

Abstract
There is increasing recognition that traumatic brain injury (TBI) may initiate long-term neurodegenerative processes, particularly chronic traumatic encephalopathy. However, insight into the mechanisms transforming an initial biomechanical injury into a neurodegenerative process remain elusive, partly as a consequence of the paucity of informative pre-clinical models. This study shows the functional, whole brain imaging and neuropathological consequences at up to one year survival from single severe TBI by controlled cortical impact in mice. TBI mice displayed persistent sensorimotor and cognitive deficits. Longitudinal T2 weighted magnetic resonance imaging (MRI) showed progressive ipsilateral (il) cortical, hippocampal and striatal volume loss, with diffusion tensor imaging demonstrating decreased fractional anisotropy (FA) at up to one year in the il-corpus callosum (CC: −30%) and external capsule (EC: −21%). Parallel neuropathological studies indicated reduction in neuronal density, with evidence of microgliosis and astrogliosis in the il-cortex, with further evidence of microgliosis and astrogliosis in the il-thalamus. One year after TBI there was also a decrease in FA in the contralateral (cl) CC (−17%) and EC (−13%), corresponding to histopathological evidence of white matter loss (cl-CC: −68%; cl-EC: −30%) associated with ongoing microgliosis and astrogliosis.
These findings indicate that a single severe TBI induces bilateral, long-term and progressive neuropathology at up to one year after injury. These observations support this model as a suitable platform for exploring the mechanistic link between acute brain injury and late and persistent neurodegeneration.
Keywords: traumatic brain injury, long-term outcome, neurodegeneration, white matter damage, neuroimaging, inflammation
Graphical Abstract
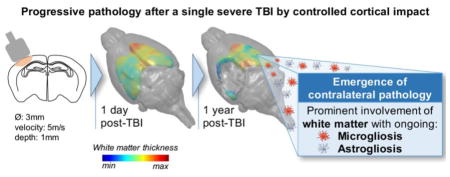
Introduction
There is increasing evidence that, rather than a single, acute, self-limiting event, for many individuals traumatic brain injury (TBI) can trigger a chronic, sometimes life-long disease process.1 Particular attention has focused on the relation between TBI and increased risk of late neurodegenerative disease, such as chronic traumatic encephalopathy (CTE).2 Originally described in clinical studies of boxers as the ‘punch-drunk’ syndrome,3 the associated neuropathology later became recognised as dementia pugilistica, more recently CTE.4 However, in the past decade increasing descriptions of this pathology in non-boxer athletes exposed to repetitive brain injury5,6 and in individuals surviving a year or more from single moderate or severe TBI7 suggest it is exposure to TBI, independent of the circumstance, that predisposes to neurodegenerative pathology.8,9
The neuropathology of late survival from TBI is complex and multifaceted and includes abnormalities in tau, amyloid β, TDP-43, neuroinflammation, axonal degeneration, neuronal loss and white matter degradation.10 Although there have been advances in describing these pathologies, our understanding of the processes linking acute phase TBI to late neurodegenerative pathology is still incomplete. As such, in the absence of candidate pathways driving late poor outcome, progress in identifying strategies for intervention has been limited partly because of the paucity of relevant pre-clinical models for late TBI survival, with the overwhelming majority of models defining ‘late’ follow-up as two months post-injury.11,12
Limited evidence assessing outcomes one year after experimental TBI across models and injury severities indicates that sensorimotor and cognitive deficits persist up to one year after severe fluid percussion injury (FPI)13,14 with progressive tissue loss, white matter damage and associated ongoing axonal pathology in the ipsilateral hemisphere.13–16 Persistence of functional deficits17–19 has also been reported after focal brain injury by controlled cortical impact (CCI) in mice, with progressive tissue loss, reduction in cerebral blood flow19,20 and persistent neuroinflammatory processes, again in the ipsilateral (il) cortex, corpus callosum (cc) and thalamus.21 Similarly, models of repetitive mild TBI report long-term cognitive deficits associated with late pathologies,22,23 with variable results depending on the inter-injury interval.24,25 However, while there have been notable descriptions of persisting and devolving pathologies adjacent to the site of injury and in the ipsilateral hemisphere, there has been little insight into remote and contralateral pathologies so far.
In this work, we have longitudinally analysed the behavioral outcomes of a single severe TBI. Using in vivo quantitative magnetic resonance imaging (MRI) techniques and parallel neuropathological studies we describe the long-term and progressive, bilateral hemispheric consequences of TBI. We show a progression of pathology to regions remote from the original injury and into contralateral structures at one year after injury, with ongoing white matter pathology, including active neuroinflammation.
Materials and methods
Animals
C57BL/6 mice (Harlan Laboratories, Italy) were housed in a specific pathogen free vivarium at a constant temperature (21±1°C) with a 12h light–dark cycle and free access to food and water. The IRCCS-Istituto di Ricerche Farmacologiche Mario Negri (IRFMN) adheres to the principles set out in the following laws, regulations, and policies governing the care and use of laboratory animals: Italian Governing Law (D.lgs 26/2014; Authorization n.19/2008-A issued March 6, 2008 by Ministry of Health); Mario Negri Institutional Regulations and Policies providing internal authorization for persons conducting animal experiments (Quality Management System Certificate – UNI EN ISO 9001:2008 – Reg. No. 6121); the NIH Guide for the Care and Use of Laboratory Animals (2011 edition) and EU directives and guidelines (EEC Council Directive 2010/63/UE).The Statement of Compliance (Assurance) with the Public Health Service (PHS) Policy on Human Care and Use of Laboratory Animals has been recently reviewed (9/9/2014) and will expire on September 30, 2019 (Animal Welfare Assurance #A5023-01). All efforts were made to minimize animal suffering and reduce the number of animals used.
Experimental design
Sham and TBI mice (n=8/group) were longitudinally analyzed following the experimental plan shown in Fig. 1A. Sensorimotor deficits were evaluated by neuroscore (1, 4, 6, 8 and 10 weeks, 3 and 12 months) and beam walk tests (1, 2, 4, 6, 8 and 10 weeks, 3, 6 and 12 months) by the same operator for the entire duration of the study. Cognitive deficits were evaluated by Morris water maze test at 3 and 6 months. Imaging studies were done as T2 weighted (T2w) magnetic resonance imaging (MRI) (1, 7 days, 1, 3, 12 months) and diffusion tensor imaging (DTI) MRI (1, 7 days, 1, 3, 6, 12 months). All mice were sacrificed at 12 months for histopathological analysis. Body weight at all time points was recorded in all mice (Fig. 1B). TBI did not result in any acute mortality; one mouse died three months after injury.
Figure 1. Experimental design and weights.

Mice were subjected to controlled cortical impact (CCI) or sham injury. Functional deficits and MRI analysis were assessed at multiple intervals up to one year post injury/sham at the time points indicated (A). Both sham and TBI mice gained body weight over time (p<0.0001), with no significant difference between sham and TBI weight at any time. (B). Data are mean±SD, n=7–8.
Experimental traumatic brain injury
Eight-week-old male mice were anesthetized with isoflurane inhalation (induction 3%; maintenance 1.5%) in an N2O/O2 (70%/30%) mixture and placed in a stereotaxic frame. Rectal temperature was maintained at 37°C. Mice were then subjected to craniectomy followed by induction of CCI brain injury as previously described.26–29 Briefly, the injury was induced using a 3 mm diameter rigid impactor driven by a pneumatic piston rigidly mounted at an angle of 20° from the vertical plane and applied vertically to the exposed dura mater, between bregma and lambda, over the left parieto-temporal cortex (antero-posteriority: −2.5 mm, laterality: −2.5 mm), at impactor velocity of 5m/s and deformation depth 1 mm, resulting in a severe level of injury.30,31 The craniotomy was then covered with a cranioplasty and the scalp sutured. Sham-operated mice received identical anesthesia and surgery without brain injury.
Behavioural tests
Sensorimotor deficits were evaluated by neuroscore and beam walk tests, and cognitive deficits were evaluated by the Morris water maze test as previously described.26–29,32
Neuroscore
Mice were scored from 4 (normal) to 0 (severely impaired) for each of the following: (1) forelimb function during walking on the grid and flexion response during suspension by the tail; (2) hindlimb function during walking on the grid and extension during suspension by the tail; (3) resistance to lateral right and left push. The best score is 12.
Beam walk
The beam walk test measures the number of foot faults of the mouse walking twice on an elevated, narrow wooden beam (5 mm wide and 100 cm long). Before each test, mice are trained in three habituation trials. Data are expressed as the sum of the number of foot faults during the two tests. The best score is 0.
Morris water maze (MWM)
A circular pool (1 m diameter) filled with water (18–20°C) made opaque with a nontoxic white paint, with a fixed submerged platform (1 cm below the water surface) was used. The learning task consisted of eight trials/day for four consecutive days for a total of 32 trials. Latencies to reach and climb onto the platform were recorded for each trial with a maximum of 60 sec per trial. Cognitive performance was obtained by averaging the latencies of each day (MWM learning task) and the latencies of 32 trials over the four days (all trials). Five days after the learning task, mice were tested for their ability to remember the location of the submerged platform (memory task). Animals were allowed to swim for 60sec with the platform removed, and their swim paths were recorded using a computerized video analysis system (Ethovision XT 5.0; Noldus Information Technology, Wageningen, The Netherlands). A memory score was calculated to grade memory retention deficits in the different groups.33 Mice were tested twice (at 3 and 6 months) in the MWM, with the same protocol. The platform was placed in the southwest quadrant of the pool at three months and northeast at six months.
MRI studies
Mice were anesthetized with isoflurane inhalation (induction 3%; maintenance 1.5%) in an N2O/O2 (70%/30%) mixture. Body temperature was maintained at 37°C by warm water circulated in a heating cradle. Imaging was done on a 7T small-bore animal scanner (Bruker Biospec, Ettlingen, Germany). Two actively decoupled radio frequency (RF) coils were used: a volume coil of 7.2 cm diameter used as the transmitter and a surface coil as the receiver.32
Acquisition
Brain anatomical changes were evaluated in sagittal, 2D, T2-weighted RARE sequences. The morphological images were obtained with a voxel size of 117 x 147 x 300 μm (FOV 3 x 1.5 cm, matrix 256 x 102, slice thickness 300 μm), repetition time 5000 ms, effective echo time 36 ms with a RARE factor of 8, for 8 averages.
Diffusion tensor data were acquired in coronal DTI echo-planar imaging (EPI) sequences acquired with an in-plane image resolution of 107 x 107 μm (FOV 1.5 x 1.5 cm, acquisition matrix 140 x 140, slice thickness 1 mm); repetition time 3000 ms, echo time 36 ms. Four averages were used to boost the signal-to-noise ratio. For diffusion, encoding b factors of 0 (b0 image) and 800 mm2/s were used and diffusion-sensitizing gradients were applied along 19 isotropic directions of three-dimensional space.
Analysis
Diffusion tensor analysis
The diffusion tensor was computed using the freely available software FSL.34–36 The software reconstructs the diffusion tensor generating the three eigenvalue λ1, λ2, λ3 maps and the associated eigenvectors (i.e. the three principal diffusion directions). The DTI indices fractional anisotropy (FA) and mean diffusivity (MD, 10−3mm2/s) are defined as follows:
| (1) |
| (2) |
| (3) |
| (4) |
which are mean diffusivity (MD, μm2/ms) (1), fractional anisotropy (FA) (2), axial diffusivity (AD, μm2/ms) (3) and radial diffusivity (RD, μm2/ms) (4). Regions of interest (ROI) were the corpus callosum (CC), external capsule (CE), cortex and thalamus and were selected manually by a trained expert following the Paxinos atlas.37 The whole CC and EC were selected for quantification as depicted in Fig. 5A (five coronal sections 1 mm thick). In addition, for the 12 month time point, when contralateral white matter pathology emerged, we repeated the quantification restricting the ROIs in the contralateral (cl) hemisphere so the location and volume matched the remaining (r) il-CC and EC (Suppl. Fig. 1, rCC and rEC five coronal sections 1 mm of thick).
Figure 5. Quantitative analysis of diffusion tensor imaging parameters in the white matter.

Drawings represent the rostrocaudal ROI selection in the CC (red) and EC (green) in sham (left) and TBI (right) mice (A). Quantification of FA (B–C), AD (D–E), and RD (F–G) in the CC (B, D, F) and EC (C, E, G). Compared to sham mice, TBI induced a reduction of FA in the il-CC (B) and il-EC (C). At 12 months after injury, FA was also reduced in the cl-CC (B) and cl-EC (C) compared to the corresponding areas in sham-operated mice. There were no differences observed in AD (D–E). TBI increased RD in the il-CC (F) at late time points, with no changes in the EC (G). Data are mean±SD, n=7–8. Two-way ANOVA for repeated measurements followed by Tukey’s post hoc test. *p<0.05, **p<0.01, ***p<0.001 sham il vsTBI il; °°p<0.01, °°° p<0.001 sham cl vs TBI cl.
Volume analysis
The volume measurements of structural MRI images were obtained using Java-based custom-made software. ROI were selected manually by a trained expert following the Paxinos atlas.37 To obtain a 3D-reconstruction of lesion progression, manually segmented brain masks of a representative mouse were smoothed and brain surfaces visualized using MRIcroS (a matlab-based tool freely available at https://www.nitrc.org/projects/mricros/).
Tissue processing
One year after surgery, under deep anesthesia (Ketamine 20 mg + Medetomidine 0.2 mg), animals were transcardially perfused with 20 mL of phosphate buffer saline (PBS) 0.1 mol/L, pH 7.4, followed by 50 mL of chilled paraformaldehyde (4%) in PBS. The brains were carefully removed from the skull and post-fixed for 6h at 4°C, then transferred to 30% sucrose in 0.1 mol/L phosphate buffer for 24h until equilibration. The brains were frozen by immersion in isopentane at −45°C for 3 min before being sealed into vials and stored at −80°C until use. Coronal brain cryosections twenty μm thick were cut serially (from bregma +1 mm to bregma −4 mm) at 200 μm intervals and stained with Cresyl violet or Luxol fast blue/cresyl violet (LFB/CV) using standard histological protocols.22,29 Axons were visualised using Palmgren's silver impregnation technique.38 Contusion volume was analysed as previously described.39
Immunohistochemistry
Immunohistochemistry was done on 20 μm thick coronal sections, cryosectioned at −20°C and stored at 4°C in a solution of glycerol:PBS (1:1). After washing and hydration in PBS solution, sections were treated with H2O2 1% for 10 min to inactivate endogenous peroxidase activity. Blocking solutions and incubation with primary antibodies were as follows:
IBA1: blocking was achieved by applying 2% normal horse serum (Vector Labs, Burlingame, CA) in Optimax buffer (BioGenex,San Ramon, CA) for 30 min. Endogenous IgG was blocked using an avidin/biotin blocking kit according to the manufacturer's instructions (Vector Labs). Incubation with the primary antibody was then done overnight at 4°C with monoclonal rabbit anti-mouse IBA1 (1:200; Wako, Neuss, Germany) in a solution containing normal horse serum 2% in Optimax Buffer.
CD68: blocking was achieved by applying 10% normal goat serum (Merck, Darmstadt, Germany), 0.1% Triton in PBS for 1h, followed by incubation with the primary antibody overnight at 4°C with monoclonal rat anti-mouse CD68 (1:200; Serotec, Kidlington, UK), 3% NGS, 0.1% Triton in PBS. GFAP: blocking was achieved by applying 10% fetal calf serum (Gibco, Thermofisher Scientific, Waltham, MA USA), 0.5% Triton in PBS for 1h, followed by incubation with the primary antibody overnight at 4°C with monoclonal mouse anti-mouse GFAP (1:2000; Chemicon), 10% FCS, 0.5% Triton in PBS.
After incubation in primary antibodies, sections were transferred to solutions containing secondary antibody for 1h, followed by avidin-biotin complex following manufacturer's instructions (VectaStain Kit, VectorLaboratories, Burlingame, CA, USA) before visualisation of immunoreactivty with 3,3-diaminobenzidine-tetrahydrochloride (Vector Laboratories, Burlingame, CA). For each reaction, negative controls were run without the primary antibody. In each case, no staining was observed.
Selection of region of interest for histopathological analysis
Sections were scanned at 20x using a Hamamatsu Nanozoomer 2.0-HT slide scanner, with the images viewed using the Slide Path Digital Image Hub application (Leica Microsystem). Alternatively, they were acquired at 20x by an Olympus BX-61-VS microscope, inter-faced with VS-ASW-FL software (Olympus Tokyo, Japan). Images were analysed using Fiji software (http://fiji.sc/Fiji). Regions of interest for DTI analysis (cortex, thalamus, CC and EC, as indicated in Fig.2) were selected according to the Paxinos atlas.37 The coronal section with maximum lesion size (AP −1.6 mm from bregma40) was selected. The ipsilateral (il) cortex was analyzed over an area 1 mm deep from the edge of the contusion. A corresponding area of the contralateral (cl) hemisphere and sham animals was analysed. For the thalamus the analysis was done in the areas corresponding to the latero-dorsal and latero-posterior nuclei and the lateral geniculate complex. White matter areas (CC and EC) were disrupted by the focal trauma pathology in the il-hemisphere, so only the cl-hemisphere was quantified. The whole cl-CC was selected, and the cl-EC was analysed up to the lower limit of the primary somatosensory cortex. The whole selected regions of interest were analysed.
Figure 2. Region of interest for histopathological analysis.

Drawings represent the regions of interest selected for quantification in the corpus callosum (CC; red), external capsule (EC; green), cortex (blue) or thalamus (yellow) in sham (A) or TBI (B) mice.
Neuronal cell count
Coronal sections 20 μm thick were stained with cresyl violet. The entire sections were acquired at 20x with an Olympus BX-61 Virtual Stage microscope, with pixel size 0.346 μm. Acquisition was done over 10 μm thick stacks, with a step size of 2 μm. The different focal planes were merged into a single stack by mean intensity projection to ensure consistent focus throughout the sample. Neuronal count was done using an appropriate size threshold (25 μm2) to exclude small objects not representing an entire cell, such as ramification portions from out-of-focus cells.31,40–42 Quantification was done with Fiji software and neuronal cell loss was expressed as the number of neurons per mm2.
Quantification of histological markers
For silver staining, sections were acquired at 40x with an Olympus BX-61 Virtual Stage microscope, with pixel size 0.173 μm. For IBA1, CD68 and GFAP, sections were acquired at 20x with an Olympus BX-61 Virtual Stage microscope, with pixel size 0.346 μm. Acquisition was done over 6 μm thick stacks, with step size 2 μm. The different focal planes were merged into a single stack by mean intensity projection to ensure consistent focus throughout the sample. Images were analysed using Fiji software. Data are expressed as positive pixels/total assessed pixels, and reported as the percentage stained area for subsequent statistical analysis.
Statistical analysis
Mice were allocated to surgery by a list randomizer (http://www.random.org/list). All behavioral evaluations were done blinded to injury status. Data are presented as means and standard deviation of the mean (SD). For neuroscore, beam walk, Morris water maze, volumetric measurements and DTI data, groups were compared using a two-way analysis of variance for repeated measures, followed by Tukey’s post hoc test. For the sake of clarity, all the graphs referring to longitudinal analysis (behavior and MRI) report statistical significances between sham and TBI animals only, while longitudinal intra-group differences are reported in the text. For histological analysis, groups were compared using the unpaired t-test; p-values <0.05 were considered statistically significant. Assumptions of normality were checked using the Kolmogorov–Smirnov test. Standard software packages were utilised throughout (GraphPad Prism version 6.00, La Jolla, CA, USA).
Results
TBI induces long-term functional deficits
The neuroscore indicated significant sensorimotor deficits in TBI compared to sham-operated mice at all time points (Fig. 3A). TBI mice had an initial poor score of 3.6 out of 12, with subsequent improvement over the next four weeks up to a score of 7.5 (p<0.001), but no improvement later on. Sham mice had neuroscores in the range of 11 to 12 at all times.
Figure 3. Behavioral deficits.

Sensorimotor deficits were assessed by composite neuroscore (A) and beam walk (B) tests up to one year after injury. TBI mice showed clear motor dysfunction compared with sham-operated mice for the whole duration of the study in both tests. Cognitive deficits were assessed by MWM test at three and six months after injury (C–F). Compared to sham-operated, TBI mice had a longer latency to the platform on days 1–4 of learning at three months (C) but only on day 4 at six months (D). The overall learning performance showed significant deficits at three but not six months after surgery (E), and clear deficits in memory retention at both time points in TBI compared to sham mice (F). Data are mean+SD, n=7–8, A–F: Two way ANOVA for repeated measurements, followed by Tukey’s post hoc test. *p<0.05, **p<0.01, ***p<0.001 TBI vs sham.
The beam walk test showed clear motor deficits in TBI compared to sham-operated mice at all time points (Fig. 3B). Longitudinal intra-subject analysis in TBI mice indicated an improvement from 1 to 4 weeks (p<0.001), followed by stable performance up to 3 months. After this, both TBI and sham mice had similar increases in footfault numbers (sham: 12 versus 52 weeks p<0.0001; TBI: 12 versus 52 weeks p<0.0001) suggesting that weight gain (see Fig. 1B for changes in body weight) may affect the performance in the beam walk test.
Cognitive deficits were evaluated at 3 and 6 months in the MWM test (Fig. 3C–F). TBI and sham mice showed no significant difference in swim speed throughout the testing period (data not shown). At 3 months, TBI mice showed a significant learning dysfunction compared to sham mice, as shown by the longer time spent to reach the platform over the four days of learning (Fig. 3C). At six months, mice were exposed for the second time to the same test, and TBI mice showed a significant difference in learning task only on day 4 (Fig. 3D), with overall performance similar to sham mice (Fig. 3E). The probe trial indicated persistent memory retention deficit at both three and six months in TBI compared to sham mice (Fig. 3F). TBI mice had improvements in both learning (p<0.001) and memory (p<0.01) functions at six compared to three months, while sham mice performed similarly over time.
T2w-MRI reveals lesion progression in ipsilateral hemisphere over time
3D-reconstruction of the brain after traumatic injury indicated the evolution of the lesion, increasing over time (Fig. 4A). Compared to sham mice, TBI produced a significant volume reduction already detectable on day 1 after injury in the il-cortex (Fig. 4C) and il-hippocampus(Fig. 4D) measured by T2w-MRI. At longer times (one month after TBI) the il-striatum was also affected (Fig. 4E). Cortical, hippocampal and striatal damage in the il-hemisphere expanded over time up to one year (1 day versus 12 months: p<0.001). No T2w-MRI changes were detected in the cl-hemisphere. Whole brain atrophy was also measured by conventional histology at 12 months, giving values that correlated strongly with those measured by T2w (Fig. 4F, Pearson correlation r=0.895, p<0.01).
Figure 4. Anatomical damage.

A) 3D reconstruction of lesion progression from one day up to 12 months post injury. B) Representative images for sham (upper) and TBI (lower) mice, depicting the regions of interest for analysis of structural damage by MRI in cortex (blue), hippocampus (red) and striatum (green). C–E) Quantification of lesion progression in the cortex (C), hippocampus (D) and striatum (E) up to 12 months post-injury. TBI resulted in a volume reduction in the ipsilateral cortex and hippocampus starting from day 1 and in striatum starting from one month, compared to the corresponding area in sham-operated mice. No changes were seen between TBI and sham contralateral hemispheres. F) Representative brain coronal section by MRI and by conventional histology at 12 months in the same mouse. Lesion volume quantified by MRI correlated strongly with that measured by conventional histology (Pearson correlation r=0.895, p<0.01). Data are mean±SD, n=7–8. Two-way ANOVA for repeated measurements followed by Tukey’s post hoc test. *p<0.05, **p<0.01, ***p<0.001 sham il vs TBI il.
DTI-MRI shows contralateral degeneration at chronic stages after TBI
DTI-MRI analysis was done longitudinally on white matter (CC and EC, Fig. 5) and grey matter (cortex and thalamus, Suppl. Fig. 2) up to 12 months. Compared to sham, TBI induced a significant reduction in FA in the il-CC at 1, 6 and 12 months after surgery (Fig. 5B) and in the il-EC at 1 day and 3, 6 and 12 months after injury (Fig. 5C). Notably, at 12 months, there was also a significant decrease of FA in the cl-CC (Fig. 5B) and cl-EC (Fig. 5C) of TBI compared to sham mice, documenting the spread of neurodegenerative processes to the contralateral white matter at chronic stages after TBI. At 12 months, we also restricted the ROIs in the cl-hemisphere to match the il-CC and EC remaining tissue (rCC and rEC, Suppl. Fig. 1). FA was significantly reduced in il and cl-rCC and il-rEC but not in cl-rEC compared to sham (Suppl. Fig. 1), indicating a spatial gradient in FA impairment moving away from the site of injury.
No significant differences were observed in AD (Fig. 5D–E), but there was a significant increase in RD in il-CC from six months on in TBI mice compared to sham (Fig. 5F). Analysis of the MD indicated a significant increase in il-CC at 12 months, reflecting observations for RD (data not shown).
DTI analysis of gray matter regions indicated increased FA in the il-cortex of TBI compared to sham animals only at six months (Suppl Fig. 2B), and no significant differences in the thalamus (Suppl Fig. 2C, E).
Persistent and bilateral neuropathology 12 months post CCI
Compared to sham animals, neuropathological studies on material from the il-hemisphere 12 months after CCI gave evidence of neuronal loss with reduced neuronal density in the il-cortex (−32%, Fig. 6A–C), associated with both microgliosis and astrogliosis, marked by increased area of staining for IBA1 (+176%, Fig. 6D–F) and GFAP (+158%, Fig. 6J–L). However, this was not associated with any demonstrable increase in CD68 (Fig. 6G–I).
Figure 6. Histopathology in the ipsilateral and contralateral cortex one year after TBI.

Representative images of cresyl violet stained neurons (A), IBA1 (D), CD68 (G) and GFAP (J) of sham and TBI ipsilateral and contralateral cortex one year after injury. Compared to sham-operated mice, TBI resulted in: reduced neuronal density in the il (B) but not cl (C) hemisphere; an increase in IBA1 immunoreactivity in both il (E) and cl (F) hemispheres, but no demonstrable change in CD68 expression (H, I); and increased GFAP staining in the il (K) but not cl (L) hemisphere. Bar, 50μm. Data are mean±SD, n=7–8. Unpaired t-test, **p<0.01, ***p<0.001.
Studies on the il-thalamus found evidence of ongoing microgliosis and astrogliosis with increased staining for microglial (IBA1: +68%, Fig. 7A–C; CD68: +581%, Fig. 7D–F) and glial markers (GFAP: +598%, Fig. 7G–I).
Figure 7. Histopathology in the ipsilateral and contralateral thalamus one year after TBI.

Representative micrographs of IBA1 (A), CD68 (D) and GFAP (G) and their quantification in ipsilateral (B, E, H) and contralateral thalamus (C, F, I) one year after surgery. TBI increased IBA1, CD68, and GFAP immunoreactivity in the il but not the cl thalamus, compared to the corresponding areas in sham-operated mice. Data are mean±SD, n=7–8. Unpaired t-test, **p<0.01, ***p<0.001.
In contrast to the il-hemisphere, there was no detectable neuronal loss in the cl-cortex and thalamus in CCI mice at 12 months compared to sham mice, nor was there any evidence of a neuroinflammatory reaction. However, sections of the cl-hemisphere stained for LFB showed loss of white matter, with reduced area staining in both cl-CC and cl-EC, extending from −0.4 to −2.2 mm from bregma in TBI compared to sham mice (Suppl. Fig. 3). This loss of volume peaked at −1.6 mm from bregma in TBI compared to sham mice (Fig. 8A–B) and coincided with comparable fiber density in sham and TBI animals in both cl-CC and cl-EC in silver stained sections (Fig. 8C–D), indicating axonal loss.
Figure 8. Histopathology of the contralateral white matter one year after TBI.

Luxol fast blue staining of sham and TBI brain coronal sections (A). Quantification of the Luxol fast blue stained area (B) indicated evidence of white matter loss in the cl-CC and EC one year after TBI. Representative images of Palmgren silver stained sections of the cl-EC of sham and TBI mice (C), and quantification in cl-CC and EC (D) one year after surgery. No difference in fiber density was found between sham and TBI in either CC or EC. Data are mean±SD, n=7–8. Unpaired t-test, ***p<0.001.
These changes were associated with ongoing microgliosis, marked by increased area staining in both IBA1 (cl-CC +235%, Fig. 9A; cl-EC +127%, Fig. 9B) and CD68 (cl-CC +1919%, Fig. 9C; cl-EC +255%, Fig. 9D), and astrogliosis, with increased area staining in GFAP (cl-CC +419%, Fig. 9E; cl-EC +98%, Fig.9F). A colorimetric representation of the degree of activation of markers analysed in the different ROIs is shown in Suppl. Fig. 4.
Figure 9. Inflammatory markers in the contralateral white matter one year after TBI.

Representative micrographs of IBA1 (A–B), CD68 (C–D) and GFAP (E–F), and their quantifications in cl-CC (A, C, E) and EC (B, D, F) one year after surgery. TBI increased IBA1, CD68 and GFAP immunoreactivity in both cl-CC and EC, compared to the corresponding areas in sham-operated mice. Data are mean±SD, n=7–8. Unpaired t-test, *p<0.05, **p<0.01, ***p<0.001.
Discussion
In this study we show that a single severe TBI triggers an evolving and progressive neuropathology that at chronic stages spreads to the cl-hemisphere. In vivo longitudinal MRI studies demonstrated the emergence of cl-brain pathology with late survival post-CCI, with a prominent involvement of white matter structures and ongoing neuroinflammation, demonstrated by histological studies at 12 months survival. In addition, we show widespread ipsilateral changes, with both grey and white matter damage and persistent and evolving behavioral dysfunction.
As previously shown, TBI acutely (one week) induced severe sensorimotor functional impairment after injury, followed by partial resolution in the four weeks post-TBI27–29, but with persisting deficits compared to sham mice for up to one year. While both neuroscore and beam walk tests clearly distinguished sham from TBI mice throughout the study, an injury-independent effect was observed in the beam walk test, from six months on, suggesting that neuroscore over beam walk may be preferred for long-term studies. Among factors possibly contributing to worsening of the performance in beam walk at chronic stages are the longer inter-test interval and weight gain. The increase in the inter-test interval (from 14 to 90 days) coincided with worsening of performance, suggesting that practice contributes to ability in this test. Similarly, increases of respectively 36% and 28% over their baseline weight were observed at six months in sham and TBI mice, which might contribute to worsening in their motor performance, balance and coordination32,43,44. However, a major aging effect45,46 was unlikely, since the increase in foot fault number emerged in adult (8 months old) rather than aged mice.
Cognitive deficits were longitudinally assessed in the MWM. There was clear impairment of spatial learning and memory retention in TBI compared to sham mice at three months after injury. Six months post-injury, TBI mice showed improvement of learning ability but the persistence of memory retention deficits. Thus, despite the progressive atrophy/tissue loss in cortical and hippocampal regions, as shown by our MRI data, there was partial recovery of cognitive function. Remodeling changes after injury in surviving neurons may attenuate the initial deficit and compensate the function of tissue loss.47 Caution, however, is needed when interpreting our MWM data as partial recovery of hippocampal functions. The MWM is strongly influenced by the “novelty effect”. Initial exposures to the aquatic field are associated with a variety of stress and/or anxiety behavioral responses, such as thigmotaxis, that contribute to the test performance. 48 Repeated exposure to the test leads to habituation to the novelty, reducing neophobia and thigmotaxis, factors that all help improve the test performance, limiting the possibility of repeating the test.
Little is known about the long-term progression/resolution of white matter damage in experimental TBI after a single focal CCI. At acute to subacute stages the involvement of contralateral white matter structures has been shown histologically in developing49 and adult50 mice after focal TBI, with scattered data obtained by DTI51–53 showing both persistence or resolutions of FA contralateral changes. Zhuo and colleagues51 reported decreased FA in the cl EC 2h after CCI in rats, with resolution to baseline values at seven days. Interestingly, Harris53 and colleagues reported that CCI injury resulted in a persistent, unilateral CC FA reduction up to 28 days post-injury, with cl regions of CC showing a severity-dependence relationship for FA at four weeks post-injury but not at one week, suggesting ongoing degeneration from the il to the cl hemisphere in the more severely injured rats. Notably, here we document the progressive white matter damage spreading from the ipsilateral to the contralateral hemisphere with FA values becoming significantly lower than controls in the cl-CC and cl-EC by one year after injury. FA impairment showed a spatial gradient moving away from the site of injury with only FA values in the farest region of the cl-EC no longer different from sham mice. Contralateral white matter degeneration with diffuse axonal loss was confirmed in histological sections stained for myelin (LFB), which showed a marked reduction of cl-CC and cl-EC thickness extending from −0.4 to −2.2 mm from bregma, but no demonstrable increase in fiber density in silver staining. These data are consistent with progressive white matter degeneration evolving from the ipsilateral to the contralateral side with survival from injury.
As previously shown,53–55 we also noted a progressive decrease of FA in ipsilateral white matter which progressed with survival up to one year post-CCI, together with an increase in RD in the il-CC. These results are closely correlated with clinical findings in TBI,56,57 that indicate an increase in RD rather than AD in longitudinal studies with long-term follow-up.58–61 FA and RD partially recover over time after moderate59 but not severe TBI in patients,58 indicating that the initial injury severity affects progression and/or recovery of axonal injury.
Axonal damage is a complex phenomenon starting in the acute phases post TBI, involving anterograde and retrograde degeneration processes62 accountable for the spread of axonal damage to areas distal from the primary injury. There is increasing evidence that, for some patients, these acute pathologies may not resolve after the acute phase but persist long after TBI, with ongoing axonal degeneration detectable even years after single moderate or severe TBI in human autopsy derived material.56,57 Whether these chronic axonal changes are the consequence of Wallerian degeneration or are due to secondary, evolving pathologies detrimental to axons still needs to be clarified.
Axonal damage is associated with the release of myelin debris, a strong inhibitor of axonal sprouting and regrowth. Thus, removal of this debris is essential for adequate tissue healing, a process involving phagocytosis of myelin debris by activated microglia and astrocytes.63,64 Classically activated neuroinflammatory microglia induce a subtype of reactive astrocytes named A1 that lose the ability to promote phagocytosis, and synaptogenesis, and contribute to neuronal death.65 We observed a sustained chronic inflammatory process with activated microglia and astrocytosis in the cl-white matter of TBI mice one year post-CCI supporting, the notion that active neurotoxic events driven by reactive astrogliosis are in place at this stage. Importantly, these data reflect clinical observations in moderate to severe TBI patients, where atrophy of the CC, with evidence of axonal degeneration and a florid, ongoing neuroinflammatory response was evident in approximately one third those surviving a year or more from a single TBI.66
Histopathological analysis also indicated preferential white over grey matter involvement in long-term neurodegenerative and inflammatory processes after a single CCI. When considering the perilesional cortex decreased neuronal density was associated with persistent neuroinflammatory processes characterized by active microgliosis and astrogliosis. Results were similar oin the ipsilateral thalamus, confirming previous studies on CCI mice where at two months67 or onw year21, active astro/microgliosis was associated with oxidative stress in the ipsilateral cortex and thalamus, leading to neurodegeneration. Our study adds the observation of modest involvement of the contralateral grey over white matter structures with increased microgliosis not associated with an increase in CD68 related to phagocytic activity and with no astrogliosis. No glial activation was observed in the contralateral thalamus. Thus, our data indicate that one year after a single severe TBI, white matter damage is the main histopathological feature in the contralateral hemisphere suggesting that chronic inflammatory processes do not appear randomly throughout the brain, but progress through distinct neural networks.
The potential value of these observations for human studies is suggested by growing evidence on the long-term consequences after TBI. Structural and functional alterations are documented after moderate to severe TBI68–70 and even 12–14 years after injury.70 Long-term consequences have a profound impact on patients, families and society. With the exception of a single study72 there is no proven treatment for improving recovery after TBI.
Nevertheless, caution is always required when findings from animal experiments are transferred to clinical aspects in human patients. Our current model produces a substantial focal lesion, with a contusion volume that increases from about 8% of the hemisphere at 1 day to 22% at one year. The extent and location of the tissue injury produced in this rodent model may be different from several contusional lesion patterns in patients. In humans, multiple lesions are common, with a significant risk of life-threatening evolution when the contused temporal lobe impacts the brainstem through the tentorial hiatus. Additionally, in clinical practice expanding contusions may require surgical removal and intensive care. Here we induced a single, substantial lesion but with no evidence of brainstem involvement, and our observations simply describe the temporal evolution of the initial damage without any therapeutic intervention. Importantly, this degree of injury, still producing progressive, large-scale tissue damage, is not fatal. We interpret this prolonged survival of mice capable of feeding themselves as a marker of important but not too severe damage.
All these dissimilarities, however, do not reduce the physiopathological importance of our observation, with evidence of long-term progression, and bilateral involvement, after the initial focal injury.
Conclusions
This study shows that a single severe TBI induces long-term, progressive neuropathology that not only involves the peri-lesional and ipsilateral cortex, but also extends to the contralateral hemisphere one year after injury, with particular involvement of white matter structures. The widespread nature of these chronic events underlines the clinical importance of our CCI model for reproducing distinct neurodegenerative features recapitulating features of the neuropathology of human single severe TBI. It therefore offers a reliable model of disease progression to address the mechanistic link between the acute biomechanical insult and chronic and progressive degeneration.
Supplementary Material
Highlights.
Longitudinal effects of single severe TBI by controlled cortical impact were examined
TBI triggers an evolving damage that at chronic stages spreads to the contralateral hemisphere
Contralateral pathology at one year post TBI, shows a prominent involvement of white matter with ongoing neuroinflammation
Findings support the model for studies interrogating the link between biomechanical impact and late neurodegeneration
Acknowledgments
Fundings
Partially supported by Ministero della Salute (within the framework of ERA-NET NEURON, RG86200), ESICM Rita Levi Montalcini Award to Elisa R. Zanier, and IBRO/PERC InEurope Short Stay Funding Program to Francesca Pischiutta. Francesca Pischiutta received a fellowship from “Fondazione Umberto Veronesi” (FUV, 2016). Dr William Stewart is supported by DOD grant, PT110785, NIH grants NS038104, NS094003 and by an NHS Research Scotland Career Researcher Fellowship
Abbreviations
- AD
axial diffusivity
- CC
corpus callosum
- CCI
controlled cortical impact
- cl
contralateral
- CTE
chronic traumatic encephalopathy
- CV
cresyl violet
- DTI
diffusion tensor imaging
- EC
external capsule
- EPI
echo-planar imaging
- FA
fractional anisotropy
- FPI
fluid percussion injury
- il
ipsilateral
- LFB
luxol fast blue
- MD
mean diffusivity
- MRI
magnetic resonance imaging
- MWM
Morris water maze
- r
remaining
- RD
radial diffusivity
- ROI
regions of interest
- T2w
T2 weighted
- TBI
traumatic brain injury
Footnotes
Conflict of interest
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Masel BE, DeWitt DS. Traumatic brain injury: a disease process, not an event. J Neurotrauma. 2010;27:1529–1540. doi: 10.1089/neu.2010.1358. [DOI] [PubMed] [Google Scholar]
- 2.Washington PM, Villapol S, Burns MP. Polypathology and dementia after brain trauma: Does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy? Exp Neurol. 2016;275(Pt 3):381–388. doi: 10.1016/j.expneurol.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martland HS. PUNCH DRUNK. JAMA. 1928;91:1103–1107. [Google Scholar]
- 4.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3:270–303. doi: 10.1017/s0033291700049588. [DOI] [PubMed] [Google Scholar]
- 5.McKee AC, Daneshvar DH, Alvarez VE, Stein TD. The neuropathology of sport. Acta Neuropathol. 2014;127:29–51. doi: 10.1007/s00401-013-1230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stewart W, McNamara PH, Lawlor B, Hutchinson S, Farrell M. Chronic traumatic encephalopathy: a potential late and under recognized consequence of rugby union? QJM Mon J Assoc Physicians. 2016;109:11–15. doi: 10.1093/qjmed/hcv070. [DOI] [PubMed] [Google Scholar]
- 7.Johnson VE, Stewart W, Smith DH. Widespread τ and amyloid-β pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012;22:142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol. 2013;9:211–221. doi: 10.1038/nrneurol.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP, Stewart W, Tripodis Y, Crary JF, Bieniek KF, Dams-O’Connor K, Alvarez VE, Gordon WA TBI/CTE group. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016;131:75–86. doi: 10.1007/s00401-015-1515-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hay J, Johnson VE, Smith DH, Stewart W. Chronic Traumatic Encephalopathy: The Neuropathological Legacy of Traumatic Brain Injury. Annu Rev Pathol. 2016;11:21–45. doi: 10.1146/annurev-pathol-012615-044116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gold EM, Su D, López-Velázquez L, Haus DL, Perez H, Lacuesta GA, Anderson AJ, Cummings BJ. Functional assessment of long-term deficits in rodent models of traumatic brain injury. Regen Med. 2013;8:483–516. doi: 10.2217/rme.13.41. [DOI] [PubMed] [Google Scholar]
- 12.Osier ND, Carlson SW, DeSana A, Dixon CE. Chronic Histopathological and Behavioral Outcomes of Experimental Traumatic Brain Injury in Adult Male Animals. J Neurotrauma. 2015;32:1861–1882. doi: 10.1089/neu.2014.3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pierce JE, Smith DH, Trojanowski JQ, McIntosh TK. Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 1998;87:359–369. doi: 10.1016/s0306-4522(98)00142-0. [DOI] [PubMed] [Google Scholar]
- 14.Immonen RJ, Kharatishvili I, Gröhn H, Pitkänen A, Gröhn OHJ. Quantitative MRI predicts long-term structural and functional outcome after experimental traumatic brain injury. NeuroImage. 2009;45:1–9. doi: 10.1016/j.neuroimage.2008.11.022. [DOI] [PubMed] [Google Scholar]
- 15.Smith DH, Chen XH, Pierce JE, Wolf JA, Trojanowski JQ, Graham DI, McIntosh TK. Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma. 1997;14:715–727. doi: 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- 16.Bramlett HM, Dietrich WD. Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta Neuropathol. 2002;103:607–614. doi: 10.1007/s00401-001-0510-8. [DOI] [PubMed] [Google Scholar]
- 17.Shelton SB, Pettigrew DB, Hermann AD, Zhou W, Sullivan PM, Crutcher KA, Strauss KI. A simple, efficient tool for assessment of mice after unilateral cortex injury. J Neurosci Methods. 2008;168:431–442. doi: 10.1016/j.jneumeth.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shear DA, Tate MC, Archer DR, Hoffman SW, Hulce VD, Laplaca MC, Stein DG. Neural progenitor cell transplants promote long-term functional recovery after traumatic brain injury. Brain Res. 2004;1026:11–22. doi: 10.1016/j.brainres.2004.07.087. [DOI] [PubMed] [Google Scholar]
- 19.Dixon CE, Kochanek PM, Yan HQ, Schiding JK, Griffith RG, Baum E, Marion DW, DeKosky ST. One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J Neurotrauma. 1999;16:109–122. doi: 10.1089/neu.1999.16.109. [DOI] [PubMed] [Google Scholar]
- 20.Kochanek PM, Hendrich KS, Dixon CE, Schiding JK, Williams DS, Ho C. Cerebral blood flow at one year after controlled cortical impact in rats: assessment by magnetic resonance imaging. J Neurotrauma. 2002;19:1029–1037. doi: 10.1089/089771502760341947. [DOI] [PubMed] [Google Scholar]
- 21.Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol. 2014;73:14–29. doi: 10.1097/NEN.0000000000000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mouzon BC, Bachmeier C, Ferro A, Ojo JO, Crynen G, Acker CM, Davies P, Mullan M, Stewart W, Crawford F. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol. 2014;75:241–254. doi: 10.1002/ana.24064. [DOI] [PubMed] [Google Scholar]
- 23.Winston CN, Noël A, Neustadtl A, Parsadanian M, Barton DJ, Chellappa D, Wilkins TE, Alikhani AD, Zapple DN, Villapol S, Planel E, Burns MP. Dendritic Spine Loss and Chronic White Matter Inflammation in a Mouse Model of Highly Repetitive Head Trauma. Am J Pathol. 2016;186:552–567. doi: 10.1016/j.ajpath.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meehan WP, Zhang J, Mannix R, Whalen MJ. Increasing recovery time between injuries improves cognitive outcome after repetitive mild concussive brain injuries in mice. Neurosurgery. 2012;71:885–891. doi: 10.1227/NEU.0b013e318265a439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mannix R, Meehan WP, Mandeville J, Grant PE, Gray T, Berglass J, Zhang J, Bryant J, Rezaie S, Chung JY, Peters NV, Lee C, Tien LW, Kaplan DL, Feany M, Whalen M. Clinical correlates in an experimental model of repetitive mild brain injury. Ann Neurol. 2013;74:65–75. doi: 10.1002/ana.23858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pischiutta F, D’Amico G, Dander E, Biondi A, Biagi E, Citerio G, De Simoni MG, Zanier ER. Immunosuppression does not affect human bone marrow mesenchymal stromal cell efficacy after transplantation in traumatized mice brain. Neuropharmacology. 2014;79C:119–126. doi: 10.1016/j.neuropharm.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 27.Zanier ER, Montinaro M, Vigano M, Villa P, Fumagalli S, Pischiutta F, Longhi L, Leoni ML, Rebulla P, Stocchetti N, Lazzari L, De Simoni MG. Human umbilical cord blood mesenchymal stem cells protect mice brain after trauma. Crit Care Med. 2011;39:2501–2510. doi: 10.1097/CCM.0b013e31822629ba. [DOI] [PubMed] [Google Scholar]
- 28.Zanier ER, Pischiutta F, Riganti L, Marchesi F, Turola E, Fumagalli S, Perego C, Parotto E, Vinci P, Veglianese P, D’Amico G, Verderio C, De Simoni MG. Bone marrow mesenchymal stromal cells drive protective M2 microglia polarization after brain trauma. Neurotherapeutics. 2014;11:679–695. doi: 10.1007/s13311-014-0277-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zanier ER, Marchesi F, Ortolano F, Perego C, Arabian M, Zoerle T, Sammali E, Pischiutta F, De Simoni MG. Fractalkine Receptor Deficiency Is Associated with Early Protection but Late Worsening of Outcome following Brain Trauma in Mice. J Neurotrauma. 2016;33:1060–1072. doi: 10.1089/neu.2015.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brody DL, Mac Donald C, Kessens CC, Yuede C, Parsadanian M, Spinner M, Kim E, Schwetye KE, Holtzman DM, Bayly PV. Electromagnetic controlled cortical impact device for precise, graded experimental traumatic brain injury. J Neurotrauma. 2007;24:657–673. doi: 10.1089/neu.2006.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Blasio D, Fumagalli S, Longhi L, Orsini F, Palmioli A, Stravalaci M, Vegliante G, Zanier ER, Bernardi A, Gobbi M, De Simoni MG. Pharmacological inhibition of mannose-binding lectin ameliorates neurobehavioral dysfunction following experimental traumatic brain injury. J Cereb Blood Flow Metab. 2017;37:938–950. doi: 10.1177/0271678X16647397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zanier ER, Pischiutta F, Villa P, Paladini A, Montinaro M, Micotti E, Orrù A, Cervo L, De Simoni MG. Six-month ischemic mice show sensorimotor and cognitive deficits associated with brain atrophy and axonal disorganization. CNS Neurosci Ther. 2013;19:695–704. doi: 10.1111/cns.12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith DH, Okiyama K, Thomas MJ, Claussen B, McIntosh TK. Evaluation of memory dysfunction following experimental brain injury using the Morris water maze. J Neurotrauma. 1991;8:259–269. doi: 10.1089/neu.1991.8.259. [DOI] [PubMed] [Google Scholar]
- 34.Jenkinson M, Beckmann CF, Behrens TEJ, Woolrich MW, Smith SM. FSL. NeuroImage. 2012;62:782–790. doi: 10.1016/j.neuroimage.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 35.Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TEJ, Johansen-Berg H, Bannister PR, De Luca M, Drobnjak I, Flitney DE, Niazy RK, Saunders J, Vickers J, Zhang Y, De Stefano N, Brady JM, Matthews PM. Advances in functional and structural MR image analysis and implementation as FSL. NeuroImage. 2004;23(Suppl 1):S208–219. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 36.Woolrich MW, Jbabdi S, Patenaude B, Chappell M, Makni S, Behrens T, Beckmann C, Jenkinson M, Smith SM. Bayesian analysis of neuroimaging data in FSL. NeuroImage. 2009;45:S173–186. doi: 10.1016/j.neuroimage.2008.10.055. [DOI] [PubMed] [Google Scholar]
- 37.Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. San Diego, CA, USA: Academic Press; 2004. p. 138. [Google Scholar]
- 38.Palmgren A. Specific silver staining of nerve fibres. Acta Zool. 1960;41:239–265. [Google Scholar]
- 39.Longhi L, Perego C, Ortolano F, Zanier ER, Bianchi P, Stocchetti N, McIntosh TK, De Simoni MG. C1-inhibitor attenuates neurobehavioral deficits and reduces contusion volume after controlled cortical impact brain injury in mice. Crit Care Med. 2009;37:659–665. doi: 10.1097/CCM.0b013e318195998a. [DOI] [PubMed] [Google Scholar]
- 40.Zanier ER, Fumagalli S, Perego C, Pischiutta F, De Simoni MG. Shape descriptors of the “never resting” microglia in three different acute brain injury models in mice. Intensive Care Med Exp. 2015;3:39. doi: 10.1186/s40635-015-0039-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perego C, Fumagalli S, De Simoni MG. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J Neuroinflammation. 2011;8:174. doi: 10.1186/1742-2094-8-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pischiutta F, Brunelli L, Romele P, Silini A, Sammali E, Paracchini L, Marchini S, Talamini L, Bigini P, Boncoraglio GB, Pastorelli R, De Simoni MG, Parolini O, Zanier ER. Protection of Brain Injury by Amniotic Mesenchymal Stromal Cell-Secreted Metabolites. Crit Care Med. 2016;44:e1118–e1131. doi: 10.1097/CCM.0000000000001864. [DOI] [PubMed] [Google Scholar]
- 43.Zarruk JG, Garcia-Yebenes I, Romera VG, Ballesteros I, Moraga A, Cuartero MI, Hurtado O, Sobrado M, Pradillo JM, Fernandez-Lopez D, Serena J, Castillo-Melendez M, Moro MA, Lizasoain I. Neurological tests for functional outcome assessment in rodent models of ischaemic stroke. Rev Neurol. 2011;53:607–618. [PubMed] [Google Scholar]
- 44.Brooks SP, Dunnett SB. Tests to assess motor phenotype in mice: a user’s guide. Nat Rev Neurosci. 2009;10:519–529. doi: 10.1038/nrn2652. [DOI] [PubMed] [Google Scholar]
- 45.Dean RL, Scozzafava J, Goas JA, Regan B, Beer B, Bartus RT. Age-related differences in behavior across the life span of the C57BL/6J mouse. Exp Aging Res. 1981;7:427–451. doi: 10.1080/03610738108259823. [DOI] [PubMed] [Google Scholar]
- 46.Naude PJW, Dobos N, van der Meer D, Mulder C, Pawironadi KGD, den Boer JA, van der Zee EA, Luiten PGM, Eisel ULM. Analysis of cognition, motor performance and anxiety in young and aged tumor necrosis factor alpha receptor 1 and 2 deficient mice. Behav Brain Res. 2014;258:43–51. doi: 10.1016/j.bbr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 47.Griesbach GS, Hovda DA. Cellular and molecular neuronal plasticity. Handb Clin Neurol. 2015;128:681–690. doi: 10.1016/B978-0-444-63521-1.00042-X. [DOI] [PubMed] [Google Scholar]
- 48.Perrot-Sinal TS, Kostenuik MA, Ossenkopp KP, Kavaliers M. Sex differences in performance in the Morris water maze and the effects of initial nonstationary hidden platform training. Behav Neurosci. 1996;110:1309–1320. doi: 10.1037//0735-7044.110.6.1309. [DOI] [PubMed] [Google Scholar]
- 49.Tong W, Igarashi T, Ferriero DM, Noble LJ. Traumatic brain injury in the immature mouse brain: characterization of regional vulnerability. Exp Neurol. 2002;176:105–116. doi: 10.1006/exnr.2002.7941. [DOI] [PubMed] [Google Scholar]
- 50.Hall ED, Sullivan PG, Gibson TR, Pavel KM, Thompson BM, Scheff SW. Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. J Neurotrauma. 2005;22:252–265. doi: 10.1089/neu.2005.22.252. [DOI] [PubMed] [Google Scholar]
- 51.Zhuo J, Xu S, Proctor JL, Mullins RJ, Simon JZ, Fiskum G, Gullapalli RP. Diffusion kurtosis as an in vivo imaging marker for reactive astrogliosis in traumatic brain injury. NeuroImage. 2012;59:467–477. doi: 10.1016/j.neuroimage.2011.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robinson S, Winer JL, Berkner J, Chan LAS, Denson JL, Maxwell JR, Yang Y, Sillerud LO, Tasker RC, Meehan WP, Mannix R, Jantzie LL. Imaging and serum biomarkers reflecting the functional efficacy of extended erythropoietin treatment in rats following infantile traumatic brain injury. J Neurosurg Pediatr. 2016;17:739–755. doi: 10.3171/2015.10.PEDS15554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harris NG, Verley DR, Gutman BA, Sutton RL. Bi-directional changes in fractional anisotropy after experiment TBI: Disorganization and reorganization? NeuroImage. 2016;133:129–143. doi: 10.1016/j.neuroimage.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mac Donald CL, Dikranian K, Song SK, Bayly PV, Holtzman DM, Brody DL. Detection of traumatic axonal injury with diffusion tensor imaging in a mouse model of traumatic brain injury. Exp Neurol. 2007;205:116–131. doi: 10.1016/j.expneurol.2007.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Budde MD, Janes L, Gold E, Turtzo LC, Frank JA. The contribution of gliosis to diffusion tensor anisotropy and tractography following traumatic brain injury: validation in the rat using Fourier analysis of stained tissue sections. Brain. 2011;134:2248–2260. doi: 10.1093/brain/awr161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain J Neurol. 2013;136:28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol. 2013;246:35–43. doi: 10.1016/j.expneurol.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sidaros A, Engberg AW, Sidaros K, Liptrot MG, Herning M, Petersen P, Paulson OB, Jernigan TL, Rostrup E. Diffusion tensor imaging during recovery from severe traumatic brain injury and relation to clinical outcome: a longitudinal study. Brain. 2008;131:559–572. doi: 10.1093/brain/awm294. [DOI] [PubMed] [Google Scholar]
- 59.Kumar R, Saksena S, Husain M, Srivastava A, Rathore RKS, Agarwal S, Gupta RK. Serial changes in diffusion tensor imaging metrics of corpus callosum in moderate traumatic brain injury patients and their correlation with neuropsychometric tests: a 2-year follow-up study. J Head Trauma Rehabil. 2010;25:31–42. doi: 10.1097/HTR.0b013e3181bff331. [DOI] [PubMed] [Google Scholar]
- 60.Farbota KD, Bendlin BB, Alexander AL, Rowley HA, Dempsey RJ, Johnson SC. Longitudinal diffusion tensor imaging and neuropsychological correlates in traumatic brain injury patients. Front Hum Neurosci. 2012;6:160. doi: 10.3389/fnhum.2012.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perez AM, Adler J, Kulkarni N, Strain JF, Womack KB, Diaz-Arrastia R, Marquez de la Plata CD. Longitudinal white matter changes after traumatic axonal injury. J Neurotrauma. 2014;31:1478–1485. doi: 10.1089/neu.2013.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hill CS, Coleman MP, Menon DK. Traumatic Axonal Injury: Mechanisms and Translational Opportunities. Trends Neurosci. 2016;39:311–324. doi: 10.1016/j.tins.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Buss A, Schwab ME. Sequential loss of myelin proteins during Wallerian degeneration in the rat spinal cord. Glia. 2003;42:424–432. doi: 10.1002/glia.10220. [DOI] [PubMed] [Google Scholar]
- 64.Shinjo R, Imagama S, Ito Z, Ando K, Nishida Y, Ishiguro N, Kadomatsu K. Keratan sulfate expression is associated with activation of a subpopulation of microglia/macrophages in Wallerian degeneration. Neurosci Lett. 2014;579:80–85. doi: 10.1016/j.neulet.2014.07.018. [DOI] [PubMed] [Google Scholar]
- 65.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Green REA, Colella B, Maller JJ, Bayley M, Glazer J, Mikulis DJ. Scale and pattern of atrophy in the chronic stages of moderate-severe TBI. Front Hum Neurosci. 2014;8:67. doi: 10.3389/fnhum.2014.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Onyszchuk G, LeVine SM, Brooks WM, Berman NEJ. Post-acute pathological changes in the thalamus and internal capsule in aged mice following controlled cortical impact injury: a magnetic resonance imaging, iron histochemical, and glial immunohistochemical study. Neurosci Lett. 2009;452:204–208. doi: 10.1016/j.neulet.2009.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Okie S. TBI’s Long-Term Follow-up--Slow Progress in Science and Recovery. N Engl J Med. 2016;375:180–184. doi: 10.1056/NEJMms1604272. [DOI] [PubMed] [Google Scholar]
- 69.Stocchetti N, Zanier ER. Chronic impact of traumatic brain injury on outcome and quality of life: a narrative review. Crit Care. 2016:20. doi: 10.1186/s13054-016-1318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rigon A, Duff MC, McAuley E, Kramer AF, Voss MW. Is Traumatic Brain Injury Associated with Reduced Inter-Hemispheric Functional Connectivity? A Study of Large-Scale Resting State Networks following Traumatic Brain Injury. J Neurotrauma. 2016;33:977–989. doi: 10.1089/neu.2014.3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McMillan TM, Teasdale GM, Stewart E. Disability in young people and adults after head injury: 12–14 year follow-up of a prospective cohort. J Neurol Neurosurg Psychiatry. 2012;83:1086–1091. doi: 10.1136/jnnp-2012-302746. [DOI] [PubMed] [Google Scholar]
- 72.Giacino JT, Whyte J, Bagiella E, Kalmar K, Childs N, Khademi A, Eifert B, Long D, Katz DI, Cho S, Yablon SA, Luther M, Hammond FM, Nordenbo A, Novak P, Mercer W, Maurer-Karattup P, Sherer M. Placebo-controlled trial of amantadine for severe traumatic brain injury. N Engl J Med. 2012;366:819–826. doi: 10.1056/NEJMoa1102609. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.