

Abstract
Beta‐thalassemia is one of the most common recessive genetic diseases, caused by mutations in the HBB gene. Over 200 different types of mutations in the HBB gene containing three exons have been identified in patients with β‐thalassemia (β‐thal) whereas a homozygous mutation in exon 1 causes sickle cell disease (SCD). Novel therapeutic strategies to permanently correct the HBB mutation in stem cells that are able to expand and differentiate into erythrocytes producing corrected HBB proteins are highly desirable. Genome editing aided by CRISPR/Cas9 and other site‐specific engineered nucleases offers promise to precisely correct a genetic mutation in the native genome without alterations in other parts of the human genome. Although making a sequence‐specific nuclease to enhance correction of a specific HBB mutation by homology‐directed repair (HDR) is becoming straightforward, targeting various HBB mutations of β‐thal is still challenging because individual guide RNA as well as a donor DNA template for HDR of each type of HBB gene mutation have to be selected and validated. Using human induced pluripotent stem cells (iPSCs) from two β‐thal patients with different HBB gene mutations, we devised and tested a universal strategy to achieve targeted insertion of the HBB cDNA in exon 1 of HBB gene using Cas9 and two validated guide RNAs. We observed that HBB protein production was restored in erythrocytes derived from iPSCs of two patients. This strategy of restoring functional HBB gene expression will be able to correct most types of HBB gene mutations in β‐thal and SCD. stem cells translational medicine 2018;7:87–97
Keywords: Gene therapy, CRISPR/Cas9, Gene editing, Beta‐thalassemia, Hemoglobinopathies, Stem cells
Significance Statement.
A universal strategy to direct a targeted insertion of the HBB cDNA at the endogenous HBB gene exon 1 using Cas9 and two validated guide RNAs is presented. This strategy is expected to allow correction of most types of HBB mutations and to restore functional HBB gene expression for treating β‐thalassemia and sickle cell disease. It will likely also be applicable to developing gene therapy strategies for treating other types of recessive monogenic diseases.
Introduction
Beta‐thalassemia (β‐thal) and sickle cell disease (SCD), two of the most common genetic diseases, are caused by mutations in the HBB gene encoding the postnatal form of the beta subunit of hemoglobin. After birth, hemoglobin tetramers contain two alpha subunits and two beta globins coded by the HBB gene that is expressed neonatally and after. Before that, beta globins coded by one of the two HBG genes that are expressed during the fetal stage and normally silenced after birth. While a point mutation in codon 6 (GAG > GTG, resulting in substitution of glutamic acid to valine amino acid) in the HBB gene creates a SCD trait, various mutations in HBB gene resulting in reduced or absent of HBB protein cause β‐thal starting in early childhood. Over 200 different types of mutations in the HBB gene have been identified in patients with β‐thal, which could be located anywhere within the ∼1,600 basepair (bp) DNA segment containing the three coding exons, splicing sites, and other regulatory elements 1. Patients with mutations in both HBB alleles that significantly reduce the HBB protein production (called β‐thal major or Cooley's anemia) suffer from severe anemia and skeletal abnormalities, and have a high level of mortality or shortened life expectancy if left untreated 1. Similarly, patients carrying both copies of the SCD HBB mutation, or a heterozygous SCD mutation plus a copy of a severe β‐thal mutation will make dysfunctional HBB protein that impedes hemoglobin functions 1.
Although chronic transfusion of red blood cells and some small molecules ameliorate symptoms of β‐thal and SCD patients, it is highly desirable to develop a cure for treating these monogenic diseases due to HBB gene mutations. Bone marrow transplantation (BMT) using hematopoietic stem cells (HSCs) from an allogeneic donor with the wildtype HBB gene has been explored in the past several decades for treating β‐thal and SCD. Although successful in some cases, the BMT technology is limited because of graft‐versus‐host disease and a lack of immunologically matched donors that are unrelated to the treated patients 2. An alternative approach is to insert a functional copy of the HBB gene into the patient's HSCs followed by BMT. In the past decades, scientists have overcome many hurdles in efficient delivery of a functional copy of the HBB gene ex vivo into human HSCs, which will home into patient's marrow, differentiate to erythrocytes and express a high‐level of the added HBB gene 2, 3. Currently, the best developed approach of gene therapy for treating β‐thal and SCD patients relies on using genome‐inserting lentiviral vectors that carry the HBB or related HBG coding sequence (CDS) plus shortened regulatory elements, inserting them permanently into the genome of autologous HSCs 2, 3, 4. Although ongoing clinical trials will ultimately determine the balance of efficacy and risks for treating β‐thal and SCD patients, the uncontrollable nature of lentiviral vector insertion that favors coding regions is always a potential risk especially over a long‐term 2, 3, 4, 5, 6, 7. In recent years, scientists moved back to achieve precise genome editing via homology‐directed repair (HDR) of a HBB mutation, which has been explored since 1985 but with a very low efficiency (10−6) 7, 8.
The recent advents of engineered nucleases that make a double‐stranded DNA break (DSB) greatly improved our ability to achieve HDR and other forms of DNA repair and recombination in nontransformed human cells. In addition, the availability of immortalized human stem cells harboring HBB mutations with ability to differentiate to erythrocytes significantly accelerates the development of functional correction of HBB mutations. Since 2008, it became possible to generate human induced pluripotent stem cells (iPSCs) from β‐thal and SCD patients that have unique HBB mutations 9, 10, 11, 12. During this time, engineered nucleases such as Zinc Finger Nucleases, Transcription Activator Like Effector Nucleases and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas systems have also been developed to enhance HDR and achieve precise genome editing to correct a HBB mutation in iPSCs 6, 7, 8. Although the HDR efficiency is still relatively low (<1%) in nontransformed cells, rare clones of iPSCs after HDR‐mediated gene correction can be selected, characterized, and expanded extensively. Aided by validated nucleases targeting specific locations of various specific HBB mutations, precise genome correction of the SCD point mutation in exon 1, the TCTT deletion in exon 2, or the IVS2‐654 mutation in intron 2 has been achieved when a donor DNA template specific for each type HBB mutation is also provided 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23. The ease and robustness of the CRISPR/Cas9 system has become the preferred choice in recent years for making a specific DSB in the HBB locus and achieving HDR to correct a specific HBB gene mutation 17, 18, 19, 20, 21, 22, 23.
For future clinical applications of correcting various HBB mutations, it is highly desirable to develop a universal strategy to correct most if not all of >200 types of HBB mutations by using validated CRISPR guide RNAs and one donor DNA template for HDR. To provide a proof‐of‐principle, we developed a strategy of using two validated guide RNAs (targeting at the HBB exon 1 and 3′‐un‐translated region (UTR)) and a DNA template providing all the HBB CDS. In this way, a HDR event near the guide RNA will provide a functional correction of HBB mutations not only in exon 1, but also exon 2 and 3 or any downstream sites. We used iPSC lines from two transfusion‐dependent β‐thal patients with HBB mutations in exon 2 and intron 2 as well as an exon 1 mutation to test this new and more universal strategy. To provide a simple readout, we linked a GFP reporter gene downstream to the HBB coding cDNA via the 2A self‐cleaving peptide so that the GFP reporter expression is indicative of the HBB expression from the same transcript and pro‐peptide. Our data provide evidence that this universal approach is able to correct various HBB gene mutations and restore HBB protein production. In addition, it provides an experimental system to screen bioactive molecules and to improve HBB protein expression in iPSC‐derived erythrocytes based on coexpression of GFP reporter.
Materials and Methods
Human iPSC Generation and Differentiation
The β‐thal iPSC lines, BH1 and BH2, were generated by a methodology under feeder‐free and xeno‐free culture conditions as previously described 24. Briefly, peripheral blood mononuclear cells (MNCs) were obtained from the two transfusion‐dependent, β‐thal major patients in the Third Affiliated Hospital of Sun Yat‐sen University in China, with approval from the internal review board on research ethics and informed consents. MNCs were cultured to expand and enrich erythroblasts, which were then reprogrammed via transient expression from three improved episomal vectors expressing the four Yamanaka factor plus the BCL2L1 (BCL‐xL) gene 24. At day 14 of reprogramming, live fluorescent staining of the TRA‐1‐60 pluripotency marker on emerging iPSC colonies was performed, and the TRA‐1‐60+ positive colonies were selected and pooled for further iPSC characterization. The established β‐thal iPSC lines were cultured in the Essential 8 (E8) medium (Thermo Scientific, Carlsbad, CA, #A1517001) on tissue‐culture plates coated with vitronectin (Thermo Scientific, Carlsbad, CA, #A14700) 25. The previously established normal iPSC line BC1 (as a normal control) and the TNC1 iPSC line from a sickle cell patient were cultured and differentiated similarly 12, 25. The differentiation of iPSCs was driven via embryoid bodies (EBs) on 96‐well plates in the serum‐free medium 18, 25. Fourteen days after EB formation and hematopoietic differentiation, the formed CD34+CD45+ hematopoietic stem/progenitor cells (HSPCs) released into medium in suspension were harvested and examined. To generate iPSC‐erythrocytes, the HSPCs further underwent erythroid differentiation (ED) for 10 days and terminal maturation (TM) for another 8 days consequently, as previously described 18, 25.
Whole Genome Sequencing of β‐Thal iPSC Lines
Genomic DNA from the two patient‐derived iPSC lines and their original MNCs were isolated using the DNeasy Blood & Tissue Kit (Qiagen, Germantown, MD, #69504). Library preparation using TruSeq DNA PCR‐Free Kit (Illumina, San Diego, CA) and Whole Genome Sequencing on HiSeq X using 2 × 150 bp read length, with a ×30 genome coverage on average, were performed at New York Genome Center (NYGC). Initial alignments of reads to GRCh37 (hg19) reference genome as well as variant calling using GATK were also performed by NYGC. Nonsynonymous variants in HBB gene that have been implicated in thalassemia were confirmed by Sanger sequencing. The results are shown in Table 1.
Table 1.
Human iPSC lines generated from two β‐thalassemia major patients.
| iPSC lines | Donor information | ||
|---|---|---|---|
| Sex | Transfusion dependence | HBB gene mutation | |
| BH1 | M | Yes |
β17 (AAG>TAG, rs33986703); β‐thal654 (c>t, rs34451549) |
| BH2 | F | Yes |
β17 (AAG>TAG, rs33986703); β41–42 (‐TCTT) |
Abbreviation: iPSCs, induced pluripotent stem cells.
Targeting the HBB Gene with a CRISPR/Cas9 Approach
To perform the endogenous HBB gene editing, we used the CRISPR/Cas9 system as previously described 18, 26, 27. A plasmid expressing humanized SpCas9 protein (Addgene, Cambridge, MA, plasmid #41815) and a guide RNA (gR) expression plasmid (Addgene plasmid #41824) were as previously described 26, 27. We later, we also used pCas9‐GFP (Addgene plasmid #447190) that coexpresses SpCas9 and GFP under the control of CAG promoter. The gR‐HBB‐a specifically targeting HBB exon 1 was described previously 18 and shown below. The second gR‐HBB‐UTR (5′‐AAACTGGGGGATATTATGA‐3′) targets on the 3′ UTR of the HBB gene. A donor vector providing a required DNA template for HDR is based on a plasmid (Addgene, #31938) as previously published 28, with additional DNA segments inserted by Gibson assembly. Briefly, it contains a left homology arm (560 bp), a right homology arm (880 bp), an HBB coding cDNA sequence followed by a P2A linker, an (e)GFP coding DNA segment, and a loxP‐flanked PGK‐puromycin selection cassette. A synonymous change at HBB codon 14 (Leucine) from CTG to TTA is introduced to the HBB cDNA in the donor vector, to prevent from recut by Cas9 complexed with gR‐HBB‐a that recognizes the target DNA sequence including CTG (5′‐GTCTGCCGTTACTGCCCTGT‐3′). The relevant DNA sequences of the donor vector (HBB‐GFP‐PGK‐puro‐V2) are provided as Supporting Information Figure S1.
Gene targeting in various human iPSC lines was conducted as we previously described 18, 27. Briefly, two million iPSCs were resuspended in 100 μl P3 primary cell solution and mixed with 10 μg DNA containing equal amounts of four plasmids (2.5 μg for each of two guide RNAs, pCas9‐GFP, and the donor vector), and then underwent electroporation by 4D Nucleofector (Lonza, Allendale, NJ). The locations of homologous recombination were verified by PCR using primer L1‐F and L1‐R at 5′ terminal and L2‐F and L2‐R at 3′ terminal. The positive iPSC colonies with an HDR event were identified by genomic PCR using primer gDNA‐75‐F and gDNA363‐R. To excise the loxP‐flanked PGK‐puromycin selection cassette, human iPSCs were transfected with the plasmid pCAG‐Cre‐IRES2‐GFP (Addgene, #26646) as done previously 18. After 3 days of transfection, the GFP positive cells were selected by fluorescence‐activated cell sorting on FACSAria II (BD Biosciences, San Jose, CA). The cells were plated in low density for clonal selection. Individual clones were picked and screened for excision by genomic PCR using primer L2‐F and V4193‐R. The positive colonies with excision were further confirmed by Sanger DNA sequencing 18. The relevant primer sequences are listed in Supporting Information Table S1.
MiSeq‐Based Deep Sequencing of Putative Off‐Targets by the Second HBB Guide RNA in iPSCs
The specificity of the second guide RNA gR‐HBB‐UTR that targets the HBB 3′ UTR and possibly other off‐target candidate (OTC) sites in the human genome was validated as we did previously with the first guide RNA gR‐HBB‐a 18, 27, with minor modifications. The list of the sequences of the on‐target HBB and top 19 algorithmically predicted OTC loci were listed in Supporting Information Table S2. The MiSeq data of cutting efficiency and specificity in human iPSCs are summarized in Supporting Information Figure S2A. Additional information of the MiSeq is also provided in the Supporting Information.
Immunocytochemistry
At day 14 of reprogramming, the iPSC cultures were directly incubated with fluorescence (R‐PE) conjugated antibody human TRA‐1‐60 (Miltenyi Biotec, Auburn, CA, #130‐100‐347, 1:10 dilution) in E8 medium for 1 hour 24. After thorough washing with phosphate‐buffered saline (PBS), the live staining was examined by a fluorescence microscope.
Teratoma Formation as an In Vivo Pluripotency Assay
NOD.Cg‐Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were obtained from Jackson Laboratory. The animal experiments were approved by IACUC at Johns Hopkins University School of Medicine. The care of all experimental animals was in accordance with institutional guidelines. The iPSCs were treated with TrypLE to generate single‐cell suspensions before being tested in vivo by teratoma formation 29. The cells (5 × 106) were mixed with Matrigel (BD Biosciences, 5 mg/ml) in a final volume of 50 μl, and intramuscularly injected into the hind limbs of 8‐week‐old NSG mice. The mice were sacrificed 8 weeks after iPSC injection. Teratomas were harvested, fixed for 24 hours in 10% buffered formalin and then examined by a routine wax‐embedding histological procedure. The paraffin sections of 5 μm thickness were mounted on slides and stained with H&E. The typical morphologies of endoderm, mesoderm, and ectoderm were observed under microscope (×40 magnifications).
Real‐Time Quantitative PCR Analysis
Total RNA was isolated using Quick‐RNA MiniPrep kit (Zymo Research, Irvine, CA, #R1055), including DNase I (RNase free) treatment to eliminate DNA contamination. The reverse transcriptions were undertaken with the SuperScript III First‐Strand Synthesis System (Thermo Scientific, #18080‐051) following manufacturer's instruction. Real‐time quantitative PCR was performed on StepOnePlus Real‐Time PCR System (Applied Biosystems, Inc., Foster City, CA) with SYBR Green PCR Master Mix reagent (Applied Biosystems, #4309155). The expression levels of relevant genes (OCT4, NANOG, HBB, HBG) were quantified, and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as an internal standard for normalization. The gene‐specific PCR primers are also listed in Supporting Information Table S1.
Flow Cytometric Analysis of iPSCs and Differentiated Hematopoietic Cells
Human iPSCs or derived‐EBs were dissociated to into a single‐cell suspension by TrypLE (Life Technologies, Carlsbad, CA), and washed with a buffer for FACS (PBS plus 1% FBS and 1 mM EDTA). The cells were resuspended in the FACS buffer, and labeled with fluorochrome‐conjugated anti‐human CD34‐APC (Miltenyi Biotec, #130‐090‐954), CD45‐PE (Miltenyi Biotec, #130‐080‐201), CD45‐ Brilliant Violet 605 (BioLegend, San Diego, CA, #304042), CD235a‐Pacific Blue (BioLegend, #349108) antibodies. Isotype‐matched control antibodies were used to determine the background staining. The erythrocyte enucleation was evaluated by DRAQ5 staining (Thermo Scientific, #62251, 1:2,000 dilution). To characterize human iPSCs, the primary antibodies anti‐human TRA‐1‐60 (Millipore, Billerica, MA, #MAB4360) and SSEA‐4 (Millipore, #MAB4303) were used. Their recognitions were detected by respective secondary antibodies anti‐mouse IgM‐Alexa Fluor 555 (Thermo Scientific, #A21426) and anti‐mouse IgG‐Alexa Fluor 555 (Thermo Scientific, #A21422). Flow cytometric analysis was performed on a FACSCalibur or LSR II analyzer (BD Biosciences). Data analysis was performed using FlowJo or FCS Express software.
Western Blotting
The iPSCs and erythrocytes at TM day 8 were lysed with RIPA buffer (Sigma, St. Louis, MO, #R0278) in the presence of protease/phosphatase inhibitor cocktail (Cell Signaling Technology, Danvers, MA, #5872S). The protein samples were treated and run with NuPAGE Bis‐Tris Mini Gel System (Life Technologies) following manufacturer's instruction. The Western blot was performed with Invitrogen iBlot Dry Blotting System with 7 minutes running time in P3 program. To detect HBB protein production, expression, we used a mouse monoclonal IgG1 (Santa Cruz Biotechnology, Dallas, TX, #sc‐21757) as a primary antibody, which specifically recognize HBB but not HBG proteins as previously reported 18. As a loading control, we also probed the blot after using anti‐human GAPDH antibodies (Cell Signaling Technology, #5174S, rabbit IgG). For secondary antibodies used for Western blot detection, we used peroxidase labeled anti‐mouse IgG(H + L) (Vector Laboratories, Burlingame, CA, #PI‐2000) or anti‐rabbit IgG(H + L) (Jackson ImmunoResearch Laboratories, West Grove, PA, #711‐035‐152), respectively. Positive signals were developed by ECL Primer Western Blotting Detection Reagent (GE Healthcare, Pittsburgh, PA, #RPN2232). The protein expression levels were quantified using Photoshop software based on band area and gray scale.
Statistical Analysis
Experiments were repeated three times. The data were subjected to statistical analysis by unpaired Student's two‐tailed t test. Results were presented as means ± SD. In all comparisons, p < .05 was used as the criterion for statistical significance.
Results
Derivation and Characterization of iPSC Lines from Two Beta‐Thal Patients
To develop new models and treatment for β‐thal major patients, we derived iPSCs from two patients who need chronic blood transfusion (Table 1). Peripheral blood MNCs were obtained with informed consents and expanded to generate erythrocyte populations. They were used to generate human iPSC lines by transient expression of three episomal vectors as previously described 24. The derived iPSC lines, BH1 and BH2, from these two patients showed typical morphology and markers of human pluripotent stem cells (Fig. 1). The derived iPSC lines BH1 and BH2 also have a normal karyotype (and confirmed XY and XX genotypes), and are pluripotent by in vivo and in vitro assays (data not shown). Whole genome sequencing reveals that BH1 iPSCs has the codon 17 (β17) mutation in exon 1 (AAG > TAG resulting in a stop codon; rs33986703) and the IVS2‐654 splicing mutation located in intron 2 (rs4451549). For BH2, we found the β17 mutation and a β41–42 mutation in exon 2 (TCTT deletion). The same mutations were also found in patient's MNCs, respectively. We did not detect other mutations in BH1 and BH2 that would be deleterious to cell growth or hematopoietic differentiation. The three types of HBB mutations detected were among common mutations of β‐thal major in south China 1, 30, resulting in premature termination of the HBB protein. The sequencing data are consistent with patients' clinical phenotypes of β‐thal major, which are commonly transfusion‐dependent. Sanger sequencing was used to confirm and monitor the compound heterozygous HBB mutations present in the BH1 and BH2 iPSC lines (Fig. 1D).
Figure 1.

Characterization of BH1 and BH2 induced pluripotent stem cells (iPSCs) generated from beta‐thalassemia major patients. (A): Live cell staining of iPSCs at reprogramming day 14 under feeder‐free and xeno‐free culture conditions. The iPSC colonies were incubated with anti‐human TRA‐1‐60‐PE in a cell culture incubator for 1 hour. The representative images of live staining were obtained using a fluorescence microscope. The colony morphologies were observed using light phase. Bar size = 100 μm. (B): Positive iPS cell colonies of alive TRA‐1–60 staining were picked up and pooled to establish BH1 and BH2 human iPS cell lines. The iPSCs were expanded on vitronectin‐coated plate in E8 media. The pluripotent stem cell markers, TRA‐1‐60 and SSEA‐4, were measured by flow cytometry and shown in histograms. Black lines represented isotype‐matched antibody controls. (C): Real‐time quantitative PCR analyses of OCT4 and NANOG gene expression in BH1 and BH2 iPSCs. The relative gene expression levels were normalized by housekeeping gene GAPDH. The BC1 iPSCs were used as controls. (D): The HBB gene mutations in BH1 and BH2 iPSCs were confirmed by Sanger DNA sequencing. β17 and β‐thal654 mutations were identified in BH1 iPSCs, while β17 and β41‐42 mutations were identified in BH2 iPSCs. Abbreviation: GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase.
Using the standard protocol to generate HSPCs followed by sequential formation of erythroblasts and erythrocytes 18, 25, we examined the capacity of β‐thal BH1 and BH2 iPSCs to form erythrocytes (Fig. 2A). They both had similar capacity to form CD34 + CD45+ HSPCs after 14‐day differentiation, as compared to the control BC1 iPSCs (Fig. 2B). After ED (10 days) and terminal differentiation (additional 8 days), both BH1 and BH2 also form erythrocytes that are positive for CD235a (glycophorin A) and negative for CD45 (common leukocyte antigen) (Fig. 2C). However, the BH1 and BH2 derived‐erythrocytes lacked HBB protein expression (Fig. 2D). These data are consistent with the transfusion‐dependent phenotypes of the BH1 and BH2 patients.
Figure 2.
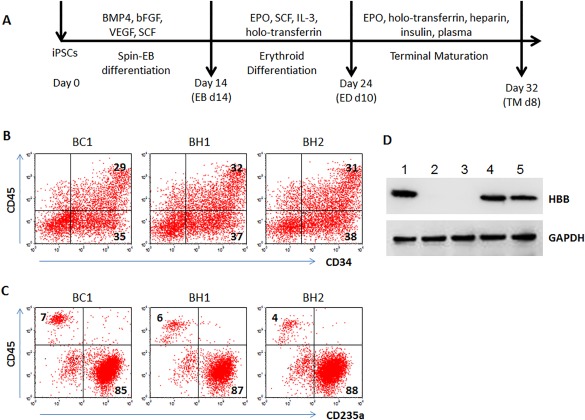
Erythrocyte differentiation of BH1 and BH2 iPSCs. (A): Diagrammatic sketch of differentiation procedure. To generate red blood cells, the iPSCs underwent three stages of spin‐EB differentiation, erythroid differentiation, and erythrocyte terminal maturation (TM) in feeder‐free and xeno‐free culture conditions. (B): Flow cytometric analyses of hematopoietic stem/progenitor cells (CD34+CD45+) cells at EB day 14. (C): Flow cytometric analyses of erythrocytes (CD235a+CD45‐) at TM day 8. The differentiation derivatives from BC1 iPSCs were used as controls. (D): Western blot to detect HBB proteins of erythrocytes from various iPSCs after terminal differentiation. GAPDH was used as loading control. Lane 1: BC1; Lane 2: BH1; Lane 3: BH2; Lanes 4, 5: two other control iPSC lines. Abbreviations: EB, embryoid bodies; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; iPSCs, induced pluripotent stem cells.
Development of a Universal HBB Gene Targeting Approach
While both BH1 and BH2 patients have the β17 mutation, and the HBB gene expression could be restored by correcting this point mutation, there are many patients who have either homozygous IVS2‐654 (in intron 2) or β41‐42 (in exon 2) mutations, or other forms of HBB mutations that severely reduce HBB protein production. In order to develop a universal strategy to correct most of HBB mutations found in SCD and β‐thal, we tested the scheme shown in Figure 3A. Briefly, we designed a strategy to achieve HDR in the exon 1 of the HBB locus with targeted insertion of the HBB coding cDNA, using a validated guide RNA that we previously used to target the SCD point mutation (codon 6) with a high‐level of specificity as well as efficiency 18. In this way, patients with HBB mutations in exon 2, intron 2, or exon 3 in both alleles will benefit from the targeted insertion of the HBB CDS as those patients with exon 1 mutations in both alleles. To facilitate testing a universal DNA donor template, we further added a GFP reporter gene downstream to the HBB CDS (linked by the P2A element). Another feature of this universal donor template, similar to what we and others have used, is a puromycin‐resistance expression cassette allowing selection of rare events of HDR at the HBB locus 18, 27, 28.
Figure 3.

CRISPR/Cas9 mediated genome editing of the human HBB gene by its cDNA. (A): A diagram of a strategy to replace the HBB genomic DNA by its CDS linked by a P2A self‐cleaving peptide with the GFP reporter gene. The two guide RNAs, gR‐HBB‐a and gR‐HBB‐UTR, were designed to target on exon 1 and 3′ UTR of HBB gene, respectively. The donor vector containing HBB CDS, a L‐HA and a R‐HA was transfected into BH1, BH2, or BC1 induced pluripotent stem cells (iPSCs) along with guide‐RNA vectors and Cas9 vector. The positive colonies with homology‐directed repair (HDR) were identified by genomic PCR using indicated primers in Supporting Information Figure S3. Next, the edited iPSCs were transfected with plasmid pCAG‐Cre‐IRES2‐GFP to excise the loxP‐flanked PGK‐puromycin expression cassette. The positive colonies with anticipated excision were identified by genomic PCR using primer L2‐F and V4193‐R. (B): Genomic PCR screening for HDR positive (either homozygous or heterozygous) colonies using primer gDNA‐75‐F and gDNA363‐R. The wildtype HBB locus gave rise to a 438 bp PCR product (WT) whereas the corrected edited locus would be a 306 bp PCR product due to the lack of the intron (132 bp) in the donor cDNA. (C): Genomic PCR screening for excision of the PGK‐Puro cassette using primer L2‐F and V4193‐R. The targeted colonies without excision (such as BC1, #6) gave rise to a 952 bp PCR product whereas the positive colonies with excision did not give rise to specific PCR product. (D): Sanger sequencing to confirm HBB gene corrections in BH1 and BH2 iPSCs. The synonymous change of β14 (Leucine) from CTG to TTA, in addition to the corrected β17 (AAG), was observed in selected clones of BH1 and BH2 iPSCs. Abbreviations: bp, basepair; CDS, coding sequence; L‐HA, left homology arm; R‐HA, right homology arm; WT, wild type.
In the initial experiments, we observed abundant HDR events in iPSCs using transient transfection of plasmids expressing SpCas9 and a guide RNA (gR‐HBB‐a) as we previously used in targeting SCD iPSCs 18. However, with this new donor vector that contains HBB CDSs identical to the exon 2 and exon 3 of the endogenous HBB locus, we often observed undesirable recombination events between the donor and the endogenous HBB at these homologous regions. To eliminate this aberrant recombination events, we used an additional guide RNA targeting the 3′ UTR immediately downstream to the stop codon in HBB exon 3 (Fig. 3A). We first vigorously tested the efficiency and specificity of this new gR‐HBB‐UTR in human iPSCs (Supporting Information Fig. S2A), similarly to what we did previously with the gR‐HBB‐a guide RNA using a transient transfection assay in human iPSCs followed by deep‐sequencing 18. With this improved HBB‐targeting strategy, we readily obtained clones from both patient‐derived iPSC lines and a control iPSC line BC1: either with an insertion (and replacement of the genomic DNA) only at one allele of the HBB locus (such as BC1‐HBB‐GFP #6, BH1‐HBB‐GFP #2, BH2‐HBB‐GFP #11) or at both alleles (BC1‐HBB‐GFP#8), as shown in Figure 3B. These selected iPSC clones were further expanded and characterized to confirm the targeted insertion by standard molecule techniques 13, 18. The characterizations include the verification of a desirable HDR event in the above‐mentioned clones by specific PCR amplification and sequencing at both junctions of the targeted insertion (Supporting Information Fig. S3). We further removed the puromycin‐selection cassette flanked by the two loxP sites using a plasmid that transiently expresses Cre in selected iPSC lines (Fig. 3A). The clones of selected iPSC lines with deletion of the puromycin‐selection cassette were identified (Fig. 3C). With further characterization of candidates, we identified iPSC clones such as BH1‐HBB‐GFP‐#2 or BH2‐HBB‐GFP‐#11, which have a targeted insertion at a single allele to correct the β17 mutation (Fig. 3D). Similarly, we obtained BC1 iPSC clones with insertion of HBB‐GFP sequence at either a single or both alleles of the HBB locus (data not shown). In addition, we confirmed that there were no sequence alterations at the 19 possible OTC sites that might be resulted in the gR‐HBB‐UTR mediated DSB (Supporting Information Fig. S2B).
Characterization of Edited iPSC Clones for HBB and GFP Reporter Gene Expression
We first differentiated the modified BC1 iPSC lines with the HBB‐GFP allele to erythrocytes as we did previously 18 with the wildtype BC1 iPSCs (Fig. 4). After the standard three‐step differentiation lasting for a total of 32 days, we generated a similar level of erythrocytes that expressed CD235a (>84%). By FACS analysis, the BC1‐HBB‐GFP #6C2 iPSC line that has a single copy of the HBB‐GFP allele showed GFP+ cells at 24%, while the BC1‐HBB‐GFP‐#8C8 clone that have two copies the HBB‐GFP allele showed GFP+ cells at 35% (Fig. 4A). Analysis of sorted cell populations based on expression of GFP, which is designed to serve as a reporter for HBB expression, demonstrated that the GFP+ population is enriched for HBB gene expression (Fig. 4B). In contrast, GFP– cells are enriched for HBG gene expression. Nearly all the GFP+ cells expressed CD235a but lacked CD45 leukocyte antigen. In addition, most of the GFP+ cells have much reduced DNA staining reflecting an active enucleation process. These data show that iPSCs with the engineered HBB (cDNA) allele coupled with a GFP gene indeed rendered GFP expression in terminally differentiated erythrocytes as designed.
Figure 4.

The GFP expression under the control of the HBB locus from induced pluripotent stem cell (iPSC)‐derived erythrocytes after genome editing. (A): Histogram of flow cytometric analyses at terminally differentiated erythrocytes from genome edited BC1 iPSCs. BC1 cells with a heterozygous integration of the HBB‐GFP allele (#6C2) or homozygous integration (#8C8) were used, together with parental BC1 iPSCs. (B): Real‐time quantitative PCR analyses of HBB and HBG gene expression. GFP positive and negative cells were isolated by fluorescence‐activated cell sorting at day 8 after terminal differentiation. The relative gene expression levels were normalized by housekeeping gene GAPDH. (C): Flow cytometric analyses of other marker expression in the GFP positive versus negative cells at day 8 of terminal differentiation of #6C2 iPSCs. CD235a and CD45 were used to characterize erythrocytes. A DNA‐staining fluorescent dye DRAQ5 was used to evaluate enucleation of erythrocytes lacking nuclear DNA. Abbreviation: GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase.
We also analyzed the selected clones of BH1 and BH2 iPSCs that have a single copy of the HBB‐GFP sequence replacing a mutated β‐thal allele. After the terminal differentiation (TM) to generate erythrocytes, we observed cell populations that expressed CD235a at a high level (Fig. 5A). We analyzed the HBB protein directly by Western blotting, using levels of the housekeeping GAPDH protein in various samples as a loading control (Fig. 5B). The differentiated erythrocytes from the parental BH1 and BH2 iPSCs derived from two β‐thal major patients did not show HBB protein production even after using the same TM procedure (Fig. 5B, 5C). However, HBB protein production is restored in the corrected BH1 and BH2 cells (with the HBB‐GFP allele).
Figure 5.

Expression of HBB protein in erythrocytes from the HBB‐corrected BH1 and BH2 iPSCs. (A): Histogram of flow cytometric analyses of erythrocytes from HBB gene targeted BH1, BH2, and BC1 iPSCs at TM day 8. Efficient erythrocyte generation was demonstrated by a high percentage of cells expressing CD235a and a low percentage of cells expressing CD45. (B): Western blot of HBB expression in erythrocytes at TM day 8. The defective beta hemoglobin expression in erythrocytes from beta thalassemia patient iPSCs was rescued by mono allelic HBB gene targeting (heterozygous). The undifferentiated iPSCs of each line were used as negative controls. (C): Quantification (mean ± SEM) of HBB protein expression based on Western blot data of multiple experiments (n = 3). Abbreviations: GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; iPSC, induced pluripotent stem cells; TM, terminal maturation.
Discussion
In this report, we describe a universal strategy to correct HBB mutations in human iPSC lines that are derived from transfusion‐dependent β‐thal patients, by achieving a targeted insertion at the 5′ end of the first exon and express a full‐length of HBB cDNA. This universal strategy enables correction of not only HBB mutations in first exon (such as SCD and β17 mutations) but also downstream mutations in exon 2, intron 2, and exon 3 found in β‐thal patients. In this study, we used the validated guide RNA that we previously employed to correct the SCD point mutation at codon 6 18, so that this universal strategy should also be applicable to patients with a single or double SCD mutations. With a guide RNA targeting a further upstream HBB DNA sequence in exon 1 (including 5′ UTR) or even at the intragenic sequence 5′ to exon 1, this strategy will be applicable to most if not all the mutations found in β‐thal and SCD patients. Although we focused on β‐thal iPSCs from two such patients in this study, we also achieved similar result using the same donor DNA and guide RNAs in TNC1 iPSCs derived from a SCD patient 12, 18 (data not shown).
One can envision that human iPSCs and derived hematopoietic progeny may provide an unlimited cell source for autologous cell therapy for β‐thal and SCD patients by two distinct strategies. In the first strategy, we will need to generate transplantable HSPCs from gene corrected iPSCs, which are able to generate erythrocytes in marrow for a long‐term and produce functional HBB proteins in circulating erythrocytes. Despite extensive studies in the past two decades, however, a robust and reproducible method is still elusive for generating long‐term transplantable HSPCs from human iPSCs as well as embryonic stem cells 31, 32, 33. This is at least partially due to the lack of culture condition to expand extensively transplantable HSPCs, whether they are from postnatal cord blood and adult marrow or human iPSCs 33. The second strategy is to produce erythrocytes ex vivo from autologous human iPSCs, and then infuse to patients who are transfusion dependent 34, 35, 36. Despite of recent progress to increase efficiencies of enucleation and HBB production from iPSC‐derived erythrocytes 18, 37, 38, further improvements are needed for this approach to be clinically relevant 34, 35, 36. Currently, the level of HBB expression in iPSC‐derived erythrocytes is approximately at 10% of the endogenous HBG expression, or of that in erythrocytes from postnatal HSPCs after terminal differentiation 18, 37, 38, 39, 40. The gene‐targeting system developed in this study may also help us to screen for bioactive molecules or culture conditions that increase the HBB protein, by simply monitoring the co‐expressed GFP level. This strategy of using a reporter gene expression under the control of HBB or HBG locus was previously used in K562 cell line (that expresses HBG but not HBB proteins) 41. By this approach, several small molecules have been identified, which stimulated HBG and HBB gene expression. However, our current study provides the first case that functional human erythrocytes expressing both HBB and genetically linked GFP reporter (albeit at a relatively a low level) can be produced consistently and in large numbers. We plan to use the HBB‐GFP marked erythrocytes to determine if previously identified small molecules 41 could improve HBB production and enucleation by iPSC‐derived erythrocytes.
The universal strategy we describe here for HBB gene correction could also be adapted to correct various HBB gene mutations in primary human postnatal HSPCs, followed by BMT. Historically, it has been very difficult to achieve HDR in human CD34+ HSPCs because the DNA delivery efficiency is low. The inability to expand extensively transplantable postnatal HSPCs further limits our capability to select and expand targeted HSPCs as we did with human iPSCs 33. Recently, several papers described approaches to improve HDR efficiency in primary human CD34+ HSPCs and correct the SCD point mutation 42, 43, 44. The reported improved efficiency was achieved at least in part by delivering the required Cas9 protein and guide RNA as a preassembled complex, and by delivering a donor template as an oligonucleotide or recombinant AAV6 viruses into CD34+ HSPCs. For this approach to be successfully used for treating β‐thal patients with various HBB gene mutations, one still has to design a universal template that can be efficiently delivered into postnatal HSPCs. It is likely that we or others will be able to combine the strength of this new delivery approach with our described strategy to correct most types of various HBB mutations using a universal donor DNA template. This strategy of inserting a cDNA (or mini‐gene) in a 5′ exon with a validate guide RNA to achieve targeted insertion and functional restoration of gene expression/function will likely be applicable to many other forms of human monogenic diseases.
Conclusion
The advent of efficient and precise human genome editing aided by improved tools such as CRISPR/Cas9 significantly broadens our horizons to treat monogenic diseases such as β‐thal and SCD. While other scientists are working on developing a universal approach of genome editing in postnatal CD34+ HSPCs to derepress the expression of HBG in erythrocytes (i.e., able to substitute the missed HBB in β‐thal or to counter‐balance the SCD HBB proteins) 45, 46, our described approach is to directly restore functional HBB proteins after genome editing at the HBB locus. Future investigations will determine if one or both approaches will be clinically effective for permanently curing β‐thal patients with various HBB mutations using an optimized gene and cell therapy.
Author Contributions
L.C. and Y.W. provided and processed blood samples from β‐thal patients in China. L.C. also performed experiments in China and US. H.B. conducted research, collected and analyzed data, and wrote the manuscript. V.M. made and tested target vectors. Y.G., C.H., Y.C.J., Y.W., R.L.P., and A.Q. performed or assisted research. Z.Y. designed research and target vectors, analyzed and interpreted data, wrote the manuscript. L.C. designed research, analyzed and interpreted data, wrote the manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Note Added in Proof
This article was published online on 21 November 2017. Minor edits have been made that do not affect data. This notice is included in the online and print versions to indicate that both have been corrected 28 December 2017.
Supporting information
Supporting Information
Acknowledgments
We thank Drs. Gabsang Lee and Yohan Oh for discussions of using gene targeting vectors. This work was supported in part by research grants from National Natural Sciences Foundation of China (81170533) and Guangdong Natural Science Foundation (S2012040006785) to Dr. Cai and her team in Guangzhou, NIH (R01‐HL130676) to Dr. Cheng, Maryland stem cell fund (2016‐MSCRFE‐2739) to Dr. Mahairaki, and a summer intern fellowship from the Foundation for Advanced Research in the Medical Sciences to RL Pan. Professor Cheng is also supported by Edythe Harris Lucas and Clara Lucas Lynn Chair in Hematology of Johns Hopkins University.
Contributor Information
Zhaohui Ye, Email: Zhaohui.Ye@fda.hhs.gov.
Linzhao Cheng, Email: Lcheng2@jhmi.edu.
References
- 1. Weatherall DJ. Phenotype‐genotype relationships in monogenic disease: Lessons from the thalassaemias. Nat Rev Genet 2001;2:245–255. [DOI] [PubMed] [Google Scholar]
- 2. Wu C, Dunbar CE. Stem cell gene therapy: The risks of insertional mutagenesis and approaches to minimize genotoxicity. Front Med 2011;5:356–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Naldini L. Ex vivo gene transfer and correction for cell‐based therapies. Nat Rev Genet 2011;12:301–315. [DOI] [PubMed] [Google Scholar]
- 4. Rivière I, Dunbar CE, Sadelain M. Hematopoietic stem cell engineering at a crossroads. Blood 2012;119:1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goodman MA, Malik P. The potential of gene therapy approaches for the treatment of hemoglobinopathies: Achievements and challenges. Ther Adv Hematol 2016;7:302–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cottle RN, Lee CM, Bao G. Treating hemoglobinopathies using gene‐correction approaches: Promises and challenges. Hum Genet 2016;135:993–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yu KR, Natanson H, Dunbar CE. Gene editing of human hematopoietic stem and progenitor cells: Promise and potential hurdles. Hum Gene Ther 2016;27:729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Porteus MH, Carroll D. Gene targeting using zinc finger nucleases. Nat Biotechnol 2005;23:967–973. [DOI] [PubMed] [Google Scholar]
- 9. Mali P, Ye Z, Hammond H et al. Improved efficiency and pace of generating induced pluripotent stem cells from human adult and fetal fibroblasts. Stem Cells 2008;26:1998–2005. [DOI] [PubMed] [Google Scholar]
- 10. Wang Y, Jiang Y, Liu S et al. Generation of induced pluripotent stem cells from human beta‐thalassemia fibroblast cells. Cell Res 2009;19:1120–1123. [DOI] [PubMed] [Google Scholar]
- 11. Ye L, Chang JC, Lin C et al. Induced pluripotent stem cells offer new approach to therapy in thalassemia and sickle cell anemia and option in prenatal diagnosis in genetic diseases. Proc Natl Acad Sci USA 2009;106:9826–9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chou BK, Mali P, Huang X et al. Efficient human iPS cell derivation by a non‐integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res 2011;21:518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zou J, Mali P, Huang X et al. Site‐specific gene correction of a point mutation in human iPS cells derived from sickle cell disease patient. Blood 2011;118:4599–4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sebastiano V, Maeder ML, Angstman JF et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells 2011;29:1717–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yan W, Smith C, Cheng L. Expanded activity of dimer nucleases by combining ZFN and TALEN for genome editing. Sci Rep 2013;3:2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ma N, Liao B, Zhang H et al. Transcription activator‐like effector nuclease (TALEN)‐mediated gene correction in integration‐free β‐thalassemia induced pluripotent stem cells. J Biol Chem 2013;288:34671–34679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xie F, Ye L, Chang JC et al. Seamless gene correction of β‐thalassemia mutations in patient‐specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res 2014;24:1526–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang X, Wang Y, Yan W et al. Corrected adult β‐globin protein production in mature erythrocytes differentiated from patient iPSCs after genome editing of the sickle point mutation. Stem Cells 2015;33:1470–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Song B, Fan Y, He W et al. Improved hematopoietic differentiation efficiency of gene‐corrected beta‐thalassemia induced pluripotent stem cells by CRISPR/Cas9 system. Stem Cells Dev 2015;24:1053–1065. [DOI] [PubMed] [Google Scholar]
- 20. Xu P, Tong Y, Liu XZ et al. Both TALENs and CRISPR/Cas9 directly target the HBB IVS2–654 (C > T) mutation in β‐thalassemia‐derived iPSCs. Sci Rep 2015;5:12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ou Z, Niu X, He W et al. The combination of CRISPR/Cas9 and iPSC technologies in the gene therapy of human β‐thalassemia in mice. Sci Rep 2016;6:32463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Niu X, He W, Song B et al. Combining single strand oligodeoxynucleotides and CRISPR/Cas9 to correct gene mutations in β‐thalassemia‐induced pluripotent stem cells. J Biol Chem 2016;291:16576–16585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang Y, Zhang X, Yi L et al. Naïve induced pluripotent stem cells generated from β‐thalassemia fibroblasts allow efficient gene correction with CRISPR/Cas9. Stem Cells Translational Medicine 2016;5:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chou BK, Gu H, Gao Y et al. A facile method to establish human iPS cells from adult blood cells under feeder‐free and xeno‐free culture conditions: A clinically compliant approach. Stem Cells Translational Medicine 2015;4:320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Y, Chou BK, Dowey SN et al. Scalable expansion of human induced pluripotent stem cells in the defined xeno‐free E8 medium under adherent and suspension culture conditions. Stem Cell Res 2013;11:1103–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mali P, Yang L, Esvelt KM et al. RNA‐guided human genome engineering via Cas9. Science 2013;339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smith C, Abalde‐Atristain L, He C et al. Efficient and allele‐specific genome editing of disease loci in human iPSCs. Mol Ther 2014;23:570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hockemeyer D, Wang H, Kiani S et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol 2011;29:731–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dowey SN, Huang X, Chou BK et al. Generation of integration‐free human induced pluripotent stem cells from postnatal blood mononuclear cells by plasmid vector expression. Nat Protoc 2012;7:2013–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cao A, Galanello R. Beta‐thalassemia. Genet Med 2010;12:61–76. [DOI] [PubMed] [Google Scholar]
- 31. Kaufman DS. Toward clinical therapies using hematopoietic cells derived from human pluripotent stem cells. Blood 2009;114:3513–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature 2012;481:295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Slukvin II. Hematopoietic specification from human pluripotent stem cells: Current advances and challenges toward de novo generation of hematopoietic stem cells. Blood 2013;122:4035–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bouhassira EE. Concise review: Production of cultured red blood cells from stem cells. Stem Cells Translational Medicine 2012;1:927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Migliaccio AR, Whitsett C, Papayannopoulou T et al. The potential of stem cells as an in vitro source of red blood cells for transfusion. Cell Stem Cell 2012;10:115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shah S, Huang X, Cheng L. Stem cell‐based approaches of red blood cell production for transfusion. Stem Cells Translational Medicine 2014;3:346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kobari L, Yates F, Oudrhiri N et al. Human induced pluripotent stem cells can reach complete terminal maturation: In vivo and in vitro evidence in the erythropoietic differentiation model. Haematologica 2012;97:1795–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Trakarnsanga K, Wilson MC, Griffiths RE et al. Qualitative and quantitative comparison of the proteome of erythroid cells differentiated from human iPSCs and adult erythroid cells by multiplex TMT labelling and nanoLC‐MS/MS. PLoS One 2014;9:e100874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. An X, Schulz VP, Li J et al. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood 2014;123:3466–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Giani FC, Fiorini C, Wakabayashi A et al. Targeted application of human genetic variation can improve red blood cell production from stem cells. Cell Stem Cell 2016;18:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Voit RA, Hendel A, Pruett‐Miller SM et al. Nuclease‐mediated gene editing by homologous recombination of the human globin locus. Nucleic Acids Res 2014;42:1365–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. DeWitt MA, Magis W, Bray NL et al. Selection‐free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci Transl Med 2016;8:360ra134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dever DP, Bak RO, Reinisch A et al. CRISPR/Cas9 β‐globin gene targeting in human haematopoietic stem cells. Nature 2016;539:384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hoban MD, Lumaquin D, Kuo CY et al. CRISPR/Cas9‐mediated correction of the sickle mutation in human CD34+ cells. Mol Ther 2016;24:1561–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Canver MC, Smith EC, Sher F et al. BCL11A enhancer dissection by Cas9‐mediated in situ saturating mutagenesis. Nature 2015;527:192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Traxler EA, Yao Y, Wang YD et al. A genome‐editing strategy to treat β‐hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med 2016;22:987–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information