

Abstract
During sepsis, systemic inflammation is observed and is associated with multiple organ failure. Activation of NF-κB is crucial for inducing inflammation, which is controlled by degradation of IκB. The ubiquitination proteasome pathway is responsible for the regulation of protein turnover. In this study, we hypothesized that administration of PYR-41, an inhibitor of ubiquitination, could reduce inflammation and organ injury in septic mice. PYR-41 prevented the reduction of IκB protein levels and inhibited release of TNF-α in mouse macrophage RAW264.7 cells at 4 h after lipopolysaccharide stimulation dose-dependently. Male C57BL/6 mice were subjected to cecal ligation and puncture (CLP) to induce sepsis. PYR-41 (5 mg/kg) or DMSO in saline (vehicle) was injected intravenously immediately after CLP. At 20 h after CLP, PYR-41 treatment significantly decreased serum levels of proinflammatory cytokines (TNF-α, IL-1β, and IL-6) and organ injury markers (AST, ALT, and LDH). PYR-41 significantly improved microscopic structure, and reduced myeloperoxidase activity, number of apoptotic cells and caspase-3 degradation in the lungs of septic mice. The reduced protein levels of IκB in the lungs after CLP were restored by PYR-41 treatment. PYR-41 inhibited the expression of cytokines (IL-1β and IL-6), chemokines (KC and MIP-2), and inflammatory mediators (COX-2 and iNOS) in the lungs of septic mice. Importantly, PYR-41 significantly increased 10-day survival in septic mice from 42% to 83%. Therefore, targeting ubiquitination by PYR-41 to inhibit NF-κB activation may represent a potential strategy of sepsis therapeutics.
Keywords: ubiquitination, IκB, inflammation, lung injury, sepsis
INTRODUCTION
Sepsis is one of the most prevalent medical conditions and accounts for 20% of admissions to intensive care units (1). In the United States, more than 750,000 people develop sepsis each year, of which at least 225,000 die (2, 3). Despite numerous efforts to advance the understanding of sepsis progression, including more than thirty failed clinical trials, there is still no effective pharmacotherapy available to treat sepsis (4–6). Clearly, there is an urgent, unmet medical need to identify and develop a new class of therapeutic agents against sepsis.
The pathophysiology of sepsis is complex. Recent consensus definitions state that sepsis is a life-threatening organ dysfunction caused by a dysregulated host response to infection (7). During the early phase of sepsis, patients present with an exaggerated, hyperinflammatory immune response, also described as a “cytokine storm” (8). Such massive cytokine production can be triggered by exogenous pathogen-associated molecular pattern molecules (PAMPs) from invading microorganisms, or by endogenous damage-associated molecular pattern molecules (DAMPs) released from injured host cells (9, 10). Both PAMPs and DAMPs bind to various types of receptors in immune cells and activate several signaling pathways, and in particular the NF-κB pathway, to stimulate the production of inflammatory mediators such as cytokines and chemokines (11).
NF-κB is a family of inducible transcription factors comprised of five structurally related proteins that form homodimers and heterodimers in mammalian cells (12). These NF-κB dimers are inactive in the cytoplasm through binding to a family of inhibitor molecules (IκB). When cells are engaged with extracellular stimuli, IκB is phosphorylated and undergoes ubiquitination and proteasomal degradation, leading to the release of NF-κB. Then, NF-κB proteins translocate to the nucleus and bind to targeted DNA sequences, upregulating the expression of a wide range of genes critical to host defense and the inflammatory response (12). Therefore, inhibiting NF-κB activity is a rational strategy for controlling the cytokine storm in sepsis.
Ubiquitination of IκB plays a key role in its degradation and subsequently the activation of NF-κB. Ubiquitination is a post-translational modification that occurs through a multistep enzymatic process that attaches a small ubiquitin protein covalently on a lysine residue of a substrate protein (13). The process is initiated by a ubiquitin-activating (E1) enzyme that uses ATP to activate ubiquitin for conjugation and transfers it to a ubiquitin-conjugating (E2) enzyme. The E2 enzyme then interacts with a ubiquitin ligase (E3) and transfers the ubiquitin to the target protein (13). The ubiquitinated proteins are then recognized, unfolded, and degraded by the proteasome enzyme complex (14). 4[4-(5-nitro-furan-2-ylmethylene)-3, -dioxo-pyrazolidin-1-yl]-benzoic acid ethyl ester (PYR-41), is a small, cell permeable chemical compound that selectively inhibits the E1 enzyme (15). In this study, we hypothesized that inhibiting IκB degradation by administration of PYR-41 would attenuate sepsis-induced inflammation and organ injury in mice.
MATERALS AND METHODS
Cell culture
Mouse macrophage RAW264.7 cells obtained from ATCC (Manassas, VA) were cultured in DMEM media (Invitrogen, Grand Island, NY) containing 10% FBS, 1% penicillin/streptomycin and 1% glutamine, and maintained in a 37°C incubator with 5% CO2.
Animal model of sepsis
Male C57BL/6 mice (20–25 g) were obtained from Taconic (Albany, NY) and sepsis was induced by cecal ligation and puncture (CLP). Before operation, mice were anesthetized by isoflurane inhalation, and the abdomen was shaved and washed with 10% povidone iodine. An incision was made and the cecum was ligated using 4-0 silk suture and double-punctured with a 22-gauge needle. Animals were administered 20% DMSO in normal saline (vehicle) or 5 mg/kg body weight PYR-41 (Tocris, Park Ellisville, MO) by intravenous injection immediately after CLP. Mice were resuscitated with 0.5 ml normal saline by subcutaneous injection immediately after surgery. At 20 h after CLP, blood and tissues were collected for various analyses. For the survival study, mice were administered the broad-spectrum antibiotic Primaxin (0.5 mg/kg; Merck, Kenilworth, NJ) subcutaneously after CLP. The animals were monitored for 10 days to record survival. All experiments were performed in accordance with the guidelines for the use of experimental animals by the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of The Feinstein Institute for Medical Research.
Measurements of cytokines and organ injury markers
TNF-α, IL-1β, and IL-6 were quantified using specific mouse ELISA kits (BD Biosciences, Franklin Lakes, NJ). Aspartate aminotransferase (AST), alanine aminotransferase (ALT), and lactate dehydrogenase (LDH) were measured using commercial assay kits (Pointe Scientific, Lincoln Park, MI).
Histopathologic examination
Lung tissues were fixed in 10% formalin, embedded in paraffin, and stained with hematoxylin and eosin. Morphologic examination of these tissues was performed under a light microscope in a blinded manner. The severity of lung injury was judged by a semiquantitative scoring system according to the following pathological features: (i) focal alveolar membrane thickening, (ii) capillary congestion, (iii) intra-alveolar hemorrhage, (iv) interstitial neutrophil infiltration, and (v) intra-alveolar neutrophil infiltration. Each feature was scored from 0 to 3 based on its absence (0) or presence to a mild (1), moderate (2), or severe (3) degree, and a cumulative total histology score was determined (16).
Myeloperoxidase (MPO) activity assay
Lung tissues were homogenized in potassium phosphate buffer containing 0.5% hexa-decyl-trimethyl-ammonium bromide. After centrifugation, the supernatant was diluted in reaction solution, and the rate of change in optical density for 2 min was measured at 460 nm to calculate MPO activity.
Western blotting analysis
Tissue and cell lysates were homogenized in lysis buffer (10 mM Tris-HCl, pH 7.5, 120 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) containing a protease inhibitor cocktail (Roche Diagnostics) by sonication. Total protein was fractionated on Bis-Tris gels (4–12%) and transferred to nitrocellulose membranes. Nitrocellulose membranes were blocked by incubation in TBST (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.1% Tween 20) containing 10% non-fat dry milk for 1 h. Membranes were incubated with anti-IκB (Santa Cruz Biotechnology, Santa Cruz, CA), anti-cleaved caspase-3, or anti-β-actin (Sigma, St Louis, MO) antibodies overnight at 4°C, followed by incubation with HRP-conjugated secondary antibody for 1 h. Specific proteins were visualized by using Pierce ECL2 Western Blotting Substrate (Thermo Scientific, Southfield, MI) or Luminata Forte™ Western HRP Substrate (Millipore, Billerica, MA). Band densities were determined using a Bio-Rad image system.
q PCR analysis
Total RNA was extracted from lung tissues using a Trizol reagent (Invitrogen, Carlsbad, CA) and was reverse-transcribed into cDNA using murine leukemia virus reverse transcriptase (Applied Biosystems, Foster City, CA). A PCR reaction was carried out in 25 μl of a final volume containing 0.08 μmol of each forward and reverse primer, cDNA, and 12.5 μl SYBR Green PCR Master Mix (Applied Biosystems). Amplification was conducted in an Applied Biosystems 7300 real-time PCR machine under the thermal profile of 50°C for 2 min, 95°C for 10 min followed by 45 cycles of 95°C for 15 sec and 60°C for 1 min. The level of mouse β-actin mRNA was used for normalization. Relative expression of mRNA was expressed as the fold change compared to the sham. The primers used for this study are listed in Table 1.
TABLE 1.
A list of primer sequences used in this study
| Name | GenBank | Forward | Reverse |
|---|---|---|---|
| IL-1β | NM_008361 | CAGGATGAGGACATGAGCACC | CTCTGCAGACTCAAACTCCAC |
| IL-6 | NM_031168 | CCGGAGAGGAGACTTCACAG | CAGAATTGCCATTGCACAAC |
| KC | NM_008176 | GCTGGGATTCACCTCAAGAA | ACAGGTGCCATCAGAGCAGT |
| MIP-2 | NM_009140 | CCCTGGTTCAGAAAATCATCCA | GCTCCTCCTTTCCAGGTCAGT |
| COX-2 | NM_011198 | CTCAGCCAGGCAGCAAATC | ACATTCCCCACGGTTTTGAC |
| iNOS | NM_010927 | GCAGGTCGAGGACTATTTCTTTCA | GAGCACGCTGAGTACCTCATTG |
| β-actin | NM_007393 | CGTGAAAAGATGACCCAGATCA | TGGTACGACCAGAGGCATACAG |
Terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) assay
Tissue slides were dewaxed and incubated with proteinase K. Slides were then stained using a TUNEL kit (Roche Diagnostics, Indianapolis, IN), counterstained with propidium iodide and examined under a fluorescence microscope. Apoptotic cells appeared green fluorescent and were counted across 10 visual fields at 200× magnification.
Statistical analysis
Data are expressed as mean ± standard error and compared with one-way analysis of variance (ANOVA) and the Student-Newman-Keuls test for multiple group analyses. The survival rate was estimated by the Kaplan-Meier method, and rates were compared using the log-rank test. Differences in values were considered significant if P < 0.05.
RESULTS
PYR-41 inhibits cytokine production in macrophages stimulated with LPS
In sepsis, release of proinflammatory cytokines from macrophages is the main cause of the “cytokine storm” (8). We first determined the effectiveness of PYR-41 in attenuating NF-κB activation in macrophages. The expression levels of IκB, an NF-κB inhibitor, in RAW 264.7 cells were decreased by 54% at 30 min after lipopolysaccharide (LPS) stimulation (Fig. 1A). With PYR-41 treatment at 10 and 20 μM, the expression levels of IκB were restored to 89% and 95% of those in the non LPS-stimulated RAW 264.7 cells, respectively (Fig. 1A). We then measured the TNF-α levels in cultured media grown with RAW 264.7 cells. After 4 h-LPS stimulation, the TNF-α levels increased from 0.03 to 1.34 ng/ml, while treatment with PYR-41 at doses of 5, 10, and 20 μM decreased TNF-α levels by 38%, 81%, and 94%, respectively (Fig. 1B). We also examined the viability of RAW 264.7 cells by MTS assay and observed no adverse effects of PYR-41 on cell viability (Fig. 1C).
FIG. 1. Effect of PYR-41 on IκB protein levels and TNF-α production in macrophages stimulated with LPS.
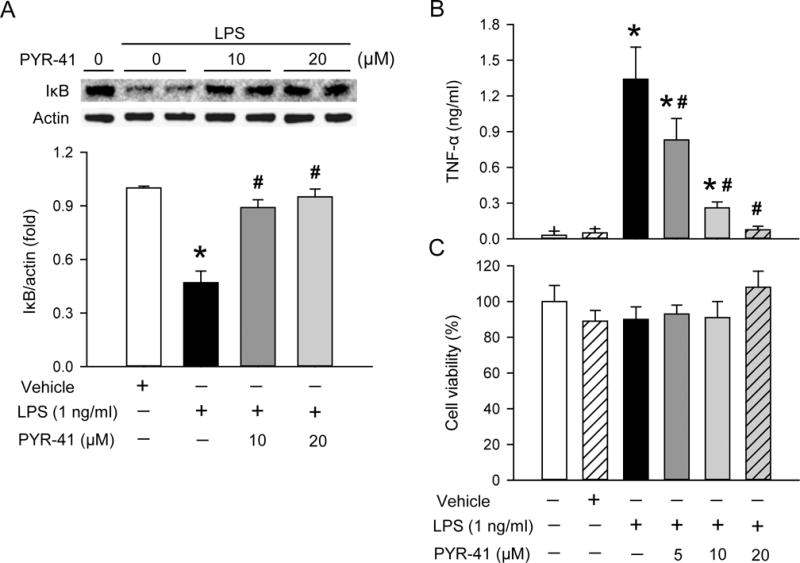
Cultured RAW 264.7 cells were pre-treated with PYR-41 at the indicated concentration for 30 min and then stimulated with LPS (1 ng/ml) for a period of time. (A) Western blot analysis of total cell lysates after 30 min of LPS stimulation. Representative blots against IκB and actin that is used as a loading control. Blots were scanned and quantified with densitometry. The levels of protein expression in the non LPS-stimulated cells are designated 1 for comparison. (B) Medium from cultured RAW 264.7 cells with 4 h of LPS-stimulation was subjected to ELISA for measurement of TNF-α levels. (C) Cell viability of RAW 264.7 cells was determined by MTS assay. The viability of untreated RAW 264.7 cells was considered to be 100%. Data expressed as mean ± standard error (n = 3–5/group). *P < 0.05 versus vehicle and #P < 0.05 versus LPS alone.
PYR-41 treatment attenuates systemic inflammation and organ injury markers in septic mice
After demonstrating PYR-41’s inhibition of cytokine production in vitro, we then administered PYR-41 to mice that underwent sepsis-induction by CLP. At 20 h after CLP, serum levels of proinflammatory cytokines TNF-α, IL-1β, and IL-6 were markedly increased compared to the sham (Fig. 2, A–C). In contrast, treatment with PYR-41 significantly reduced their levels by 79%, 77%, and 89%, respectively, compared to the vehicle (Fig. 2, A–C). Serum levels of organ injury markers AST, ALT, and LDH were also elevated after CLP, while their levels were reduced by 27%, 43%, and 52%, respectively, with PYR-41 treatment (Fig. 2, D–F).
FIG. 2. Effect of PYR-41 treatment on serum levels of cytokines and organ injury markers in mice after CLP.
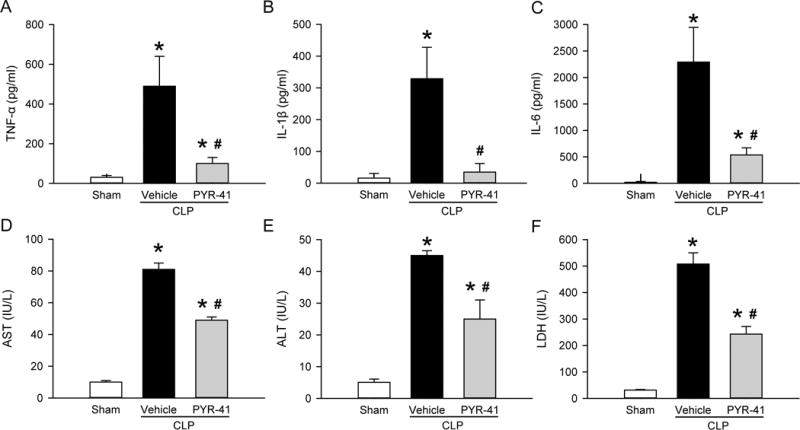
Mice were sham-operated or subjected to CLP with intravenous injection of vehicle (20% DMSO in saline) or PYR-41 (5 mg/kg) immediately after CLP. Blood samples were collected at 20 h after CLP to measure serum levels of (A) TNF-α, (B) IL-1β, and (C) IL-6 by ELISA as well as levels of (D) AST, (E) ALT, and (F) LDH by enzymatic methods. Data expressed as mean ± standard error (n = 5/group). *P < 0.05 versus sham and #P < 0.05 versus vehicle.
PYR-41 treatment prevents lung injury in septic mice
The lungs are one of the most vulnerable organs affected during sepsis (17). We first performed histologic examination of the lung tissues at 20 h after CLP and observed substantial morphologic changes in the vehicle group, including alveolar collapse, edema, hemorrhage and infiltration of inflammatory cells compared to the sham (Fig. 3A). After treatment with PYR-41, the morphologic appearance of lung tissues was improved (Fig. 3A). We further graded the severity of the lung damage and showed a 74% reduction in histology injury score in the treatment group compared to the vehicle (Fig. 3B). Excessive immune cell infiltration, especially by neutrophils, is one important factor causing lung injury in sepsis (18). Lung myeloperoxidase (MPO) activity, a marker of neutrophil infiltration (19), was markedly increased after CLP by 19-fold compared to the sham, while it was significantly reduced by 64% with PYR-41 treatment compared to the vehicle (Fig. 3C).
FIG. 3. Histologic analysis in the lungs of mice after CLP.
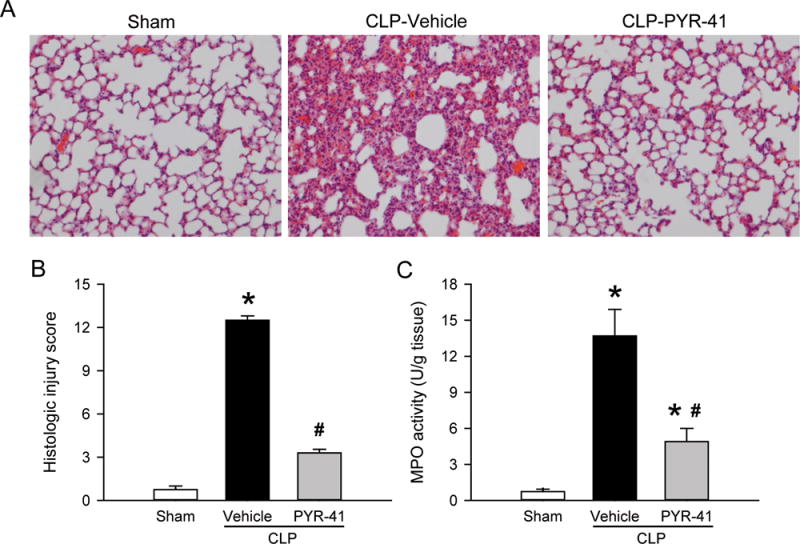
Mice were sham-operated or subjected to CLP with intravenous injection of vehicle (20% DMSO in saline) or PYR-41 (5 mg/kg) immediately after CLP. Lungs were harvested at 20 h after CLP. (A) Representative images of lung tissue sections stained with H & E under light microscope at 200× magnification. (B) Histologic injury scores of the lung in each group were graded as described in methods. (C) Myeloperoxidase (MPO) activity in lung tissue homogenates was determined spectrophotometrically. Data are expressed as mean ± standard error (n = 5/group). *P < 0.05 versus sham and #P < 0.05 versus vehicle.
We also assessed apoptosis in the lung by performing a TUNEL assay on the tissue sections. As shown in Figure 4A, the TUNEL-positive cells in the vehicle were well-detected, while they were barely seen in the sham. Treatment with PYR-41 significantly reduced the number of TUNEL-positive or apoptotic cells in the lungs (Fig. 4, A and B). In addition, we examined the expression of cleaved caspase-3, another marker of apoptosis (20), in the lungs by Western blotting. The levels of cleaved caspase-3 expression were significantly increased after CLP, but reduced with PYR-41 treatment to levels comparable to the sham (Fig. 4C).
FIG. 4. Detection of apoptosis in the lungs of mice after CLP.
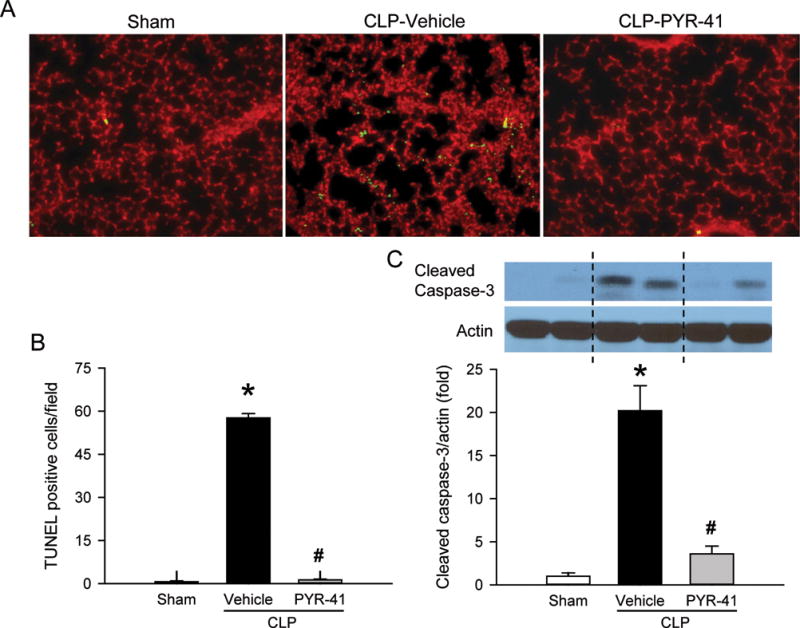
Mice were sham-operated or subjected to CLP with intravenous injection of vehicle (20% DMSO in saline) or PYR-41 (5 mg/kg) immediately after CLP. Lungs were harvested at 20 h after CLP. (A) Lung tissue sections were immunostained with terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL; green) for apoptotic cells and propidium iodide (red) staining for nuclei. Representative images of each group at 200× magnification. (B) Graphical representation of TUNEL-positive staining cells averaged over 10 microscopic fields per mouse. (C) Western blot analysis of cleaved caspase-3 in lung tissue homogenates. Actin is used as a loading control. Blots were scanned and quantified with densitometry. The levels of protein expression in the sham group are designated 1 for comparison. Data are expressed as mean ± standard error (n = 5/group). *P < 0.05 versus sham and #P < 0.05 versus vehicle.
PYR-41 treatment inhibits inflammation in the lungs of septic mice
After demonstrating the protective effect of PYR-41 on the lungs of septic mice, we examined the status of NF-κB activation by assessing the expression levels of IκB using Western blotting. The levels of IκB were reduced in the vehicle group compared to the sham, while its levels in the PYR-41 treatment group were returned to the sham level (Fig. 5). Consequently, we measured the expression of proinflammatory cytokines IL-1β and IL-6, target genes of the NF-κB signaling pathway, in the lungs. The mRNA and protein levels of both cytokines were markedly elevated after CLP, while levels of IL-1β and IL-6 were significantly reduced by PYR-41 treatment, ranging from a 62% to 87% reduction compared to the vehicle (Fig. 6, A–D).
FIG. 5. Effect of PYR-41 treatment on IκB protein levels in the lungs of mice after CLP.
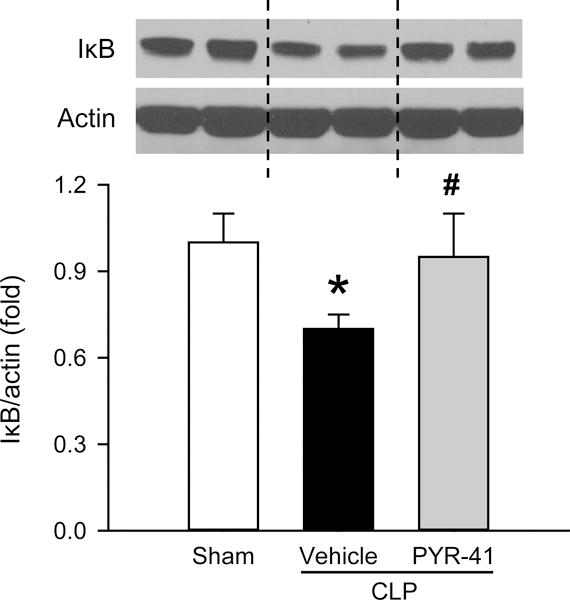
Mice were sham-operated or subjected to CLP with intravenous injection of vehicle (20% DMSO in saline) or PYR-41 (5 mg/kg) immediately after CLP. Lungs were harvested at 20 h after CLP. Western blot analysis of IκB expression in lung tissue homogenates. Representative blots against IκB and actin that is used as a loading control. Blots were scanned and quantified with densitometry. The levels of protein expression in the sham group are designated 1 for comparison. Data are expressed as mean ± standard error (n = 5/group). *P < 0.05 versus sham and #P < 0.05 versus vehicle.
FIG. 6. Effect of PYR-41 treatment on expression of cytokines and chemokines in the lungs of mice after CLP.
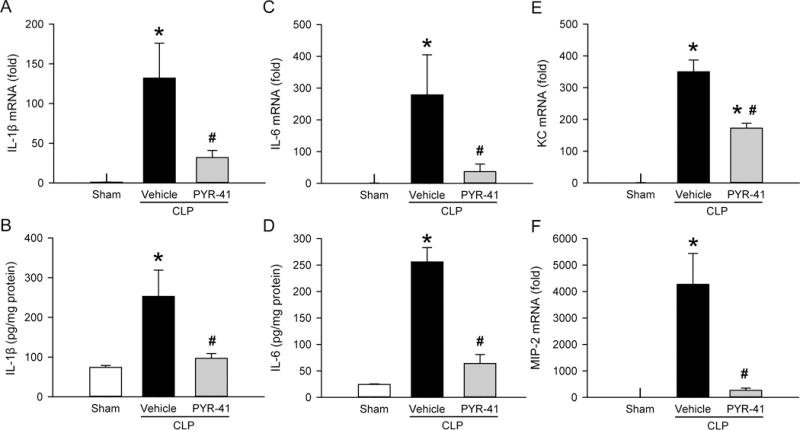
Mice were sham-operated or subjected to CLP with intravenous injection of vehicle (20% DMSO in saline) or PYR-41 (5 mg/kg) immediately after CLP. Lungs were harvested at 20 h after CLP. The levels of (A) IL-1β mRNA, (B) IL-1β protein, (C) IL-6 mRNA, (D) IL-6 protein, (E) KC mRNA, and (F) MIP-2 mRNA in lung tissue homogenates were determined by qPCR for mRNA levels and ELISA for protein levels. The results of qPCR analysis are normalized with actin as an internal control and are expressed as fold induction compared to the sham group. Data expressed as mean ± standard error (n = 5/group). *P < 0.05 versus sham and #P < 0.05 versus vehicle.
We also determined the expression of proinflammatory chemokines keratinocyte-derived chemokine (KC) and macrophage inflammatory protein 2 (MIP-2), additional target genes of the NF-κB signaling pathway. Similar to cytokines, mRNA levels of both chemokines in the lungs were increased after CLP, but inhibited with PYR-41 treatment (Fig. 6, E and F).
PYR-41 treatment decreases the expression of inflammatory mediators in the lungs of septic mice
In the inflammatory cascade, cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) are important mediators and contribute to organ injury (21). We next measured mRNA levels of both molecules in the lungs by qPCR. At 20 h after CLP, the level of COX-2 mRNA was increased by 3.4-fold compared to the sham, while its level was reduced by 56% with PYR-41 treatment (Fig. 7A). Likewise, the level of iNOS mRNA was increased by 8.6-fold compared to the sham, while its level was reduced by 72% with PYR-41 treatment (Fig. 7B).
FIG. 7. Effect of PYR-41 treatment on expression of inflammatory mediators in the lungs of mice after CLP.
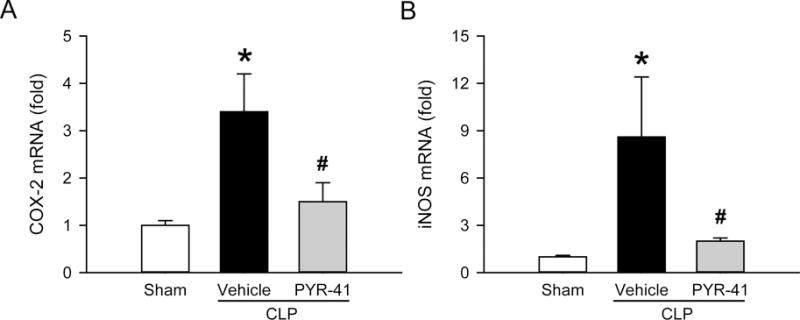
Mice were sham-operated or subjected to CLP with intravenous injection of vehicle (20% DMSO in saline) or PYR-41 (5 mg/kg) immediately after CLP. Lungs were harvested at 20 h after CLP. The mRNA levels of (A) COX-2 and (B) iNOS in lung tissue homogenates were determined by qPCR. The results of qPCR analysis are normalized with actin as an internal control and are expressed as fold induction compared to the sham group. Data expressed as mean ± standard error (n = 5/group). *P < 0.05 versus sham and #P < 0.05 versus vehicle.
PYR-41 treatment improves survival in septic mice
Lastly, we performed a 10-day survival study to evaluate the overall beneficial effect of PYR-41 in sepsis using a mouse CLP model. When administered immediately after CLP, PYR-41 significantly increased the survival rate was from 42% in the vehicle group to 83% (Fig. 8). When administered 5 h after CLP, PYR-41 increased the survival rate to 67%, although this did not reach statistical significance (Fig. 8).
FIG. 8. Effect of PYR-41 treatment on the survival of mice after CLP.
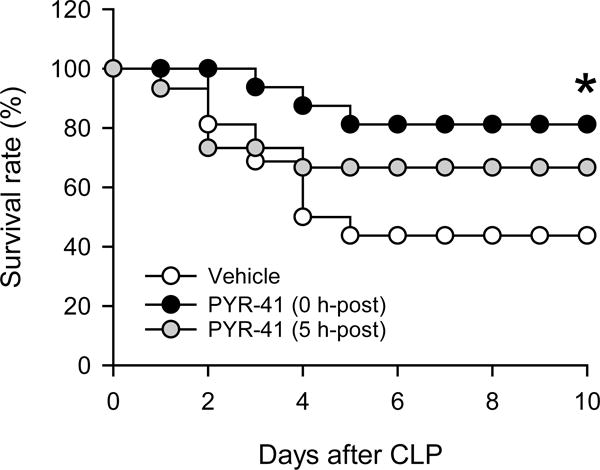
Mice were subjected to CLP with intravenous injection of vehicle (20% DMSO in saline; white circle), PYR-41 (5 mg/kg) immediately (black circle), or PYR-41 (5 mg/kg) at 5 h (gray circle) after CLP. Mice were observed for 10 days to record the mortality. Survival rates were analyzed by the Kaplan-Meier estimator using a log-rank test. *P < 0.05 versus vehicle.
DISCUSSION
Current management of septic patients is supportive in nature, and no effective pharmacotherapies are available with the exception of early administration of antibiotics. Despite the gap in the translation of laboratory findings to the clinical setting, identifying pharmacological targets remains a high priority for the development of effective therapies for sepsis. In sepsis, patients will develop hyperinflammation against infection, which leads to multiple organ dysfunction (22). It is well understood that activation of the transcription factor NF-κB plays a crucial role in regulating the production of proinflammatory cytokines and mediators during sepsis (22). In this study, we evaluated the effect of ubiquitination inhibition on inflammation and organ injury in sepsis using a CLP mouse model, which is a clinically relevant model to mimic human sepsis (23).
LPS is the major component of the outer membrane of Gram-negative bacteria and activates the NF-κB pathway in various cell types leading to the release of proinflammatory cytokines in the host (24). By using an in vitro cell culture system, we first demonstrated that PYR-41 treatment inhibits TNF-α production in LPS-stimulated macrophages in a dose-dependent manner. We also demonstrated that the degradation of IκB is inhibited in LPS-stimulated macrophages after PYR-41 treatment. In the study of Yang et al, they have used an in vitro ubiquitylation reaction assay to demonstrate the specificity of PYR-41 in inhibiting ubiqutin E1 enzyme activity (15). Consistently, they have also shown that PYR-41 treatment prevents IκB degradation in cancer cells stimulated with IL-1α and TNF-α (15). It also reports that PYR-41 treatment induces cell death in transformed cells in a dose-dependent manner, but has much less of an effect on untransformed cells (15). In this study, we did not observe the effect of PYR-41 treatment on macrophage viability, which also indicates that the reduction of TNF-α levels by PYR-41 is not due to a decrease in macrophage number in the assay.
Based on PYR-41’s effectiveness in inhibiting cytokine production in vitro, we then sought to administer it in an animal model of sepsis. By analyzing serum samples from septic mice, we showed that the levels of inflammatory cytokines TNF-α, IL-1β, and IL-6 are significantly reduced with PYR-41 treatment. The synthesis of these three cytokines is known to be mediated by the NF-κB pathway (25). This result indicates that administration of PYR-41 is effective in blocking downstream events of NF-κB activation in the inflammatory response in animals. Subsequently, we demonstrated that several serum organ injury markers (AST, ALT and LDH) are also attenuated in septic mice after PYR-41 treatment, validating the link between hyperinflammation and multiple organ injury in sepsis.
At the organ level, we examined the lung in septic mice. After CLP, the lung tissues demonstrated severe damage on histologic analysis. With PYR-41 treatment, the structural integrity of the lung was better preserved, with statistically significant improvement in histology score. Apoptosis is an important contributor to lung injury, and inflammation is a major inducer of apoptosis (26). By using TUNEL assay and detection of caspase-3 cleavage, we demonstrated apoptosis in the lungs of mice after CLP. PYR-41 treatment effectively decreased the number of apoptotic cells and caspase-3 cleavage in the lungs, reflecting an improvement in lung damage after CLP. Unlike its role in inducing apoptosis in cancer cells (15), PYR-41 treatment has not been associated with induction of apoptosis in septic animals. Whether PYR-41 treatment can improve pulmonary function in septic mice requires further investigation.
Furthermore, we demonstrated that PYR-41 treatment restored expression levels of IκB in the lungs of septic mice. The decrease of IκB levels may be due to its degradation triggered by the ubiquitination. It has been reported a marked increase in accumulation of ubiquitinated proteins in the lungs of septic mice (27). Therefore, PYR-41 treatment can effectively reduce the IκB ubiquitination and subsequently its degradation, leading to inhibition of NF-κB activation. Similarly to systemic inflammation, we also demonstrated that mRNA and protein levels of IL-1β and IL-6 in the lungs are significantly reduced with PYR-41 treatment. NF-κB plays a key role in controlling lung injury. A study has also shown that targeting the NF-κB pathway by administering an IκB kinase inhibitor can reduce the severity and prevent the progression of lung injury in mice in an LPS pump model (28).
In addition to cytokines, we also examined the levels of chemokines KC and MIP-2 in the lungs. The transcriptional regulation of both chemokines can be mediated through NF-κB (29). Consistently, we demonstrated that PYR-41 treatment significantly inhibits the elevation of KC and MIP-2 mRNA levels in septic mice. It has also been reported that KC and MIP-2 are sufficient to induce neutrophil recruitment to the lung (30, 31). Indeed, we observed an increase in neutrophil infiltration (MPO activity) in the lungs of septic mice, which was reduced by PYR-41 treatment. Due to the release of proteolytic enzymes and reactive oxygen species, excessive infiltration of neutrophils disrupts the endothelial barrier and causes tissue damage (18, 32). COX-2 and iNOS are additional inflammatory mediators that we have examined in the lungs. The promoters of COX-2 and iNOS contain NF-κB binding sites (33, 34). We also observed upregulation of mRNA levels of these mediators in the lungs of septic mice, whereas their levels were significantly inhibited by PYR-41 treatment. It has also been reported that administration of a selective COX-2 inhibitor ameliorates inflammation, injury, and sepsis-associated mortality in an animal model (35). Elevation of iNOS expression can lead to increased production of nitric oxide and formation of highly reactive peroxynitrite, which causes tissue damage and organ failure (36).
In addition to ubiquitin-tagging, protein turnover is also regulated by proteasome degradation (37). These two successive processes are known as the ubiquitin-proteasome pathway (UPP) (37). Several inhibitors targeting the proteasome have been developed as therapeutic agents, including bortezomib, which is approved by the Food and Drug Administration for the treatment of multiple myeloma (38). When using a proteasome inhibitor to study UPP, IκB was the first substrate to be identified (39). Thus, the potential of using these proteasome inhibitors for treating various inflammatory diseases has been evaluated (40), including the use of MG132 for sepsis treatment in the CLP model (41). The success of proteasome inhibitors for clinical use holds promise for the further development of ubiquitination inhibitors as therapeutic agents to target UPP. However, ubiquitination is a basic mechanism for controlling protein turnover in various types of cells. The toxicity and adverse effects of targeting ubiquitination need to be carefully evaluated for clinical use. Furthermore, whether PYR-41 has targets other than the NF-κB pathway requires further investigation. Another concern is that the use of anti-inflammatory drugs may lead to susceptibility to secondary infection and worsen wound healing in septic patients. However, careful control of the dose and timing of anti-inflammatory drug administration may be achieved to balance immune system functions and provide an overall beneficial outcome for patients.
In summary, by administering PYR-41 to restore the expression of IκB, an NF-κB inhibitor, we have demonstrated its effect on attenuating systemic inflammation and general organ injury in septic mice. At the organ level, we have shown an improvement in morphologic integrity, a reduction in proinflammatory cytokine and chemokine levels, and an inhibition of neutrophil infiltration in the lungs with PYR-41 treatment. Finally, we have demonstrated that administration of PYR-41 immediately and at 5 h after CLP improve the overall survival of septic mice. Although there is a gap in the translation of animal studies to clinical outcomes, targeting UPP by PYR-41 inhibition of the NF-κB pathway as demonstrated here is an initial step in the development of novel therapeutic agents to treat sepsis.
Acknowledgments
We thank Dr. Alexandra C. Bolognese for critically reviewing the manuscript.
The work was supported by National Institute of Health grants R35GM118337, R01GM053008, and R01GM057468 (P.W.).
Footnotes
SM performed the experiments, analyzed the data and drafted the manuscript. AS conducted the experiment. PW interpreted the data and reviewed the manuscript. W-LY designed the study, analyzed the data and revised the manuscript.
The authors report no conflicts of interest.
References
- 1.Levy MM, Dellinger RP, Townsend SR, Linde-Zwirble WT, Marshall JC, Bion J, Schorr C, Artigas A, Ramsay G, Beale R, et al. The Surviving Sepsis Campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Crit Care Med. 2010;38(2):367–374. doi: 10.1097/CCM.0b013e3181cb0cdc. [DOI] [PubMed] [Google Scholar]
- 2.Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med. 2013;41(5):1167–1174. doi: 10.1097/CCM.0b013e31827c09f8. [DOI] [PubMed] [Google Scholar]
- 3.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369(21):2063. doi: 10.1056/NEJMc1312359. [DOI] [PubMed] [Google Scholar]
- 4.Ulloa L, Brunner M, Ramos L, Deitch EA. Scientific and clinical challenges in sepsis. Curr Pharm Des. 2009;15(16):1918–1935. doi: 10.2174/138161209788453248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Opal SM, Dellinger RP, Vincent JL, Masur H, Angus DC. The next generation of sepsis clinical trial designs: what is next after the demise of recombinant human activated protein C? Crit Care Med. 2014;42(7):1714–1721. doi: 10.1097/CCM.0000000000000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fink MP, Warren HS. Strategies to improve drug development for sepsis. Nat Rev Drug Discov. 2014;13(10):741–758. doi: 10.1038/nrd4368. [DOI] [PubMed] [Google Scholar]
- 7.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) JAMA. 2016;315(8):801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420(6917):885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 9.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 10.Oppenheim JJ, Yang D. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol. 2005;17(4):359–365. doi: 10.1016/j.coi.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11(5):373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 12.Napetschnig J, Wu H. Molecular basis of NF-kappaB signaling. Annu Rev Biophys. 2013;42:443–468. doi: 10.1146/annurev-biophys-083012-130338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 14.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y, Kitagaki J, Dai RM, Tsai YC, Lorick KL, Ludwig RL, Pierre SA, Jensen JP, Davydov IV, Oberoi P, et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007;67(19):9472–9481. doi: 10.1158/0008-5472.CAN-07-0568. [DOI] [PubMed] [Google Scholar]
- 16.Murao Y, Loomis W, Wolf P, Hoyt DB, Junger WG. Effect of dose of hypertonic saline on its potential to prevent lung tissue damage in a mouse model of hemorrhagic shock. Shock. 2003;20(1):29–34. doi: 10.1097/01.shk.0000071060.78689.f1. [DOI] [PubMed] [Google Scholar]
- 17.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, Legall JR, Morris A, Spragg R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149(3 Pt 1):818–824. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 18.Yang WL, Sharma A, Zhang F, Matsuo S, Wang Z, Wang H, Wang P. Milk fat globule epidermal growth factor-factor 8-derived peptide attenuates organ injury and improves survival in sepsis. Crit Care. 2015;19:375. doi: 10.1186/s13054-015-1094-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmekel B, Karlsson SE, Linden M, Sundstrom C, Tegner H, Venge P. Myeloperoxidase in human lung lavage. I. A marker of local neutrophil activity. Inflammation. 1990;14(4):447–454. doi: 10.1007/BF00914095. [DOI] [PubMed] [Google Scholar]
- 20.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376(6535):37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 21.Murakami A, Ohigashi H. Targeting NOX, INOS and COX-2 in inflammatory cells: chemoprevention using food phytochemicals. Int J Cancer. 2007;121(11):2357–2363. doi: 10.1002/ijc.23161. [DOI] [PubMed] [Google Scholar]
- 22.Abraham E, Singer M. Mechanisms of sepsis-induced organ dysfunction. Crit Care Med. 2007;35(10):2408–2416. doi: 10.1097/01.ccm.0000282072.56245.91. [DOI] [PubMed] [Google Scholar]
- 23.Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov. 2005;4(10):854–865. doi: 10.1038/nrd1854. [DOI] [PubMed] [Google Scholar]
- 24.Schmid JA, Birbach A. IkappaB kinase beta (IKKbeta/IKK2/IKBKB)–a key molecule in signaling to the transcription factor NF-kappaB. Cytokine Growth Factor Rev. 2008;19(2):157–165. doi: 10.1016/j.cytogfr.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt EP, Tuder RM. Role of Apoptosis in Amplifying Inflammatory Responses in Lung Diseases. J Cell Death. 2010;2010(3):41–53. doi: 10.4137/JCD.S5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bodas M, Min T, Vij N. Early-age-related changes in proteostasis augment immunopathogenesis of sepsis and acute lung injury. PLoS One. 2010;5(11):e15480. doi: 10.1371/journal.pone.0015480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Everhart MB, Han W, Sherrill TP, Arutiunov M, Polosukhin VV, Burke JR, Sadikot RT, Christman JW, Yull FE, Blackwell TS. Duration and intensity of NF-kappaB activity determine the severity of endotoxin-induced acute lung injury. J Immunol. 2006;176(8):4995–5005. doi: 10.4049/jimmunol.176.8.4995. [DOI] [PubMed] [Google Scholar]
- 29.Orlichenko LS, Behari J, Yeh TH, Liu S, Stolz DB, Saluja AK, Singh VP. Transcriptional regulation of CXC-ELR chemokines KC and MIP-2 in mouse pancreatic acini. Am J Physiol Gastrointest Liver Physiol. 2010;299(4):G867–876. doi: 10.1152/ajpgi.00177.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194(4):519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizgerd JP. Molecular mechanisms of neutrophil recruitment elicited by bacteria in the lungs. Semin Immunol. 2002;14(2):123–132. doi: 10.1006/smim.2001.0349. [DOI] [PubMed] [Google Scholar]
- 32.Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31(4 Suppl):S195–199. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 33.Juan YS, Lee YL, Long CY, Wong JH, Jang MY, Lu JH, Wu WJ, Huang YS, Chang WC, Chuang SM. Translocation of NF-kappaB and expression of cyclooxygenase-2 are enhanced by ketamine-induced ulcerative cystitis in rat bladder. Am J Pathol. 2015;185(8):2269–2285. doi: 10.1016/j.ajpath.2015.04.020. [DOI] [PubMed] [Google Scholar]
- 34.Xie QW, Kashiwabara Y, Nathan C. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem. 1994;269(7):4705–4708. [PubMed] [Google Scholar]
- 35.Ang SF, Sio SW, Moochhala SM, MacAry PA, Bhatia M. Hydrogen sulfide upregulates cyclooxygenase-2 and prostaglandin E metabolite in sepsis-evoked acute lung injury via transient receptor potential vanilloid type 1 channel activation. J Immunol. 2011;187(9):4778–4787. doi: 10.4049/jimmunol.1101559. [DOI] [PubMed] [Google Scholar]
- 36.Alvarez S, Evelson PA. Nitric oxide and oxygen metabolism in inflammatory conditions: sepsis and exposition to polluted ambients. Front Biosci. 2007;12:964–974. doi: 10.2741/2117. [DOI] [PubMed] [Google Scholar]
- 37.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82(2):373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 38.Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov. 2006;5(7):596–613. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- 39.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell. 1994;78(5):773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 40.Wang J, Maldonado MA. The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases. Cell Mol Immunol. 2006;3(4):255–261. [PubMed] [Google Scholar]
- 41.Safranek R, Ishibashi N, Oka Y, Ozasa H, Shirouzu K, Holecek M. Modulation of inflammatory response in sepsis by proteasome inhibition. Int J Exp Pathol. 2006;87(5):369–372. doi: 10.1111/j.1365-2613.2006.00490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]