

Highlights
-
•
We modeled host specificity of hepatitis E virus, genotypes 3 and 4 (HEV3 & HEV4).
-
•
Epistasis among HEV protein sites was modeled using Bayesian Networks.
-
•
Host-specific dependencies among coevolving HEV protein sites were observed.
-
•
Coevolving sites associating HEV3 & HEV4 strains to host of origin are reported.
-
•
Host specificity is a convergent and abundant trait in HEV3 & HEV4.
Keywords: Hepatitis E virus, Prediction, HEV ORFs, Adaptation, Bayesian network, Coevolution
Abstract
Hepatitis E virus (HEV) causes epidemic and sporadic cases of hepatitis worldwide. HEV genotypes 3 (HEV3) and 4 (HEV4) infect humans and animals, with swine being the primary reservoir. The relevance of HEV genetic diversity to host adaptation is poorly understood. We employed a Bayesian network (BN) analysis of HEV3 and HEV4 to detect epistatic connectivity among protein sites and its association with the host specificity in each genotype. The data imply coevolution among ∼70% of polymorphic sites from all HEV proteins and association of numerous coevolving sites with adaptation to swine or humans. BN models for individual proteins and domains of the nonstructural polyprotein detected the host origin of HEV strains with accuracy of 74–93% and 63–87%, respectively. These findings, taken together with lack of phylogenetic association to host, suggest that the HEV host specificity is a heritable and convergent phenotypic trait achievable through variety of genetic pathways (abundance), and explain a broad host range for HEV3 and HEV4.
1. Introduction
Hepatitis E virus (HEV), a member of the Hepeviridae family, has a positive-sense, single-stranded RNA genome of about 7.2 kb with a 5′-methylguanine cap and a 3′-poly(A) tail. The HEV genome contains three partially overlapping reading frames (ORFs) (Emerson et al., 2004). The ORF1 codes for a large polyprotein containing several functional domains responsible for viral replication. These include, from the amino to carboxyl terminus, the viral methyltransferase (Mt, pfam 01660), the Y domain, the papain-like cysteine protease C41 (Plp, pfam 05417), a region of unknown function (Unk), the polyproline region (Pp, pfam 12526) (Purdy et al., 2012b), the Appr-1”-p processing enzyme/macro domain (Md, pfam 01661) (Han et al., 2011), the UvrD/REP helicase (Hel, pfam 01443) and the RNA-dependent RNA polymerase (Pol, pfam 00978). ORF2 codes for the viral capsid, and ORF3 for a regulatory protein (Ahmad et al., 2011).
HEV causes epidemic and sporadic hepatitis in humans, for which there are no specific therapeutic options. Usually, hepatitis E is a self-limiting disease similar to hepatitis A (Khuroo, 2011). HEV was originally assumed to be transmitted only through a fecal-oral route of transmission, as humans were the only recognized host for the virus (Khuroo, 2011, Viswanathan, 1957, Zhuang et al., 1991). Initially, two genotypes, 1 and 2 (HEV1 and HEV2), of human HEV were identified. It was not until 1997 that a new genotype was isolated. This genotype, 3 (HEV3), infects humans and swine (Meng et al., 1997). Two years later, a fourth genotype (HEV4) was identified (Wang et al., 1999). Unlike HEV1 and HEV2, these new genotypes were more permissive with respect to their host range. The expanded host range included deer, wild boar and mongoose (Meng, 2011). Additional research showed that these new animal genotypes could be transmitted to humans zoonotically (Meng, 2011, Tei et al., 2003). More recently, two or three additional putative genotypes have been isolated: from rabbits (Zhao et al., 2009) and wild boar (Smith et al., 2013, Takahashi et al., 2011), although there is controversy about the exact classification of these viruses. A serosurvey of humans and swine in Bolivia showed that HEV3 may be transmitted from humans to humans (Purdy et al., 2012a) and evidence from changing epidemiological patterns for HEV in China suggests human-to-human transmission of HEV4 (Krawczynski et al., 2000).
Viruses endemic to a reservoir host species that acquire capacity to be transmitted among new host populations can pose a threat to public health. One remarkable example of the threat to public health posed by such viruses is the severe acute respiratory syndrome-coronavirus (SARS-CoV) 2003 outbreak in Asia (Tsang et al., 2003). Zoonotic strains of HEV have the potential to cause serious disease and mortality (Aggarwal, 2011, Mizuo et al., 2005, Patra et al., 2007) in infected patients or change into phenotypes that may become more transmissible among humans (Krawczynski et al., 2000, Purdy et al., 2012a). The need to assess the risk of HEV outbreaks calls for genetic surveillance of the emerging zoonotic strains in their reservoir hosts.
Genetic variation and covariation are important molecular mechanisms for genomic diversification and adaptation of viruses during intra-host evolution. Epistasis plays a crucial role in viral evolution (Bonhoeffer et al., 2004, Sanjuan et al., 2004). Epistatic interactions among sites along the viral genome are widespread (Campo et al., 2008, Donlin et al., 2012) and frequently observed in the form of coordinated (Campo et al., 2008) and compensatory substitutions (Khudyakov, 2010, Yi et al., 2007). The pervasive nature of coevolution and its association with adaptation suggest the use of coevolving genomic sites as genetic markers of important viral phenotypic traits such as drug resistance (Lara and Khudyakov, 2012, Lara et al., 2011b) and virulence (Khudyakov, 2012). Coordinated substitutions among genomic sites were shown to be associated with response to combined interferon and ribavirin therapy among hepatitis C virus (HCV) infected patients (Aurora et al., 2009, Lara et al., 2011b) and resistance to lamivudine among hepatitis B virus infected patients (Thai et al., 2012). Host factors such as gender, ethnicity and age have also been linked to coordination among HCV genomic substitutions (Lara et al., 2011a), suggesting host specificity of viral evolution.
Although coevolution among HEV sites was noted (Donlin et al., 2012), no association between coordinated substitutions and HEV phenotypic traits has been explored. With HEV infecting a broad range of host species (Meng, 2011, Meng et al., 1997, Takahashi et al., 2011, Tei et al., 2003, Zhao et al., 2009), this virus offers an opportunity to assess the viral genetic contribution to host specificity and provides an important model for understanding emerging infectious diseases. This study evaluated host-specific coevolution among protein sites in HEV3 and HEV4. For this purpose, a BN approach was used to: (i) model epistatic connectivity among amino acid (aa) sites from proteins of HEV3 and HEV4 strains identified in swine and humans; (ii) examine the strength of association between the aa substitutions and host origin, and (iii) identify genetic markers of HEV host specificity.
2. Materials and methods
2.1. Data
Full-length consensus genomic sequences of HEV3 (n = 65) and HEV4 (n = 55) recovered from human and swine hosts were obtained from GenBank. Sequences from deer, wild boar, mongoose and rabbits were removed from the dataset because there are too few strains characterized from each of these animals to construct host-specific models. The HEV nucleotide sequences for all three ORFs were translated into respective aa sequences. The generated sequences were connected into a single concatenated polyprotein sequence for each HEV strain and aligned using MUSCLE (ver. 3.6) (Edgar, 2004).
After sequence alignment, the respective host source was assigned to each HEV sequence according to GenBank annotations. Residue site numbering was based on reference sequences with the GenBank accession numbers EU723514 for HEV3 and AB220971 for HEV4. Conserved aa sites and gaps were excluded from analyses. Only polymorphic sites for all proteins obtained from human and swine strains were analyzed. Analyses were carried out on datasets of polymorphic sites from all proteins or from ORF1-protein domains. Sites that fall outside the functionally characterized boundaries of protein domains in ORF1 are herein denoted as Orf1(x). The HEV3 dataset consisted of 29 swine and 36 human strains and the HEV4 dataset consisted of 16 swine and 39 human strains (GenBank accession numbers in Supplementary Material).
Since all full-size sequences were used for modeling, only short sequences from GenBank were available for validation. The validation datasets for testing classifiers consisted of 3 swine and 16 human sequences of HEV3 Pol, 4 swine sequences of HEV4 Pp and 7 human sequences of HEV4 Pol (GenBank accession numbers in Supplementary Material).
2.2. BN learning
BN is a probabilistic graphical model, where nodes in the graph represent random variables and directed arcs between the nodes represent relationships (Jensen, 2001, Neapolitan, 2004). Directed arcs define parenthood ordering among variables and encode the probability distributions in data. Given a finite set S = {Xi,…, Xn} of random variables, where Xi can take any value in S, a BN is an annotated directed acyclic graph (DAG) G = {V, E} that encodes the joint probability distribution over S. The nodes (V) of G correspond to random variables {Xi,…, Xn}. The edges (E) in G represent direct dependencies between variables. Each node Xi is associated with a conditional probability distribution (CPD) P(Xi|Pa(Xi)) that quantifies the effect of the parents on the node, where Pa(Xi) denotes the parents of Xi in G. The pair (G, CPD) encodes the joint probability distribution P(Xi,…, Xn) given G. The joint probability distribution over S from G is factorized as:
The HEV full-length polyprotein sequence alignment data were used to learn BN, G = {V, E}, where nodes in the graph represent polymorphic residue sites (Xi,…,Xn) in the sequence alignment and the CPD associated to a node encode the prior distribution of observed residue states in X i. For a host-virus dependency representation, an additional 2-state variable X i (where X i = human or swine host) was included in BN to associate the host source of the HEV sequence as annotated in GenBank.
Because BN provides a complete model of the probabilistic distribution for variables and their relationships, models can be used to answer probabilistic queries about the state of a subset of features when other features (evidence features) are observed. The process of computing the posterior distribution of features is achieved in BN by computing marginal probabilities for each unobserved node (target node) given information on the states of a set of observed nodes, a process known as probabilistic inference. In the absence of any observations, this computation is based on a priori probabilities and, when observations are given, the information is integrated into BN and all probabilities are updated accordingly.
Here, an unsupervised technique was used to automatically learn BN from data, which was then used to conduct probabilistic inference. Because learning BN from data has been proven to be NP-hard (Chickering et al., 2004) and with available sample size being relatively small, a heuristic score-and-search-based approach was adopted. This approach has two components: a scoring function, used to evaluate how well the learned BN fits the data, and a search strategy, which consists of a learning algorithm to identify BN structure(s) with high scores among the possible structures in BN space.
The Minimum description length (MDL) score (Bouckaert, 1993, Rissanen, 1986) was used for the scoring function. The MDL score is a criterion based on information theory that favors BN which provides the shortest description of the data. The MDL score has been shown to have better performance than other scoring methods in BN structure learning tasks (Bouckaert, 1993). Also, this score is conservative and returns by default highly significant relationships. Given BN = (G, CPD), and a training dataset D, the MDL score of BN is defined as ScoreMDL(BN|D) = MDL(BN) + MDL(D|BN). The first term of the MDL score is the description length of BN (number of bits required to encode BN parameters – structural complexity) and second term is the negative log likelihood of BN model given D (gives the number of bits necessary to describe D with BN – data likelihood). Structural complexity (SC) was pre-set prior to the start of BN learning. This threshold was set to a structural coefficient = 2.0 (except for the HEV3 BNSwine, where SC threshold was set at 1.0).
For the task of BN structure searching, an unsupervised learning algorithm, the EQ method (Munteanu and Bendou, 2001), was used to identify the best BN model. This method, which is based on searching the equivalent BN classes (structures representing the same conditional dependencies), has been shown to be efficient for finding optimal BN models of the data (Jouffe and Munteanu, 2001, Munteanu and Bendou, 2001). The cycle of exploration in BN space continued until no further improvement in the MDL score was observed.
Several unsupervised learned BN models were generated to represent the whole set of probabilistic relationships in the HEV data: BN of the HEV3 (BNHEV3) and HEV4 (BNHEV4) to represent interdependencies among polymorphic residue sites and association to the host. Also, host-based BN of the HEV (BNHuman and BNSwine) to represent host-specific interrelationships among the coevolving residue sites, where the sequence data of each HEV genotype was stratified by host of origin to derive respective BN.
BN learning and BN analyses (probabilistic inference, quality assessment, etc.) presented in this study were conducted using the BayesiaLaB™ software version 5.0 (Bayesia SAS, Laval, France). Details of BN analyses are described in Supplementary Material.
2.3. Classification tests
2.3.1. Feature selection (FS)
FS was performed on HEV data to identify and select residue site markers in order to maximize accuracy performance of the classifiers. FS was based on selecting subsets of features highly correlated with host origin of HEV sequences while having low inter-correlation between them (Hall, 1999). Protein sequence alignments of selected variables from each HEV ORF or individual ORF1 domains were labeled according to host of origin. These alignments comprised the data used for the training and cross-validation of classifiers. Herein, two machine-learning methods were used to generate classifier models: one based on a BN method and another on a linear projection method.
2.3.2. Bayesian network classifier (BNC)
A set of BNC were developed for the task classifying HEV strains by host of origin based on the primary structure information of each sequence of selected features. The standard 1-letter representation of aa was used to encode selected features of HEV strains for the training/testing of BNC, which took into account the interdependency among aa sites and aa composition of sequences. BNC representing HEV ORFs were tested to evaluate their individual contribution and relevance for association to host origin. Conversely, the same was done for the individual domains of ORF1.
2.3.3. Physicochemical mappings
The physicochemical space of HEV strains was mapped by transforming the standard 1-letter aa representation into numerical values encoding an aa physicochemical property. Protein sequences were transformed into N × 5 dimensional numerical vectors, where N is the sequence length and 5 represents the number of physicochemical values assigned to each residue site in the sequence alignment. Five statistically-derived factors for polarity, secondary structure, molecular volume, aa composition and electrostatic charge (Atchley et al., 2005) were used to represent HEV strains. Host-specific probabilistic mapping of HEV strains were developed using a visual machine-learning technique in the form of a linear projection (LP) (Demsar et al., 2005).
2.3.4. Classifiers performance evaluations
Performances of models was evaluated by 10-fold cross-validation (10-fold CV) during the training phase. Two measures were used to evaluate the classification performance of classifier models: overall classification accuracy (CA) and the harmonic mean of precision and recall (F-measure). The overall CA was measured as [(no. correctly classified instances/total no. of instances) × 100]. The F-measure was computed as: (2 ∗ TP)/(2 ∗ TP + FP + FN), where TP is the number of true positives; FP is the number of false positives; and FN is the number of false negatives.
In addition, validation trails were conducted by testing classifier models on new data of HEV sequences retrieved from GenBank and measuring the CA performance (see Supplementary Material). Since all available whole-genome sequences were used for construction of models, short sequences of Pol and Pp obtained from GenBank were used for validation tests. In total, 19 and 7 Pol sequences were available for HEV3 and HEV4, respectively, and 4 Pp sequences for HEV4.
3. Results
3.1. Phylogenetic association with host specificity
The degree of correlation between host specificity and shared ancestry of HEV variants was quantified from a posterior set of trees (PST) created for each genotype using a Bayesian Markov chain Monte Carlo method (see Supplementary Material for details and Fig. S1). The Bayesian Tip-significance analysis (Befi-BaTS) results show that only about half of the indices used to measure phenotype/phylogenetic relatedness are statistically significant (Table S1 in Supplementary Material). The association index (Wang et al., 2001), parsimony score statistic (Slatkin and Maddison, 1989) and net relatedness index (Webb, 2000) were statistically significant for both genotypes (Table S1 in Supplementary Material). The unique fraction metric (Lozupone and Knight, 2005) and the monophyletic clade size metric for the swine host (Parker et al., 2008) were not statistically significant for both genotypes. The nearest taxa index (Webb, 2000), phylogenetic diversity (Hudson et al., 1992) and the MC metric for the human host (Parker et al., 2008) gave ambiguous results. Those metrics that examine host specificity across the PST as a whole tend to be statistically significant and indicate that the host specificity trait is not randomly distributed across the tips of each genotype tree; however, the monophyletic clade size statistic, which analyzes the host specificity by host, tend not to be statistically significant indicating that host specificity is randomly distributed across each genotype PST.
3.2. Broad coordination among protein sites
A BN was used to model epistatic connectivity among sites of all HEV proteins. This approach allows for identification of coordinated changes at protein sites using conditional probabilities and visualization of the identified associations among sites in the form of a network, thus providing a general framework for exploring epistatic connectivity among HEV sites and its potential linkage to phenotypic traits. Analysis was conducted using proteins encoded by all 3 ORFs of full-genome sequences obtained from HEV3 and HEV4 strains. Since both genotypes are zoonotic, HEV sequences sampled from humans or swine were selected for analysis.
The learned BN showed a broad interdependence among protein sites for both genotypes (Fig. 1 ). All 3 proteins contributed sites to the BNs, with 68% and 74% of polymorphic sites being involved in BNHEV3 and BNHEV4, respectively. BNHEV3 comprised 153 arcs connecting 147 sites. Each site had from 2 to 11 states (average, 3.2), with the number of connections varying from 1–22 (average, 2.1). BNHEV4 comprised 174 arcs connecting 163 sites. Each site had from 2 to 9 states (average, 3.1) and was connected to 2–9 other sites (average, 2.1). The ORF2- and ORF3-encoded proteins contributed only 17.0% and 6.8% of sites to BNHEV3, respectively, and 14.7% and 9.2% of sites to BNHEV4, respectively (Fig. 2 A). The ORF1-protein contributed 76.2% and 76.1% of sites to BNHEV3 and BNHEV4, respectively. The Pp, Pol and Plp domains contributed 65% and 71% of the ORF1-protein sites to BNHEV3 and BNHEV4, respectively. The major difference observed between the two genotypes was in the number of sites contributed by the Pp domain. This domain alone provided 21.8% and 30.7% of all sites to BNHEV3 and BNHEV4, respectively (Fig. 2A). Dependencies among all sites involved in both BNs were statistically significant (χ2; p ⩽ 0.0003) (details in Supplementary Material).
Fig. 1.
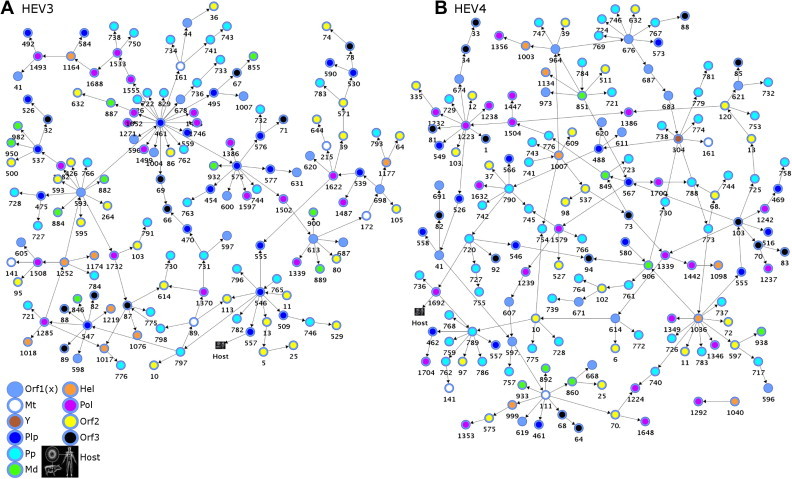
Coordination among HEV protein sites modeled with BNs. The BN models show genome-wide epistatic connectivity among aa sites (a structural coefficient threshold = 2.0) and association to host variable (a structural coefficient threshold = 0.95). Nodes represent polymorphic aa sites and arcs between them represent dependency. Nodes are color-coded according to the ORF and ORF1-domains and numbered according to the aa positions in the respective ORFs. Orf1(x) encompasses UNK and denotes aa sites that fall outside known ORF1 domains (n = 16 in BNHEV3 and n = 18 in BNHEV4). (A) A learned BN of HEV3 sequences (n = 65) and (B) A learned BN of HEV4 sequences (n = 55).
Fig. 2.
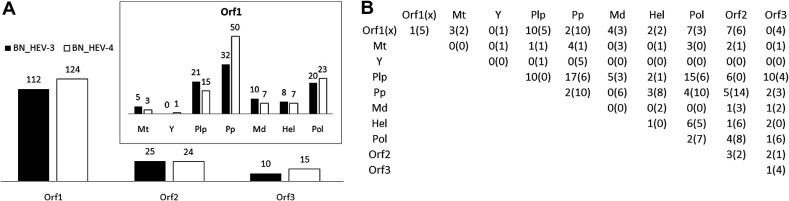
Contribution of proteins and ORF1-domains to BNHEV3 and BNHEV4. (A) Bar charts show number of aa sites involved in BNs for each protein and ORF1-domain. (B) Number of links among aa sites from all proteins and ORF1-domains observed in BNs. Numbers outside and inside of parenthesis are for BNHEV3 and BNHEV4, respectively.
3.3. Inter- and intra-protein coordination
To estimate the contribution of inter- and intra-protein relationships to the global coordination among sites, the ratio of intra- to inter-protein links were examined for each ORF (Fig. 2B). This ratio was 2.48, 0.11 and 0.06 in BNHEV3, and 1.66, 0.05 and 0.19 in BNHEV4 for ORF1-, ORF2- and ORF3-proteins, respectively. Thus, only the ORF1-protein sites were involved in substantial intra-protein coordination, while sites from ORF2- and ORF3-proteins showed predominance of inter-protein coordination. The ORF1-protein sites had 146 arcs in BNHEV3 and 173 arcs in BNHEV4, with only 28.8% and 37.6% of the arcs connecting to sites from other proteins in these 2 BNs, respectively. Therefore, the ORF1-protein sites in BNHEV4 coordinated their states with 1.5-times more sites in ORF2- and ORF3-proteins than BNHEV3, indicating a greater dependence of the HEV4 evolution on inter-protein coordination. Only 2 arcs connected sites from the ORF2- and ORF3-proteins in BNHEV3 and 1 arc in BNHEV4 (Fig. 2B).
The ORF1-encoded protein is multifunctional and divided into 7 functional domains (Koonin et al., 1992). Among the domains, only Plp, Pp and Pol had up to 23% of intra-domain links, while Mt, Y and Md had links only to other domains, with Hel having a single intra-domain link in HEV3 (Fig. 2B). This finding indicates extensive coordination among the ORF1 domains in both HEV3 and HEV4. Domains Plp and Pp showed a dramatically different distribution of links in HEV3 and HEV4. While the HEV3 Plp had 10 intra-domain links, the HEV4 Plp had none. However, the HEV3 Pp had only 2 intra-domain links but HEV4 Pp had 10. Both domains had many links connecting each other and Pol. The ORF1 region with unassigned function, Unk, also had a number of links to Plp, Pp and Pol as well as internal links (Fig. 2B).
3.4. Strength of influences among ORFs
KL-divergence was calculated for each link in BNs to estimate the strength of influences that aa variations at one site have on the state of another site (see Supplementary Material). Fig. 3 shows sums of the KL-divergence values calculated for all links (global value) as well as for outgoing and incoming links for each protein. In BNHEV3, the Plp domain had the strongest overall global influence (KL-divergence = 53.3) over the entire BN, followed by Pol (KL-divergence = 25.4), Unk (KL-divergence = 23.7) and Pp (KL-divergence = 22.9). In BNHEV4, Pp (KL-divergence = 49.6), Pol (KL-divergence = 25.3), Unk (KL-divergence = 23.5) and the ORF2 protein (KL-divergence = 18.3) showed the greatest influence over the entire BN. The overall global influence of Plp in BNHEV3 was associated primarily with strong outgoing links directed to other proteins and ORF1 domains; whereas for Pp in BNHEV4, this influence was associated primarily with strong incoming links directed from other proteins and ORF1 domains (Fig. 3A).
Fig. 3.
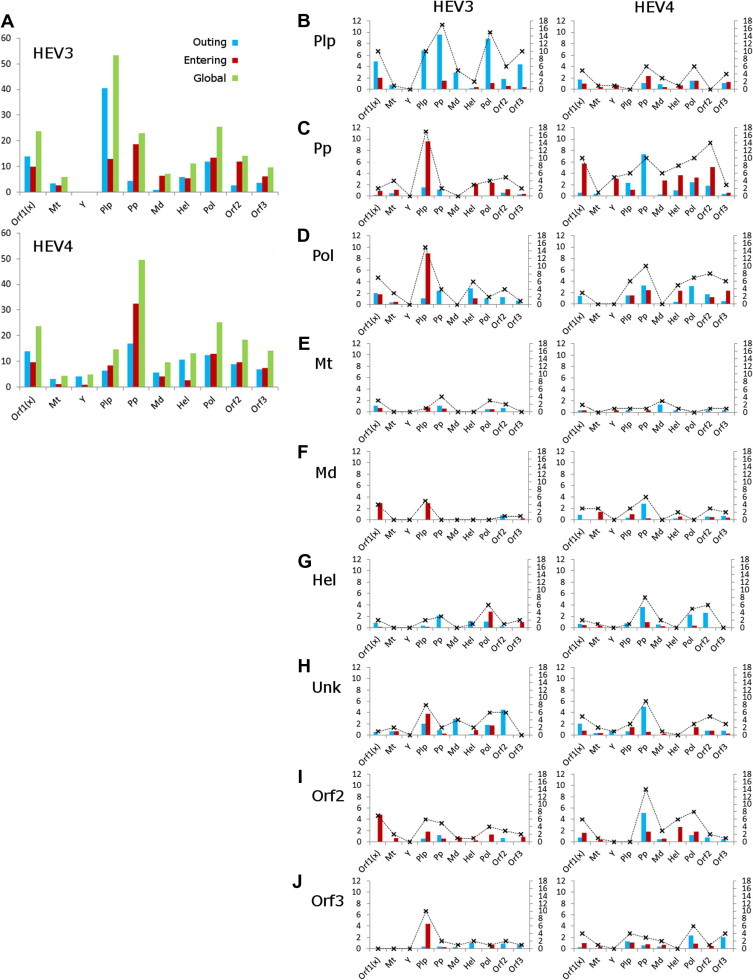
Strengths of epistatic influences. Strength of linkages (primary y-axis) and number of links (secondary y-axis; crosses joined with dashed lines) among HEV proteins and ORF1-domains (x-axis) was computed from learned BNHEV3 and BNHEV4. (A) Overall strengths measured for each protein or ORF1-domain in entire BNs. Directionality of influences is color coded. (B–J) Strength and number of links to all proteins and ORF1-domains in BNHEV3 and BNHEV4 observed for each protein and ORF1-domain.
A more detailed analysis of the strength of influences among proteins and ORF1 domains showed further differences in the strength of epistatic signal associated with Plp and Pp in HEV3 and HEV4. In BNHEV3, Plp sites had a strong influence over sites in Pp, Pol, Unk and ORF3, whereas Plp had a very limited effect on the states of sites in other proteins and ORF1 domains in BNHEV4 (Fig. 3B). The strongest epistatic signal came to Pp from Plp in BNHEV3. In BNHEV4, the state of sites in Pp was strongly influenced by Unk, Y, Md, Hel, Pol and ORF2, while Plp had a very limited effect on this domain (Fig. 3C). Collectively, these observations emphasize important differences in the structure of epistatic connectivity among aa sites in the 2 HEV genotypes.
3.5. Most influential sites
Using the degree of connectivity and KL-divergence values as criteria for finding sites that impose the greatest influence on the state of the entire network (Fig. 1), we identified the most influential sites (degree k ⩾ 5 and KL-divergence ⩾ 2.0) in BNHEV3 (n = 9) and BNHEV4 (n = 16) (Fig. 4 ). The ORF1 sites 461 and 593 were responsible for 34.1% and 40.9% of the overall global influences of Plp and Unk in BNHEV3, respectively. In BNHEV4, the ORF1 sites 789 and 1036 were responsible for 12.1% and 67.9% of the overall global influence of Pp and Hel, respectively.
Fig. 4.
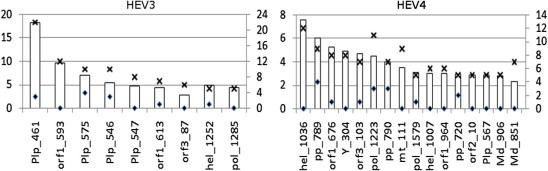
The most interconnected (n ⩾ 5) and influential (global KL-divergence ⩾ 2.0) nodes in BNHEV3 and BNHEV4. Bars show global strength of influence (primary y-axis) and number of links (secondary y-axis) for aa sites identified in BNs (Fig. 1). The numbers of total and intra-protein (or intra-domain) links are shown with crosses and rhombi, respectively.
3.6. Unk contribution
The region of the ORF1-encoded protein at positions 593–706 (HEV3 and HEV4), designated Unk, has not been assigned any function (Koonin et al., 1992). However, sites from this region were among most influential in BNs (Fig. 3); e.g., sites 593 and 613 in BNHEV3, and 676 in BNHEV4 (Fig. 4). In general, significant dependencies (KL-divergence ⩾ 0.80) were detected for the Unk sites, with many of them having outgoing links to other proteins and ORF1 domains (Fig. 3H). In BNHEV3, the ORF1 sites 1252, 882, 766 and 475 and the ORF2 sites 264, 426, and 593 were linked to site 593 in Unk; the ORF1 site 461 was linked to sites 596 and 678 in Unk, and the ORF1 site 575 to the Unk site 600. In BNHEV4, sites 621 and 676 in Unk were linked to the ORF1 sites 732 and 746, and sites 683 and 687 were interlinked in Unk.
3.7. Association to the host
The finding of the variable representing the host origin of HEV strains in learned BNHEV3 and BNHEV4 (Fig. 1) suggests that the coevolution among aa sites has association to the host. Site 557 from Plp in BNHEV3 and 1692 from Pol in BNHEV4 had direct links to the host variable, which were found to be significant (X 2, p = 0.0002 and p = 0.0001, respectively).
Evaluation of the BN models (see Target analysis in Supplementary Material) showed that BNHEV3 and BNHEV4 associate HEV strains to the host (swine or human) with the mean accuracy of 72.3% and 78.2%, respectively. However, despite such high accuracy observed in the target analysis, none of the HEV3 and HEV4 proteins contained any individual aa site with strong MI directly to the host (Fig. 5 ), suggesting that the virus-host dependency is contingent on the overall concerted effects of several aa sites from HEV proteins. Thus, association to the host through site 557 (BNHEV3) and 1692 (BNHEV4) should be considered in conjunction with aa heterogeneity at other sites in the network. Aa sites, which as a group were found to notably affect probability distributions of the host variable in BN are identified in Table 1 (based on target analysis; see Supplementary Material for details). It is important to note that there were many sites from all 3 proteins in BNHEV3 and BNHEV4 that together had a measurable effect on the state of the host variable. The Pp region, positions 721–796 and 720–790, constituted the largest fraction of ORF1-protein sites reflecting the host dependency in both HEV3 and HEV4, respectively.
Fig. 5.
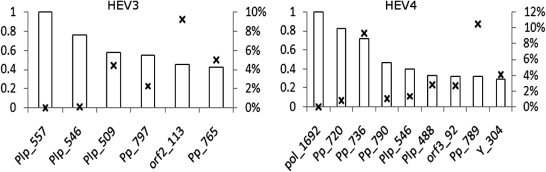
HEV3 and HEV4 protein sites most associated with host specificity. Bars show relative MI values (primary y-axis) for aa sites identified in BNs (Fig. 1). MI for the Plp site 557 (MI = 0.16) in BNHEV3 and Pol site 1692 (MI = 0.19) in BNHEV4 were assigned a relative value of 1. P values for each site are identified with crosses and shown as percentage (secondary y-axis).
Table 1.
Protein sites with relevant effects on the BN host variable.
| Genotype | ORF | Protein sitesa | Standardized total effectsb |
|---|---|---|---|
| HEV3 | ORF 1 | 89, 113, 509, 546, 547, 555, 557, 598, 605, 721, 746, 765, 782, 784, 796, 846, 1017, 1018, 1219, 1252, 1285, 1370 and 1508 | 0.459–0.0784 |
| ORF 2 | 5, 11, 13, 25 and 529 | 0.177–0.069 | |
| ORF 3 | 82, 88 and 89 | 0.188–0.125 | |
| HEV4 | ORF 1 | 161, 304, 462, 469, 488, 516, 546, 555, 557, 566, 573, 611, 620, 676, 683, 687, 720, 727, 736, 738, 740, 742, 743, 745, 755, 759, 762, 767, 769, 773, 774, 779, 781, 783, 786, 789, 790, 906, 938, 964, 1003, 1007, 1036, 1237, 1242, 1346, 1349, 1356, 1632, 1692 and 1704 | 0.5140–0.081 |
| ORF 2 | 11, 37, 39, 537, 597, 609 and 632 | 0.182–0.081 | |
| ORF 3 | 70, 73, 92, 94 and 103 | 0.298–0.081 | |
3.8. Host-specificity of epistatic connectivity
The finding of genetic associations to host origin (Fig. 5 and Table 1) suggests that epistatic connectivity among aa sites in HEV3 and HEV4 is specific to the host. However, the host specificity of epistatic connectivity is not explicitly obvious in the learned BNHEV3 and BNHEV4 (Fig. 1). To examine the host-specific coevolution among the HEV aa sites, additional models BNSwine and BNHuman were generated using HEV sequences obtained from swine and humans, respectively (Fig. 6 ). To determine the level of host specificity of epistatic connectivity in learned BN we measured the degree of accuracy with which the modeled epistatic connectivity among aa sites reflects the host origin of HEV variants.
Fig. 6.
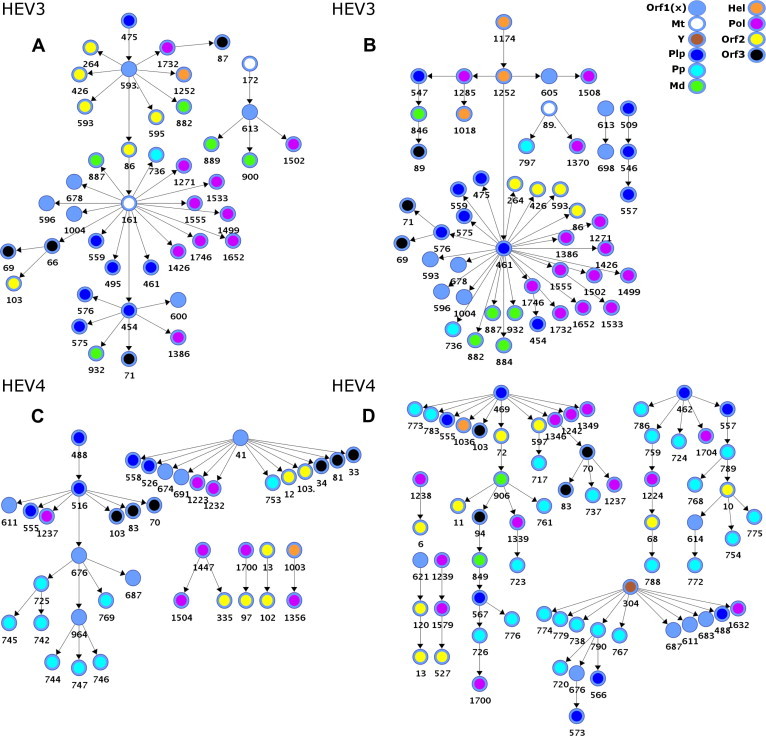
HEV3 and HEV4 host-specific epistatic motifs. (A) HEV3-BNSwine contains 40 arcs connecting 42 aa sites; (B) HEV3-BNHuman contains 44 arcs connecting 48 aa sites; (C) HEV4-BNSwine contains 34 arcs connecting 40 aa sites; and (D) HEV4-BNHuman contains 60 arcs connecting 66 aa sites. All links are statistically significant (p ⩽ 10−5) and highly correlated (avg. r = 0.9326 and r = 0.8791 – A and B, respectively; r = 0.8075 and r = 0.7934 – C and D, respectively). Color coding and numbering are as in Fig. 1.
The log-likelihoods of BN (see Supplementary Material for details of computations) were compared to evaluate accuracy with which learned BNs (Fig. 6) represented data distribution of HEV variants originating from same or different host species. The log-likelihood values for BNSwine or BNHuman tested on swine or human data, respectively, were only 15%-30% different, while cross-tests on human or swine data, respectively, resulted in ∼3.0–5.5-fold differences (Table 2 ). This finding indicate that BNs shown in Fig. 6 have a structure that accurately represents the unseen data obtained from HEV strains recovered from the same hosts but does not fit as well data obtained from HEV strains recovered from different hosts. In addition, relationships among variables observed in BNSwine and BNHuman were highly conserved among sets of BNs learned from 15 re-samples of the HEV data. Arc confidence analysis by jackknife method (Supplementary Material) showed that >88% of all arcs are present in >73% of the k-samples, indicating robustness of the modeled epistatic connectivity in the host-specific BNs (Table 3 ).
Table 2.
Quality assessment of BNHEV3 and BNHEV4. Comparisons between log-likelihood values (within shaded and unshaded row pairs).
 |
a Statistical tests were performed on networks shown in Fig. 6.
b BN learned using data of HEV variants sampled from humans (BNHuman) were tested on HEV data sampled from swine (DSwine).
c BN learned from swine data (BNSwine) was tested on HEV data sampled from humans (DHuman).
Table 3.
Validation of host-specific dependency among aa sites.
| HEV genotype | Host-specific networkb | Cross-validation testa |
|
|---|---|---|---|
| (e)c | (f) | ||
| HEV3 | BNHuman (44) | 9 | 100% |
| 30 | 80.0–93.3% | ||
| 5 | 0% | ||
| BNSwine (40) | 20 | 100% | |
| 18 | 73.3–93.3% | ||
| 2 | 0–60% | ||
| HEV4 | BNHuman (60) | 29 | 100% |
| 27 | 73.3–93.3% | ||
| 4 | 46.7–60.0% | ||
| BNSwine (34) | 2 | 100% | |
| 28 | 73.3–93.3% | ||
| 4 | 46.7–66.7% | ||
Cross-validation tests were performed by jackknife method to determine percent frequency (f) with which edges (e) appeared in sampled networks relative to the corresponding reference BN (Fig. 6).
Values shown in parenthesis denote the total arc counts in reference BNs.
Values represent edge counts between aa sites observed for a given (f).
Inspection of BNSwine and BNHuman graphs showed differences in the modeled epistatic connectivity between swine and human strains of HEV3 and HEV4 (Fig. 6). For HEV3, both BNs shared 30 aa sites, which represent 63% and 71% of all sites involved in BNHuman and BNSwine, correspondingly. For HEV4, however, BNs shared only 9 sites, representing only 14% and 23% of all sites in BNHuman and BNSwine, correspondingly. Taken together, these observations suggest that epistatic connectivity captured by the BNs strongly reflects host specificity.
3.9. Host specificity of different regions
The specificity with which genetic diversity is coordinated through the epistatic connectivity among polymorphic sites associates host origin of HEV strains was further examined by evaluating performance of classifier models. Aa sites found relevant for improving classification performance of models are identified in Table 4, Table 5 (also see Table S2, in Supplementary Material). BNCs showed 73.8–92.7% accuracy of classification into swine and human strains in the 10-fold CV for variants of all three ORF proteins of both genotypes, with greatest accuracy being achieved by using aa sequence information of sites from the ORF1-encoded protein (Table 4). Although all 3 ORF proteins had sites with epistatic connectivity specific to swine or humans (Fig. 6), sequence variation in the ORF1-encoded protein of both genotypes was most strongly associated with the host origin of strains. In 10-fold CV, classification accuracy of >80% was observed with BNCs using sites from the ORF1-domains Pp and Pol of both genotypes (Table 5). Except for the Mt-domain, such level of accuracy was achieved by BNCs derived from all other ORF1-domains only in HEV4.
Table 4.
Overall performance of BNC constructed for each ORF-encoded protein.
| ORFs | HEV3 |
HEV4 |
||||
|---|---|---|---|---|---|---|
| Protein sitesa | F-measure Swine/Human | CAb (%) | Protein sitesa | F-measure Swine/Human | CAb (%) | |
| ORF1 | 514, 557, 643, 719, 720, 724, 728, 764, 775, 783, 795, 836, 855, 1005, 1234, 1449, 1506, 1507, 1599 and 1612 | 0.86/0.89 | 87.7 | 17, 462, 523, 531, 546, 560, 562, 574, 650, 683, 732, 733, 742, 756, 772, 779, 802, 804, 1096 and 1456 | 0.88/0.95 | 92.7 |
| ORF2 | 2, 12, 25, 30, 34, 48, 53, 67, 76, 103, 113, 149, 158, 188, 356, 473, 501, 511, 527, 529, 554, 571, 609, 649, 651 and 652 | 0.79/0.84 | 81.5 | 38, 39, 46, 78, 96, 98, 119, 146, 175, 318, 521, 527, 546, 609 and 614 | 0.73/0.88 | 83.6 |
| ORF3 | 3, 4, 7, 31, 33, 36, 38, 41, 56, 70, 76, 78, 81, 83, 85, 93, 94 and 99 | 0.59/0.81 | 73.8 | 2, 17, 29, 32, 42, 53, 67, 73, 79, 82, 84, 90 and 92 | 0.60/0.85 | 78.2 |
Numbering represent protein positions in respective ORFs.
Overall classification accuracy of classification by 10-fold CV.
Table 5.
Overall performance of BNC’s for different ORF1-domains.
| Domain | HEV3 |
HEV4 |
||||
|---|---|---|---|---|---|---|
| Protein sitesa | F-measure Swine/Human | CAb (%) | Protein sitesa | F-measure Swine/Human | CAb (%) | |
| Mt | 70, 81, 89, 129, 137, 141, 151, 152, 158, 161, 189, 192, 200 and 206 | 0.51/0.72 | 64.6 | 61, 72, 75, 89, 122, 139, 148, 150, 189 and 204 | 0.0/0.83 | 70.9 |
| Y | 219, 240, 246, 248, 302, 323, 355, 363, 393, 399 and 400 | 0.29/0.75 | 63.1 | 239, 248, 274, 277, 304, 306, 332, 335, 338, 340, 356, 357, 359, 363, 413, 423, 428 and 429 | 0.56/0.87 | 80.0 |
| Plp | 468, 495, 502, 509, 512, 514, 517, 530, 542, 557 and 589 | 0.69/0.77 | 73.8 | 462, 546, 560, 562 and 574 | 0.79/0.91 | 87.3 |
| Pp | 719, 720, 724, 728, 740, 749, 764, 775, 783, 790, 791, 795 and 797 | 0.83/0.86 | 84.6 | 707, 718, 732, 733, 742, 756, 760, 767, 772 and 779 | 0.77/0.90 | 85.5 |
| Md | 843, 855, 873, 876, 879, 914, 915, 941, 949, 959, 972, 985 and 992 | 0.52/0.77 | 69.2 | 803, 804, 811, 817, 835, 836, 846, 869, 874, 876, 902, 942 and 952 | 0.31/1.0 | 80.0 |
| Hel | 1016, 1024, 1043, 1076, 1101, 1146, 1160, 1233, 1234 and 1242 | 0.59/0.72 | 66.2 | 977, 981, 983, 984, 1003, 1007, 1044, 1047, 1064, 1094, 1096, 1100, 1117, 1120, 1124, 1136 and 1196 | 0.64/0.88 | 81.8 |
| Pol | 1370, 1285, 1508, 1599, 1732, 1386, 1612, 1283, 1481, 1533, 1746, 1426, 1652, 1499, 1638 and 1555 | 0.79/0.83 | 81.5 | 1235, 1236, 1247, 1266, 1303, 1355, 1360, 1447, 1456, 1572, 1632, 1648, 1652, 1692, 1693 and 1704 | 0.67/0.89 | 83.6 |
Site numbering based on polyprotein positions.
Overall classification accuracy of classification by 10-fold CV.
Likewise, genetic host specificity of HEV strains was also supported by BNC classification performance on validation datasets (Table S2). Selected sites from domains Pol (n = 16) of the ORF1-encoded protein of both genotypes and Pp (n = 10) of HEV4 are also listed in Table S2. Furthermore, the distribution of physicochemical properties for these selected aa sites was evaluated using LP models. The clustering of 65 HEV3 and 55 HEV4 strains in LP models using the selected aa sites was found to be strongly associated with the host origin of strains (Fig. 7 ). On validation trails, accuracy performance of LP model of the HEV3 Pol aa sites (Fig. 7A) was 84.0%, while for LP models constructed for HEV4 Pol (Fig. 7B) and Pp (Fig. 7C) were 100% accurate. Because of the lack of additional data, the HEV3 Pp model could not be evaluated.
Fig. 7.
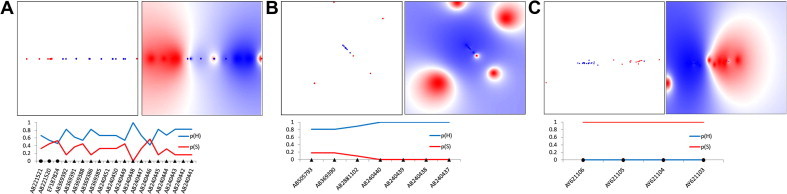
Host-specific separation of HEV3 and HEV4 strains in LP-modeled physicochemical space. Shown are LP plots of physicochemical properties for aa sites from Pol and Pp (Table 4). Probability mapping of human and swine strains is color-coded, with human space shown in blue and swine in red. Color density is proportional to probability values. (A) LP map of HEV3 variants (n = 65) using Pol aa physicochemical properties or markers (n = 16); (B) LP map of HEV4 variants (n = 55) using Pol markers (n = 16) and (C) LP map of HEV4 variants (n = 55) using the Pp markers (n = 10). Below the mappings are line charts showing the prediction results (probability scores) on validation datasets from the above corresponding LP maps; y-axis represents probability [0–1]; p(H) and p(S) are probabilities of the human (blue line) and swine (red line) origin of a strain, respectively. GenBank accession numbers (x-axis) are shown for each test sequence; black triangles and circles identify HEV strains obtained from humans and swine, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
4.1. Lack of phylogenetic separation by host among HEV3 and HEV4 strains
A strong phylogenetic association between HEV strains and host range is clearly established, with the HEV1 and HEV2 strains infecting humans, whereas HEV3 and HEV4 strains infect animals and humans (Purdy and Khudyakov, 2011, Krawczynski et al., 2000). However, no ancestral associations with host specificity were found among the HEV3 or HEV4 strains despite many attempts to identify a phylogenetic linkage of individual strains to the host origin (Bouquet et al., 2012a, Purdy et al., 2012b, Smith et al., 2012). Although the host-specific distribution of HEV3 subtypes was observed in a small rural community in southeastern Bolivia, it was most probably caused by the high prevalence of the infection rather than adaptation of different viral lineages to swine or humans (Purdy et al., 2012a). Complex patterns of HEV transmission among hosts of different species generate conditions for maintenance of significant heterogeneity among HEV strain, which cannot be adequately represented in short genomic regions usually used for phylogenetic inference (Purdy et al., 2012a). However, the ineffective representation of genealogical relationships with short genomic regions cannot explain elusiveness of ancestral connections to the host origin of HEV strains since analysis of the HEV whole-genome sequences is also unsuccessful in detecting the host-specific clustering of HEV3 or HEV4 lineages derived from different host species (Bouquet et al., 2012a, Purdy et al., 2012b).
Phylogenetic analysis conducted in this study strongly supports previous observations of phylogenetic intermixing among HEV isolates from different hosts. Several measures for examining the phylogenetic relatedness of phenotypic characteristics have been developed over the past 20 years. Parker et al. (2008) created a software platform, Befi-BaTS, that calculates seven of these metrics to analyze the degree to which phenotypic characteristics are correlated with shared ancestry. Befi-Bats was used to analyze the relatedness of host specificity to HEV ORF1 PSTs for HEV3 and HEV4. Befi-BaTS uses a Bayesian PST to estimate the significance of the taxon-phenotypic character associations. Table S1 (Supplementary Material) shows that only some of these metrics were significant. Those metrics, which examine all traits together across the PST under investigation, tended to be statistically significant, while the monophyletic clade size, which examines each trait individually, were statistically insignificant. More test metrics were not statistically significant for the HEV4 PST as compared to HEV3 (Table S1). This lack of agreement among all methods is difficult to interpret as there is no definitive guide for comparing these metrics. The distribution of sequences from the swine host appears to be random in the PSTs from both genotypes. As noted by Gittleman and Luh (1992), “if phylogentic correlation is not observed, then comparative method procedures should not be adopted.” For this reason we chose to use Bayesian networks to elucidate the relationship between host specificity and genome sequence.
4.2. Genetic coordination
Analyses conducted here indicated a broad coevolution among sites of proteins encoded by all 3 HEV ORFs (Fig. 1). Coordinated variations were observed for ∼70% of all polymorphic aa sites in both HEV3 and HEV4 strains. Owing to its length, the ORF1-encoded protein contributed ∼76% of sites to BNHEV3 and BNHEV4. In both BNs, the ORF2- and ORF3-protein sites were linked predominantly to sites from the ORF1-protein, while ∼65–70% of ORF1-protein sites had intra-protein links. The ORF1-protein contains 7 functional domains (Koonin et al., 1992) and links were detected predominantly among these domains. Plp, Pp and Pol had 28% of intra-domain links, with Hel having a single intra-domain link in HEV3. It is important to note that these 3 domains, Plp, Pp and Pol, contributed 65–71% of the ORF1-sites to BNs (Fig. 1), which suggest an important role in HEV protein evolution. Coordination between the ORF1-sites, on one side, and ORF2- and ORF3-sites, on the other, was more extensive for HEV4 than for HEV3 (Fig. 2B), suggesting a greater dependence of the HEV4 evolution on the inter-protein coordination.
4.3. Genetic association with host origin
One of the most important observations made in this study is the strong association of the modeled epistatic connectivity among HEV3 and HEV4 aa sites with the host origin of HEV strains. This observation implies host-specific coevolution among HEV protein sites. Although only a single site is linked to the host variable in both BNs (Fig. 1), host association is not encoded at any single protein position. Rather, many sites contribute to the host-specific epistatic connectivity. This inference is supported by the observation that not a single site had a strong MI with the host origin (Fig. 5). The host association of any aa site should be considered in conjunction with many other sites in BNs (Fig. 5). Sites, collectively affecting the distribution of the host probability (Fig.1), were identified for each HEV3 and HEV4 protein (Table 1), indicating association between genetic heterogeneity of these groups of sites with host specificity. However, this association was most measurable for the ORF1-encoded protein of both genotypes (Table 1, Table 4), with the ORF2- and ORF3-encoded proteins containing only small groups of sites producing a smaller effect on the host variable in BNs (Table 1).
4.4. Contribution of ORF1 domains
Analysis of the log-likelihood values for BNSwine and BNHuman (Table 2) and jackknife CV tests (Table 3) showed that all BNs in Fig. 6 had a strong host-specific structure, indicating genetic differences between HEV strains recovered from swine or human hosts. These host-specific genetic differences were predominantly established among ORF1-domains (Fig.6). The genetic composition of domains Pp and Pol was found to be strongly associated with host specificity in both HEV3 and HEV4 (Table 5). Analysis of the LP models constructed using protein physicochemical properties provided additional support of host-specific genetic variations in domains Pp and Pol (Fig. 7).
The association between Pp and the host range of HEV3 and HEV4 strains was suggested earlier (Purdy et al., 2012b). This domain was shown to belong in a class of intrinsically disordered regions or proteins, which play important regulatory roles facilitated by their propensity to highly specific interactions with numerous intra-cellular ligands (Uversky, 2011). Accordingly, domain Pp was found to contain many ligand binding sites, supporting its potential regulatory functions. These findings in conjunction with the extensive homoplasy of the Pp sites identified along the major HEV3 and HEV4 phylogenetic lineages suggest a role for Pp in the intra-host adaptation (Purdy et al., 2012b) and, taken together with observations made in this study, strongly support the prominent role of this domain in adaptation to the broad host range of these 2 HEV genotypes.
The Unk region located at positions 593–706 of the ORF1-protein has not been assigned any function (Koonin et al., 1992). Here, the data indicated that this region imposes a considerable influence on the state of BNHEV3 and BNHEV4. Many sites from this region had outgoing links to other proteins and ORF1-domains (Fig. 3H). Such wide-ranging participation in epistatic connectivity modeled using BN suggests that Unk potentially has an important, but yet to be recognized, function or role in evolution of HEV3 and HEV4.
4.5. Host specificity is a convergent and abundant trait
Observations of the high extent of homoplasy along the HEV3 and HEV4 genomes compared to HEV1 (Purdy et al., 2012b) and the association between the HEV aa sites and host origin identified here suggest that host specificity is a convergent trait, which originates independently among HEV3 and HEV4 lineages. Identification of the phylogenetic connection to host would indicate heritable reduction in the host range for individual strains, similar to that observed in HEV1 and HEV2. For HEV3 and HEV4 lineages, convergence implies a certain genetic plasticity of host adaptation. Host specificity seems to be an abundant phenotype, which can be established by many HEV3 and HEV4 genetic variants. The BN models indicated coevolution among ∼70% of all polymorphic aa sites (Fig. 1), suggesting involvement of the entire HEV genome in host adaptation. However, each HEV protein and ORF1 domain had a strong independent association with the host. Identification of the various groups of aa sites associated with host origin (Fig. 6; Table 1, Table 4, Table 5) indicates that HEV may efficiently achieve this adaptation via many genetic pathways, each requiring small but specific genetic adjustments rather than global genetic changes across the entire genome. The lack of a clear phylogenetic separation among HEV3 and HEV4 lineages by hosts (Fig. S1 in Supplementary Material) further suggests that each HEV strain achieves adaptation to the host using different small subsets of coevolving aa sites rather than hardwiring host specificity into a small number of invariant aa sites. The presence of certain minimal genetic changes seems to be sufficient for establishing effective infection in a different host and renders all other genetic changes across the genome, though also associated with host specificity, redundant.
The HEV3 and HEV4 genetic composition allows for many strains to replicate in different hosts (Meng, 2011, Purdy et al., 2012a, Takahashi et al., 2011, Tei et al., 2003, Zhao et al., 2009). We hypothesize that there are various ways for achieving host adaptation, each requiring a few genetic changes. These changes may be generated rapidly during HEV infection (Bouquet et al., 2012b). Accumulation of substitutions leads to diverse intra-host HEV population in swine and humans (Bouquet et al., 2012b, Grandadam et al., 2004). Thus, host-specific substitutions may preexist among intra-host variants in the previous host (Borucki et al., 2013). Taking into consideration the zoonotic nature of HEV3 and HEV4, it is conceivable that the swine intra-host HEV variants have a much greater range of replication rates when introduced to humans, with only a fraction of the swine variants capable of replicating efficiently in human hosts.
Such consideration implies that, with swine being the primary hosts, HEV3 and HEV4 strains have greater intra-host heterogeneity in swine than in humans. Indeed, a lower variability among human than swine intra-host HEV variants was recently reported after the experimental transmission of a single human HEV strain to swine (Bouquet et al., 2012b), suggesting differences in selection pressures acting on HEV in different hosts that result in variation in genetic heterogeneity. Thus, the HEV variants replicating efficiently in humans may represent a subset of swine variants. The previously reported dose dependence of establishing HEV infection and clinical manifestation of the infection (Aggarwal et al., 2001, Takahashi et al., 2012) is consistent with this hypothesis, which further implies that HEV transmission from swine should be most effective when achieved in bulk. Such transmission is frequently associated with consumption of raw or under-cooked meat from infected animals (Lewis et al., 2010, Li et al., 2005, Tei et al., 2003, Teo, 2010). In these cases, exposure to the large HEV quantity is expected while low-dose transmission would lead to subclinical infection and explain the high rates of seroprevalence seen in many industrialized countries (Purdy and Khudyakov, 2011).
4.6. Comparison between HEV3 and HEV4
Discordance between the identified swine and human aa motifs in HEV3 and HEV4 strains suggests that these 2 genotypes adopt different genetic pathways for host adaptation. HEV4 strains employ a greater number of aa sites than HEV3 (n = 97 for HEV4 vs. n = 60 for HEV3 in BNs shown in Fig. 6) for adaptation. There were ∼2.5–3.0-times more aa sites, which were not shared by BNSwine and BNHuman, for HEV4 than for HEV3 (Fig. 6). These differences resulted in a very low correlation (r = 0.0313) between features of BNSwine and BNHuman for HEV4, whereas this correlation was r = 0.7004 for HEV3. Coordination of sites from the Pp domain was especially genotype-specific (Fig. 3). Although the HEV3 host motifs contained only 1 Pp site, there were 8 and 23 sites from this domain involved in the HEV4 BNSwine and BNHuman, respectively, with none of them shared by both BNs (Fig. 6). Thus, the data indicate that HEV4 has more complex genetic requirements for the efficient replication in different host species than HEV3, which is reflected in a more accurate performance of all models generated using HEV4 sequences (Table 4, Table 5, Fig. 7 and Table S2).
We speculate that specific requirement for coordination of heterogeneity among many aa sites (Fig. 6) renders the swine HEV4 less prone than HEV3 to the rapid acquisition of a particular genetic composition favorable for the efficient propagation in human hosts and, as a consequence, potentially generates a greater genetic disparity among swine HEV4 than HEV3 strains for establishing productive infections in human hosts. The estimated low number of symptomatic HEV infections in China (Wedemeyer and Pischke, 2011), where HEV4 is endemic (Liu et al., 2012), suggests that HEV4 is less virulent and/or may only be transmitted infrequently in a dose sufficient for causing the manifestation of clinical symptoms. Additionally, the host specific separation between HEV genotypes has been reported in India, where HEV4 was found infecting animals while HEV1 infections were detected among humans (Arankalle et al., 2002, Shukla et al., 2007). Although the molecular and epidemiological mechanisms underlying these phenomena are not known (Purdy and Khudyakov, 2011), both observations are consistent with the suggested low infectivity of HEV4 to humans. However, once the specific genetic composition is acquired; e.g., through continuous passaging among humans, HEV4 should attain a greater capacity for establishing human infection and, as a result, become more virulent. We speculate that the increased detection of HEV4 infections in China observed over the last decade (Liu et al., 2012, Zhang et al., 2011) is related to such adaptation of HEV4 to humans.
4.7. Association with virulence
Although the data presented in this study do not have direct implications for HEV virulence, it is intriguing to note that sites 605 in UNK, 1017 and 1252 in Hel, which have been associated with severe hepatitis in HEV3-infected patients (Takahashi et al., 2009), were among the most influential sites of the HEV3 ORF1-encoded protein (Table 1). All 3 sites were involved in BNHEV3 (Fig. 1). Additionally, site 1252 was included in the HEV3 host-specific motifs, with site 605 being a part of the human motif (Fig. 6). These observations suggest the possibility that the host-specific coevolution among protein sites has association with HEV virulence.
In conclusion, emerging infectious diseases are frequently associated with host shift for infectious agents (Purdy and Khudyakov, 2011). Understanding of the extent of heritability of host specificity and genetic factors facilitating zoonotic transmission is important for the efficient control of emerging infectious diseases. Findings made in this study indicate that HEV is uniquely suitable for assessing these parameters of viral infections. The HEV capacity to infect humans is not uniformly distributed among HEV strains. It is strongly encoded in genetic composition of HEV1 and HEV2, whereas closely related strains of HEV3 and HEV4 vary in their capacity to establish infection in humans. Such breadth of genetic associations to host adaptation presents a valuable opportunity for exploring heritability of host specificity and understanding genetic mechanisms responsible for emerging viral diseases.
Here, the extensive coevolution among aa sites was shown to be associated with the host adaptation of HEV3 and HEV4. This finding, taken together with phylogenetic intermixing among human and swine lineages, suggests that HEV host specificity is a heritable, convergent and abundant phenotypic trait, which can be achieved independently by various HEV3 and HEV4 strains through many genetic pathways. Such genetic host specificity warrants further investigation, leading not only to understanding the epidemiology of HEV3 and HEV4 infections, but also to prediction of future patterns of transmission, morbidity and virulence, and formulation of appropriate public health control measures.
Author contributions
JL, MAP and YEK conceived the study. All authors contributed to study design. JL and MAP analyzed the data. All authors wrote and edited the manuscript.
Acknowledgments
Disclaimer: The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention/the Agency for Toxic Substances and Disease Registry.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.meegid.2014.03.011.
Appendix A. Supplementary data
This document contains Supplementary Material.
References
- Aggarwal R. Clinical presentation of hepatitis E. Virus Res. 2011;161:15–22. doi: 10.1016/j.virusres.2011.03.017. [DOI] [PubMed] [Google Scholar]
- Aggarwal R., Kamili S., Spelbring J., Krawczynski K. Experimental studies on subclinical hepatitis E virus infection in cynomolgus macaques. J. Infect. Dis. 2001;184:1380–1385. doi: 10.1086/324376. [DOI] [PubMed] [Google Scholar]
- Ahmad I., Holla R.P., Jameel S. Molecular virology of hepatitis E virus. Virus Res. 2011;161:47–58. doi: 10.1016/j.virusres.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arankalle V.A., Chobe L.P., Joshi M.V., Chadha M.S., Kundu B., Walimbe A.M. Human and swine hepatitis E viruses from Western India belong to different genotypes. J. Hepatol. 2002;36:417–425. doi: 10.1016/s0168-8278(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Atchley W.R., Zhao J., Fernandes A.D., Druke T. Solving the protein sequence metric problem. Proc. Natl. Acad. Sci. USA. 2005;102:6395–6400. doi: 10.1073/pnas.0408677102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurora R., Donlin M.J., Cannon N.A., Tavis J.E. Genome-wide hepatitis C virus amino acid covariance networks can predict response to antiviral therapy in humans. J. Clin. Invest. 2009;119:225–236. doi: 10.1172/JCI37085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhoeffer S., Chappey C., Parkin N.T., Whitcomb J.M., Petropoulos C.J. Evidence for positive epistasis in HIV-1. Science. 2004;306:1547–1550. doi: 10.1126/science.1101786. [DOI] [PubMed] [Google Scholar]
- Borucki M.K., Allen J.E., Chen-Harris H., Zemla A., Vanier G., Mabery S., Torres C., Hullinger P., Slezak T. The role of viral population diversity in adaptation of bovine coronavirus to new host environments. PLoS One. 2013;8:e52752. doi: 10.1371/journal.pone.0052752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert R. Probabilistic network construction using the minimum description length principle. In: Clarke M., Kruse R., Moral S., editors. Symbolic and Quantitative Approaches to Reasoning and Uncertainty. Springer; Berlin/Heidelberg: 1993. pp. 41–48. [Google Scholar]
- Bouquet J., Cherel P., Pavio N. Genetic characterization and codon usage bias of full-length Hepatitis E virus sequences shed new lights on genotypic distribution, host restriction and genome evolution. Infect. Genet. Evol. 2012;12:1842–1853. doi: 10.1016/j.meegid.2012.07.021. [DOI] [PubMed] [Google Scholar]
- Bouquet J., Cheval J., Rogee S., Pavio N., Eloit M. Identical consensus sequence and conserved genomic polymorphism of hepatitis E virus during controlled interspecies transmission. J. Virol. 2012;86:6238–6245. doi: 10.1128/JVI.06843-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campo D.S., Dimitrova Z., Mitchell R.J., Lara J., Khudyakov Y. Coordinated evolution of the hepatitis C virus. Proc. Natl. Acad. Sci. USA. 2008;105:9685–9690. doi: 10.1073/pnas.0801774105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chickering D.M., Heckerman D., Meek C. Large-sample learning of Bayesian networks is NP-hard. J. Mach. Learn. Res. 2004;5:1287–1330. [Google Scholar]
- Demsar, J., Leban, G., Zupan, B., 2005. FreeViz – an intelligent vizualization approach for class-labeled multidimensional data sets. In: Intelligent Data Analysis in Medicine and Pharmacology (IDAMAP 2005). Aberdeen, Scotland, UK, pp. 61–66.
- Donlin M.J., Szeto B., Gohara D.W., Aurora R., Tavis J.E. Genome-wide networks of amino acid covariances are common among viruses. J. Virol. 2012;86:3050–3063. doi: 10.1128/JVI.06857-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson S.U., Anderson D., Arankalle A., Meng X.J., Purdy M.A., Schlauder G.G., Tsarev S.A. Hepevirus. In: Fauquet C.M., Mayo M.A., Maniloff J., Desselberger U., Ball L.A., editors. Virus Taxonomy. Eight Report of the International Committee on Taxonomy of Viruses. Elsevier/Academic Press; London: 2004. pp. 853–857. [Google Scholar]
- Gittleman J.L., Luh H.K. On comparing comparative methods. Annu. Rev. Ecol. Syst. 1992;23:383–404. [Google Scholar]
- Grandadam M., Tebbal S., Caron M., Siriwardana M., Larouze B., Koeck J.L., Buisson Y., Enouf V., Nicand E. Evidence for hepatitis E virus quasispecies. J. Gen. Virol. 2004;85:3189–3194. doi: 10.1099/vir.0.80248-0. [DOI] [PubMed] [Google Scholar]
- Hall, M.A., 1999. Correlation-based Feature Subset selection for Machine Learning. PhD thesis, University of Waikato, Hamilton, New Zealand.
- Han W., Li X., Fu X. The macro domain protein family: structure, functions, and their potential therapeutic implications. Mutat. Res. 2011;727:86–103. doi: 10.1016/j.mrrev.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson R.R., Boos D.D., Kaplan N.L. A statistical test for detecting geographic subdivision. Mol. Biol. Evol. 1992;9:138–151. doi: 10.1093/oxfordjournals.molbev.a040703. [DOI] [PubMed] [Google Scholar]
- Jensen F. Springer; New York: 2001. Bayesian Networks and Decision Graphs. [Google Scholar]
- Jouffe, L., Munteanu, P., 2001. New Search Strategies for Learning Bayesian Networks. In: Proceedings of the 10th International Symposium on Applied Stochastic Models and Data Analysis, Compiègne, France, pp. 217–245.
- Khudyakov Y. Coevolution and HBV drug resistance. Antivir. Ther. 2010;15:505–515. doi: 10.3851/IMP1515. [DOI] [PubMed] [Google Scholar]
- Khudyakov Y. Molecular surveillance of hepatitis C. Antivir. Ther. 2012;17:1465–1470. doi: 10.3851/IMP2476. [DOI] [PubMed] [Google Scholar]
- Khuroo M.S. Discovery of hepatitis E: the epidemic non-A, non-B hepatitis 30 years down the memory lane. Virus Res. 2011;161:3–14. doi: 10.1016/j.virusres.2011.02.007. [DOI] [PubMed] [Google Scholar]
- Koonin E.V., Gorbalenya A.E., Purdy M.A., Rozanov M.N., Reyes G.R., Bradley D.W. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: delineation of an additional group of positive-strand RNA plant and animal viruses. Proc. Natl. Acad. Sci. USA. 1992;89:8259–8263. doi: 10.1073/pnas.89.17.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczynski K., Aggarwal R., Kamili S. Hepatitis E. Infect. Dis. Clin. North Am. 2000;14:669–687. doi: 10.1016/s0891-5520(05)70126-4. [DOI] [PubMed] [Google Scholar]
- Lara J., Khudyakov Y.E. Epistatic connectivity among hepatitis C virus genomic sites as genetic marker of interferon resistance. Antivir. Ther. 2012;17:1471–1475. doi: 10.3851/IMP2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara J., Tavis J.E., Donlin M.J., Lee W.M., Yuan H.J., Pearlman B.L., Forbi J.C., Xia G.L., Khudyakov Y.E. Coordinated evolution among hepatitis C virus genomic sites is coupled to host factors and resistance to interferon. In Silico Biol. 2011;11:213–224. doi: 10.3233/ISB-2012-0456. [DOI] [PubMed] [Google Scholar]
- Lara J., Xia G., Purdy M.A., Khudyakov Y.E. Coevolution of the hepatitis C virus polyprotein sites in patients on combined pegylated interferon and ribavirin therapy. J. Virol. 2011;85:3649–3663. doi: 10.1128/JVI.02197-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis H.C., Wichmann O., Duizer E. Transmission routes and risk factors for autochthonous hepatitis E virus infection in Europe: a systematic review. Epidemiol. Infect. 2010;138:145–166. doi: 10.1017/S0950268809990847. [DOI] [PubMed] [Google Scholar]
- Li T.C., Chijiwa K., Sera N., Ishibashi T., Etoh Y., Shinohara Y., Kurata Y., Ishida M., Sakamoto S., Takeda N., Miyamura T. Hepatitis E virus transmission from wild boar meat. Emerg. Infect. Dis. 2005;11:1958–1960. doi: 10.3201/eid1112.051041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P., Li L., Wang L., Bu Q., Fu H., Han J., Zhu Y., Lu F., Zhuang H. Phylogenetic analysis of 626 hepatitis E virus (HEV) isolates from humans and animals in China (1986–2011) showing genotype diversity and zoonotic transmission. Infect. Genet. Evol. 2012;12:428–434. doi: 10.1016/j.meegid.2012.01.017. [DOI] [PubMed] [Google Scholar]
- Lozupone C., Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microb. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X.J. From barnyard to food table: the omnipresence of hepatitis E virus and risk for zoonotic infection and food safety. Virus Res. 2011;161:23–30. doi: 10.1016/j.virusres.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X.J., Purcell R.H., Halbur P.G., Lehman J.R., Webb D.M., Tsareva T.S., Haynes J.S., Thacker B.J., Emerson S.U. A novel virus in swine is closely related to the human hepatitis E virus. Proc. Natl. Acad. Sci. USA. 1997;94:9860–9865. doi: 10.1073/pnas.94.18.9860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuo H., Yazaki Y., Sugawara K., Tsuda F., Takahashi M., Nishizawa T., Okamoto H. Possible risk factors for the transmission of hepatitis E virus and for the severe form of hepatitis E acquired locally in Hokkaido, Japan. J. Med. Virol. 2005;76:341–349. doi: 10.1002/jmv.20364. [DOI] [PubMed] [Google Scholar]
- Munteanu, P., Bendou, M., 2001. The EQ framework for learning equivalence classes of Bayesian networks. In: Proceedings 2001 IEEE International Conference on Data Mining, pp. 417–424.
- Neapolitan R.E. Pearson/Prentice Hall; Upper Saddle River, NJ: 2004. Learning Bayesian Networks. [Google Scholar]
- Parker J., Rambaut A., Pybus O.G. Correlating viral phenotypes with phylogeny: accounting for phylogenetic uncertainty. Infect. Genet. Evol. 2008;8:239–246. doi: 10.1016/j.meegid.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Patra S., Kumar A., Trivedi S.S., Puri M., Sarin S.K. Maternal and fetal outcomes in pregnant women with acute hepatitis E virus infection. Ann. Intern. Med. 2007;147:28–33. doi: 10.7326/0003-4819-147-1-200707030-00005. [DOI] [PubMed] [Google Scholar]
- Purdy M.A., Dell’Amico M.C., Gonzales J.L., Segundo H., Tolari F., Mazzei M., Bartoloni A., Khudyakov Y.E. Human and porcine hepatitis E viruses, southeastern Bolivia. Emerg. Infect. Dis. 2012;18:339–340. doi: 10.3201/eid1802.111453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdy M.A., Khudyakov Y.E. The molecular epidemiology of hepatitis E virus infection. Virus Res. 2011;161:31–39. doi: 10.1016/j.virusres.2011.04.030. [DOI] [PubMed] [Google Scholar]
- Purdy M.A., Lara J., Khudyakov Y.E. The hepatitis E virus polyproline region is involved in viral adaptation. PLoS One. 2012;7:e35974. doi: 10.1371/journal.pone.0035974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissanen J. Stochastic complexity and modeling. Ann. Stat. 1986;14:1080–1100. [Google Scholar]
- Sanjuan R., Moya A., Elena S.F. The contribution of epistasis to the architecture of fitness in an RNA virus. Proc. Natl. Acad. Sci. USA. 2004;101:15376–15379. doi: 10.1073/pnas.0404125101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla P., Chauhan U.K., Naik S., Anderson D., Aggarwal R. Hepatitis E virus infection among animals in northern India: an unlikely source of human disease. J. Viral Hepat. 2007;14:310–317. doi: 10.1111/j.1365-2893.2006.00815.x. [DOI] [PubMed] [Google Scholar]
- Slatkin M., Maddison W.P. A cladistic measure of gene flow inferred from the phylogenies of alleles. Genetics. 1989;123:603–613. doi: 10.1093/genetics/123.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.B., Purdy M.A., Simmonds P. Genetic variability and the classification of hepatitis E virus. J. Virol. 2013;87:4161–4169. doi: 10.1128/JVI.02762-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.B., Vanek J., Ramalingam S., Johannessen I., Templeton K., Simmonds P. Evolution of the Hepatitis E virus hypervariable region. J. Gen. Virol. 2012;93(pt 11):1842–1853. doi: 10.1099/vir.0.045351-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H., Tanaka T., Jirintai S., Nagashima S., Takahashi M., Nishizawa T., Mizuo H., Yazaki Y., Okamoto H. A549 and PLC/PRF/5 cells can support the efficient propagation of swine and wild boar hepatitis E virus (HEV) strains: demonstration of HEV infectivity of porcine liver sold as food. Arch. Virol. 2012;157:235–246. doi: 10.1007/s00705-011-1153-2. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Okamoto H., Abe N., Kawakami M., Matsuda H., Mochida S., Sakugawa H., Suginoshita Y., Watanabe S., Yamamoto K., Miyakawa Y., Mishiro S. Virulent strain of hepatitis E virus genotype 3, Japan. Emerg. Infect. Dis. 2009;15:704–709. doi: 10.3201/eid1505.081100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M., Nishizawa T., Sato H., Sato Y., Jirintai, Nagashima S., Okamoto H. Analysis of the full-length genome of a hepatitis E virus isolate obtained from a wild boar in Japan that is classifiable into a novel genotype. J. Gen. Virol. 2011;92:902–908. doi: 10.1099/vir.0.029470-0. [DOI] [PubMed] [Google Scholar]
- Tei S., Kitajima N., Takahashi K., Mishiro S. Zoonotic transmission of hepatitis E virus from deer to human beings. Lancet. 2003;362:371–373. doi: 10.1016/S0140-6736(03)14025-1. [DOI] [PubMed] [Google Scholar]
- Teo C.G. Much meat, much malady: changing perceptions of the epidemiology of hepatitis E. Clin. Microbiol. Infect. 2010;16:24–32. doi: 10.1111/j.1469-0691.2009.03111.x. [DOI] [PubMed] [Google Scholar]
- Thai H., Campo D.S., Lara J., Dimitrova Z., Ramachandran S., Xia G., Ganova-Raeva L., Teo C.G., Lok A., Khudyakov Y.E. Convergence and coevolution of hepatitis B virus drug resistance. Nat. Commun. 2012;3:789. doi: 10.1038/ncomms1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang K.W., Ho P.L., Ooi G.C., Yee W.K., Wang T., Chan-Yeung M., Lam W.K., Seto W.H., Yam L.Y., Cheung T.M., Wong P.C., Lam B., Ip M.S., Chan J., Yuen K.Y., Lai K.N. A cluster of cases of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003;348:1977–1985. doi: 10.1056/NEJMoa030666. [DOI] [PubMed] [Google Scholar]
- Uversky V.N. Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 2011;43:1090–1103. doi: 10.1016/j.biocel.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Viswanathan R. Infectious hepatitis in New Delhi (1955–56): a critical study. Indian J. Med. Res. 1957;45:1–29. [Google Scholar]
- Wang T.H., Donaldson Y.K., Brettle R.P., Bell J.E., Simmonds P. Identification of shared populations of human immunodeficiency virus type 1 infecting microglia and tissue macrophages outside the central nervous system. J. Virol. 2001;75:11686–11699. doi: 10.1128/JVI.75.23.11686-11699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Ling R., Erker J.C., Zhang H., Li H., Desai S., Mushahwar I.K., Harrison T.J. A divergent genotype of hepatitis E virus in Chinese patients with acute hepatitis. J. Gen. Virol. 1999;80(Pt 1):169–177. doi: 10.1099/0022-1317-80-1-169. [DOI] [PubMed] [Google Scholar]
- Webb C.O. Exploring the Phylogenetic Structure of Ecological Communities: An Example for Rain Forest Trees. Am. Nat. 2000;156:145–155. doi: 10.1086/303378. [DOI] [PubMed] [Google Scholar]
- Wedemeyer H., Pischke S. Hepatitis: Hepatitis E vaccination – is HEV 239 the breakthrough? Nat. Rev. Gastroenterol. Hepatol. 2011;8:8–10. doi: 10.1038/nrgastro.2010.207. [DOI] [PubMed] [Google Scholar]
- Yi M., Ma Y., Yates J., Lemon S.M. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 2007;81:629–638. doi: 10.1128/JVI.01890-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Wang J., Yuan Q., Ge S., Zhang J., Xia N., Tian D. Clinical characteristics and risk factors of sporadic Hepatitis E in central China. Virol. J. 2011;8:152. doi: 10.1186/1743-422X-8-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C., Ma Z., Harrison T.J., Feng R., Zhang C., Qiao Z., Fan J., Ma H., Li M., Song A., Wang Y. A novel genotype of hepatitis E virus prevalent among farmed rabbits in China. J. Med. Virol. 2009;81:1371–1379. doi: 10.1002/jmv.21536. [DOI] [PubMed] [Google Scholar]
- Zhuang H., Cao X.Y., Liu C.B., Wang G.M. Epidemiology of hepatitis E in China. Gastroenterol. Jpn. 1991;26(Suppl. 3):135–138. doi: 10.1007/BF02779283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This document contains Supplementary Material.