fluorophenyl)-1 h-pyrazol-4-yl]pyridine (GG5) | 239 |
| 3b | 4f9w | mitogen-activated protein kinase | 4-[3-(4-fluorophenyl)-1 h-pyrazol-4-yl]pyridine (GG5) | 239 |
| 4 | 3cpa | carboxy-peptidase | GY | 256 |
| 5 | 1qcf | haematopoetic cell kinase (HCK) | 1-ter-butyl-3-p-tolyl-1 h-pyrazolo[3,4-d]pyrimidin- 4-ylamine (PP1) | 281 |
| 6 | 1h61 | pentaerythritol tetranitrate reductase | Prednisone® | 358 |
| 7 | 2bal | mitogen-activated protein kinase | [5-amino-1-(4- Fluorophenyl)-1H-Pyrazol-4- yl] [3-(piperidin-4-yloxy) phenyl]methanone | 380 |
| 8 | 1hvy | thymidylate synthase | Ralitrexed® | 459 |
| 9 | 3g5d | tyrosine-protein kinase Src | Dasatinib® | 488 |
| 10 | 1be9 | PDZ-domain | KQTSV | 544 |
aPDB ID of the holo X-ray structure
bMolecular weight of the ligand
Systems and test metrics
A diverse set of ten target-ligand systems were selected (Table 1) and prepared (Additional file 2: Table S1) as test cases of WnS. Challenging systems with multiple (prerequisite or allosteric) binding sites were included (Table 1). Our selection contains both small ligands and bulky, flexible ones. Apo protein structures were used as targets except System 8. In the case of System 5 another protein tyrosine-protein kinase was used as apo structure similar to a previous study [33].
Three standard metrics were used to quantify the results of tests. (1) root mean squared deviation (RMSD) measures structural precision of WnS results by comparison of atomic positions of ligand conformations produced by WnS and those of the crystallographic reference. Prior to calculation of RMSD, a structural alignment (Additional file 2: Table S10) was performed on the holo and apo target residues surrounding the ligand within 5 Å similarly to a previous work [33]. (2) Shaker Rate (SR = N/n) is a ratio of counts of the N ligand copies residing on the target surface (N) after Wrapper and the n final cluster representatives (n) produced by Shaker. The larger the SR, the more efficiently Shaker eliminated ligand copies from the target surface. (3) Rank serial number (#Rank) is calculated using relative ligand-target interaction energies corresponding to the docked ligand positions. WnS ranks docked ligand copies by their interaction energies with the target. The smaller the #Rank, the stronger the target-ligand interaction is at a ligand position. The #Rank of the docked ligand copy of the lowest RMSD is listed for all systems in Table 2. In the final rank lists, docked ligand copies with small RMSD, i.e. close to the crystallographic conformations should be preferably placed at the top of the rank lists, with small #Rank values.
Table 2.
Results for the test systems
| # | Na | CLSb | #Rankc | nd | SRe | |
|---|---|---|---|---|---|---|
| MDBSA | MDF | |||||
| 1a | 68 | 6 | 1 | 1 | 6 | 11 |
| 1bg | 74 | 5 | 1 | – | 4 | 19 |
| 1cg | 71 | 6 | 1 | – | 5 | 14 |
| 2 | 300 | 18 | 2 | 4 | 13 | 23 |
| 3a | 222 | 46 | 3 | 4 | 21 | 11 |
| 3b | 222 | 46 | 9 | 12 | 21 | 11 |
| 4 h | 155 | 12 | 1 | 1 | 8 | 19 |
| 5 | 143 | 25 | 2 | 1 | 12 | 12 |
| 6i | 116 | 26 | 1 | 2 | 12 | 10 |
| 7 | 123 | 26 | 4 | 4 | 12 | 10 |
| 8 | 106 | 25 | 1 | 1 | 10 | 11 |
| 9j | 92 | 23 | 2 | 1 | 10 | 9 |
| 10 | 49 | 11 | 2 | 1 | 4 | 12 |
aTotal count of ligand copies after Wrapper
bCount of ligands surviving the Shaker, after MDBSA
cRank serial number of the structure with the best RMSD value, after MDBSA and after MDF
dCount of cluster representatives (final solutions) Shaker
eShaker Rate
fTotal computational time required for MDB, MDBSA and MDF, as explained in Additional file 2: Table S12
gFor System 1, WnS was performed with different seeds for data reproduction purposes
hFinal clustering was done using van der Waals and Coulomb interactions due to interactions of zinc ion with the ligand
iWrapper process was done, using the LJ interaction as a scoring function, instead of AD4 (Additional file 2: Table S13)
jFinal clustering was done with 6 Å distance limit between clusters
Results and discussion
Association or dissociation?
Encouraged by results of pioneering MD studies [31, 33, 34], association of ligand benzamidine to bovine trypsin was followed in three MD simulations. Benzamidine is an easy case for docking and it has also been used in tests of recent approaches [44]. The present MD simulations were 1-µs-long and benzamidine was placed at three different starting positions (Fig. 3, Additional file 2: Table S11), at various distances (Fig. 3a) from the crystallographic binding site.
Fig. 3.
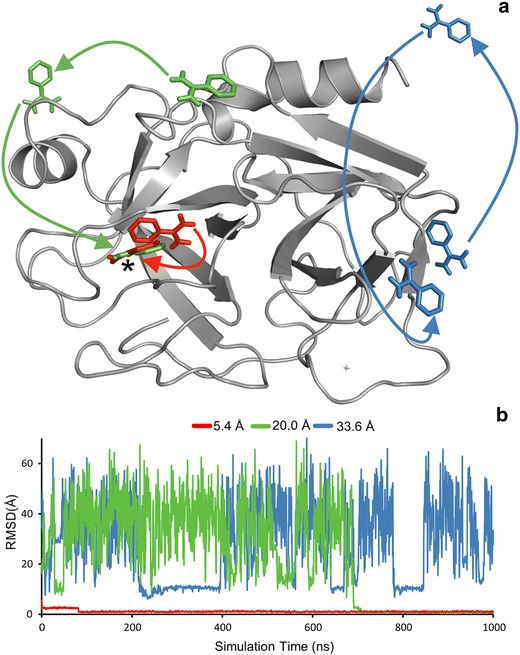
Pilot molecular dynamics simulations. Benzamidine ligand (sticks) started the MD simulations from three positions at different distances (as indicated in the legend) from the native binding site on the trypsin target (grey cartoon). Arrows in a point from starting (t = 0 ns) to final (t = 1000 ns) ligand positions. Only two of the three 1000 ns-long simulations with the closest starting position succeeded in finding the reference binding pose (*) known from the crystallographic structure (3ptb). b Time-dependence of root mean squared deviation (RMSD) of the ligand measured from its reference pose
Analysis of the trajectories shows that the crystallographic binding position was found in two out of the three simulations after 81 and 690 ns simulation time (drop of red and green lines in Fig. 3b), respectively. In the 3rd case with the largest starting distance, 1 µs was not enough to dock to the native site by association (blue line). Thus, the usefulness of association MD runs for docking strongly depends on the starting ligand position even in the easy case of benzamidine. MD needs a simulation time comparable to the real association time of the ligand (Fig. 3b). This can be considerable, as migration of the ligand is hindered by friction in the surrounding water. Previous studies [33, 36, 45], have also reported simulations of several hundreds of nanoseconds for navigation of the ligand to the desired binding pocket.
All-in-all, the necessary time for successful docking by association MD depends on the actual starting position of the ligand, the size and shape of the target, ligand etc. To overcome such uncertainties on simulation length and still use the benefits of MD we elaborated a new strategy, the Wrap ‘n’ Shake (WnS, Fig. 1). Instead of simulating the association process, WnS is based on the dissociation of the ligand. Dissociation is fast and reproducible at binding sites of low stability.
A systematic approach
Naturally, a dissociation approach requires a set of ligand copies bound to the target. Systematic mapping of all possible ligand positions (sites) cannot be guaranteed in a single BD cycle (Introduction) even if it contains hundreds of fast BD trials [17]. A truly systematic algorithm should completely wrap the entire surface of the target in a monolayer of copies of the ligand molecule. Our initial guess of such a Wrapper algorithm was based on a previous finding [17] that the coverage of the target can be increased with several, successive fast BD cycles where accumulated docked ligand copies from the previous cycle are considered as part of the target in the next cycle. However, additional experiments with such successive BD cycles showed that previously and newly docked ligand copies can easily form multi-layer aggregates with each-other instead of the target (Additional file 2: Table S2). The formation of such aggregates hinders wrapping of the target surface into the desired monolayer of ligand copies.
During the wrapping process, parts of the target surface already covered with ligand copies has to be excluded from interactions with ligand copies docked in a next BD cycle. This task is not trivial as potential functions of the docking force fields normally cannot distinguish between target sites unbound and covered with ligands. After extensive experimentation including an optimization of the force field (“Wrapper” section, Additional file 2: Table S3, Appendix 1) we arrived at a new algorithm called Wrapper (Figs. 2, 4). Wrapper performs a systematic coverage of the target surface in several, consecutive fast blind docking cycles (Fig. 4). The algorithm continuously monitors the status of coverage of target surface (Fig. 4a) and results in the desired monolayer of N ligand copies not interacting with each-other. Figure 4b shows an example of such a monolayer. Ligands constituting the monolayer have physically realistic arrangement (Fig. 4c), maximized interactions with the target and no contacts with each-other. Thus, the target is systematically and rapidly wrapped in a monolayer of N (Table 2) ligands.
Fig. 4.
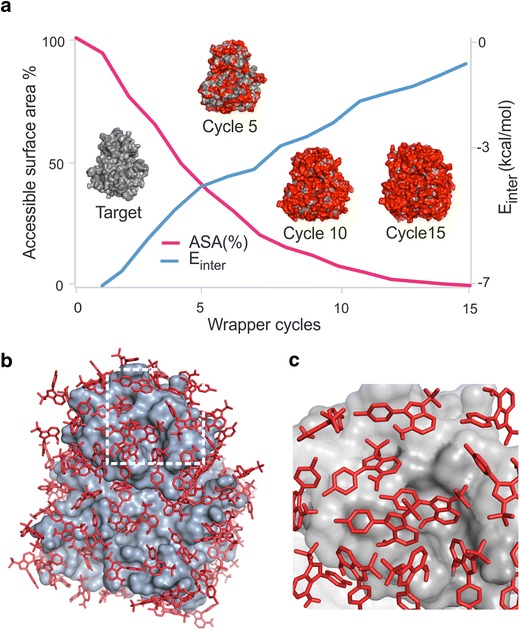
Wrapping tyrosine-protein kinase Src target into a mono-layer of ligand copies (System 5). a Unbound (ligand free) accessible surface area (ASA) of the target and the lowest Einter of the cluster representatives in consecutive wrapping cycles. Target-ligand interaction energy (Einter) increases with increasing number of cycles finding strong binding sites in the first few cycles, before the final, saturation region. ASA finally decreases below 1%. Structural images show the wrapping of the target (grey surface) with ligands (red). b The monolayer arrangement of the ligands (red sticks) wrapping the entire target surface (grey) after the final cycles. c A close-up of a section of the monolayer showing that the ligand copies are evenly arranged without overlap
Having a realistic input geometry, the resulting target-ligandN complex is transferred to the Shaker including MD simulation(s) with explicit water (“Shaker” section), filtering, and clustering steps. These steps eliminate ligands dissociated during MD and result in a strong binder at each pocket (Additional file 2: Table S7). Final results are shown in Table 2 using test metrics described in “Systems and test metrics”. Parameter SR characterizes efficiency of removal of loose binders. SR values of Table 2 indicate that a considerably large part of the weak binders were efficiently removed at all test systems beyond the default ECS of 75% (SR = 4). Other important metrics are RMSD and #Rank. In most of the systems analyzed, ligand conformations with the lowest RMSD were placed into the first two ranks (Table 2, Fig. 5, and Additional file 2: Table S8). For stable ligand copies, good structural matches to the corresponding reference conformations (Fig. 5 and Additional file 2: Table S8), as well as low #Rank values (Table 2) were found. Fair results were obtained for challenging cases too (Systems 2 and 3). The somewhat lower rank in these cases may be explained by the relatively high B-factor of the ligands of these systems (Additional file 2: Table S1) suggesting an increased mobility and a less stable target-ligand interaction.
Fig. 5.
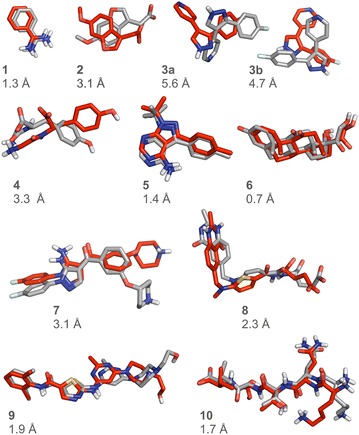
Structural fits quantified as root mean squared deviation (RMSD) with values given in Å. Ligand conformations after Shaker (grey) compared to the crystallographic references (red sticks). System# is bold
For example, B-factors of measured atomic positions of ligand MSo (System 2) vary in a range between 54 and 95 Å2 (Additional file 2: Table S1). During MDF simulations we found that the RMSD varied between 2.5 and 5.1 Å (Additional file 2: Table S8), and a final #Rank of 4 and an RMSD of 3.1 Å were obtained. Considering the above high B-factor values, it is realistic to assume that ligand MSo adopts various conformations when bound to farnesyl phosphate synthase (System 2) including the one close to the assigned position found with an RMSD of 2.5 Å. This conformational variability of the bound MSo is probably due to its carboxylate group with the highest B-factor of 95 Å2. This group is hydrated by bulk water molecules, helping the dissociation of MSo from the target (Fig. 2d). At the same time, MD simulations with explicit water molecules also account for a hydrophobic, anchoring interaction between the benzofuran part of MSo (no waters present, Fig. 2d) and the target. This example shows the necessity of use of explicit water model during the shaking process in order to account for all, even antagonistic interactions.
In our pilot study (“Association or dissociation?” section) it was demonstrated that MD methods following the association pathways often need large amount of computational time and/or a fortunate starting conformation in order to find the primary site correctly for System 1. WnS yielded the correct solution for this system (Additional file 2: Table S8) in a 5-ns-long MDB simulation which is at least one order of magnitude shorter than the lengthy association times discussed in “Association or dissociation?” section. Elimination of ligand excess (dissociation of ligand copies) (Tables S14 and S15) at an SR of 11 was facilitated by hydrogen bonding with explicit water molecules [46, 47]. Thermal motion of water molecules also contributed to fast “shake off” of the ligand copies especially in the cases of Systems with small ligands with the application of the simulated annealing protocol (MDBSA, see an SR of 23 in case of System 2 in Table 2).
A case with a small ligand
WnS was tested on tyrosine protein kinase target with a pyrazolopyrimidine 1 ligand (PP1, System 5). Regulation of kinase activity is important in numerous human diseases [48, 49]. At the same time, this kinase is a challenging test target for WnS as it has multiple sites including an allosteric one identified in previous studies [50, 51]. The native, PP1 site was found (Fig. 5) at an excellent RMSD agreement (1.4 Å, Fig. 5) with the crystallographic position. Besides obtaining very good RMSD (Fig. 5), the #Rank was improved from second to first place (Table 2) during the final MDF simulation (Additional file 2: Table S16). Apart from the primary site, our goal was to find other, prerequisite binding sites, as well. As described in a previous MD study [33], such sites correspond to poses on the binding pathway leading to the primary site. WnS found both low- and high energy prerequisite sites described previously [33] (Fig. 6). Besides structural matches, #Rank and the corresponding energy values are also comparable to the previous results. Notably, WnS can predict multiple binding sites beyond experimentally observable ones. These binding sites can be considered as prerequisite or allosteric binding sites. Previous MD results [33, 52] concluded, that finding prerequisite binding sites is a substantial advantage of the MD simulations.
Fig. 6.
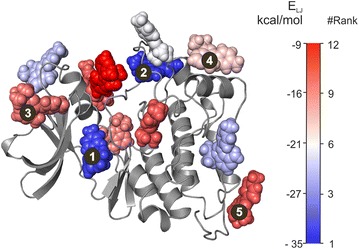
Haematopoetic cell kinase (HCK, System 5) with ligand copies remaining after Shaker. Ligand copies are colored by the calculated target-ligand interaction energy E, and the #Rank assigned. The previously reported pockets 1(ATP), 2(A-loop), 3(PIF site), 4(G-loop) and 5(MYR) are numbered by their increasing ELJ
Cases with large ligands
Tyrosine kinase also binds dasatinib (System 9), a bulky ligand, for which an SR of 9 was obtained (Table 2), after six simulated annealing cycles (Additional file 2: Table S12). The same four binding pockets were found for dasatinib as for the above PP1 (Additional file 2: Table S17). After the final MDF step, local conformational refinement of dasatinib was observed, improving the RMSD from 2.3 to 1.9 Å. Similar to PP1, this could be partially explained by the role of the water molecules and the enhanced target motion during MDBSA. WnS was further tested on the challenging System 10 with a pentapeptide ligand with twenty-three flexible torsions. The correct binding position of the ligand was obtained after the MDF stage of Shaker with an improvement of RMSD from 6.8 to 1.7 Å (Fig. 7, Additional file 3: Supporting Movie 2).
Fig. 7.
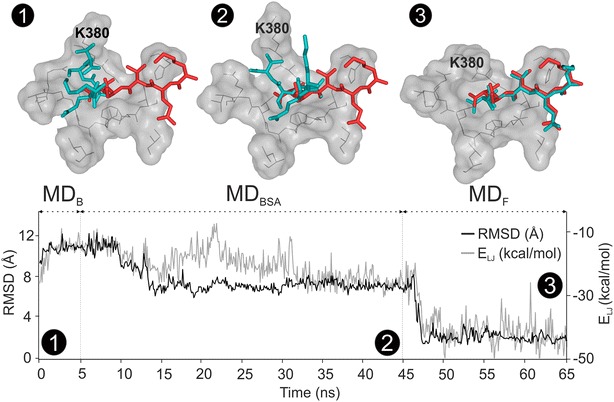
During Shaker, conformational changes of the pentapeptide KQTSV are observed, while remains bound to its pocket on the PDZ domain (System 10). Red sticks represent the native ligand conformation from PDB (1be9). Teal sticks represent ligand conformations at different Shaker stages starting with the conformation right after Wrapper (1), and continuing with conformation after MDBSA (2), and after MDF (3). The changes of target-ligand interaction energy (ELJ) and the RMSD during the MD stages in the Shaker protocol are plotted below the structural snapshots. See also Additional file 3: Supporting Movie 2 for further details of conformational changes
A re-ranking (Table 2) from Rank 2 to Rank 1 was also observed after MDF. For comparison, the wrapped target-ligandN complex of System 10 was subjected directly to an MDF simulation skipping the MDB and MDBSA steps of Shaker. In this case, an RMSD of 11.3 Å (Line 10b in Additional file 2: Table S8) was obtained which was worse than the RMSD obtained with the complete Shaker protocol (1.7 Å, Fig. 5). This demonstrates that both MDB and MDBSA steps of Shaker are necessary to find the correct position. After Wrapper, the pentapeptide was in a closed, cyclic conformation (Fig. 7, Snapshot 1). This unrealistic arrangement was opened up (Snapshots 2 and 3) by interacting water molecules. It can be also observed that limited protein flexibility during MDB and MDBSA allowed only moderate reduction of the ligand RMSD by improvement of the target-ligand interactions. Most of the RMSD and interaction energy improvement was achieved after MDF, and rearrangement of K380 inside the pocket was necessary, to improve the conformation of the simulated ligand (Fig. 7). All-in-all, MD steps including target flexibility have a significant influence on the results of WnS for large ligands. Introduction of MDF considerably improved structural precision, in the above case studies of large ligands (Systems 9 and 10).
Conclusions
In the present study, a systematic strategy, the Wrap ‘n’ Shake was introduced for exploration of multiple binding sites and modes of drugs on their macromolecular targets. Wrap ‘n’ Shake systematically wraps the target into a monolayer of ligand copies using a modified blind docking approach and selects stable positions by shaking off loose binders. The method offers a computationally feasible solution for the present problems of the field (Introduction). Wrapper requires only fast blind docking cycles with a program package such as AutoDock 4.2.3. The Shaker process is fairly short and can be performed by available MD packages. Shaker is further accelerated by simulated annealing and uses all benefits of explicit water model and target flexibility. Wrap ‘n’ Shake is suitable to study interactions of protein targets with even large peptide ligands. We have started the extension of the method towards protein ligands using a fragment-based approach with post hoc reconstruction of the ligand. In future applications, Wrap ‘n’ Shake could be also used for general pocket search, besides docking of individual ligands. We envision that Wrap ‘n’ Shake can become the tool of choice for systematic exploration of multiple binding sites and modes of ligands in drug design and structural biology.
Additional files
Additional file 1. Supporting Movie 1 featuring the processes of Wrapper and Shaker in the case of System 5. The first part presents the results of 15 wrapping cycles. The second part contains MDB and two MDBSA cycles of Shaker. Final cluster representatives are the outputs of WnS. Additional refinement steps are shown in Supporting Movie 2 (Additional file 3).
Additional file 2. Supporting Tables S1–S17 and Appendix 1–4 with detailed methods and results.
Additional file 3. Supporting Movie 2 featuring conformational changes of pentapeptide KQTSV, bound to PDZ-domain (System 10) during 65 ns simulations performed Shaker. The binding pocket of KQTSV on the PDZ domain is presented with grey surface. The simulated and crystallographic reference structures of KQTSV are presented as teal and red sticks.
Authors’ contributions
MB, CH, NJ, and IH performed research. MB, CH, and DvdS designed research, and wrote the manuscript. All authors formulated the research. All authors read and approved the final manuscript.
Acknowledgements
We acknowledge a grant of computer time from CSCS Swiss National Supercomputing Centre, and NIIF Hungarian National Information Infrastructure Development Institute. We acknowledge that the results of this research have been achieved using the DECI resource Archer based in the UK at the National Supercomputing Service with support from the PRACE aisbl.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
A software package is released under the GNU GPL, freely accessible with examples and a manual at http://www.wnsdock.xyz.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
The work was supported by the K123836, K112807, K120391 grants from the National Research, Development, and Innovation Office, Hungary. The University of Pécs is acknowledged for a grant PTE ÁOK_KA/2017 and also support in the frame of “Viral Pathogenesis” Talent Centre program. We are thankful to the Gedeon Richter Pharmaceutical Plc. for a pre-doctoral scholarship to N.J.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- BD
blind docking
- ECW
exit criterion of Wrapper
- ECS
exit criterion of Shaker
- Einter
intermolecular interaction energy
- ELJ
Lennard-Jones potential
- MDB
molecular dynamics with backbone position restraints
- MDBSA
molecular dynamics with backbone position restraints and simulated annealing
- MDF
molecular dynamics without restraints (flexible simulation
- PDB
protein data bank
- RMSD
root mean squared deviation
- SR
Shaker Rate
- WnS
Wrap ‘n’ Shake
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13321-017-0255-6) contains supplementary material, which is available to authorized users.
Contributor Information
Mónika Bálint, Email: monibalint18@gmail.com.
Norbert Jeszenői, Email: jeszenoi.norbert@gmail.com.
István Horváth, Email: horvathi@gmx.de.
David van der Spoel, Email: david.vanderspoel@icm.uu.se.
Csaba Hetényi, Email: csabahete@yahoo.com.
References
- 1.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 2.Fischer M, Coleman RG, Fraser JS, Shoichet BK. Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat Chem. 2014;6:575–583. doi: 10.1038/nchem.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hou XB, Li KS, Yu X, Sun JP, Fang H. Protein flexibility in docking-based virtual screening: discovery of novel lymphoid-specific tyrosine phosphatase inhibitors using multiple crystal structures. J Chem Inf Modeling. 2015;55:1973–1983. doi: 10.1021/acs.jcim.5b00344. [DOI] [PubMed] [Google Scholar]
- 4.Pan AC, Borhani DW, Dror RO, Shaw DE. Molecular determinants of drug–receptor binding kinetics. Drug Discov Today. 2013;18:667–673. doi: 10.1016/j.drudis.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Halperin I, Ma BY, Wolfson H, Nussinov R. Principles of docking: an overview of search algorithms and a guide to scoring functions. Proteins Struct Funct Genet. 2007;47:409–443. doi: 10.1002/prot.10115. [DOI] [PubMed] [Google Scholar]
- 6.Brooijmans N, Kuntz ID. Molecular recognition and docking algorithms. Annu Rev Biophys Biomol Struct. 2003;32:335–373. doi: 10.1146/annurev.biophys.32.110601.142532. [DOI] [PubMed] [Google Scholar]
- 7.Iorga B, Herlem D, Barre E, Guillou C. Acetylcholine nicotinic receptors: finding the putative binding site of allosteric modulators using the “blind docking” approach. J Mol Modeling. 2006;12:366–372. doi: 10.1007/s00894-005-0057-z. [DOI] [PubMed] [Google Scholar]
- 8.Othman R, Kiat TS, Khalid N, Yusof R, Newhouse EI, Newhouse JS, et al. Docking of noncompetitive inhibitors into dengue virus type 2 protease: understanding the interactions with allosteric binding sites. J Chem Inf Modeling. 2008;48:1582–1591. doi: 10.1021/ci700388k. [DOI] [PubMed] [Google Scholar]
- 9.Mancera RL. Molecular modeling of hydration in drug design. Curr Opin Drug Discov Dev. 2007;10:275–280. [PubMed] [Google Scholar]
- 10.Jeszenoi N, Bálint M, Horváth I, Van Der Spoel D, Hetényi C. Exploration of interfacial hydration networks of target–ligand complexes. J Chem Inf Modeling. 2016;56:148–158. doi: 10.1021/acs.jcim.5b00638. [DOI] [PubMed] [Google Scholar]
- 11.Jeszenoi N, Horvath I, Balint M, van der Spoel D, Hetenyi C. Mobility-based prediction of hydration structures of protein surfaces. Bioinformatics. 2015;31:1959–1965. doi: 10.1093/bioinformatics/btv093. [DOI] [PubMed] [Google Scholar]
- 12.Hetenyi C, van der Spoel D. Toward prediction of functional protein pockets using blind docking and pocket search algorithms. Protein Sci. 2011;20:880–893. doi: 10.1002/pro.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmad M, Helms V, Lengauer T, Kalinina OV. Enthalpy–entropy compensation upon molecular conformational changes. J Chem Theory Comput. 2015;11:1410–1418. doi: 10.1021/ct501161t. [DOI] [PubMed] [Google Scholar]
- 14.Ahmad M, Kalinina O, Lengauer T. Entropy gain due to water release upon ligand binding. J Cheminform. 2014;6(1):P35. doi: 10.1186/1758-2946-6-S1-P35. [DOI] [Google Scholar]
- 15.Rastelli G, Pacchioni S, Sirawaraporn W, Sirawaraporn R, Parenti MD, Ferrari AM. Docking and database screening reveal new classes of plasmodium falciparum dihydrofolate reductase inhibitors. J Med Chem. 2003;46:2834–2845. doi: 10.1021/jm030781p. [DOI] [PubMed] [Google Scholar]
- 16.Hetenyi C, van der Spoel D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 2002;11:1729–1737. doi: 10.1110/ps.0202302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hetenyi C, van der Spoel D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006;580:1447–1450. doi: 10.1016/j.febslet.2006.01.074. [DOI] [PubMed] [Google Scholar]
- 18.Grinter SZ, Zou X. Challenges, applications, and recent advances of protein-ligand docking in structure-based drug design. Molecules. 2014;19:10150–10176. doi: 10.3390/molecules190710150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuriev E, Holien J, Ramsland PA. Improvements, trends, and new ideas in molecular docking: 2012–2013 in review. J Mol Recognit. 2015;28:581–604. doi: 10.1002/jmr.2471. [DOI] [PubMed] [Google Scholar]
- 20.Yuriev E, Ramsland PA. Latest developments in molecular docking: 2010–2011 in review. J Mol Recognit. 2013;26:215–239. doi: 10.1002/jmr.2266. [DOI] [PubMed] [Google Scholar]
- 21.Hocker HJ, Rambahal N, Gorfe AA. LIBSA—a method for the determination of ligand-binding preference to allosteric sites on receptor ensembles. J Chem Inf Model. 2014;54:530–538. doi: 10.1021/ci400474u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schindler CE, de Vries SJ, Zacharias M. Fully blind peptide-protein docking with pepATTRACT. Structure. 2015;23:1507–1515. doi: 10.1016/j.str.2015.05.021. [DOI] [PubMed] [Google Scholar]
- 23.Whalen KL, Tussey KB, Blanke SR, Spies MA. Nature of allosteric inhibition in glutamate racemase: discovery and characterization of a cryptic inhibitory pocket using atomistic MD simulations and pK(a) calculations. J Phys Chem B. 2011;115:3416–3424. doi: 10.1021/jp201037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia-Sosa AT, Sild S, Maran U. Design of multi-binding-site inhibitors, ligand efficiency, and consensus screening of avian influenza H5N1 wild-type neuraminidase and of the oseltamivir-resistant H274Y variant. J Chem Inf Modeling. 2008;48:2074–2080. doi: 10.1021/ci800242z. [DOI] [PubMed] [Google Scholar]
- 25.Roumenina L, Bureeva S, Kantardjiev A, Karlinsky D, Andia-Pravdivy JE, Sim R, et al. Complement C1q-target proteins recognition is inhibited by electric moment effectors. J Mol Recognit. 2007;20:405–415. doi: 10.1002/jmr.853. [DOI] [PubMed] [Google Scholar]
- 26.Bugatti A, Giagulli C, Urbinati C, Caccuri F, Chiodelli P, Oreste P, et al. Molecular interaction studies of HIV-1 matrix protein p17 and heparin: identification of the heparin-binding motif of p17 as a target for the development of multitarget antagonists. J Biol Chem. 2013;288:1150–1161. doi: 10.1074/jbc.M112.400077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kovacs M, Toth J, Hetenyi C, Malnasi-Csizmadia A, Sellers JR. Mechanism of blebbistatin inhibition of myosin II. J Biol Chem. 2004;279:35557–35563. doi: 10.1074/jbc.M405319200. [DOI] [PubMed] [Google Scholar]
- 28.Agarwal T, Annamalai N, Khursheed A, Maiti TK, Bin Arsad H, Siddiqui MH. Molecular docking and dynamic simulation evaluation of Rohinitib—Cantharidin based novel HSF1 inhibitors for cancer therapy. J Mol Graph Modelling. 2015;61:141–149. doi: 10.1016/j.jmgm.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 29.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ganesan A, Coote ML, Barakat K. Molecular dynamics-driven drug discovery: leaping forward with confidence. Drug Discov Today. 2017;22:249–269. doi: 10.1016/j.drudis.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Dror RO, Dirks RM, Grossman J, Xu H, Shaw DE. Biomolecular simulation: a computational microscope for molecular biology. Annu Rev Biophys. 2012;41:429–452. doi: 10.1146/annurev-biophys-042910-155245. [DOI] [PubMed] [Google Scholar]
- 32.Durrant JD, McCammon JA. Molecular dynamics simulations and drug discovery. BMC Biol. 2011;9:71. doi: 10.1186/1741-7007-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shan Y, Kim ET, Eastwood MP, Dror RO, Seeliger MA, Shaw DE. How does a drug molecule find its target binding site? J Am Chem Soc. 2011;133:9181–9183. doi: 10.1021/ja202726y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buch I, Giorgino T, De Fabritiis G. Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc Natl Acad Sci USA. 2011;108:10184–10189. doi: 10.1073/pnas.1103547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Limongelli V, Bonomi M, Parrinello M. Funnel metadynamics as accurate binding free-energy method. Proc Natl Acad Sci USA. 2013;110:6358–6363. doi: 10.1073/pnas.1303186110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shan Y, Gnanasambandan K, Ungureanu D, Kim ET, Hammaren H, Yamashita K, et al. Molecular basis for pseudokinase-dependent autoinhibition of JAK2 tyrosine kinase. Nat Struct Mol Biol. 2014;21:579–584. doi: 10.1038/nsmb.2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jensen MØ, Jogini V, Borhani DW, Leffler AE, Dror RO, Shaw DE. Mechanism of voltage gating in potassium channels. Science. 2012;336(6078):229–233. doi: 10.1126/science.1216533. [DOI] [PubMed] [Google Scholar]
- 38.Borhani DW, Shaw DE. The future of molecular dynamics simulations in drug discovery. J Comput Aided Mol Des. 2012;26:15–26. doi: 10.1007/s10822-011-9517-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casasnovas R, Limongelli V, Tiwary P, Carloni P, Parrinello M. Unbinding kinetics of a p38 MAP kinase type II inhibitor from metadynamics simulations. J Am Chem Soc. 2017;139:1480–4788. doi: 10.1021/jacs.6b12950. [DOI] [PubMed] [Google Scholar]
- 40.Kuzmanic A, Sutto L, Saladino G, Nebreda AR, Gervasio FL, Orozco M. Changes in the free-energy landscape of p38α MAP kinase through its canonical activation and binding events as studied by enhanced molecular dynamics simulations. eLife. 2017;6:e22175. doi: 10.7554/eLife.22175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prakash P, Hancock JF, Gorfe AA. Binding hotspots on K-ras: consensus ligand binding sites and other reactive regions from probe-based molecular dynamics analysis. Proteins Struct Funct Bioinform. 2015;83:898–909. doi: 10.1002/prot.24786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 43.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, et al. A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J Am Chem Soc. 1995;117:5179–5197. doi: 10.1021/ja00124a002. [DOI] [Google Scholar]
- 44.Soderhjelm P, Tribello GA, Parrinello M. Locating binding poses in protein-ligand systems using reconnaissance metadynamics. Proc Natl Acad Sci USA. 2012;109:5170–5175. doi: 10.1073/pnas.1201940109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan YB, et al. Pathway and mechanism of drug binding to G-protein-coupled receptors. Biophys J. 2012;102:410. doi: 10.1016/j.bpj.2011.11.2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van der Spoel D, van Maaren PJ, Larsson P, Timneanu N. Thermodynamics of hydrogen bonding in hydrophilic and hydrophobic media. J Phys Chem B. 2006;09(110):4393–4398. doi: 10.1021/jp0572535. [DOI] [PubMed] [Google Scholar]
- 47.Schmidtke P, Luque FJ, Murray JB, Barril X. Shielded hydrogen bonds as structural determinants of binding kinetics: application in drug design. J Am Chem Soc. 2011;133:18903–18910. doi: 10.1021/ja207494u. [DOI] [PubMed] [Google Scholar]
- 48.Cohen P. Protein kinases—the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 49.Shukla D, Meng Y, Roux B, Pande VS. Activation pathway of Src kinase reveals intermediate states as targets for drug design. Nat Commun. 2014;5:3397. doi: 10.1038/ncomms4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Foda ZH, Seeliger MA. Kinase inhibitors: an allosteric add-on. Nat Chem Biol. 2014;10:796–797. doi: 10.1038/nchembio.1630. [DOI] [PubMed] [Google Scholar]
- 51.Sadowsky JD, Burlingame MA, Wolan DW, McClendon CL, Jacobson MP, Wells JA. Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proc Natl Acad Sci USA. 2011;108:6056–6061. doi: 10.1073/pnas.1102376108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tiwary P, Limongelli V, Salvalaglio M, Parrinello M. Kinetics of protein-ligand unbinding: predicting pathways, rates, and rate-limiting steps. Proc Natl Acad Sci USA. 2015;112:E386–E391. doi: 10.1073/pnas.1424461112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Supporting Movie 1 featuring the processes of Wrapper and Shaker in the case of System 5. The first part presents the results of 15 wrapping cycles. The second part contains MDB and two MDBSA cycles of Shaker. Final cluster representatives are the outputs of WnS. Additional refinement steps are shown in Supporting Movie 2 (Additional file 3).
Additional file 2. Supporting Tables S1–S17 and Appendix 1–4 with detailed methods and results.
Additional file 3. Supporting Movie 2 featuring conformational changes of pentapeptide KQTSV, bound to PDZ-domain (System 10) during 65 ns simulations performed Shaker. The binding pocket of KQTSV on the PDZ domain is presented with grey surface. The simulated and crystallographic reference structures of KQTSV are presented as teal and red sticks.
Data Availability Statement
A software package is released under the GNU GPL, freely accessible with examples and a manual at http://www.wnsdock.xyz.