

Abstract
DNA methylation (DNAm) has been found to show robust and widespread age-related changes across the genome. DNAm profiles from whole blood can be used to predict human aging rates with great accuracy. We sought to test whether DNAm-based predictions of age are related to phenotypes associated with type 2 diabetes (T2D), with the goal of identifying risk factors potentially mediated by DNAm. Our participants were 43 women enrolled in the Women’s Health Initiative. We obtained methylation data via the Illumina 450K Methylation array on whole blood samples from participants at three timepoints, covering on average 16 years per participant. We employed the method and software of Horvath, which uses DNAm at 353 CpGs to form a DNAm-based estimate of chronological age. We then calculated the epigenetic age acceleration, or Δage, at each timepoint. We fit linear mixed models to characterize how Δage contributed to a longitudinal model of aging and diabetes-related phenotypes and risk factors. For most participants, Δage remained constant, indicating that age acceleration is generally stable over time. We found that Δage associated with body mass index (p = 0.0012), waist circumference (p = 0.033), and fasting glucose (p = 0.0073), with the relationship with BMI maintaining significance after correction for multiple testing. Replication in a larger cohort of 157 WHI participants spanning 3 years was unsuccessful, possibly due to the shorter time frame covered. Our results suggest that DNAm has the potential to act as a mediator between aging and diabetes-related phenotypes, or alternatively, may serve as a biomarker of these phenotypes.
Electronic supplementary material
The online version of this article (10.1007/s11357-017-0001-z) contains supplementary material, which is available to authorized users.
Keywords: Aging, diabetes, DNA methylation, BMI, biomarker, biological age
Introduction
Worldwide, the population aged 65 years and older is growing rapidly, with a 150% expansion projected over the next few decades (He et al. 2016). Despite these recent global gains in life expectancy, age-related disease burden and the incidence of chronic disabilities remain high (Burch et al. 2014). The healthspan, or years spent in good health, among the aging population remains highly variable, with some maintaining good health throughout their lives while others fall ill (Kennedy et al. 2014). Age itself is the leading risk factor for the development of most diseases and conditions that drive morbidity and mortality and contribute to limited healthspan (Kaeberlein et al. 2015; Kennedy et al. 2014). In many countries, age-related diseases like cardiovascular disease, diabetes, cancer, and neurodegenerative disorders are among the predominant health problems faced by the population (Niccoli and Partridge 2012).
A particularly widespread age-related disease adversely impacting the healthspan of millions worldwide is type 2 diabetes (T2D), which is now considered a global epidemic (International Diabetes Federation 2015). Due to population growth, increased longevity, and urbanization (which can promote physical inactivity and an unhealthy diet) (Hu 2011), the global burden of T2D is expected to worsen over time as the prevalence increases from 415 million living with the disease in 2015 to an estimated 642 million in 2040 (International Diabetes Federation 2015; Shaw et al. 2010). There are many well-documented risk factors associated with the development of T2D, including weight gain (Ford et al. 1997), high body mass index (BMI) (Chan et al. 1994), high waist circumference (Koh-Banerjee et al. 2004), ethnicity (Shai et al. 2006), smoking status (Hu et al. 2001), high fasting glucose (Nathan et al. 2007), high fasting insulin (Weyer et al. 2000), and age (Mokdad et al. 2003; Stamler et al. 1993). Diabetes contributed to approximately 5 million deaths globally in 2015 (International Diabetes Federation 2015) and is itself a risk factor for numerous other comorbidities. Globally, ~ 50% of diabetic individuals are unaware of their condition, and subsequently are unaware of their increased risk of diabetes-related complications. Thus, a better marker of early T2D risk could provide mechanistic insights and facilitate earlier identification of high-risk individuals most likely to benefit from targeted lifestyle interventions (International Diabetes Federation 2015).
Differential susceptibility to age-related diseases can be attributed to biological differences between individuals, which work to modify disease risk (reviewed in Feinberg 2007). Among these biological differences are epigenetic changes, which arise without changes to the underlying DNA sequence and have the potential to modify disease risk through their regulatory influence on gene expression (Goldberg et al. 2007). Additionally, because the major risk factors for T2D are lifestyle factors, such as diet and exercise behavior (Pan et al. 1997), an epigenetic mechanism in which these factors can modify underlying genetic predisposition to disease incidence is highly plausible. DNA methylation (DNAm), the presence of a methyl group on the cytosine within a CpG dinucleotide, is the most studied epigenetic modification. The robust and genome-wide changes to DNAm observed with age make it an ideal biomarker of aging (Alisch et al. 2012; Bollati et al. 2009; Christensen et al. 2009; Teschendorff et al. 2010; Xu and Taylor 2014). Biomarkers of aging are indicators of the biological age of an organism that predict its physiological functioning and disease susceptibility better than its chronological age alone (Baker and Sprott 1988; Johnson 2006). Recently, highly accurate biomarkers of aging have been developed that capitalize on age-related changes to DNAm at a subset of CpGs across the genome to predict chronological age (Hannum et al. 2013; Horvath 2013). The approach of Horvath (Horvath 2013) uses methylation data from just 353 CpGs to form a multi-tissue, DNAm-based estimate of chronological age (DNAm age). Using DNAm age as a measure of biological age, the difference between a participants’ DNAm age and their chronological age can be calculated. This measure is termed the participants’ epigenetic age acceleration (Δage) and may proxy for the general health or rate of aging of the individual (Horvath 2013). Instances in which the Δage term is positive indicate an epigenetic age that is higher than the participant’s chronological age.
Many studies support the hypothesis that epigenetic Δage is associated with negative health outcomes, including increased risk of premature mortality (Chen et al. 2016; Christiansen et al. 2016; Marioni et al. 2015a; Perna et al. 2016; Zheng et al. 2016), early onset of age-related disease (Breitling et al. 2016; Levine et al. 2015), and changes in physical and cognitive fitness (Marioni et al. 2015b). These findings indicate that Δage contributes more predictive information about these health outcomes than chronological age alone. This is consistent with the possibility that Δage may be acting to mediate the health outcome or risk of disease onset, but also with the possibility that DNAm age may be marking another biological process that is acting as a mediator. Consistent with the adverse health outcomes associated with positive Δage, a negative Δage can predict positive outcomes: centenarians in an Italian population and their offspring tended to have a DNAm age that was lower than their chronological age (Horvath et al. 2015). Taken together, these results support that epigenetics can be important in predicting both negative health outcomes and healthy aging.
Previous studies (Hidalgo et al. 2014; Nilsson et al. 2014; Ronn and Ling 2015) have reported associations between site-specific methylation differences and T2D as well as related phenotypes across several cell types and tissues. Our study aims to assess the potential of 5mC as a mediator between aging and age-related T2D risk phenotypes. To model age-related 5mC patterns, we focus on a well-studied methylation-based biomarker of aging (Horvath 2013) which identified 353 CpG sites as being the most predictive in modeling chronological age. We take advantage of a longitudinal study spanning 16 years to (1) characterize the changes to participants’ Δage over time, and (2) characterize the contribution of DNAm age and Δage in modeling T2D susceptibility. Given that many T2D risk factors (including high BMI, waist circumference, and fasting glucose and insulin levels) reflect age-related changes, a measure of biological aging may help predict which participants are at a higher risk of T2D incidence throughout the study. Though we do not have the power to model incidence of clinical T2D in our sample, the longitudinal nature of this study allows us to model changes to phenotypes intermediate between age and disease risk. We will use the DNAm-based measure of biological age as a proxy for genome-wide DNAm and other age-related biological processes that may underlie age-related disease risk. We aim to inform future studies by assessing the utility of genome-wide methylation changes and other biological processes as potential mediators between age and risk factors for and indicators of T2D (subsequently referred to as “diabetes-related phenotypes”).
Methods
Study population and study design
Participants are a subsample from the 68,132 women who took part in the Women’s Health Initiative (WHI) Clinical Trials (CT) Cohort. The WHI was a national study which sought to investigate interventions and treatments for the prevention and management of common causes of morbidity and mortality among older women (The Women’s Health Initiative Study Group 1998). All WHI participants were post-menopausal women, aged 50 to 79 years at the time of enrollment, with minority women recruited at the same proportion found in the US population at the time (Hays et al. 2003). Women in the WHI were also more likely to be overweight, with three quarters of the women overweight or obese at the time of enrollment (Hays et al. 2003).
The study began in 1993 with participants completing questionnaires detailing their sociodemographic information (including their age and race), current health behaviors (including weekly physical activity and smoking behavior), and current health status (any disease diagnosis and medications or supplements currently prescribed). Participants also attended scheduled clinic visits in which anthropometric measurements were assessed, including the following: weight, height, and waist circumference; from these measures, body mass index was calculated. Additionally, a 6% minority oversample of participants had blood drawn during these clinic visits from which insulin, glucose, triglyceride, and high-density lipoprotein cholesterol concentrations were measured and buffy coat was archived. “Epigenetic Mechanisms of PM-Mediated CVD Risk” (WHI-EMPC) measured DNAm on a genome-wide scale using DNA extracted from the archived buffy coat in a stratified, random sample (N = 2200) of the participants who were examined between 1993 and 2001. Among a subset (N = 200) of the 2200 participants, WHI-EMPC also measured DNAm in buffy coat archived at a second timepoint on average 3.3 years later. Subsequently, a “Longitudinal Study of DNA Methylation as a Mediator between Age and Cardiovascular Risk” (AS #534) measured DNAm in buffy coat archived at the third timepoint, on average 16.1 years after the first, for a subset (N = 43) of the 200 participants who were followed up as part of the Long Life Study (LLS). These 43 participants are included in our study and described in Table 1.
Table 1.
Demographic and clinical characteristics of study population (N = 43)
| Variable | Mean +/− SD or percentage | |||
|---|---|---|---|---|
| Baseline (SD) | Follow-up (SD) | LLS (SD) | # missing obs. | |
| Chronological age (years) | 61.52 (6.94) | 65.05 (6.77) | 77.48 (6.48) | 0 |
| DNAm age (years) | 58.05 (8.05) | 60.19 (6.93) | 72.85 (7.92) | 0 |
| Δage (years) | − 3.47 (4.36) | − 4.86 (4.47) | − 4.56 (5.20) | 0 |
| BMI (kg/m2) | 29.02 (5.23) | 29.32 (4.47) | 28.34 (5.88) | 4 |
| Fasting glucose (mg/dL) | 94.88 (8.47) | 95.47 (14.22) | 100.19 (18.76) | 0 |
| Fasting insulin (μIU/mL) | 12.00 (5.21) | 13.04 (7.58) | 18.94 (15.10)a | 6 |
| HOMA-IR | 2.82 (1.23) | 3.17 (2.37) | 5.01 (4.56)a | 6 |
| TG/HDL-C ratio | 3.09 (1.65) | 2.92 (1.91) | 2.08 (1.19) | 1 |
| TyG index | 8.85 (0.47) | 8.84 (0.46) | 8.57 (0.49) | 0 |
| Waist circumference (cm) | 87.64 (12.29) | 88.87 (11.55) | 89.20 (13.37) | 10 |
aLLS insulin measures were obtained from a different analyzer from the baseline and follow-up measures and units were converted from picomole/liter to micro IU/milliliter. We observe higher values and standard errors for this measure. These observed differences between timepoints could reflect a true increase in fasting insulin with age, or could be due to differences in measurement
Data cleaning
Chronological age of the participants was approximated at each timepoint as participant’s self-reported age at screening (in years) + 0.5 + number of days between screening and blood sampling / 365.25. Phenotypic measures include the following: BMI measured as weight (kg) divided by the square of height (m2), waist circumference (cm), fasting glucose (mg/dL), and fasting insulin (μIU/mL). Homeostasis Model Assessment of Insulin Resistance, termed HOMA-IR, was calculated using the following equation: Insulin (μU/mL) × Glucose (mg/dL) / 405 (Yokoyama et al. 2003). The ratio of plasma triglycerides (mg/dL) to high-density lipoprotein cholesterol concentration (mg/dL), termed TG/HDL-C ratio, was calculated (McLaughlin et al. 2003). Lastly, the triglyceride-glucose index, termed the TyG index, was calculated using the following equation: ln[Triglycerides / (Fasting glucose / 2)] (Simental-Mendía et al. 2008). Both TG/HDL-C and TyG were included as markers of insulin resistance.
Three unrealistic data points believed to be entered in error were removed. These included BMI measures below 15 kg/m2, or above 55 kg/m2; these values were > 2 SD away from the participant’s mean throughout the study and were flanked by more moderate values measured within 4 years. Additionally, a waist measurement above 150 cm was removed, as it was 1.7 SD from the participant’s mean and was flanked by more moderate measurements within 6 years. Phenotypic data collected from within 30 days of a blood draw were assumed to approximate data that would have been collected at the time of the draw. Additionally, several waist circumference measurements originally recorded in inches were converted to centimeters, with 1 in. equivalent to 2.54 cm. Insulin measures at the first and second timepoints were ascertained using different but similar methods. All insulin testing for the first timepoint used the radioimmunoassay (RIA) method. For some participants, the second timepoint used an automated ES300 analyzer. Because ES300 and RIA methods gave comparable results at insulin levels below 60 μIU/mL, and because all participants had insulin levels below this cutoff for the first two timepoints, the insulin results were combined into a single variable. The method for measuring insulin concentrations changed again for the third timepoint with the Roche Elecsys 2010 Immunoassay analyzer being used. Measures from the third timepoint were recorded in picomole/liter and were converted to micro IU/milliliter, with 6 pmol/L equivalent to 1 μIU/mL (Heinemann 2010). Self-reported smoking behavior, originally recorded as “Never Smoked,” “Past Smoker,” and “Current Smoker,” was recoded to “Never Smoked” and “Smoked” due to only one participant being classified as a “Current Smoker.”
Alcohol intake, total caloric intake, and family history of diabetes were self-reported at the start of the study. Alcohol intake reported was weekly intake of alcoholic beverages. This includes the number of servings per weeks of beer, wine, and/or liquor based on a serving size of 12 oz for beer, 6 oz for wine, and 1.5 oz for liquor. Entries ranged from 0 to 12.4 drinks per week (mean = 1.5) with missing data for one participant. Total caloric intake was reported in kilocalories per day, ranging from 660.1 to 3455.2 (mean = 1487.4) with data missing for one participant. Participants whose energy intake estimates suggested that they were not properly completing the food frequency questionnaire (i.e., those with daily intake less than 600 kcal or greater than 3500 kcal) were excluded (N = 2) (Patterson et al. 1999). In characterizing family history, participants were asked: “Did your mother, or father, or full-blooded sisters, full-blooded brothers, daughters, or sons ever have sugar diabetes or high blood sugar that first appeared as an adult?” Participants’ responses were as follows: “Yes” (11 participants), “No” (31 participants), or “Unsure” (1 participants). For the model, participants who answered either “No” or “Unsure” were combined into “No or Unsure.” Incident diabetes and incident diabetes treatment occurring within the study period were also characterized as part of the sensitivity analysis. Incident diabetes was defined, according to standards set by the American Diabetes Association (Association 2016), as anyone who fasted for eight or more hours and has a glucose measure ≥ 126 mg/dL, or anyone who fasted for fewer than 8 h and has a glucose measure ≥ 200 mg/dL (4 participants). Timepoints occurring after a participant indicated they were prescribed medication to treat diabetes were considered incident treatment with an antidiabetic agent (4 participants).
DNA methylation data
DNA was extracted from buffy coat from participants at each timepoint. DNA (500 ng) was used for the bisulfite conversion with the EZ-96 DNA Methylation Kit (Zymo Research, Irvine, CA, USA), following the manufacturer’s protocol. Once converted and amplified, DNA (15 μL) was fragmented, and hybridized to the Infinium HumanMethylation450 Bead Chip (Illumina Inc., CA, USA). DNAm profiles of > 485,000 cytosine-guanine (CpG) sites were measured using the Infinium HumanMethylation450 BeadChip at the Northwestern University Genomics Core Facility in two batches, with the first two timepoints run as part of WHI-EMPC and the third run as part of AS #534. DNA methylation was subject to quality controls: excluding probes targeting CpG sites on the Y chromosome, probes with detection p values > 0.01 in > 10% of samples, and samples with detection p values > 0.01 across in > 1% of probes; 484,220 CpG sites passed this quality control step and were eligible for further analysis. Two control DNA samples on each BeadChip were used to assess reproducibility, and duplicates from the first batch were run with the second to account for batch effects. Methylated (M) and unmethylated (U) signals were used to compute estimates of the methylation proportion, β-values, (β = M/(U + M)). Next, beta-mixture quantile normalization (BMIQ) was performed to reduce technical variation and intra-array bias between differing types of probes (Teschendorff et al. 2013). Lastly, ComBat, which employs an empirical Bayes method to adjust for batch effects, was used to adjust for differences between the two batches (Leek et al. 2012).
Measures of DNA methylation age and Δage
DNAm age at each timepoint was calculated using the methylation profiles from 353 CpGs and the R pipeline detailed in Horvath (2013). The difference between DNAm predicted age and the chronological age of each participant at each of the three timepoints, termed “age acceleration” (Δage), was calculated at each point.
Testing for association between age and DNA methylation
Using the R package CpGassoc (Barfield et al. 2012), we performed an epigenome-wide association study (EWAS) to test for association between chronological age and DNAm. For each CpG site, we fit a linear mixed model that included a random effect for each participant to account for the repeated measures within participants, and self-reported ethnicity and Illumina chip and row as covariates.
Testing for association between Δage and diabetes-related phenotypes
Phenotypes analyzed included seven diabetes-related phenotypes: fasting insulin and glucose, HOMA-IR, BMI, waist circumference, TG/HDL-C ratio, and TyG index. Using the R package nlme (Pinheiro et al. 2016), we fit longitudinal, mixed effect models with the phenotype as the outcome and a random effect for participants. For each phenotype, two models were fit: the first regressed each phenotype on chronological age and relevant covariates, while the second regressed each phenotype on both chronological age and Δage, in addition to other covariates. Our goal was to assess whether the additional term Δage associates independently with the phenotype, indicating that Δage contributes to our ability to model the phenotype. Thus, for participant (i) at timepoint (j), the following models were fit:
Model 1:
Model 2:
where ∆ageij represents age acceleration for individual i at time j, ν i represents a random effect (individual-specific error term) for individual i, and ε ij represents the error term for individual i and timepoint j. Significance of the age acceleration coefficient γ in the second model was taken to suggest that the relationship between chronological age and that phenotype could potentially be mediated by methylation or a related biological process, or that Δage could serve as a biomarker for this phenotype. To adjust for potential confounding, ethnicity, cigarette smoking, and fasting hours (where relevant) were included as covariates. Sensitivity analysis were performed with several well-known T2D risk factors added individually as covariates, including total energy expenditure, total caloric intake, alcohol intake, and family history of diabetes.
Estimation of blood cell proportions based on DNA methylation
A complication in analysis of whole blood samples in aging studies is that cell proportions in whole blood change with age (Fagnoni et al. 2000; Houseman et al. 2012), and different subpopulations of blood cells feature different methylation patterns (Reinius et al. 2012). Together, these can confound the relationship between DNAm and aging, since it is difficult to distinguish DNAm changes in whole blood with age from DNAm changes in blood with disease development if the model does not explicitly account for differences in cell proportions (Adalsteinsson et al. 2012). Houseman’s regression-based method (Houseman et al. 2012) was used to estimate the composition of white blood cells in whole blood using DNAm array data. This tool uses DNAm data from 500 CpGs found to be most informative of white blood cell (WBC) type in whole blood. The tool constrains the sum of the six blood type proportions (CD4+ helper T cells, CD8+ cytotoxic T cells, granulocytes, monocytes, natural killer cells, B cells) to 100%, then fits a regression model to the DNAm data at the 500 sites. This allows for the estimation of the six WBC proportions, which were then included as covariates in the comparisons of Models 1 and 2, with granulocyte proportions excluded as the reference category.
Testing for change in Δage over time
A mixed effects model, with the year of the participant’s clinic visit as a fixed effect and a random effect for participants, was used to test whether there was significant change in Δage over time.
Results
Sample characteristics
The sample characteristics of our population are detailed in Table 1. Participants were 43 post-menopausal women, between 50 and 76 years of age at enrollment, with a mean age of 61.5 years (sd = 6.9). A plurality of our sample was non-Hispanic White (41.9%), about a third were African American (32.6%), and about a quarter were Hispanic or Latino (25.6%). Self-reported smoking behavior indicated that 23 participants (54.8%) were either current or previous smokers; 19 participants report having never smoked (45.2%), while one participant failed to respond. Longitudinal DNAm data were available for three timepoints with the second and third timepoints occurring on average 3.3 and 16.1 years after the first, respectively. At baseline, none of the participants were being treated for diabetes. The distributions of the seven diabetes-related phenotypes in our population are shown in Supplementary Fig. 1.
DNA methylation changes with chronological age
Using DNAm array data, we performed a longitudinal epigenome-wide association study as proof of concept that many CpGs display differential methylation associated with participants’ estimated chronological ages, a pattern which has been well-established in many other datasets (e.g., Alisch et al. 2012; Bollati et al. 2009; Christensen et al. 2009; Teschendorff et al. 2010; Xu and Taylor 2014). In our data, 232 sites showed significant changes with age according to the Holm step-down Bonferroni procedure (p < 1.0E − 7), while 3064 sites were found significant by the Benjamini–Hochberg procedure (FDR < .05). Top CpGs are listed in Supplementary Table 1. Supplementary Fig. 2 features a Manhattan plot of p values reflecting the association between methylation and chronological age. Our results appear consistent with those reported by Xu and Taylor (2014), who identified 749 high confidence age-related CpGs in > 1000 individuals. Supplementary Fig. 3 demonstrates a high correlation (r = 0.74) between t-statistics across the two studies. Additionally, 11 of our significant sites overlap with the 353 CpGs that make up the epigenetic clock (Horvath 2013).
DNA methylation age estimates over time
Participants’ chronological ages show high correlation with their predicted DNAm ages (r = 0.89) (Fig. 1). The difference between this predicted age and the chronological age of each participant at each of the three timepoints, termed Δage, is calculated at each point. DNAm age at enrollment ranges from 43.2 to 84.5, while Δage, at enrollment ranges from − 12.3 to 9.0. The median Δage value across participants is − 4.5. Δage is negative for 109 of the 129 measurements (84.5%), which is consistent with previous reports showing that women tend to have lower Δage than men (Hannum et al. 2013; Horvath et al. 2016). The average Δage at the first timepoint is − 3.5 (sd = 4.4), − 4.9 (sd = 4.5) at the second timepoint, and − 4.6 (sd = 5.2) at the third timepoint (Table 1, Supplementary Fig. 4). According to a Shapiro-Wilk normality test, Δage is normally distributed at timepoints 1 (p = 0.16) and 2 (p = 0.87), but not timepoint 3 (p = 0.0033). However, with the removal of a single individual with an extreme Δage, values for timepoint 3 are consistent with a normal distribution (p = 0.94).
Fig. 1.
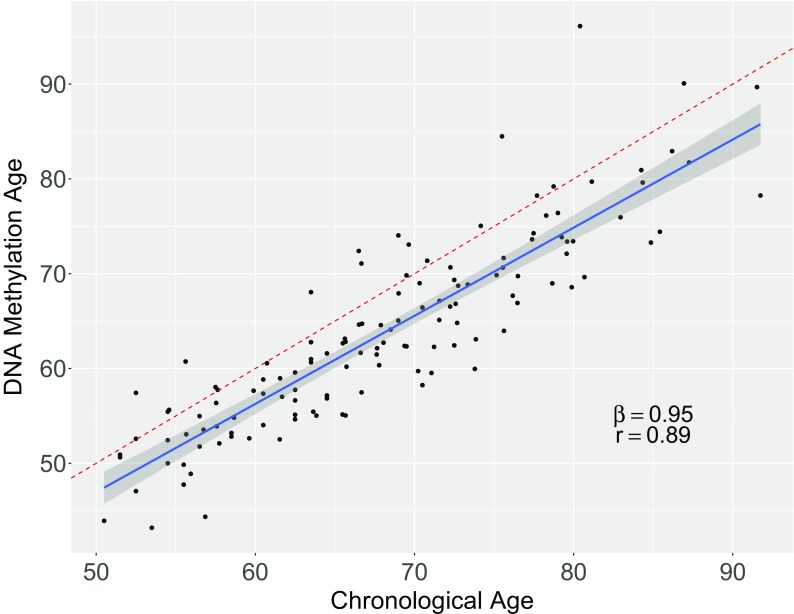
Chronological Age (x-axis) vs. DNA methylation Age (y-axis). Each point shows chronological age and DNAm age for one participant at one of the three timepoints. The dotted red line represents the equivalence line, meaning there would be perfect agreement between the computed DNAm age and the approximated chronological age. The blue line represents the regression line obtained from a regression of DNAm age on chronological age with random effects to account for repeated measures within subjects. The shaded gray region around the blue line represents a 95% confidence interval of the regression line
Δage is not significantly associated with smoking status (p = 0.51) in our data. It is also not significantly associated with chronological age (r = − 0.14, p = 0.13) (Supplementary Fig. 5), though the negative correlation is consistent with previous reports (Chen et al. 2016; Christiansen et al. 2016; Marioni et al. 2015b). It does vary by ethnicity, with the Hispanic/Latino group having a smaller Δage, but this difference is not statistically significant in our sample (p = 0.39). This observation agrees with recent findings that Hispanic/Latina women participating in the WHI study have a lower Δage compared to WHI Caucasians (Horvath et al. 2016), though our study did not have power to detect a significant difference.
Stability of Δage
Within individuals, very little change in Δage is observed over time, suggesting that the value of age acceleration remains roughly constant over time among our participants (Fig. 2). On average, Δage showed a 0.041 decrease each year, which does not differ significantly from a change of zero (p = 0.25) (Supplementary Fig. 6). To identify individuals whose Δage changed significantly during the study, each of the 43 participants’ DNAm age was regressed on their chronological age. The mean slope of this regression was close to 1 (mean = 0.96, SD = 0.29), suggesting that on average, DNAm age increases at a similar rate to chronological age. Five participants (10, 26, 27, 33, and 34) were at least 1.5 standard deviations from the mean, with slope values of 0.46, 1.41, 0.52, 1.71, and 2.02, respectively. To assess whether these changes in Δage could be influenced by changes in blood cell proportions, we regressed each of six estimated cell type proportions onto the year of the participant’s visit, and found that cell proportions did not change significantly over the course of the study (Supplementary Fig. 7).
Fig. 2.
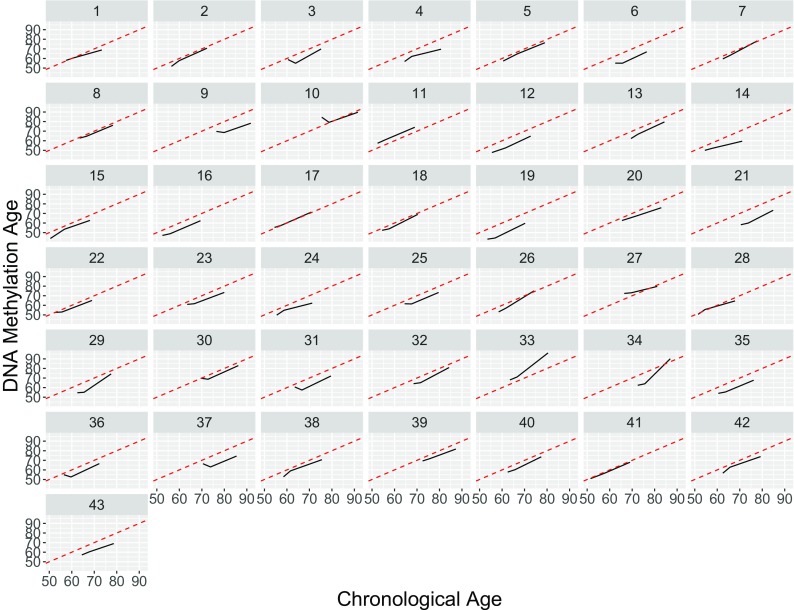
Chronological Age (x-axis) vs. DNA methylation Age (y-axis) for each participant. Each subplot represents one participant; a solid black line connects the participants’ three measures of Δage across the three timepoints. The dotted red line represents a line of slope = 1, reflecting perfect agreement between DNAm age and chronological age. A participant’s black line being nearly parallel to the dotted red line indicates little change in Δage within a participant over the course of the study, while a black line with a slope other than 1 would reflect changes in Δage over time. Figure 1 provides a composite view of these data combined across all 43 subjects
DNAm age acceleration associates with several diabetes-related phenotypes
Results from our models of diabetes-related phenotypes are listed in Table 2. Δage has a significant positive association with fasting glucose (p = 0.0073), BMI (p = 0.0012), and waist circumference (p = 0.033). Using a Bonferroni-corrected α of 0.0071 to adjust for the seven phenotypes tested, the association remains significant for BMI and near-significant for glucose. To assess the robustness of our results to inclusion of covariates, we performed sensitivity analyses that added the following covariates to the model: alcohol intake, total caloric intake, family history of diabetes, incident diabetes during follow-up, and incident treatment with antidiabetic agents. Supplementary Table 2 shows that the addition of each covariate produces similar results to our baseline model. Furthermore, inclusion of a covariate for participants taking medication for incident diabetes suggests that, in addition to Δage contributing significantly to modeling of BMI, it also contributes significantly (p < .0071) to modeling fasting glucose among our participants.
Table 2.
Multivariate regression analysis of diabetes-related phenotypes on age and biological age acceleration
| Phenotype | Model 1 Coefficients on chronological age |
Model 2 Coefficients on chronological age |
Model 2 Coefficients on Δage |
|||
|---|---|---|---|---|---|---|
| Est. (SE) | p value | Est. (SE) | p value | Est. (SE) | p value | |
| BMI | − 0.046 (0.030) | 0.13 | − 0.032 (0.029) | 0.27 | 0.29 (0.087) | 0.0012* |
| Fasting glucose | 0.24 (0.14) | 0.081 | 0.30 (0.13) | 0.027 | 0.97 (0.34) | 0.0073 |
| Fasting insulin | 0.28 (0.10) | 0.0078 | 0.30 (0.10) | 0.0050* | 0.26 (0.24) | 0.28 |
| HOMA-IR | 0.084 (0.028) | 0.0043* | 0.091 (0.029) | 0.0022* | 0.089 (0.065) | 0.18 |
| TG/HDL-C ratio | − 0.051 (0.015) | 0.0013* | − 0.048 (0.016) | 0.0029* | 0.048 (0.040) | 0.24 |
| TyG index | − 0.014 (0.0045) | 0.0023* | − 0.013 (0.0046) | 0.0055* | 0.021 (0.012) | 0.073 |
| Waist circumference | 0.10 (0.077) | 0.18 | 0.13 (0.076) | 0.082 | 0.48 (0.22) | 0.033 |
The model includes the following covariates for the 43 participants: ethnicity, smoking history, age, and estimated cell type proportions. p values marked with an asterisk (*) are significant at our Bonferroni-corrected α of 0.0071
Supplementary Fig. 8 reflects measurements of BMI over the 16-year study period for our participants. Of the five participants with extreme Δage slope values, three participants (10, 34, and, to a lesser extent, 27) also had extreme changes in BMI during the study. This BMI fluctuation could, perhaps, be linked to changes in DNAm and Δage. To test whether the relationship between Δage and BMI, fasting glucose, and waist circumference were driven by these five participants, we removed them in a sensitivity analysis. Supplementary Table 3 includes the results of this analysis in which it appears that our findings are driven by the participants with dynamic Δage, since the effect sizes decrease substantially upon their removal compared to the original results in Table 2. This loss of an association with the removal of the most dynamic participants suggests that the association may be driven by within-person changes in Δage and BMI, rather than static differences between individuals.
Replication study in a second WHI subsample
A subset of 200 women from a stratified, random sample of 2200 WHI-CT participants had two DNAm measurements assessed as part of WHI-EMPC. Our 43 participants with three DNAm timepoints are part of this subset of 200; we attempt to replicate our findings in the remaining 157 participants who had two DNAm timepoints on average 3.7 years apart. The replication cohort’s ethnic make-up is fairly similar to our participants, with 55.4% non-Hispanic, White, 19.6% Black or African American, 15.92% Hispanic/Latino, 4.46% Asian or Pacific Islander, 3.18% American Indian or Alaska Native, and 1.27% Other. Smoking behavior had a high rate of missingness (85.7% of participants did not provide data on their smoking habits), and thus was not included in regression models. The sample characteristics of our replication population are detailed in Supplementary Table 4. The replication cohort mirrored our finding of female participants having lower DNAm age than their chronological age (mean Δage is − 4.30 years in our data and − 3.87 in the replication cohort). However, while the correlation between Δage and chronological age was not significant in our analysis of 43 participants (r = − 0.14, p = 0.13), analysis of this larger sample yielded a significant negative correlation (r = − 0.20, p = 3.9E − 6, Supplementary Fig. 9).
Results of the regression of diabetes-related phenotypes on age and Δage are shown in Supplementary Table 5. We found that Δage did not contribute significantly to models of our seven diabetes-related phenotypes in our replication group. To test whether the significant findings in the original dataset were due to its longer timespan relative to the replication data, we censored the original dataset so that only the first two timepoints were included in the regression. In Supplementary Table 6, we see that the originally reported associations with BMI and glucose disappear when only two timepoints are used, marked by a substantial drop in the estimated effect size. This suggests that these results may depend on the ability to observe individual changes over a sufficiently long time period.
Discussion
This study supports previous findings on the utility of DNAm-based biomarkers of age in modeling health outcomes. We analyzed longitudinal DNAm data in order to capture the relationship between participants’ changes in DNAm age over time and diabetes-related phenotypes. We found that age acceleration contributes significantly to models of diabetes-related phenotypes among our 43 participants. Epigenetic age acceleration is positively associated with longitudinal changes in participants’ body mass index. Additionally, epigenetic age acceleration shows a suggestive association with longitudinal changes to participants’ glucose, narrowly missing our Bonferroni cutoff for significance. Glucose does in fact reach significance in our sensitivity analysis in which a covariate for incident T2D treatment is included (p = 0.0054). This indicates that age acceleration may contribute to longitudinal models of fasting glucose and that more research should be done with a larger sample. Age acceleration does not appear to significantly contribute to longitudinal models of waist circumference, insulin, HOMA-IR measurements, TG/HDL-C ratio, or TyG index. These findings give us leads into which aspects of diabetes-related phenotypes may feature an important epigenetic component. The utility of epigenetic-based biomarkers is that they can offer a more personalized model of an individual’s health status than age alone, though this may not be true for all phenotypes. This is evident in the result that Δage contributes to models of BMI and fasting glucose but that chronological age appears to be a better predictor of fasting insulin, HOMA-IR, TG/HDL-C ratio, and TyG index.
An intriguing finding is that, for most of our participants, a DNAm-based measure of age acceleration remains stable over the course of the study. This indicates that participants who displayed accelerated biological age at the start of the study were likely to display the same degree of epigenetic age acceleration 16 years later. The dynamics of Δage over time have not been extensively characterized, but this observed stability of Δage over time among adults is consistent with findings in previous longitudinal studies of age acceleration (Kananen et al. 2016; Marioni et al. 2015b). Additionally, we found that Δage exhibits a negative correlation with chronological age, which is consistent with previous reports (Chen et al. 2016; Christiansen et al. 2016; Marioni et al. 2015b). While this relationship was not significant in our initial sample, it reaches significance in our larger replication cohort. While this could suggest a non-linear relationship between DNAm age and chronological age over the life course, Δage did not change significantly over time for the majority of individuals in our study. Thus, the negative correlation appears to result from between-individual differences, and may reflect a selection bias due to biologically “younger” individuals being more likely to survive to old age (Christiansen et al. 2016).
A recent study reported that Hispanic/Latinos from the WHI feature a significantly lower epigenetic age acceleration compared to Caucasians (Horvath et al. 2016). In our study, Hispanic/Latinos also featured a lower Δage compared to Caucasians and African Americans, but this was not significant due to our small sample size. Additionally, our findings, that Δage did not associate significantly with several diabetes-related phenotypes, have been corroborated by another study of Δage among WHI participants; however, in contrast to our findings, this study did not find a significant association between Δage and BMI or glucose (Horvath et al. 2016). Reasons for this difference could lie in our use of longitudinal data over 16 years, while most previous studies of epigenetic age acceleration have relied on cross-sectional data.
A recent publication, which used longitudinal data from an overlapping set of subjects within the WHI, observed a significant association of age acceleration with individual changes in BMI over a 3-year study period (Quach et al. 2017). Another study, using longitudinal methylation data, found that an increase in the BMI is significantly associated with an increase in age acceleration (Nevalainen et al. 2017). These findings suggest that a longitudinal approach to modeling diabetes-related phenotypes may allow for the detection of associations previously not possible with a cross-sectional study. The increased ability to detect association between DNAm and the phenotypes tested can be attributed to the length of time between repeated measures. The 3-year study period may explain why our replication sample, though larger, did not reflect the associations between age acceleration and diabetes-related phenotypes noted in our 16-year study.
Longitudinal studies provide a powerful means to identify phenotypic changes associated with within-person changes in DNA methylation, while avoiding potential confounding due to between-person differences. Sensitivity analyses revealed that our observed association between BMI and Δage was driven by within-individual differences in the participants with the most dynamic Δage and BMI over the time period studied. We also noted that Δage was relatively stable over time for most individuals. Based on these observations, to maximize within-person variation in predictors and phenotypes, future longitudinal studies of DNAm and age-related phenotypes should strive to focus on the age ranges that are most dynamic with respect to the phenotypes of interest, and incorporate the widest possible study duration within the relevant age range. In addition, a previous finding that events like menopause can accelerate biological aging in blood (Levine et al. 2016) imply that perhaps studies of DNAm and/or biological aging could benefit from focusing on post-menopausal women.
Our study had several limitations. Our population of only post-menopausal women potentially limits the generalizability of our findings. More research into the contribution of Δage to health outcomes in both men and women, and in participants across different age groups is necessary. Furthermore, a disproportionally high number of participants enrolled in the WHI are obese, potentially limiting generalizability to non-obese populations. Additionally, data on smoking behavior, alcohol consumption, exercise habits, and ethnicity were self-reported and thus could be biased, potentially affecting our results. Data on time spent exercising per week was unavailable for the third timepoint, and was thus not included in our models. Because physical activity is known to protect against the development of diabetes (Colberg et al. 2010), this may inflate the importance in the contribution of DNAm to disease development. Lastly, T2D incidence was included as a covariate in the sensitivity analysis and not analyzed as an outcome because only 4 participants in our study developed T2D over the 16-year time period—which would limit our power to detect associations with disease incidence. Because of this limitation, our focus was on phenotypes associated with the incidence of T2D rather than the incidence itself.
Finally, while our study benefits from a longitudinal design with DNAm spanning an average of 16 years within subjects, the number of subjects is small. Larger studies will be needed to confirm the associations reported here and to investigate mechanisms underlying the associations. Our results are consistent with a scenario in which the relationship between age and these diabetes-related phenotypes may be mediated by DNAm or a related process. However, much larger studies are required to tease out causality in the relationship between epigenetic aging rates and phenotypes associated with diabetes such as high BMI. Recent cross-sectional publications have used Mendelian randomization approaches to assess causality between DNAm and obesity from whole blood (Mendelson et al. 2017; Wahl et al. 2017). Their findings suggest that the majority of obesity-associated differences in DNAm patterns may be a result, rather than a cause, of the development of obesity. Regardless of the direction of causality, our results and others support the potential of DNAm and epigenetic factors as candidates to develop biomarkers for diabetes-related phenotypes.
Conclusions
Diabetes is associated with genetic, lifestyle, and environmental factors, suggesting that the epigenome may be important in determining both susceptibility and progression of the disease. While numerous past studies have noted small-scale DNAm changes that accompany diabetes risk and progression, our findings speak to the utility of genome-wide methylation changes in modeling phenotypes associated with diabetes. This contribution of Δage in modeling diabetes phenotypes also speaks to the ability of DNAm to serve as a potential mediator of the relationship between aging and the phenotypes associated with age-related disease, or alternatively as a biomarker. We believe this pilot study can inform future studies of DNAm-based biomarkers and their potential to predict phenotypes associated with disease.
Electronic supplementary material
(DOCX 33842 kb)
Funding information
We gratefully acknowledge funding from “Longitudinal study of DNA methylation as a mediator between age and cardiovascular risk” (National Heart, Lung, and Blood Institute (NHLBI) Contract #HHSN268201100046C, to K.C.) and “Epigenetic Mechanisms of PM-Mediated CVD Risk” (National Institute of Environmental Health Sciences (NIEHS) R01-ES020836, to E.W.), P30ES009089 to A.B., National Institute on Aging (NIA) U34-AG051418 to K.C. and A.B., and DGE-1444932 (National Science Foundation Graduate Research Fellowships Program (NSF GRFP), to C.G.). This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. (DGE-1444932). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201600018C, HHSN268201600001C, HHSN268201600002C, HHSN268201600003C, and HHSN268201600004C. All contributors to WHI science are listed at https://www.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Long%20List.pdf.
Footnotes
Electronic supplementary material
The online version of this article (10.1007/s11357-017-0001-z) contains supplementary material, which is available to authorized users.
References
- Adalsteinsson BT, et al. Heterogeneity in white blood cells has potential to confound DNA methylation measurements. PLoS One. 2012;7:e46705. doi: 10.1371/journal.pone.0046705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, Conneely KN, Warren ST. Age-associated DNA methylation in pediatric populations. Genome Res. 2012;22:623–632. doi: 10.1101/gr.125187.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Association AD (2016) Erratum. Classification and diagnosis of diabetes. Sec. 2. In Standards of Medical Care in Diabetes-2016. Diabetes Care 2016;39(Suppl. 1):S13-S22 Diabetes Care 39:1653 doi:10.2337/dc16-er09 [DOI] [PubMed]
- Baker GT, Sprott RL (1988) Biomarkers of aging Exp Gerontol 23:223–239 doi:10.1016/0531-5565(88)90025-3 [DOI] [PubMed]
- Barfield RT, Kilaru V, Smith AK, Conneely KN. CpGassoc: an R function for analysis of DNA methylation microarray data. Bioinformatics. 2012;28:1280–1281. doi: 10.1093/bioinformatics/bts124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollati V, et al. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev. 2009;130:234–239. doi: 10.1016/j.mad.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitling PL, Saum K-U, Perna L, Schöttker B, Holleczek B (2016) Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clin Epigenetics:8.1: 21. 10.1186/s13148-016-0186-5 [DOI] [PMC free article] [PubMed]
- Burch JB, et al. Advances in geroscience: impact on healthspan and chronic disease. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S1–S3. doi: 10.1093/gerona/glu041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JM, Rimm EB, Colditz GA, Stampfer MJ, Willett WC. Obesity, fat distribution, and weight gain as risk factors for clinical diabetes in men. Diabetes Care. 1994;17:961–969. doi: 10.2337/diacare.17.9.961. [DOI] [PubMed] [Google Scholar]
- Chen BH, et al. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging (Albany NY) 2016;8:1844–1865. doi: 10.18632/aging.101020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen BC, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5:e1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen L, Lenart A, Tan Q, Vaupel JW, Aviv A, McGue M, Christensen K. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell. 2016;15:149–154. doi: 10.1111/acel.12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colberg SR, et al. Exercise and type 2 diabetes: American College of Sports Medicine and the American Diabetes Association: joint position statement. Exercise and type 2 diabetes. Med Sci Sports Exerc. 2010;42:2282–2303. doi: 10.1249/MSS.0b013e3181eeb61c. [DOI] [PubMed] [Google Scholar]
- Fagnoni FF, et al. Shortage of circulating naive CD8(+) T cells provides new insights on immunodeficiency in aging. Blood. 2000;95:2860–2868. [PubMed] [Google Scholar]
- International Diabetes Federation (2015) IDF Diabetes Atlas, 7th edn. Brussels, Belgium: International Diabetes Federation
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- Ford ES, Williamson DF, Liu S. Weight change and diabetes incidence: findings from a national cohort of US adults. Am J Epidemiol. 1997;146:214–222. doi: 10.1093/oxfordjournals.aje.a009256. [DOI] [PubMed] [Google Scholar]
- Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- The Women’s Health Initiative Study Group (1998) Design of the Women’s Health Initiative clinical trial and observational study. Controlled Clin Trials 19.1 (1998): 61–109 [DOI] [PubMed]
- Hannum G, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays J et al. (2003) The Women’s Health Initiative recruitment methods and results. Ann Epidemiol 13.9 (2003): S18–S77 [DOI] [PubMed]
- He W, D. Goodkind, P. Kowal (2016) An aging world: 2015. International Population Reports P95/09–1
- Heinemann L (2010) Insulin assay standardization: leading to measures of insulin sensitivity and secretion for practical clinical care response to Staten et al. Diabetes Care 33:e83. 10.2337/dc10-0034 [DOI] [PubMed]
- Hidalgo B, et al. Epigenome-wide association study of fasting measures of glucose, insulin, and HOMA-IR in the genetics of lipid lowering drugs and diet network study. Diabetes. 2014;63:801–807. doi: 10.2337/db13-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, et al. Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging (Albany NY) 2015;7:1159–1170. doi: 10.18632/aging.100861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, et al. An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease. Genome Biol. 2016;17:171. doi: 10.1186/s13059-016-1030-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman EA, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu FB. Globalization of diabetes: the role of diet, lifestyle, and genes. Diabetes Care. 2011;34:1249–1257. doi: 10.2337/dc11-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu FB, Manson JE, Stampfer MJ, Colditz G, Liu S, Solomon CG, Willett WC. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N Engl J Med. 2001;345:790–797. doi: 10.1056/NEJMoa010492. [DOI] [PubMed] [Google Scholar]
- Johnson TE. Recent results: biomarkers of aging. Exp Gerontol. 2006;41:1243–1246. doi: 10.1016/j.exger.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, Rabinovitch PS, Martin GM. Healthy aging: the ultimate preventative medicine. Science. 2015;350:1191–1193. doi: 10.1126/science.aad3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kananen L, et al. The trajectory of the blood DNA methylome ageing rate is largely set before adulthood: evidence from two longitudinal studies. Age (Dordr) 2016;38:65. doi: 10.1007/s11357-016-9927-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK, et al. Geroscience: linking aging to chronic disease. Cell. 2014;159:709–713. doi: 10.1016/j.cell.2014.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh-Banerjee P, Wang Y, FB H, Spiegelman D, Willett WC, Rimm EB. Changes in body weight and body fat distribution as risk factors for clinical diabetes in US men. Am J Epidemiol. 2004;159:1150–1159. doi: 10.1093/aje/kwh167. [DOI] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M et al. (2015) DNA methylation age of blood predicts future onset of lung cancer in the Women’s Health Initiative. Aging (Albany NY) 7:690-700. 10.18632/aging.100809 [DOI] [PMC free article] [PubMed]
- Levine ME, et al. Menopause accelerates biological aging. Proc Natl Acad Sci U S A. 2016;113:9327–9332. doi: 10.1073/pnas.1604558113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni RE, et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015;16:25. doi: 10.1186/s13059-015-0584-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni RE, et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int J Epidemiol. 2015;44:1388–1396. doi: 10.1093/ije/dyu277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin T, Abbasi F, Cheal K, Chu J, Lamendola C, Reaven G (2003) Use of metabolic markers to identify overweight individuals who are insulin resistant. Ann Intern Med 139:802–809. 10.7326/0003-4819-139-10-200311180-00007 [DOI] [PubMed]
- Mendelson MM, et al. Association of body mass index with DNA methylation and gene expression in blood cells and relations to cardiometabolic disease: a Mendelian randomization approach. PLoS Med. 2017;14:e1002215. doi: 10.1371/journal.pmed.1002215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokdad AH, Ford ES, Bowman BA, Dietz WH, Vinicor F, Bales VS, Marks JS (2003) Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 289:76–79. 10.1001/jama.289.1.76 [DOI] [PubMed]
- Nathan DM, et al. Impaired fasting glucose and impaired glucose tolerance: implications for care. Diabetes Care. 2007;30:753–759. doi: 10.2337/dc07-9920. [DOI] [PubMed] [Google Scholar]
- Nevalainen T, et al. Obesity accelerates epigenetic aging in middle-aged but not in elderly individuals. Clin Epigenetics. 2017;9:20. doi: 10.1186/s13148-016-0301-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niccoli T, Partridge L. Ageing as a risk factor for disease. Curr Biol. 2012;22:R741–R752. doi: 10.1016/j.cub.2012.07.024. [DOI] [PubMed] [Google Scholar]
- Nilsson E, et al. Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes. 2014;63:2962–2976. doi: 10.2337/db13-1459. [DOI] [PubMed] [Google Scholar]
- Pan XR, et al. Effects of diet and exercise in preventing NIDDM in people with impaired glucose tolerance. The Da Qing IGT and Diabetes Study. Diabetes Care. 1997;20:537–544. doi: 10.2337/diacare.20.4.537. [DOI] [PubMed] [Google Scholar]
- Patterson RE, Kristal AR, Tinker LF, Carter RA, Bolton MP, Agurs-Collins T. Measurement characteristics of the Women’s Health Initiative food frequency questionnaire. Ann Epidemiol. 1999;9:178–187. doi: 10.1016/S1047-2797(98)00055-6. [DOI] [PubMed] [Google Scholar]
- Perna L, Zhang Y, Mons U, Holleczek B, Saum KU, Brenner H. Epigenetic age acceleration predicts cancer, cardiovascular, and all-cause mortality in a German case cohort. Clin Epigenetics. 2016;8:64. doi: 10.1186/s13148-016-0228-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro J, Bates D, DebRoy S, Sarkar D, Team RC (2016) nlme: linear and nonlinear mixed effects models. vol 3.1–128. R package version
- Quach A et al (2017) Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging (Albany NY) 9:419-446. 10.18632/aging.101168 [DOI] [PMC free article] [PubMed]
- Reinius LE, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7:e41361. doi: 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronn T, Ling C. DNA methylation as a diagnostic and therapeutic target in the battle against type 2 diabetes. Epigenomics. 2015;7:451–460. doi: 10.2217/epi.15.7. [DOI] [PubMed] [Google Scholar]
- Shai I, Jiang R, Manson JE, Stampfer MJ, Willett WC, Colditz GA, FB H. Ethnicity, obesity, and risk of type 2 diabetes in women: a 20-year follow-up study. Diabetes Care. 2006;29:1585–1590. doi: 10.2337/dc06-0057. [DOI] [PubMed] [Google Scholar]
- Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87:4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Simental-Mendía LE, Rodríguez-Morán M, Guerrero-Romero F. The product of fasting glucose and triglycerides as surrogate for identifying insulin resistance in apparently healthy subjects. Metab Syndr Relat Disord. 2008;6:299–304. doi: 10.1089/met.2008.0034. [DOI] [PubMed] [Google Scholar]
- Stamler J, Vaccaro O, Neaton JD, Wentworth D. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the multiple risk factor intervention trial. Diabetes Care. 1993;16:434–444. doi: 10.2337/diacare.16.2.434. [DOI] [PubMed] [Google Scholar]
- Teschendorff AE, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–446. doi: 10.1101/gr.103606.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29:189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl S, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541:81–86. doi: 10.1038/nature20784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer C, Hanson RL, Tataranni PA, Bogardus C, Pratley RE (2000) A high fasting plasma insulin concentration predicts type 2 diabetes independent of insulin resistance: evidence for a pathogenic role of relative hyperinsulinemia. Diabetes 49:2094–2101. 10.2337/diabetes.49.12.2094 [DOI] [PubMed]
- Xu Z, Taylor JA. Genome-wide age-related DNA methylation changes in blood and other tissues relate to histone modification, expression and cancer. Carcinogenesis. 2014;35:356–364. doi: 10.1093/carcin/bgt391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama H et al (2003) Quantitative insulin sensitivity check index and the reciprocal index of homeostasis model assessment in normal range weight and moderately obese type 2 diabetic patients. Diabetes Care 26:2426–2432. 10.2337/diacare.26.8.2426 [DOI] [PubMed]
- Zheng Y, et al. Blood epigenetic age may predict cancer incidence and mortality. EBioMedicine. 2016;5:68–73. doi: 10.1016/j.ebiom.2016.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 33842 kb)