

Abstract
Ectopic cAMP signaling is pathologic in polycystic kidney disease; however, its spatiotemporal actions are unclear. We characterized the expression of phosphorylated Creb (p-Creb), a target and mediator of cAMP signaling, in developing and cystic kidney models. We also examined tubule-specific effects of cAMP analogs in cystogenesis in embryonic kidney explants. In wild-type mice, p-Creb marked nephron progenitors (NP), early epithelial NP derivatives, ureteric bud, and cortical stroma; p-Creb was present in differentiated thick ascending limb of Henle, collecting duct, and stroma; however, it disappeared in mature NP-derived proximal tubules. In Six2cre;Frs2αFl/Fl mice, a renal cystic model, ectopic p-Creb stained proximal tubule–derived cystic segments that lost the differentiation marker lotus tetragonolobus lectin. Furthermore, lotus tetragonolobus lectin–negative/p-Creb–positive cyst segments (re)-expressed Ncam1, Pax2, and Sox9 markers of immature nephron structures and dedifferentiated proximal tubules after acute kidney injury. These dedifferentiation markers were co-expressed with p-Creb in renal cysts in Itf88 knockout mice subjected to ischemia and Six2cre;Pkd1Fl/Fl mice, other renal cystogenesis models. 8-Br-cAMP addition to wild-type embryonic kidney explants induced proximal tubular cystogenesis and p-Creb expression; these effects were blocked by co-addition of protein kinase A inhibitor. Thus p-Creb/cAMP signaling is appropriate in NP and early nephron derivatives, but disappears in mature proximal tubules. Moreover, ectopic p-Creb expression/cAMP signaling marks dedifferentiated proximal tubular cystic segments. Furthermore, proximal tubules are predisposed to become cystic after cAMP stimulation.
Polycystic kidney disease (PKD) is characterized by expansion of tubular-derived cysts and interstitial fibrosis that replace the renal parenchyma, usually leading to end-stage kidney disease.1, 2 In human patients with autosomal-dominant PKD, approximately 85% have mutations in the PKD1 gene that encodes polycystin 1 and approximately 15% have mutations in the PKD2 gene that encodes polycystin 2.3, 4 In addition, there have been many mouse models of cystic kidney disease, including those with Pkd mutations as well as in genes that encode for proteins that associate with cilia.5, 6, 7, 8, 9, 10 Our laboratory also recently found that Six2cre-mediated conditional deletion of Frs2α, a major docking protein for fibroblast growth factor receptors, in mouse nephron progenitors (Six2creFrs2αKO), leads to embryonic kidneys with a loss of nephron progenitors and postnatal kidneys with rapid and progressive renal cyst growth.11, 12 Although the precise cellular and molecular mechanisms by which these mutations lead to cystogenesis are unclear, these varied cystic models share many common features, such as ectopic canonical Wnt activation, ectopic sonic hedgehog activation, and inflammation/macrophage infiltration.13, 14, 15, 16, 17
Ectopic cAMP signaling, which activates multiple signaling pathways including direct phosphorylation of the transcription factor Creb by protein kinase A (PKA), is associated strongly with cystogenesis in both human and mouse models; however, the precise temporal and spatial actions of cAMP/Creb signaling in cystic kidney disease are not completely clear.18, 19, 20 Whether ectopic cAMP signaling has any role in Six2creFrs2αKO mice also is unknown. The severity of cystogenesis in Pkd1 mutant mice is increased by concomitant deletion of phosphodiesterase isoforms leading to higher levels of cAMP.21 Also, the addition of 8-Br-cAMP, a cAMP analog, is sufficient to induce cyst formation in wild-type rodent embryonic kidney explants. Moreover, cyst formation in embryonic kidney explants harboring Pkd1 (or other cystogenic) mutations also requires addition of cAMP agonists.22 In addition, 8-Br-cAMP–induced cyst formation in wild-type explants can be partially rescued by inhibiting PKA activity and its associated increase in Creb phosphorylation.22, 23 Together, these observations suggest a critical role of cAMP/PKA/p-Creb axis in cystogenesis, although its temporal and tubular-specific effectors of the pathway that drive cyst formation are unclear in humans and animal models of PKD, including in Six2creFrs2αKO mice.
Although cAMP/p-Creb signaling is known to play critical roles in the development of retina, lung, central nervous system, and neural crest–derived tissues, little is known about its role in kidney development, particularly in nephron progenitors.24, 25, 26, 27, 28 Kidney development occurs via reciprocal signaling between the metanephric mesenchyme (containing nephron and stromal progenitors) and the ureteric bud.29 The ureteric bud gives rise to collecting ducts and stromal progenitors generate vascular pericytes, vascular smooth muscle, subsets of endothelia, mesangial cells, and interstitial cells.30, 31 Nephron progenitors first transition to immature epithelial structures (renal vesicles, comma-shaped bodies, and S-shaped bodies) that finally develop into mature nephron epithelia, the largest volume of which becomes mature lotus tetragonolobus lectin (LTL)-expressing proximal tubules.29 A previous study has shown that cAMP/PKA signaling regulates branching of the ureteric bud by modulating bone morphogenetic protein signaling.32 PKA also has been shown to regulate renal tubulogenesis in a Wnt-dependent manner.33 However, a comprehensive assessment of p-Creb expression (ie, cAMP signaling) in the early stages of kidney development is unknown.
In the current study, we ascertained the developmental expression patterns of p-Creb to determine temporal and specific sites of cAMP signaling in normal embryonic and postnatal renal tissues. We then determined whether aberrantly activated cAMP signaling appears in Six2creFrs2αKO mice and compared these findings with other established models of cystic kidney disease. Finally, we determined which renal tubular segments become cystic in wild-type embryonic kidney explants treated with exogenous cAMP analogs. Collectively, these data suggest that ectopic cAMP-mediated signaling through Creb activation contributes to cyst development and progression by causing dedifferentiation of proximal tubules.
Materials and Methods
Mice
Six2creEGFP;Frs2αfl/fl [Six2creFrs2α knock-out (KO)] mice were generated by breeding Frs2αfl/fl mice with transgenic Six2creEGFP mice, as previously described,11, 34, 35 on a mixed genetic background (predominantly Friend leukemia virus B). Pkd1fl/fl mice were bred with transgenic Six2creEGFP mice to generate Six2creEGFP;Pkd1fl/fl (Six2crePkd1KO) mice on a mixed 129S1 and CD1 background.36 Cre-negative littermates were used as controls for both Six2creFrs2αKO and Six2crePkd1KO mice. As described, Ift88 fl/fl mice were bred with CAGG-CreERT2 transgenic mice to generate CAGG-CreERT2;Ift88 fl/fl animals and were maintained on a C57BL/6J background; induction of Cre and thus global deletion of Ift88 in adult CAGGcreER;Ift88 fl/fl (Ift88KO) was achieved by once-daily injection of tamoxifen (6 mg/40 g body weight) for 3 consecutive days.37 Ift88KO and Cre-negative littermates were subjected to 30 minutes of left renal ischemia by pedicle clamping followed by 3 weeks of reperfusion (IR). Right sham kidneys underwent identical treatment without clamping of the renal pedicle.37 Six2creEGFP transgenic mice were bred with Ai9 (RCL-tdTomato) mice (stock 007909; The Jackson Laboratory, Bar Harbor, ME), which were maintained on a C56BL/6J background to generate Six2creTG/+CAG-tdTomato+. Animals used in all of the experiments were maintained and euthanized according to the principles and procedures described in the NIH Guide for the Care and Use of Laboratory Animals.38 All animal experiments were conducted with approval by the University of Pittsburgh Institutional Animal Care and Use Committee.
Immunofluorescence Microscopy
Immunofluorescence microscopy approaches used in the current study were described previously.11, 12, 39, 40 Briefly, 4-μm, paraformaldehyde-fixed, paraffin-embedded kidney sections were deparaffinized, rehydrated, and subjected to antigen retrieval in citrate buffer (10 mm citrate, 0.1% Tween-20, pH 6.0) at 95°C for 30 minutes and then kept at room temperature for 30 minutes. The sections were blocked for 1 hour in donkey serum at room temperature and incubated for 12 to 18 hours with primary antisera or lectins as follows: aquaporin-1 (1:500, AQP11-A; Alpha Diagnostic Intl. Inc., San Antonio, TX), p-Creb (1:500, 9188; Cell Signaling, Danvers, MA), Creb (1:1000, 82630; Cell Signaling), Ncam1 (1:2000; Sigma-Aldrich, St. Louis, MO), Pax2 (1:1000, 71-6000; Thermo Fisher, Rockford, IL), Six2 (1:1000, 11562-1-AP; Protein Tech, Rosemont, IL), Sox9 (1:500, 9104; Cell Signaling), Tamm-Horsfall Protein (1:500, MAB5175; R&D Systems, Minneapolis, MN), LTL (1:500, FL-1321; Vector Biolab, Burlingame, CA), or dolichos biflorus agglutinin (DBA) (1:500, FL-1031; Vector Biolab). Alexa Fluor 594 and Alexa Fluor 488 secondary antibodies were used and nuclei were stained with 4′6′-diamidino-2-phenylindole. Staining was detected using a Leica DM 2500 fluorescence microscope (Leica, Wetzlar, Germany). Immunofluorescence images shown from Six2creFrs2αKO and CAGGCreER;Ift88 fl/fl strains are representative from at least three kidneys isolated from three mice of each genotype, whereas for Six2crePkd1KO four kidneys, each isolated from two control and two mutant mice, were analyzed. Immunofluorescence images of explants generated from the wild-type or Six2creTg/+CAG-tdTomato+ are representative of at least three kidneys per treatment group.
Embryonic Kidney Explant Culture
Kidneys were harvested from wild-type or Six2creTg/+CAG-tdTomato+ E13.5 embryos and cultured at liquid–air interface on hydrophilic polycarbonate nucleopore filters floating on Dulbecco's modified Eagle's medium/F12 medium for 5 days either in the presence or absence of the cAMP agonist, 8-Br-cAMP (100 μmol/L). Media with and without 8-Br-cAMP were replaced every 24 hours. Wild-type kidney explants were photographed with a Leica M165 FC stereo microscope every 24 hours, and after 5 days were fixed in 4% paraformaldehyde and embedded in paraffin. Sections (4 μm) were subjected to immunofluorescence microscopy analysis and imaging as described earlier. Embryonic kidney explants cultured from Six2creTg/+CAG-tdTomato+ were fixed with 4% paraformaldehyde for 30 minutes and embedded in OCT, and 8-μm cryosections were obtained and images were captured on a Leica DM 2500 fluorescence microscope.
Results
Normal p-Creb Expression in Control and Six2creFrs2αKO Embryonic Kidneys
The temporal and spatial expression of p-Creb (as a readout of cAMP signaling) was determined in developing kidneys and it was ascertained whether perturbations in p-Creb expression patterns preceded cystogenesis in Six2creFrs2αKO mice. Sections obtained from control and Six2creFrs2αKO mice at embryonic day 14.5 (E14.5) were co-immunostained with p-Creb and Ncam1, a marker of nephron progenitors and immature descendants (Figure 1). In E14.5 control and Six2creFrs2αKO mutant kidney sections, p-Creb and Ncam1 were co-expressed in nephron progenitors and immature epithelial derivatives such as S-shaped bodies (Figure 1, A and B). p-Creb signal also was present in ureteric bud epithelia that did not express Ncam1. Similar results were observed when comparing p-Creb immunofluorescence in serial E14.5 control or mutant sections with Six2, a nephron progenitor–specific marker, and Sox9, which marks both immature epithelial NP derivatives and ureteric bud (Supplemental Figure S1). At E18.5, p-Creb staining was absent in LTL-expressing mature proximal tubules in both control and Six2creFrs2αKO kidneys (Figure 1, C and D), whereas p-Creb expression persisted in DBA-positive ureteric epithelia/collecting ducts (Figure 1, E and F) and in the interstitial cells derived from stromal progenitors in both genotypes. Thus, in both control and Six2creFrs2αKO embryonic kidneys, p-Creb is expressed in nephron progenitors and early epithelial derivatives, but is absent in mature proximal tubular cells (suggesting physiologic cAMP signaling in nephron progenitors and immature derivatives). Conversely, ureteric and stromal derivatives persistently express p-Creb. Finally, in precystic Six2creFrs2αKO embryonic kidneys, cAMP signaling appears normal.
Figure 1.

In control and Six2creFrs2α knock-out (KO) embryos, phosphorylated Creb (p-Creb) signal is present in nephron progenitors and early epithelial derivatives but absent in mature proximal tubules. A and B: Representative co-immunofluorescence images for p-Creb (red) and Ncam1 (green) in embryonic day (E)14.5 control (A) and Six2creFrs2αKO (B) kidneys shows p-Creb expression in Ncam1-positive nephron progenitors (concave arrowheads) and their immature epithelial derivative S-shaped bodies (arrows), as well as in Ncam1-negative ureteric bud epithelia (arrowheads). C and D: Co-immunofluorescence for p-Creb (red) and lotus tetragonolobus lectin (LTL) (green) in E18.5 control (C) and Six2creFrs2αKO (D) kidneys shows that p-Creb signal is absent in LTL-positive mature proximal tubules (concave arrowheads), but persistent in LTL-negative ureteric bud/collecting duct cells (UB) and stromal-derived interstitial cells (arrows). E and F: Co-immunofluorescence for p-Creb (red) and dolichos biflorus agglutinin (DBA) (green) in E18.5 control (E) and Six2creFrs2αKO (F) kidneys confirms a persistent p-Creb signal in the DBA-positive ureteric bud (arrowheads). Scale bars: 50 μm.
Ectopic p-Creb Expression in LTL-Negative Segments of Proximal Tubule–Derived Cysts in Postnatal Six2creFrs2αKO Kidneys
To determine whether Creb phosphorylation is mis-regulated in the cystic kidneys of Six2creFrs2αKO mice, we co-stained kidney sections obtained from postnatal day 7 and postnatal day 21 control and Six2creFrs2αKO mice with p-Creb antibody and markers of mature renal tubular segments. In control kidneys, p-Creb signal was present in a few glomerular cells, DBA-positive collecting ducts, subsets of Tamm-Horsfall protein–positive thick ascending limbs of the Henle Loop and stroma, but was never observed in LTL-positive mature proximal tubules, similar to E18.5 kidneys (Figure 2, A and C, Supplemental Figure S2, A and C, and not shown). In both P7 and P21 Six2creFrs2αKO kidneys, nondilated LTL-positive tubules and LTL-positive cells within cysts were p-Creb negative (similar to controls); however, many cysts contained segments that had lost LTL expression and had become p-Creb–positive (Figure 2, B and D). The other mutant tubular segments examined (thick limb and collecting ducts) expressed p-Creb as in controls, and none of the cysts in Six2creFrs2αKO mice were Tamm-Horsfall protein– or DBA-positive (Supplemental Figure S2, B and D). Finally, immunofluorescence with an antibody that recognizes nonphosphorylated (total) Creb showed expression throughout all segments of control and Six2creFrs2αKO kidneys, including mutant cyst lining cells (Supplemental Figure S3). Together, these data show that the cysts in Six2creFrs2αKO mice arise from proximal tubules and that segments of the cyst lining cells that had lost LTL staining expressed p-Creb ectopically.
Figure 2.
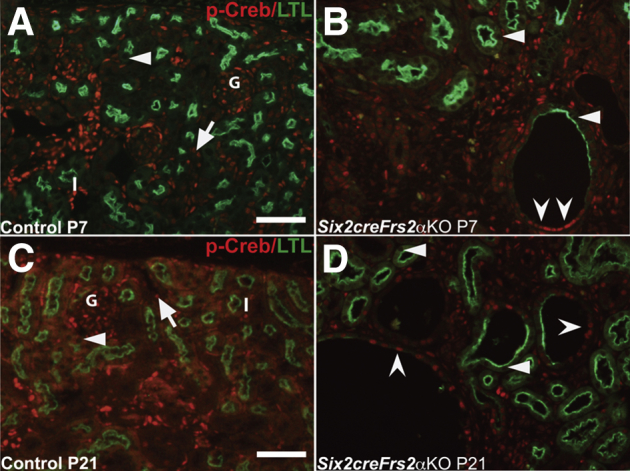
P7 and P21 Six2creFrs2α knock-out (KO) kidneys ectopically express phosphorylated Creb (p-Creb) in lotus tetragonolobus lectin (LTL)-negative segments of proximal tubule–derived cysts. A–D: Representative co-immunofluorescence images for p-Creb (red) and LTL (green) in P7 and P21 control and Six2creFrs2αKO kidneys. In P7 and P21 controls (A and C), p-Creb signal is present in stromally derived interstitium (I) and glomeruli (G) and in nondilated LTL-negative tubules (arrows), but is absent in LTL-positive proximal tubules (A and C, arrowheads). B and D: In Six2creFrs2αKO kidneys, p-Creb is absent in LTL-positive cells in both nondilated tubules and in cysts (arrowheads); however, it is expressed ectopically in LTL-negative cystic segments (concave arrowheads) and present in many nonproximal tubular cells (likely interstitium and collecting ducts). Scale bars: 50 μm.
It was unclear why these LTL-negative mutant cystic segments lost LTL expression and gained p-Creb expression. Loss of LTL expression in proximal tubules is seen in cells that have undergone dedifferentiation after acute kidney injury.41 To examine the possibility that the mutant LTL-negative/p-Creb–positive cells represented a population of dedifferentiated proximal tubular cells, p-Creb and LTL co-immunofluorescence was compared on P21 control and mutant kidney serial sections with Ncam1, Pax2, or Sox9 (three markers of immature/dedifferentiated nephron derivatives) and LTL (Figures 3 and 4). As expected, LTL-expressing control P21 proximal tubules were p-Creb and Ncam1 negative (Figure 3, A and C). In contrast P21 Six2creFrs2αKO kidneys had LTL-negative regions within proximal tubule–derived cysts that expressed both p-Creb and Ncam1 ectopically, whereas all LTL-positive cells, including cystic cells, remained p-Creb and Ncam1 negative (Figure 3, B and D). Co-staining of P21 kidney sections with Pax2/LTL showed that control LTL-positive proximal tubules were Pax2 negative, whereas nondilated LTL-negative tubules (presumably collecting ducts) expressed Pax2 (Figure 4A). In contrast, mutant P21 kidneys showed ectopic Pax2 staining in LTL-negative cystic cells as well as normal Pax2 expression in what are likely collecting ducts, but no staining in LTL-positive cells, including those within cysts (Figure 4B). Similarly, co-staining of P21 kidney sections with Sox9/LTL showed that control LTL-positive proximal tubules were Sox9 negative (Figure 4C), whereas mutant P21 kidneys showed ectopic Sox9 staining in cystic segments with reduced or absent LTL (Figure 4D). The same p-Creb, Ncam1, and Pax2 staining patterns were observed in P7 control and Six2creFrs2αKO kidneys (not shown). Together, p-Creb is co-expressed ectopically with Ncam1, Pax2, and Sox9 in regions of proximal tubule–derived cysts that were losing LTL expression, consistent with dedifferentiation of these epithelial cells.
Figure 3.

Ectopic Ncam1 expression in lotus tetragonolobus lectin (LTL)-negative/phosphorylated Creb (p-Creb)–positive proximal tubule–derived cyst segments in P21 Six2creFRS2α knock-out (KO) kidneys. A–D: Co-immunofluorescence for p-Creb (red) and LTL (green) or Ncam1 (red) and LTL (green) on serial sections obtained from P21 control and Six2creFrs2αKO kidneys. A: In controls, p-Creb signal is present in LTL-negative cells that likely include collecting ducts (arrows), but absent in LTL-positive proximal tubules (arrowheads). B: In Six2creFrs2αKO kidneys, p-Creb is absent in LTL-positive segments of nondilated tubules and cyst cells (arrowheads), but is expressed ectopically in LTL-negative cystic segments (concave arrowheads) and is present in many nonproximal tubule–derived cells similar to controls (arrow). C: In controls, Ncam1 staining is absent, including in LTL-positive proximal tubules (arrowhead). D: In Six2creFrs2αKO kidneys, Ncam1 expression is absent in LTL-positive nondilated tubules and LTL-positive cystic cells (arrowheads), whereas it is expressed ectopically in the LTL-negative cystic segments, overlapping with p-Creb (concave arrowheads) (compare B and D). Scale bars: 50 μm.
Figure 4.

Ectopic Pax2 and Sox9 expression in lotus tetragonolobus lectin (LTL)-negative proximal tubule–derived cystic segments in P21 Six2creFRS2α knock-out (KO) kidneys. A and B: Co-immunofluorescence for Pax2 (red) and LTL (green) in P21 control and Six2creFrs2αKO kidneys. A: In controls, Pax2 expression is absent in LTL-positive proximal tubules (arrowhead) and present in LTL-negative tubules (likely collecting ducts) (arrow). B: In Six2creFrs2αKO kidneys, Pax2 again is absent in all LTL-positive segments, both in nondilated tubules and cysts, but is expressed ectopically in LTL-negative cystic segments (concave arrowheads) and expressed in nondilated LTL-negative tubules (likely collecting ducts) (arrowheads). Arrow indicates Pax2 expression in LTL-negative tubules (likely collecting ducts). C and D: Co-immunofluorescence for Sox9 (red) and LTL (green) in P21 control and Six2creFrs2αKO kidneys. C: In controls, Sox9 expression is absent in LTL-positive proximal tubules (arrowhead). D: In Six2creFrs2αKO kidneys, Sox9 is absent in nondilated tubules and cyst lining cells that robustly express LTL (arrowheads), but is expressed ectopically in cystic segments with reduced or absent LTL staining (concave arrowheads). Scale bars: 50 μm.
Ectopic p-Creb Expression in LTL-Negative Proximal Tubule–Derived Cystic Cells in Ift88 Conditional KO Mice Subjected to Ischemia/Reperfusion
To determine whether the aberrant p-Creb/cAMP signaling observed in Six2creFrs2αKO occurred in other cystic kidney disease models, kidneys of mice in which Ift88, a gene encoding for primary ciliary intraflagellar transport and ciliary biogenesis is inducibly deleted throughout the kidney at P30, were examined. The kidneys were subjected to 30 minutes of unilateral renal ischemia-reperfusion injury (IRI) and allowed to recover for 3 weeks. Although deletion of Ift88 in P30 kidneys results in loss of cilia, it is only after IRI that the kidneys develop rapid and severe cystic kidney disease (as is true for postnatal deletion of many PKD-associated genes).37 As expected, cysts were observed only in the Ift88KO/IRI kidneys (and not in controls subjected to IR or sham surgery or in Ift88KO subjected to sham surgery). In all conditions, p-Creb immunostaining was observed in both collecting ducts and interstitium, and Pax2 staining was observed in collecting ducts (Figure 5 and not shown). Similar to Six2creFrs2αKO mice, immunofluorescence in Ift88KO/IRI kidneys also showed many cysts that had LTL-positive and LTL-negative regions; LTL-negative segments expressed p-Creb, Pax2, Ncam1, and Sox9 (Figure 5, J–L and Supplemental Figure S4D). In control mice subjected to IR, Pax2 (Figure 5E), Ncam1 (Figure 5F), or Sox9 (Supplemental Figure S4B) were not observed in LTL-positive proximal tubules. Rare LTL-expressing proximal tubules that also had p-Creb staining were observed (Figure 5D; this was consistent with fading p-Creb/cAMP signal in proximal tubular cells that have been redifferentiating after IRI). Sham-operated control and Ift88KO mice had no p-Creb, Pax2, Ncam1, or Sox9 expression in LTL-positive proximal tubular cells (Figure 5, A–C and G–I and Supplemental Figure S4, A and C).
Figure 5.

Ectopic phosphorylated Creb (p-Creb), Pax2, and Ncam1 expression in proximal tubule–derived cystic segments with decreased or absent lotus tetragonolobus lectin (LTL) staining in adult Ift88 knock-out (KO) kidneys 3 weeks after ischemia-reperfusion (IR). A, D, G, and J: Representative co-immunofluorescence images for p-Creb (red) and LTL (green) in control and Ift88KO, 3 weeks after IR injury or sham surgery. A: No p-Creb signal is present in control kidneys subjected to sham surgery (arrowhead), but it is present in non–LTL-positive cells that are likely interstitium (I) and collecting ducts (arrow). D: In control kidneys subjected to IR injury, p-Creb signal is observed in few LTL-positive proximal tubules (concave arrowheads) and in LTL-negative tubules (likely collecting ducts) (arrow), but absent in most LTL-positive proximal tubules (arrowheads). G: In Ift88KO kidneys subjected to sham surgery, no p-Creb signal is observed in LTL-positive proximal tubules (arrowhead), but is present in LTL-negative interstitium (I) and tubules (likely collecting ducts) (arrow). J: In Ift88KO/IR kidneys, p-Creb is absent in LTL-positive tubules and cystic cells (arrowheads), but is expressed ectopically in cystic segments with reduced or absent LTL staining (concave arrowheads). Arrow indicates p-Creb expression in interstitial cells. B, E, H, and K: Representative co-immunofluorescence images for Pax2 (red) and LTL (green). In the kidneys of control mice subjected to sham or IR injury surgery and Ift88KO kidneys subjected to sham surgery, Pax2 expression was seen only in LTL-negative tubules (likely collecting ducts) (arrows) and not in any LTL-positive tubules (arrowheads) (B, E, H). K: In Ift88KO/IR injury kidneys, Pax2 is absent in LTL-positive nondilated tubules and cystic cells (arrowheads), but is expressed ectopically in LTL-negative regions of cysts (concave arrowheads). Arrow indicates Pax2 expression in LTL-negative tubule (likely collecting duct). C, F, I, and L: Representative co-immunofluorescence images for Ncam1 (red) and LTL (green). In the kidneys of control mice subjected to sham or IR injury surgery and Ift88KO kidneys subjected to sham surgery, Ncam1 is not expressed in any LTL-positive tubules (arrowheads) (C, F, I). L: In Ift88KO/IR kidneys, ectopic Ncam1 signal is observed in LTL-negative regions of proximal tubule–derived cysts (concave arrowhead). Inset: Magnified view of the Ncam1-positive cell from boxed area. Ncam1 is not expressed in any LTL-positive tubules (arrowhead). Scale bars: 50 μm.
Ectopic p-Creb Expression in LTL-Negative Proximal Tubule–Derived Cysts in Six2crePkd1KO Kidneys
P-Creb expression then was interrogated in another bona fide PKD model, in which Six2cre was used to conditionally delete Pkd1, the gene encoding for polycystin 1, which is the most frequently mutated gene seen in patients with autosomal-dominant PKD.42, 43 Hematoxylin and eosin staining of the kidneys of Six2crePkd1KO mice showed earlier onset and more rapid progression of cysts than Six2creFrs2αKO mice; some cysts were observed at E15.5, cysts virtually replacing the renal parenchyma by P1, and larger cysts throughout the kidney at P5 (Supplemental Figure S5). Immunofluorescence in P1 Six2crePkd1KO mice showed proximal tubule–derived cysts with ectopic p-Creb staining, mostly in regions that lost LTL expression, but in occasional cells with reduced LTL staining; as predicted, control LTL-positive proximal tubules never stained for p-Creb (Figure 6, A and B). Immunostaining for Pax2, Ncam1, and Sox9 in Six2crePkd1KO mutants also showed ectopic expression in proximal tubule–derived cysts, mostly in LTL-negative regions, but in some cells with reduced LTL expression (Figure 6, C–F and Supplemental Figure S6). Thus, (re)-expression of p-Creb in dedifferentiating cells of proximal tubule–derived cysts is a feature in Six2crePkd1KO kidneys, as is true for the other two cystic models.
Figure 6.

Ectopic phosphorylated Creb (p-Creb), Pax2, and Ncam1 expression in proximal tubule–derived cystic segments with decreased or absent lotus tetragonolobus lectin (LTL) staining in P1 Six2crePKD1 knock-out (KO) kidneys. A–F: Representative co-immunofluorescence images for p-Creb (red) and LTL (green) (A and B), Pax2 (red) and LTL (green) (C and D), or Ncam1 (red) and LTL (green) (E and F) in control and Six2crePkd1KO kidneys. A: In controls, p-Creb signal is absent in all LTL-positive proximal tubules (arrowhead) and is present in LTL-negative interstitium (I) and tubules (likely collecting ducts) (arrow). B: In Six2crePkd1KO kidneys, p-Creb is absent in most cells with strong LTL staining, but is expressed ectopically in proximal tubule–derived cystic segments with reduced or absent LTL expression (concave arrowheads) and in LTL-negative interstitium (arrow). C: In controls, Pax2 is absent in LTL-positive proximal tubules (arrowhead) and present in LTL-negative tubules (likely collecting ducts) (arrow) (yellow-labeled cells are red blood cells). D: In Six2crePkd1KO kidneys, Pax2 is expressed ectopically in cystic cells with low or absent LTL expression (concave arrowhead). E: In controls, Ncam1 is present in early epithelial nephron derivatives (arrow), but absent in LTL-positive proximal tubules (arrowhead). F: In Six2crePkd1KO kidneys, Ncam1 is expressed ectopically in cystic tubules that have reduced or absent LTL staining (concave arrowheads), and present in normal early epithelial nephron derivatives (arrow). Scale bars: 50 μm.
Wild-Type Embryonic Kidney Explants Treated with 8-Br-cAMP Develop Proximal Tubular–Derived Cysts with Ectopic p-Creb and Pax2 Staining
Treatment of wild-type embryonic kidney explants with cAMP analogs including 8-Br-cAMP is known to result in cystogenesis.22 To determine whether 8-Br-cAMP stimulation induced dedifferentiation and stimulated p-Creb expression in proximal tubules, E13.5 kidney explants were cultured with 100 μmol/L 8-Br-cAMP. We first noted cysts/dilatations in cAMP analog-treated explants on day 3 that increased in size and number by day 5, whereas explants cultured with vehicle alone developed no cysts (Supplemental Figure S7). To determine the origin of the cysts and whether proximal tubule–derived cysts lost differentiation markers, co-immunofluorescence was performed in serially sectioned explants with Aqp1 (proximal tubule marker) and DBA lectin (ureteric/collecting duct marker) (Figure 7, A and B) or Aqp2 (collecting duct marker) and LTL lectin (proximal tubular marker) (Figure 7, C and D). Similar to Six2creFrs2αKO and the other PKD models tested, cAMP-treated kidney explants had cysts with both LTL-positive and LTL-negative segments (Figure 7D), and LTL-negative segments have reduced Aqp1 (Figure 7B) staining as well, suggesting that 8-Br-cAMP induced dedifferentiation of proximal tubules. Surprisingly, most of the cysts induced by 8-Br-cAMP had segments that were Aqp1 or LTL positive (Figure 7, B and D), and that none of the cysts were DBA or Aqp2 positive (Figure 7, B and D); this suggests that the majority of the cysts originated from nephron-derived proximal tubules and not from ureteric bud–derived collecting ducts.
Figure 7.

Exogenous 8-Br-cAMP in embryonic kidney explants results in cysts that are derived mostly from proximal tubules. Co-immunostaining with Aqp1 (red) and dolichos biflorus agglutinin (DBA) (green) or Aqp2 (red) and lotus tetragonolobus lectin (LTL) (green) in serial sections of vehicle or 8-Br-cAMP–treated embryonic day (E)13.5 kidney explants cultured for 5 days. A: Explants cultured in media and vehicle have normal Aqp1 (proximal tubule marker) and DBA (ureteric/collecting duct marker) staining with no cysts. B: Explants cultured in media and 8-Br-cAMP develop cysts that frequently co-label with Aqp1 (asterisks); although some cysts have partial loss of Aqp1 staining (concave arrowheads) and many Aqp1-expressing tubules are not dilated (arrowheads). None of the cysts in the 8-Br-cAMP–treated explants label for DBA (arrows). C: Explants cultured in media and vehicle have normal Aqp2 (collecting duct marker) and LTL (proximal tubule marker) staining with no cysts. D: Explants cultured in media and 8-Br-cAMP develop cysts that frequently co-label with LTL (asterisks), although some cysts have partial loss of LTL staining (concave arrowheads) and many LTL-expressing tubules are not dilated (arrowheads). None of the cysts in the 8-Br-cAMP–treated explants label for Aqp2 (arrows). Scale bars: 100 μm.
Although a minority of cysts induced by 8-Br-cAMP were Aqp1/LTL negative (and Aqp2/DBA-negative), we suspected that the cysts may have originated from proximal tubules that had completely lost both differentiation markers. To clarify whether the cysts originated from nephron progenitors (which give rise to proximal tubules) a Six2creTg/+CAG-tdTomato+ mouse strain was generated in which Six2cre-mediated recombination permanently labels nephron progenitors and their derivatives with tdTomato. Cryosections from vehicle and 8-Br-cAMP–treated kidney explants isolated from this transgenic line showed that all but a few of the cysts labeled with tdTomato (Figure 8). None of the of the cysts (including the tdTomato-negative cyst lining cells) labeled with DBA (not shown). Taken with the earlier data, virtually all of the cysts induced by exogenous cAMP originate from nephron progenitors (and likely are proximal tubule in origin).
Figure 8.

Lineage tracing shows that virtually all cysts induced by 8-Br-cAMP in embryonic kidney explants come from nephron progenitors. A–F: Fluorescent (A and B), brightfield (C and D), and merged images (E and F) of cryosections obtained from embryonic day (E)13.5 Six2creTg/+CAG-tdTomato+ kidney explants treated with vehicle or 8-Br-cAMP. A, C, and E: Vehicle-treated explants have no cysts. B, D, and F: Explants cultured in 8-Br-cAMP–containing media develop cysts, almost all of which express td-Tomato (red, asterisks). Scale bar = 100 μm.
It then was ascertained whether 8-Br-cAMP–induced proximal tubule–derived cystic segments ectopically expressed p-Creb or Pax2 (Figure 9). The cystic segments with reduced or no LTL staining expressed p-Creb and Pax2 (Figure 9, B and D). Few subpopulations of vehicle-treated explant proximal tubules (mostly LTL-negative segments) that also expressed p-Creb and Pax2 also were observed, consistent with tubular segments that had not yet fully differentiated in culture (Figure 9, A and C). Together with the data showing loss of proximal tubule differentiation markers, the ectopic p-Creb and Pax2 staining in segments of the cysts are consistent with dedifferentiation of these cystic segments.
Figure 9.

Ectopic phosphorylated Creb (p-Creb) and Pax2 expression in proximal tubule–derived cysts induced by 8-Br-cAMP in wild-type kidney explants. Representative co-immunofluorescence images for p-Creb (red) and lotus tetragonolobus lectin (LTL) (green) (A and B) or Pax2 (red) and LTL (green) (C and D) on sections obtained from embryonic day (E)13.5 kidney explants cultured in media and vehicle or media and 8-Br-cAMP for 5 days. A: In vehicle-treated explants, p-Creb largely is absent in LTL-positive proximal tubule (arrowheads), although it is expressed in few LTL-positive tubules (concave arrowheads), in nephron progenitors (arrow), and a glomerulus (G). B and D: In 8-Br-cAMP–treated explants, p-Creb–positive (B) and Pax2-positive (D) cells are found in patterns similar to vehicle-only explants, but also are expressed ectopically in proximal tubule–derived cystic cells with decreased or absent LTL staining (concave arrowheads). C: Pax2 also largely is absent in LTL-positive tubules (arrowheads), but is found in a few LTL-positive cells (concave arrowhead) and in LTL-negative tubules (likely collecting ducts) (arrow). Scale bars: 50 μm.
Inhibition of PKA Blocks 8-Br-cAMP–Mediated Cystogenesis and Creb Phosphorylation in Kidney Explants
To determine whether 8-Br-cAMP–induced cystogenesis and Creb phosphorylation is mediated by PKA, the effect of concomitant addition of the PKA inhibitor H-89 with 8-Br-cAMP in kidney explants (Supplemental Figure S8) was examined. Stereomicroscopic imaging (Supplemental Figure S8, A and B) of the whole explants and hematoxylin and eosin staining of the sections (Supplemental Figure S8, C and D) showed that H-89 blocked 8-Br-cAMP–induced cystogenesis. Co-immunofluorescence of the sections with p-Creb and LTL (Supplemental Figure S8, E and F) showed that H-89 addition clearly blocked both cyst formation and ectopic p-Creb staining in LTL-positive tubules (above the limited baseline p-Creb/LTL co-staining seen in vehicle-treated explants: compare Figure 9A with Supplemental Figure S8F). Together, these data suggest that 8-Br-cAMP activates PKA to phosphorylate Creb and drive cystogenesis.
Discussion
In the current study, the expression pattern of p-Creb, which likely reflects cyclic AMP/PKA signaling in the normal developing kidney and in several cystic kidney disease models, was first evaluated. p-Creb staining was present in nephron progenitors and early differentiating nephron structures and was absent in mature LTL-positive proximal tubular nephron derivatives in both wild-type and Six2creFrs2αKO embryos. Notably, temporal activation of cAMP/p-Creb signaling is known to regulate development in other systems, including myogenesis and somite development.27, 28, 44 Based on our study, cAMP/p-Creb signaling appears to have a role in the development or differentiation of nephron progenitors and other early developing renal tissues.
Our study also showed insights into temporal and spatial expression and actions of cAMP in models of renal cystogenesis. In the Six2creFrs2αKO model, ectopic p-Creb expression was observed in the LTL-negative regions of proximal tubule–derived cysts. This appears largely true in the Ift88KO/IRI and the Six2crePkd1KO mice as well. Several lines of evidence have suggested that these p-Creb–positive cells mark dedifferentiation of the proximal tubule–derived cells, including: i) as noted, p-Creb is expressed in nephron progenitors and their early derivative structures but is absent in mature proximal tubules, ii) p-Creb–positive proximal tubular–derived cysts have either dramatically reduced or absent LTL staining indicative of a partial or complete loss of the brush border, a microvilli-covered surface (ie, a well-defined marker of differentiation), and iii) co-immunoflourescence of serial sections obtained from kidneys of all three models showed that p-Creb–positive cells express Pax2, Ncam1, and Sox9, which also are expressed in nephron progenitors and/or immature derivatives and are well-established markers of dedifferentiation in proximal tubules subjected to acute kidney injury (AKI).45, 46 Given that aberrant/ectopic cAMP/PKA signaling is known to have a pathogenic role in PKD progression, it appears likely that in nephron-derived cysts, this pathway has a pathogenic role in driving dedifferentiation in segments of the cysts.
Interestingly, reactivation of p-Creb appears necessary for regeneration of muscle after acute injury may physiological roles after acute kidney injury.47 Dedifferentiated proximal tubular cells in mice subjected to AKI are known to re-express genes that are crucial during kidney development such as Pax2, Ncam1, and Sox9. p-Creb also is activated in distal tubules of the kidneys subjected to AKI and in a cultured proximal tubule cell line incubated with cisplatin.48, 49 Phosphorylation of Creb in proximal tubules may promote dedifferentiation and expansion of proximal tubular cells after exposure to these injury stimuli (followed by redifferentiation). Although phosphorylation of Creb has not been defined as a feature of dedifferentiation after AKI, p-Creb was detected in LTL-positive proximal tubules of the kidney of control mice 3 weeks after the induction of AKI; this suggests that Creb is phosphorylated during the recovery phase of AKI. In the context of Ift88 deletion, persistent p-Creb expression correlates with a dedifferentiation phenotype that has been identified as a feature of PKD that contributes to its pathogenesis50, 51 Further studies are required to determine whether inhibition of Creb phosphorylation in proximal tubules in Six2creFrs2αKO or Six2crePkd1KO mice would attenuate cyst progression.
Impaired terminal differentiation of proximal tubular cells also may be responsible for the LTL-negative/p-Creb–positive segments in Six2creFrs2αKO mice; however, some observations argue against this possibility. First, despite exhaustive efforts, no LTL-positive/p-Creb–positive cells were detected in E18.5 Six2creFrs2αKO mice, implying that normal differentiation of Frs2α-deficient nephron progenitors into proximal tubules occurred. In addition, Ift88KO/IRI mice have normal proximal tubular differentiation before the induced deletion of Ift88 at P30, and yet develop LTL-negative/p-Creb–positive segments in the renal cysts. Moreover, most patients with autosomal-dominant PKD develop cysts postnatally, making it unlikely that there are early renal tubular developmental/differentiation defects in these patients.52 Finally, Pkd mouse models do not appear to have defects in differentiation of tubular segments and yet Six2crePkd1KO mice have ectopic p-Creb expression in cells with reduced or absent LTL expression.3, 53, 54 It remains to be determined whether the appearance of LTL-negative/p-Creb–positive segments in cysts in different models of PKD share underlying mechanisms and stimuli.
Finally, we show that exogenous 8-Br-cAMP, which is known to be sufficient to cause cyst formation in wild-type embryonic kidney explants, induces the loss of LTL (and, to a lesser degree, Aqp1) and the re-expression of p-Creb in cystic proximal tubules; this indicates that Creb is one of the direct or indirect downstream targets of cAMP signaling in proximal tubules. Pax2 expression also was observed in cystic proximal tubules of embryonic kidney explants treated with 8-Br-cAMP. Notably, cAMP stimulation is known to increase in Pax2 expression in epithelial cells of the collecting duct.23 The cysts induced in explants stimulated with 8-Br-cAMP appeared to be derived from proximal tubules whereas, as expected, but surprisingly, no dilatations/cysts were observed in DBA- or Aqp2-positive ureteric/collecting duct cells. Although there were a few cysts in the cAMP-treated explants that did not label for either proximal tubule or collecting duct markers, virtually all of the cysts were marked by tdTomato, in the Six2creTg/+CAG-tdTomato+ lineage tracing studies, suggesting that the proximal tubules as the main target of 8-Br-cAMP induced cystogenesis in embryonic kidney explants. Notably, our results contrast to the previously reported findings in which the 8-Br-cAMP stimulation of Pkd1 KO explants led to cyst development that originated from both LTL and DBA expressing epithelium.22 One possibility for the differences is that wild-type explant collecting ducts may have a different threshold for cystogenic response than those lacking Pkd1. The apparent differential cystic response of proximal tubules versus ureteric epithelia to cAMP analog in wild-type mice may be owing to differences in their endogenous level of Creb activation. Although p-Creb is absent in mature proximal tubules, it persists in mature collecting duct cells. Thus, the collecting duct may be more resistant to cystogenesis driven by cAMP. Perhaps with age, normal p-Creb levels may decrease, rendering aged collecting ducts more susceptible to cAMP-mediated cystogenesis in patients with PKD or other mutations. Whether collecting duct cystogenesis, as occurs in autosomal-dominant PKD, is less dependent on cAMP than in other regions of the kidney such as the proximal tubule remains to be determined.
Acknowledgments
We thank Drs. Sunder Sims-Lucas and Jacqueline Ho for their helpful suggestions.
Footnotes
Supported by NIH grants 4R01DK095748-04 (C.M.B.) and P30 DK074038 (B.K.Y.).
Disclosures: None declared.
A guest editor acted as the Editor-in-Chief for this manuscript. No one at the University of Alabama at Birmingham was involved in the peer-review process or final disposition of this article.
Current Address of P.P., Department of Biomedical Sciences, College of Veterinary Medicine, Tuskegee University, Tuskegee, AL.
Supplemental material for this article can be found at https://doi.org/10.1016/j.ajpath.2017.09.015.
Contributor Information
Pawan Puri, Email: ppuri@tuskegee.edu.
Carlton M. Bates, Email: batescm@upmc.edu.
Supplemental Data
Supplemental Figure S1.

In embryonic day (E)14.5 control and Six2creFrs2α knock-out (KO) embryos, phosphorylated Creb (p-Creb) signal is present in Six2-positive nephron progenitors and Sox9-positive early nephron progenitor (NP) epithelial derivatives. A–F: Co-immunofluorescence for Six2 (red), p-Creb (red), or Sox9 (red) with E-cadherin (E-cadh) (green) on serial sections obtained from E14.5 control and Six2creFrs2αKO kidneys. A and B: In controls and Six2creFrs2αKO kidneys, Six2 signal is present in nephron progenitors (arrowheads) and E-cadherin stains the ureteric bud (arrows). C and D: In both control and Six2creFrs2αKO kidneys, p-Creb is present in Six2-positive nephron progenitors (arrowheads) in the E-cadherin–positive ureteric bud (arrows) and in the early epithelial nephron structures (concave arrowheads). E and F: Sox9 co-localizes with E-cadherin in the ureteric bud (arrowhead) in control and Six2creFrs2αKO kidneys, and also stains the early nephron epithelial structures (arrow). Scale bars: 50 μm.
Supplemental Figure S2.

In P21 control and Six2creFrs2α knock-out (KO) kidneys, phosphorylated Creb (p-Creb) is present in the thick ascending limbs of the Henle loop and collecting ducts. Co-immunofluorescence for p-Creb (red) with Tamm-Horsfall protein (THP) (green) or dolichos biflorus agglutinin (DBA) (green) on serial sections from P21 control and Six2creFrs2αKO kidneys. A and B: In both control and Six2creFrs2αKO kidneys, comparable p-Creb signal is present in subsets of THP-positive cells of the thick ascending limbs of the Henle loop (arrows). C and D: In both control and Six2creFrs2αKO kidneys, comparable p-Creb signal is present in DBA-positive collecting ducts (arrows). Note that the p-Creb staining cyst lining cells (B and D, concave arrowheads) are neither THP nor DBA positive and were found by lotus tetragonolobus lectin (LTL) staining to be proximal tubule in origin (data not shown). Arrrowhead indicates p-Creb–positive tubules that are THP-positive in A. Scale bars: 50 μm.
Supplemental Figure S3.

Although phosphorylated Creb (p-Creb) expression in mutant proximal tubules is restricted to lotus tetragonolobus lectin (LTL)-negative cystic segments, total (unphosphorylated) Creb is expressed in LTL-positive Six2creFrs2α knock-out (KO) cells. A–D: Representative co-immunofluorescence images for p-Creb (red) or unphosphorylated (total) Creb (red) with LTL (green) on serial sections obtained from P21 control and Six2creFrs2αKO kidneys. A and B: P21 controls. In controls, p-Creb and total Creb both are present in stromally derived interstitium (arrows with black borders) and glomeruli (G), and in LTL-negative tubules (likely collecting ducts) (arrows); however, although p-Creb signal is absent in LTL-positive proximal tubules (A, arrowheads), total Creb signal is present in LTL-positive proximal tubules (B, arrowheads). C and D: In Six2creFrs2αKO kidneys, p-Creb is expressed ectopically in LTL-negative cyst lining cells (concave arrowhead) and is absent in LTL-positive cells in nondilated tubules (arrowhead) and in cyst lining cells (concave arrow); however, unphosphorylated Creb is expressed in both LTL-negative and LTL-positive cells (arrows indicate Creb staining in many nonproximal tubular cells). Arrows with black borders indicate p-Creb and Creb signal in stromal cells. Scale bars: 50 μm.
Supplemental Figure S4.

Ectopic Sox9 expression in proximal tubular–derived cystic segments with decreased lotus tetragonolobus lectin (LTL) staining in adult Ift88 knock-out (KO) kidneys 3 weeks after ischemia-reperfusion (IR).Representative co-immunofluorescence images for Sox9 (red) and LTL (green) in control and Ift88KO, 3 weeks after IR injury or sham surgery. A and B: No Sox9 signal is present. LTL-positive cells in control kidneys subjected to sham surgery (A, arrowhead) or to IR injury (B, arrowhead). C: In Ift88KO kidneys subjected to sham surgery, no Sox9 signal is observed in LTL-positive proximal tubules (arrowhead). D: In Ift88KO/IR kidneys, Sox9 is absent in LTL-positive tubules and cystic cells (arrowhead), but is expressed ectopically in cystic segments with reduced LTL staining (concave arrowheads). Scale bar = 50 μm.
Supplemental Figure S5.

Tissue sections show rapid progressive cyst formation in Six2crePkd1 knock-out (KO) kidneys. Hematoxylin and eosin–stained sections of control (A–C) and Six2crePkd1KO (D–F) kidneys show that Six2crePkd1KO have few dilated tubules at embryonic day (E)15.5 (A versus D), develop cysts that are widespread at P1 (B versus E), and have many larger cysts replacing all of the parenchyma by P5 (C versus F). Scale bar = 100 μm.
Supplemental Figure S6.

Ectopic Sox9 expression in proximal tubule–derived cystic segments with reduced or absent lotus tetragonolobus lectin (LTL) staining in P1 Six2crePkd1 knock-out (KO) kidneys. A and B: Representative co-immunofluorescence images for Sox9 (red) and LTL (green) in P1 control and Six2crePkd1KO kidneys (A). In controls, Sox9 was present in early epithelial nephron derivatives (arrow), but was absent in LTL-positive proximal tubules (arrowhead). B: In Six2crePkd1KO kidneys, Sox9 is expressed ectopically in cystic tubules that have reduced or absent LTL staining (concave arrowheads), but is absent in tubules with robust LTL staining (arrowhead). Scale bar = 50 μm.
Supplemental Figure S7.
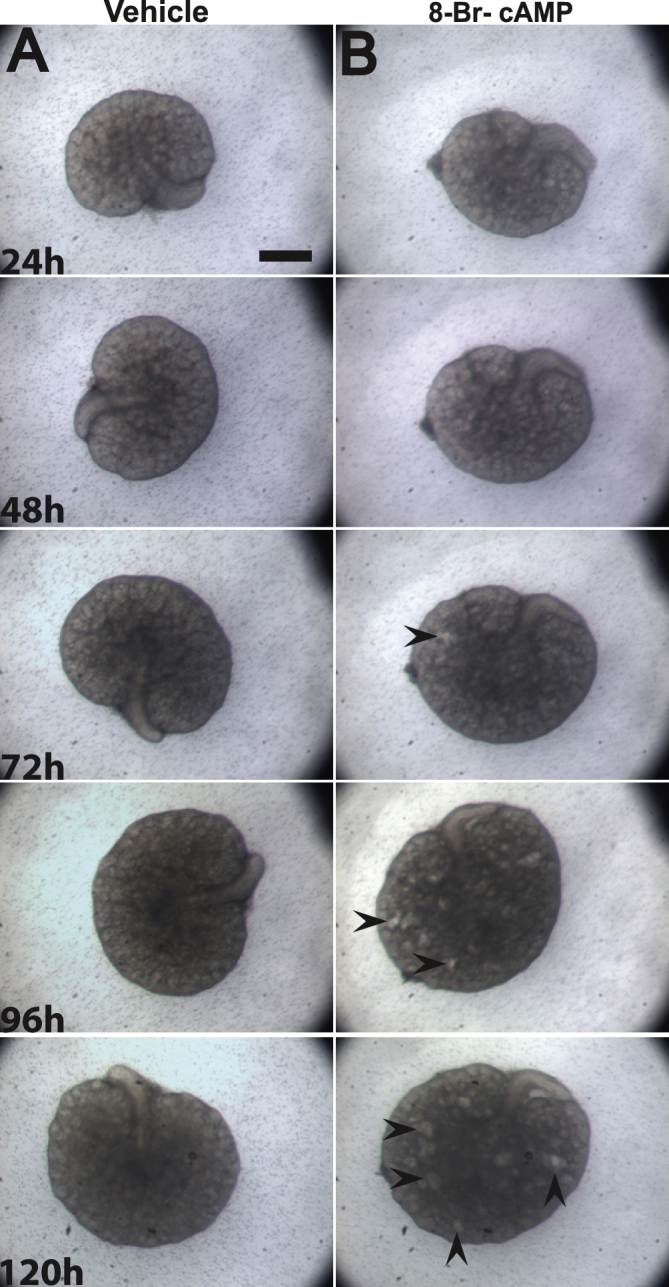
Embryonic day (E)13.5 kidney explants cultured in the presence of 8-Br-cAMP develop cysts. A and B: Representative stereomicroscopic images of kidney explants cultured in vehicle (A) or media containing 100 μmol/L 8-Br-cAMP (B) for 5 days. Cysts (concave arrowheads) can be first seen after 72 hours and continue to increase in size and number in the next 48 hours in the presence of 8-Br-cAMP. Scale bar = 500 μm.
Supplemental Figure S8.

Protein kinase A (PKA) inhibitor H-89 blocks 8-Br-cAMP–mediated cystogenesis and Creb phosphorylation in proximal tubules. A and B: Representative stereomicroscopic images of kidney explants cultured for 5 days (120 hours) in media containing 100 μmol/L 8-Br-cAMP or 100 μmol/L 8-Br-cAMP + 10 μmol/L H-89. Cysts are present in explants treated with 8-Br-cAMP alone (concave arrowhead), but are absent in the 100 μmol/L 8-Br-cAMP + 10 μmol/L H-89 treatment group (B). C and D: Representative hematoxylin and eosin images of sections obtained from kidney explants cultured with 100 μmol/L 8-Br-cAMP or 100 μmol/L 8-Br-cAMP + 10 μmol/L H-89 at 120 hours. Cysts (concave arrowhead) can be seen in explants treated with 8-Br-cAMP alone (C), but are absent in the 100 μmol/L 8-Br-cAMP + 10 μmol/L H-89 treatment group (D). E and F: Representative co-immunofluorescence images for p-Creb (red) and lotus tetragonolobus lectin (LTL) (green) on sections obtained from kidney explants cultured with 100 μmol/L 8-Br-cAMP or 100 μmol/L 8-Br-cAMP + 10 μmol/L H-89 for 5 days. In 8-Br-cAMP–treated explants, p-Creb is present in cysts with reduced LTL staining (E, concave arrowheads); however, in the 100 μmol/L 8-Br-cAMP + 10 μmol/L H-89 treatment group, p-Creb signal (F, concave arrowheads) largely is absent in LTL-positive tubules. Arrowheads indicate p-Creb staining in nephron progenitors. Scale bars: 500 μm (A and B); 100 μm (C and D); 50 μm (E and F).
References
- 1.Tan Y.C., Blumenfeld J., Rennert H. Autosomal dominant polycystic kidney disease: genetics, mutations and microRNAs. Biochim Biophys Acta. 2011;1812:1202–1212. doi: 10.1016/j.bbadis.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Harris P.C., Torres V.E. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest. 2014;124:2315–2324. doi: 10.1172/JCI72272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris P.C. What is the role of somatic mutation in autosomal dominant polycystic kidney disease? J Am Soc Nephrol. 2010;21:1073–1076. doi: 10.1681/ASN.2010030328. [DOI] [PubMed] [Google Scholar]
- 4.Mochizuki T., Wu G., Hayashi T., Xenophontos S.L., Veldhuisen B., Saris J.J., Reynolds D.M., Cai Y., Gabow P.A., Pierides A., Kimberling W.J., Breuning M.H., Deltas C.C., Peters D.J., Somlo S. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 5.Kim I., Ding T., Fu Y., Li C., Cui L., Li A., Lian P., Liang D., Wang D.W., Guo C., Ma J., Zhao P., Coffey R.J., Zhan Q., Wu G. Conditional mutation of Pkd2 causes cystogenesis and upregulates beta-catenin. J Am Soc Nephrol. 2009;20:2556–2569. doi: 10.1681/ASN.2009030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoder B.K. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2007;18:1381–1388. doi: 10.1681/ASN.2006111215. [DOI] [PubMed] [Google Scholar]
- 7.Hildebrandt F., Otto E. Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat Rev Genet. 2005;6:928–940. doi: 10.1038/nrg1727. [DOI] [PubMed] [Google Scholar]
- 8.Jonassen J.A., SanAgustin J., Baker S.P., Pazour G.J. Disruption of IFT complex A causes cystic kidneys without mitotic spindle misorientation. J Am Soc Nephrol. 2012;23:641–651. doi: 10.1681/ASN.2011080829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winyard P., Jenkins D. Putative roles of cilia in polycystic kidney disease. Biochim Biophys Acta. 2011;1812:1256–1262. doi: 10.1016/j.bbadis.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 10.Marciano D.K., Brakeman P.R., Lee C.Z., Spivak N., Eastburn D.J., Bryant D.M., Beaudoin G.M., 3rd, Hofmann I., Mostov K.E., Reichardt L.F. p120 catenin is required for normal renal tubulogenesis and glomerulogenesis. Development. 2011;138:2099–2109. doi: 10.1242/dev.056564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Giovanni V., Walker K.A., Bushnell D., Schaefer C., Sims-Lucas S., Puri P., Bates C.M. Fibroblast growth factor receptor-Frs2alpha signaling is critical for nephron progenitors. Dev Biol. 2015;400:82–93. doi: 10.1016/j.ydbio.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puri P., Bushnell D., Schaefer C.M., Bates C.M. Six2creFrs2alpha knockout mice are a novel model of renal cystogenesis. Sci Rep. 2016;6:36736. doi: 10.1038/srep36736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lancaster M.A., Gleeson J.G. Cystic kidney disease: the role of Wnt signaling. Trends Mol Med. 2010;16:349–360. doi: 10.1016/j.molmed.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tran P.V., Talbott G.C., Turbe-Doan A., Jacobs D.T., Schonfeld M.P., Silva L.M., Chatterjee A., Prysak M., Allard B.A., Beier D.R. Downregulating hedgehog signaling reduces renal cystogenic potential of mouse models. J Am Soc Nephrol. 2014;25:2201–2212. doi: 10.1681/ASN.2013070735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L., Zhou X., Fan L.X., Yao Y., Swenson-Fields K.I., Gadjeva M., Wallace D.P., Peters D.J., Yu A., Grantham J.J., Li X. Macrophage migration inhibitory factor promotes cyst growth in polycystic kidney disease. J Clin Invest. 2015;125:2399–2412. doi: 10.1172/JCI80467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swenson-Fields K.I., Vivian C.J., Salah S.M., Peda J.D., Davis B.M., van Rooijen N., Wallace D.P., Fields T.A. Macrophages promote polycystic kidney disease progression. Kidney Int. 2013;83:855–864. doi: 10.1038/ki.2012.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X., Magenheimer B.S., Xia S., Johnson T., Wallace D.P., Calvet J.P., Li R. A tumor necrosis factor-alpha-mediated pathway promoting autosomal dominant polycystic kidney disease. Nat Med. 2008;14:863–868. doi: 10.1038/nm1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalez G.A., Montminy M.R. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 19.Calvet J.P. The role of calcium and cyclic AMP in PKD. Polycystic Kidney Disease, ed 1. In: Li X., editor. Codon Publications; Brisbane, Australia: 2015. pp. 169–196. [PubMed] [Google Scholar]
- 20.Hanaoka K., Guggino W.B. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J Am Soc Nephrol. 2000;11:1179–1187. doi: 10.1681/ASN.V1171179. [DOI] [PubMed] [Google Scholar]
- 21.Ye H., Wang X., Sussman C.R., Hopp K., Irazabal M.V., Bakeberg J.L., LaRiviere W.B., Manganiello V.C., Vorhees C.V., Zhao H., Harris P.C., van Deursen J., Ward C.J., Torres V.E. Modulation of polycystic kidney disease severity by phosphodiesterase 1 and 3 subfamilies. J Am Soc Nephrol. 2016;27:1312–1320. doi: 10.1681/ASN.2015010057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magenheimer B.S., St John P.L., Isom K.S., Abrahamson D.R., De Lisle R.C., Wallace D.P., Maser R.L., Grantham J.J., Calvet J.P. Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na(+),K(+),2Cl(−) Co-transporter-dependent cystic dilation. J Am Soc Nephrol. 2006;17:3424–3437. doi: 10.1681/ASN.2006030295. [DOI] [PubMed] [Google Scholar]
- 23.Qin S., Taglienti M., Cai L., Zhou J., Kreidberg J.A. c-Met and NF-kappaB-dependent overexpression of Wnt7a and -7b and Pax2 promotes cystogenesis in polycystic kidney disease. J Am Soc Nephrol. 2012;23:1309–1318. doi: 10.1681/ASN.2011030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masai I., Yamaguchi M., Tonou-Fujimori N., Komori A., Okamoto H. The hedgehog-PKA pathway regulates two distinct steps of the differentiation of retinal ganglion cells: the cell-cycle exit of retinoblasts and their neuronal maturation. Development. 2005;132:1539–1553. doi: 10.1242/dev.01714. [DOI] [PubMed] [Google Scholar]
- 25.Sakai D., Suzuki T., Osumi N., Wakamatsu Y. Cooperative action of Sox9, Snail2 and PKA signaling in early neural crest development. Development. 2006;133:1323–1333. doi: 10.1242/dev.02297. [DOI] [PubMed] [Google Scholar]
- 26.Xu J., Tian J., Grumelli S.M., Haley K.J., Shapiro S.D. Stage-specific effects of cAMP signaling during distal lung epithelial development. J Biol Chem. 2006;281:38894–38904. doi: 10.1074/jbc.M609339200. [DOI] [PubMed] [Google Scholar]
- 27.Mantamadiotis T., Papalexis N., Dworkin S. CREB signalling in neural stem/progenitor cells: recent developments and the implications for brain tumour biology. Bioessays. 2012;34:293–300. doi: 10.1002/bies.201100133. [DOI] [PubMed] [Google Scholar]
- 28.Mantamadiotis T., Lemberger T., Bleckmann S.C., Kern H., Kretz O., Martin Villalba A., Tronche F., Kellendonk C., Gau D., Kapfhammer J., Otto C., Schmid W., Schutz G. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet. 2002;31:47–54. doi: 10.1038/ng882. [DOI] [PubMed] [Google Scholar]
- 29.Little M.H., McMahon A.P. Mammalian kidney development: principles, progress, and projections. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a008300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li W., Hartwig S., Rosenblum N.D. Developmental origins and functions of stromal cells in the normal and diseased mammalian kidney. Dev Dyn. 2014;243:853–863. doi: 10.1002/dvdy.24134. [DOI] [PubMed] [Google Scholar]
- 31.Sims-Lucas S., Schaefer C., Bushnell D., Ho J., Logar A., Prochownik E., Gittes G., Bates C.M. Endothelial progenitors exist within the kidney and lung mesenchyme. PLoS One. 2013;8:e65993. doi: 10.1371/journal.pone.0065993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gallegos T.F., Kouznetsova V., Kudlicka K., Sweeney D.E., Bush K.T., Willert K., Farquhar M.G., Nigam S.K. A protein kinase A and Wnt-dependent network regulating an intermediate stage in epithelial tubulogenesis during kidney development. Dev Biol. 2012;364:11–21. doi: 10.1016/j.ydbio.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gupta I.R., Piscione T.D., Grisaru S., Phan T., Macias-Silva M., Zhou X., Whiteside C., Wrana J.L., Rosenblum N.D. Protein kinase A is a negative regulator of renal branching morphogenesis and modulates inhibitory and stimulatory bone morphogenetic proteins. J Biol Chem. 1999;274:26305–26314. doi: 10.1074/jbc.274.37.26305. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi A., Valerius M.T., Mugford J.W., Carroll T.J., Self M., Oliver G., McMahon A.P. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 2008;3:169–181. doi: 10.1016/j.stem.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin Y., Zhang J., Zhang Y., Wang F. Generation of an Frs2alpha conditional null allele. Genesis. 2007;45:554–559. doi: 10.1002/dvg.20327. [DOI] [PubMed] [Google Scholar]
- 36.Piontek K.B., Huso D.L., Grinberg A., Liu L., Bedja D., Zhao H., Gabrielson K., Qian F., Mei C., Westphal H., Germino G.G. A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J Am Soc Nephrol. 2004;15:3035–3043. doi: 10.1097/01.ASN.0000144204.01352.86. [DOI] [PubMed] [Google Scholar]
- 37.Sharma N., Malarkey E.B., Berbari N.F., O'Connor A.K., Vanden Heuvel G.B., Mrug M., Yoder B.K. Proximal tubule proliferation is insufficient to induce rapid cyst formation after cilia disruption. J Am Soc Nephrol. 2013;24:456–464. doi: 10.1681/ASN.2012020154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Committee for the Update of the Guide for the Care and Use of Laboratory Animals; National Research Council . National Academies Press; Washington, DC: 2011. Guide for the Care and Use of Laboratory Animals: Eighth Edition. [Google Scholar]
- 39.Sims-Lucas S., Di Giovanni V., Schaefer C., Cusack B., Eswarakumar V.P., Bates C.M. Ureteric morphogenesis requires Fgfr1 and Fgfr2/Frs2alpha signaling in the metanephric mesenchyme. J Am Soc Nephrol. 2012;23:607–617. doi: 10.1681/ASN.2011020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puri P., Walker W.H. The tyrosine phosphatase SHP2 regulates Sertoli cell junction complexes. Biol Reprod. 2013;88:59. doi: 10.1095/biolreprod.112.104414. [DOI] [PubMed] [Google Scholar]
- 41.Chang-Panesso M., Humphreys B.D. Cellular plasticity in kidney injury and repair. Nat Rev Nephrol. 2017;13:39–46. doi: 10.1038/nrneph.2016.169. [DOI] [PubMed] [Google Scholar]
- 42.Peters D.J., Breuning M.H. Autosomal dominant polycystic kidney disease: modification of disease progression. Lancet. 2001;358:1439–1444. doi: 10.1016/S0140-6736(01)06531-X. [DOI] [PubMed] [Google Scholar]
- 43.Fain P.R., McFann K.K., Taylor M.R., Tison M., Johnson A.M., Reed B., Schrier R.W. Modifier genes play a significant role in the phenotypic expression of PKD1. Kidney Int. 2005;67:1256–1267. doi: 10.1111/j.1523-1755.2005.00203.x. [DOI] [PubMed] [Google Scholar]
- 44.Chen A.E., Ginty D.D., Fan C.M. Protein kinase A signalling via CREB controls myogenesis induced by Wnt proteins. Nature. 2005;433:317–322. doi: 10.1038/nature03126. [DOI] [PubMed] [Google Scholar]
- 45.Villanueva S., Cespedes C., Vio C.P. Ischemic acute renal failure induces the expression of a wide range of nephrogenic proteins. Am J Physiol Regul Integr Comp Physiol. 2006;290:R861–R870. doi: 10.1152/ajpregu.00384.2005. [DOI] [PubMed] [Google Scholar]
- 46.Abbate M., Brown D., Bonventre J.V. Expression of NCAM recapitulates tubulogenic development in kidneys recovering from acute ischemia. Am J Physiol. 1999;277:F454–F463. doi: 10.1152/ajprenal.1999.277.3.F454. [DOI] [PubMed] [Google Scholar]
- 47.Stewart R., Flechner L., Montminy M., Berdeaux R. CREB is activated by muscle injury and promotes muscle regeneration. PLoS One. 2011;6:e24714. doi: 10.1371/journal.pone.0024714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arany I., Herbert J., Herbert Z., Safirstein R.L. Restoration of CREB function ameliorates cisplatin cytotoxicity in renal tubular cells. Am J Physiol Renal Physiol. 2008;294:F577–F581. doi: 10.1152/ajprenal.00487.2007. [DOI] [PubMed] [Google Scholar]
- 49.Arany I., Megyesi J.K., Reusch J.E., Safirstein R.L. CREB mediates ERK-induced survival of mouse renal tubular cells after oxidant stress. Kidney Int. 2005;68:1573–1582. doi: 10.1111/j.1523-1755.2005.00569.x. [DOI] [PubMed] [Google Scholar]
- 50.Takakura A., Contrino L., Zhou X., Bonventre J.V., Sun Y., Humphreys B.D., Zhou J. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum Mol Genet. 2009;18:2523–2531. doi: 10.1093/hmg/ddp147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weimbs T. Polycystic kidney disease and renal injury repair: common pathways, fluid flow, and the function of polycystin-1. Am J Physiol Renal Physiol. 2007;293:F1423–F1432. doi: 10.1152/ajprenal.00275.2007. [DOI] [PubMed] [Google Scholar]
- 52.Bergmann C. ARPKD and early manifestations of ADPKD: the original polycystic kidney disease and phenocopies. Pediatr Nephrol. 2015;30:15–30. doi: 10.1007/s00467-013-2706-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weimbs T. Third-hit signaling in renal cyst formation. J Am Soc Nephrol. 2011;22:793–795. doi: 10.1681/ASN.2011030284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takakura A., Contrino L., Beck A.W., Zhou J. Pkd1 inactivation induced in adulthood produces focal cystic disease. J Am Soc Nephrol. 2008;19:2351–2363. doi: 10.1681/ASN.2007101139. [DOI] [PMC free article] [PubMed] [Google Scholar]