

Abstract
Gene therapy, cell therapy, and tissue engineering have the potential to revolutionize the treatment of disease and injury. Attaining marketing authorization for such advanced therapy medicinal products (ATMPs) requires a rigorous scientific evaluation by the European Medicines Agency—authorization is only granted if the product can fulfil stringent requirements for quality, safety, and efficacy. However, many ATMPs are being provided to patients under alternative means, such as “hospital exemption” schemes. Holoclar (ex vivo expanded autologous human corneal epithelial cells containing stem cells), a novel treatment for eye burns, is one of the few ATMPs to have been granted marketing authorization and is the first containing stem cells. This review highlights the differences in standards between an authorized and unauthorized medicinal product, and specifically discusses how the manufacture of Holoclar had to be updated to achieve authorization. The result is that patients will have access to a therapy that is manufactured to high commercial standards, and is supported by robust clinical safety and efficacy data. stem cells translational medicine 2018;7:146–154
Keywords: Adult stem cells, Autologous stem cell transplantation, Cellular therapy, Stem/progenitor cell, Tissue regeneration, Tissue‐specific stem cells
Significance Statement.
In February 2015, Holoclar was the first stem cell‐based medicine to receive authorization for commercial use throughout the European Union. Injury to the eye can destroy limbal stem cells (LSCs) leading to loss of vision as the cornea is invaded by conjunctival cells. Holoclar uses a patient's own LSCs from the unaffected eye to form a graft grown outside the body. Graft transplantation into the injured eye restores the LSC population allowing a normal transparent corneal surface to be regenerated. This is a significant development in the field of regenerative medicine—no other stem cell therapy has demonstrated sufficient quality standards and clinical successes to achieve authorization status.
Introduction
Increasing numbers of advanced‐therapy medicinal products (ATMPs), comprising gene therapies, cell therapies and tissue engineered products, are being administered to patients throughout the European Union (EU). Authorization to market ATMPs in the EU is regulated by the “ATMP Regulation” (Regulation (EC) 1394/2007) and requires an application to be made through the European Medicines Agency (EMA). However, as yet, very few ATMPs have applied for, or achieved, this authorization. This may be due, at least in part, to the fact that the pathway to regulatory approval takes a substantial amount of expertise, time and investment to navigate. Chiesi Farmaceutici S.p.A. made this investment together with Academia, to bring the product, Holoclar, through the central authorization process. In February 2015, Holoclar became the first stem cell‐based ATMP to be approved by the European Commission. This paper considers the improvements in product quality that had to be made to achieve this goal, and their relevance to patients.
Application for marketing authorization in the EU requires submission of a comprehensive product dossier to the EMA. This dossier is then subjected to an extensive and thorough scientific assessment by the EMA's specialist experts in the Committee for Advanced Therapies (CAT). The assessment is to determine if the requirements for quality, safety, and efficacy have been met, so that patients and practitioners can have high confidence in the product. Table 1 highlights the key differences in quality requirements between a product granted marketing authorization compared to one that may be used clinically under other legal provisions. Although this article focusses on product quality, it should also be noted that authorization of a medicinal product brings other additional requirements that support the product's widespread clinical use, including provisions of instructions for use (which may be supplemented with detailed training materials), product information for patients, organized safety reporting of any adverse events (pharmacovigilance), and any obligations for further clinical studies to increase the safety and efficacy data sets.
Table 1.
Key differences in quality requirements for advanced therapy medicinal products between those with and without marketing authorization in Europe
| Aspects of manufacture |
Product with central European Union marketing authorization (e.g., Holoclar) |
Other products |
|---|---|---|
| Regulatory oversight | Extensively reviewed and approved by European Medicines Agency | National and/or local approval (in most of the cases) |
| Relationship between product quality and clinical outcome | Consistent product quality correlating with extensive clinical experience | Unlikely to have extensive clinical experience or an established manufacturing process assuring consistent product quality |
| Manufacturing quality | Full GMP for commercial product | GMP level sufficient for initial clinical use |
| Manufacturing processes | Process must be validated for consistency | Process validation is not required |
| Process materials and excipients | Quality and safety must be assured | Local control of selection of materials |
| Testing |
Rigorous testing for critical quality attributes that are correlated with clinical outcome Test procedures must be validated |
Extent of testing is less regulated, and not likely to be able to be correlated with significant clinical experience Test procedure validation is not required |
| Shelf‐life | Based on stability studies with multiple batches | Unlikely to have the same level of assurance |
| Safety from infectious agents | Microbial and viral safety are subject to rigorous assessment | Microbial and viral safety are subject to local assessment |
| End use | Instructions for use including training to health care professionals approved and validated to maintain quality attributes for clinical efficacy | Instructions for use are locally controlled |
Abbreviation: GMP, good manufacturing practice.
Holoclar (manufactured by Holostem Terapie Avanzate S.R.L. and originally developed by Professors Graziella Pellegrini and Michele De Luca) is a cell‐based tissue engineered therapy that replaces epithelial cells in a damaged cornea. Renewal and repair of this surface depends on limbal stem cells (LSCs) which reside in the limbus—a narrow zone between the cornea and the bulbar conjunctiva. Ocular burns (chemical or physical) may destroy the limbus causing limbal stem cell deficiency (LSCD), a condition that allows bulbar conjunctival cells to invade the corneal surface in an attempt to reform an epithelium. This process of conjunctivalization results in neovascularization, inflammation, stromal scarring, and corneal opacity leading to loss of vision 1. Holoclar uses the patient's own (autologous) LSCs as a treatment.
There are three types of clonogenic keratinocytes defined as holoclones, meroclones, and paraclones 2, 3. The regenerative potential of Holoclar mainly relies upon the highly proliferative and self‐renewing properties of holoclones which, as stem cells, have the greatest growth potential of all keratinocytes. Holoclones generate transiently amplifying (TA) cells—meroclones—which have much lower proliferative capacity but frequently divide. Paraclones are also TA cells generated from meroclones and have even lower proliferative capacity 4, 5, 6. The purpose of both these TA cell types is to form a stratified squamous epithelium 7. This process is illustrated in Figure 1, where holoclones are identified by high level of expression of the marker, p63 (see “Efficacy” section).
Figure 1.
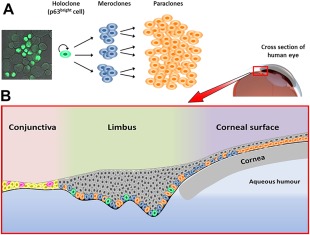
The role of clonogenic keratinocytes in generation and renewal of the corneal epithelium. (A): The holoclone differentiation process from highly proliferative self‐renewing holoclones to transiently amplifying cells (meroclones and paraclones). A confocal microscopy image of holoclone stem cells is on the left showing high expression of ΔNp63α, an isoform of the p63 transcription factor. Due to this characteristic, these cells are also referred to as p63bright cells. (B): Stem cells from (A) in their ocular context. Holoclones, meroclones, and paraclones are found in the basal layer of the limbus with holoclones having the least abundance (10%–15%). The basal layer of the cornea is populated by meroclones and paraclones at the periphery, and only paraclones in the central cornea. All suprabasal layers of the limbus and corneal surface contain terminally differentiated cells which have no capacity for self‐renewal or proliferation (shown in gray). The conjunctival surface is composed of epithelial cells (yellow) and a low proportion of goblet cells (magenta) that are interspersed throughout.
The manufacturing process, shown in Figure 2, is designed to maintain the content of holoclones in the final administered Holoclar product, and is subject to rigorous safety controls throughout the process. The starting material is a small biopsy taken from the limbus of the patient's less affected or unaffected eye. Cells isolated from this sample are enzymatically dissociated and seeded as a single primary culture onto a layer of irradiated mouse feeder cells, with growth factors, that promote LSC growth. Following primary culture expansion in media containing antibiotics, all cells are recovered and cryopreserved as an “intermediate cell bank” (ICB). This holding step has several advantages: (a) it provides time for sufficient testing of the sample; (b) it enables timing of the manufacture of the drug product to be aligned with patient, surgeon, and hospital facility availability; and (c) it can allow (dependent on the quantity available) a contingency sample of patient cells to be saved to be used in a second manufacturing round in case of incomplete result or failure with the first Holoclar graft. After thawing an ICB, it is seeded onto a fibrin matrix (secondary culture stage) which also contains the same type of mouse feeder cells; however, the media is changed to one that is antibiotic‐free but still contains growth factors. The fibrin matrix provides a solid support on which the cells can be administered into the eye. After culture expansion, support discs made of fibrin carrying the autologous cells are prepared and packaged. Holoclar is then shipped and administered to the patient by grafting into the injured eye thus restoring the LSC reservoir 8.
Figure 2.
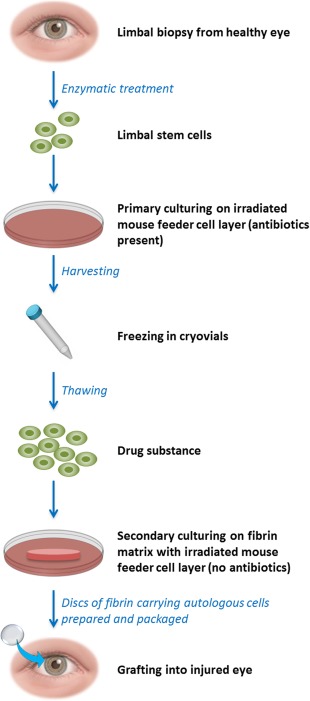
Key steps in the manufacture of Holoclar. The figure shows a simplified flow chart of the manufacture of Holoclar. The term “drug Substance” is used to denote the cell suspension of epithelial cells obtained after thawing from the frozen primary culture, and which is ready to be plated on the fibrin matrix, which will yield the final material for transplant (the “drug product”).
A number of stem cell‐based therapies have been used across Europe with the same aim as Holoclar, and some are manufactured using similar techniques (for reviews see [9, 10]). However, none of them have been demonstrated to have the quality controls required for a centralized marketing authorization. Principally from a quality perspective, this paper examines the advantages that Holoclar has to offer, having undergone the rigors of the authorization process. We explain how, since its inception, the manufacture of Holoclar has been substantially upgraded to meet the requirements of a marketed product, and the benefits that this brings to patients.
Efficacy
Throughout extensive clinical experience, it was found that the clinical success of Holoclar treatment had a positive correlation with the percentage of holoclones present in the limbal culture 8, 11. Holoclones can be reliably identified because they have a specific dimensional range and express high levels of ΔNp63α, the main isoform of the transcription factor p63 12, 13 expressed in limbal cells, sustaining the regenerative potential of these cells 13, 14, 15, 16. The percentage of p63bright cells was calculated in 91 primary cultures used to seed graftable secondary cultures, and statistical analysis showed that this parameter, was significantly associated with clinical success (p = .0114). The percentage of cells that are p63bright at this stage was therefore designated as a critical specification as a marker of product potency.
Using immunohistochemical analysis, the Company screens all limbal cultures for the abundance of p63α, and those cells with a strong signal (termed p63bright cells) within a pre‐defined dimensional range are identified as holoclones. This assay has been fully validated, and used to ensure that all cultures of Holoclar contain a sufficient proportion of p63bright cells prior to preparation for administration. This specification is based on the average result for batches yielding successful outcomes in the pivotal clinical efficacy study plus/minus two SD. Batch cultures which met this specification were associated with successful outcomes, whereas preparations with lower p63bright cell content were less likely to be clinically effective 8, 11. The test therefore saves patients from undergoing an invasive procedure only to be given a graft with insufficient holoclones, and therefore increases the prospects of beneficial effect. The assay has also allowed optimization of the ex vivo culture conditions to maintain in culture the p63bright cells so that the vast majority of Holoclar preparations contain sufficient numbers of holoclones for clinical efficacy. This is achieved through use of the mouse feeder cells (cell line 3T3‐J2), the combination of growth factors provided in the culture medium, and accurate monitoring of culture conditions.
For the authorized product, a standardized training procedure is made available to surgeons to ensure that the product is applied in the optimally effective manner, as demonstrated by previous clinical experience.
Safety
An essential purpose of the regulatory agency review is to assure the safety of medicinal products granted marketing authorization. It is described below how Holoclar has been able to achieve these safety standards through design and refinement of the manufacturing and testing strategy. The review of clinical experience is another vital element in the safety assessment of a product, but is outside of the scope of this article.
Infectious Agents
Critical for the manufacture of any biological product is the safety from risks of infectious agents that may enter the process from the source biopsy material, other materials used in the process (especially those of biological origin) or contamination from the environment of manufacture. The infectious agent risks that had to be addressed for Holoclar included the potential for contamination with bacteria, fungi, viruses, or transmissible spongiform encephalopathies (TSEs). Certainly there is a risk of microbial contamination associated with the source material taken from a biopsy of the eye, which is inherently non‐sterile as a consequence of the environmental exposure of the ocular surface. Holoclar is protected from this danger through extensive dilution by culture medium and the use of bactericidal (not merely bacteriostatic) antibiotics in the first phase of the manufacture. As it is undesirable for the product administered to patients to contain antibiotics, their use is limited to the early stages of the process (primary culture and cryopreservation of the ICB). The culture performed after cryopreservation (secondary culture) and subsequent product preparation steps are performed without antibiotics. At this stage, without antibiotics, the protection from new adventitious microbial contamination depends on the good manufacturing practice (GMP) facilities and rigorous control procedures required of a commercial manufacturer. Contaminations that have spread through the culture can be easily visualized, but Holostem have used sensitive sterility testing to validate that the manufacture of Holoclar can be maintained free from microbial contamination after antibiotic removal.
With regard to possible viral contamination of the source material, the concern is not just for the quality and safety of the product but also to assure that there is no danger of cross‐contamination of materials from other patients being processed in the facility. Up to 30 days before biopsy procurement and on the day of procurement, the patient is serologically screened for a series of pathogens, namely hepatitis A, B and C viruses, HIV1, HIV2, human T‐lymphotropic virus type I, and West Nile virus. This allows material from potentially infected patients to be processed with additional isolation from other patient materials through use of dedicated production rooms and equipment. As a result, there is no need to refuse treatment to patients on the basis of a positive serology test result.
In order to obtain marketing authorization for Holoclar, a comprehensive microbial testing strategy also needed to be implemented throughout the manufacturing process. It must be noted that after preparation the final product must be used within 36 hours, and this does not allow enough time for sterility or mycoplasma testing. It was therefore recognized by the EMA that the testing strategy had to depend on in process testing before final product production. Microbial testing has therefore been implemented at multiple steps in the production:
Prior to trypsinization of the biopsy in preparation for its primary culture stage, the biopsy transport media is tested for mycoplasma and microbial contamination. The species of bacterial flora of the healthy eye are well known, as they are described in the control population of typical bacterial infections of the eye 17, 18. Therefore, the identification of any particular species can be assessed to determine whether the species are considered “normal” or may indicate a particular eye infection or another source of contamination. The results are reported back to the treating surgeon for possible treatment of an infection but, as antibiotics are used, the primary culture processing of the biopsy can continue. In contrast, a positive mycoplasma test indicates that the cell culture would be compromised and therefore results in refusal and no further processing of the biopsy.
A sample is tested for sterility at the end of the primary culture stage. At this point, the test can use the 14‐day protocol according to the methodology of the European Pharmacopoeia. The cryopreserved hold step provides enough time for the result to be available before release of the product.
A sterility test is performed on a sample from the beginning of secondary culture. At this stage, there is not enough time for the full 14‐day European Pharmacopoeial method, so the manufacturer uses a more rapid method that was validated to demonstrate sensitivity that is comparable to that method. The sample tested represents the latest point in the process at which the sterility test result can be available for product release, and also confirms that the process was free of endogenous microbial contamination at the point where antibiotics were removed.
A second pharmacopoeial sterility test is performed on a sample taken at the final preparation stage of the Holoclar drug product which is taken just before it is administered. In this case, the result is not available by the time of product release, but serves as a confirmation of the absence of product contamination.
The other potential sources of infection with infectious agents are the raw materials. With regard to risks from viral or TSE‐causing agents, the main concerns are materials of animal or human origin. The manufacturing process for Holoclar uses bovine serum, porcine trypsin, human fibrinogen, and plasminogen together with an array of growth factors to promote proliferation of LSCs. In order to recommend marketing authorization, the CAT needed to be convinced that all such materials were appropriately sourced and tested to comply with applicable guidelines and regulations. In some cases, Holostem was required to upgrade reagents from grades that had been acceptable for clinical use before marketing authorization, and in other cases additional testing was introduced. Bovine serum was required to be irradiated, in order to inactivate any potential viral contamination. Another biological material used in the manufacture that is critical for the assessment of safety from infectious risk is the layer of feeder cells that are discussed below.
Feeder Cells
Ex vivo culture of stem cells frequently requires conditions that approximate the in vivo microenvironment, or “niche” of a given stem cell which includes growth factors, cell‐cell interactions, and cell‐matrix adhesions that allow the stem cells to remain undifferentiated and retain their characteristics of self‐renewal and pluripotency 19. The manufacture of Holoclar uses the mouse fibroblast 3T3‐J2 cell line to form a feeder layer on which the LSCs can be maintained as such and proliferate. The 3T3‐J2 cell line was established in 1962 20 and has a well‐documented history for its use as a feeder layer in stem cell culture and for manufacture of cell products with clinical application in extensive burns. Nevertheless, there are important safety issues to be addressed in the context of a product to be manufactured for use under a marketing authorization.
First, the feeder cells must themselves be assured for absence of potential to transmit infectious agents to the recipient or to their cells in Holoclar production cultures. This was achieved by manufacturing a GMP compliant master cell bank, which was fully tested for potential viral and other infectious agents according to established guidelines. By expansion from the master cell bank a working cell bank was derived, providing a strategy whereby all future Holoclar manufacture could be conducted with consistent source cells without a need to ever remanufacture the master cell bank. Validation studies demonstrated that the performance and safety characteristics were maintained even when the 3T3‐J2 cells were cultivated to passage levels beyond those used in Holoclar production.
The second risk to address was that residual lethally irradiated 3T3‐J2 cells could persist as impurities in the product. The issue is then whether these may survive administration to the patient and, if so, whether there could be any safety consequences. The level of contamination with mouse cell material is controlled through release testing. The risk that any live cells could persist in the recipient is controlled by irradiating the cells (60 Grays, equivalent to 6,000 rads) before use to ensure that they have no long‐term viability. Rendering cells mitotically incompetent by irreversible lethal irradiation means they cannot survive beyond a few rounds of passaging 21. For the CAT, it was a major safety concern that the irradiation must be rigorously validated to ensure that no mouse cells were left that retained capability for long‐term survival. Holostem was able to demonstrate that irradiated cells lose proliferative capability, while retaining the capability to function effectively as a metabolically active feeder layer for the LSCs (it may be noted that such an irradiation dose is insufficient to impact significantly on the viral safety considerations noted above). Furthermore, the company demonstrated that the irradiated 3T3‐J2 cells showed no anchorage‐independent in vitro growth potential that could have been characteristic of tumorigenic cells. Also, analysis from 26 patients that underwent a keratoplasty post Holoclar treatment (ranging from 9 months to almost 8 years after the treatment) found that no 3T3‐J2 cells could be detected in the removed samples of corneal tissue or basal lamina 11. Additional supportive evidence of the safety of irradiated 3T3‐J2 cells may be inferred from the history of their use in other cell‐based clinical products, such as the epidermal cell product Epicel that is authorized in the U.S. for treatment of skin burns.
LSC autografts that are available without marketing authorization in some countries as an alternative to Holoclar frequently do not use 3T3‐J2 cells for the feeder layer. The most widely used culture method is to grow explant cultures on human amniotic membrane (HAM) as a substratum 22, 23, and consequently the technique has been used in several products to treat LSCD 9, 10. Although donors of HAM should be screened for communicable diseases, the opportunity for testing such primary human tissue is limited and the assurance of absence of contamination with infectious agents is less than it is for a fully tested cell line. Furthermore, donor‐to‐donor variability of HAM may affect consistency of the manufacturing process (see “Quality” section). Similar considerations apply for the alternative approach of using autologous fibroblasts to form the feeder layer. In controlled comparisons, we have found that the total clonogenicity and the p63bright LSC content was markedly greater in keratinocyte populations cultured on 3T3‐J2 cells than equivalent cultures grown on feeder layers of human fibroblasts (see Fig. 3).
Figure 3.
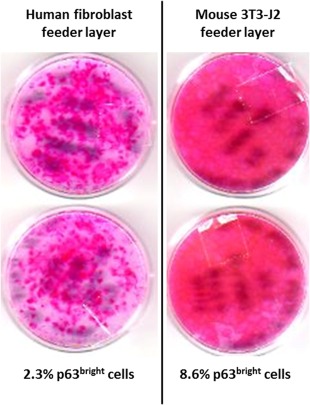
Comparison of human fibroblast or mouse 3T3‐J2 cells as feeder layers to support human keratinocyte proliferation. Both types of feeder layer cells were seeded at equal density (∼700,000 cells per plate) in duplicate after lethal irradiation. Equal numbers of human keratinocytes (1,000 cells) were then added to each plate. Following 12 days of keratinocyte culturing, plates were stained and the colonies counted. Whole plate staining allowed macroscopic comparison of total keratinocyte proliferation between the two conditions. After 6 days in culture, the parallel mass cultures were labeled and analyzed in parallel by fluorescent microscopy to determine the percentage of p63bright cells (holoclones).
Tumorigenicity
Assurance was also needed concerning the behavior and characteristics of the autologous cells in Holoclar. Therefore, the possible risks of inducing tumorigenesis due to neoplastic transformation of cells were considered. Keratinocytes from the Holoclar drug product were cultured in the presence or absence of growth factors, with or without a 3T3‐J2 cell feeder layer, to measure the growth factor dependence with a tumor cell line as positive control. For cell adhesion analysis the same cells were cultured in a semi‐solid medium (soft agar). In this environment, colony formation is considered to be indicative of anchorage‐independent growth—a hallmark of tumorigenic and metastatic potential. The results of all these assays to investigate the possible tumorigenic potential of Holoclar showed that the Holoclar manufacturing process does not transform the patient‐derived cells and proliferation is active only in “physiologic” conditions. Karyotype analysis revealed a normal phenotype with samples displaying a modal chromosome numbers of 46, indicating that they all possessed a normal human diploid karyotype (i.e., no genetic instability). Cells were entirely dependent on growth factors and the presence of the 3T3‐J2 cell feeder layer, and were unable to form colonies when grown in soft agar. This in vitro evidence of the absence of tumorigenic potential is also supported by clinical experience from over 100 patients with 10 years of follow up showing an absence of tumorigenicity in vivo 8. The risk for tumorigenicity is low because of the absence of genetic transformation. Animal studies would have been uninformative because of (a) the human origin of Holoclar, (b) its ocular administration, and (c) the differences in physiology and anatomy between human and animal eyes.
Quality
Any exploratory (unauthorized) medicine has inherent risks owing to its limited clinical experience, but quality standards are required to ensure that the manufacturing process does not introduce additional risks, notably adventitious contamination. European Union Directive 2001/20/EC stipulates that the principles of GMP should apply to manufacture of investigational medicinal products for use in clinical trials. GMP compliance is also required for manufacture of products to be used under hospital exemption or in compassionate use programmes. This does not mean that such products are expected to have the same quality standards as a product that has been granted marketing authorization. In that case, the manufactured product must also be assured to be consistent with that which was used in the pivotal clinical studies, as these were the basis for the demonstration of safety and efficacy on which the marketing authorization was granted.
For a marketed product the whole process must have validated status conferred by its demonstration of sufficient robustness and capacity to yield a consistent product. A meaningful process validation is in turn dependent on the quality of the analytics used to characterize the final product and to monitor the process, and the understanding of how the results of those tests relate to the ultimate performance of the product. Put another way, there needs to be the capability to detect anomalous batches of product that may not meet the efficacy and/or safety performance of the authorized product and which accordingly should not be administered to patients. This is particularly important for an autologous product such as Holoclar—intrinsic variability of the starting material has the consequence that final product consistency is not only dependent upon the reproducibility of the processing steps. The manufacturer therefore needs to understand and measure the quality attributes that are indicative of efficacy and safety, and accordingly set quantitative specifications that the product must meet. Clinicians and patients can then be confident that the product will be as indicated on the label, and will perform as described in the associated authorized product information.
In contrast, for a non‐commercially authorized product the manufacturer can only aim to demonstrate consistency with that used in previous non‐clinical studies, such as toxicology work, and other exploratory clinical experience. This exercise will generally not be able to define the limits of a safe and effective product and therefore may fail to detect a product that is aberrant. For products intended for use under hospital exemption, the amended ATMP regulation (EC/1394/2007) states that manufacturing quality standards should be “equivalent” to those expected of an authorized product. However, this should not be understood to indicate a requirement for process consistency and validation, which would not be feasible within the qualifying criteria for hospital exemption which include:
“….prepared on a non‐routine basis according to specific quality standards, and used within the same Member State in a hospital under the exclusive professional responsibility of a medical practitioner, in order to comply with an individual medical prescription for a custom‐made product for an individual patient.”
(EC/1394/2007 Article 28, 2).
To achieve marketing authorization, the manufacturer must demonstrate validation of the entire manufacturing process and associated analytical methods. This higher level of quality assurance is recognized in the GMP certifications granted to manufacturing organizations that normally specify whether the facility is authorized to manufacture medicinal products for commercial use, or other uses, such as clinical trials. For either type of certification, within the EU the GMP facility inspections will generally be conducted by the responsible national agency. National agencies are also normally responsible for authorizing an application to perform a clinical trial. However, it should be noted that the manufacturing information that is reviewed, from the content of the Investigational Medicinal Product Dossier, is normally much less than that given in the detailed dossier (known as the Common Technical Document) that must be reviewed by the EMA for an application for marketing authorization.
Extensive experience from batches manufactured for clinical application was used to set the product release specifications. Holostem was also able to undertake specifically designed validation studies using samples taken from donated cadaveric corneas (due to the long‐term post‐mortem epithelial viability) that were shown to be representative of biopsies from living donors. The feasibility and validity of the latter approach was important, as the biopsy material from living patient donors had to be exclusively reserved for use in treatment of those patients. It would also have been unethical to biopsy healthy donors for this purpose. The first round of validation studies using cadaveric corneas showed the consistency of production, including multiple batches prepared from the same source material which demonstrated manufacturing consistency independent of donor‐to‐donor variability. A particularly high degree of consistency was found between replicate secondary cultures derived from the same primary culture (Table 2).
Table 2.
Validation study showing consistency between cultures from the same or different primary culture
| Primary culture | Secondary culture | Test | ||||
|---|---|---|---|---|---|---|
| p63bright (%) | K3+ (%) | CFE (%) | Viable cells (%) | Cell yield | ||
| 1 | a | 8.6 | 90.3 | 14.2 | 74.6 | 315,000 |
| b | 8.4 | 83.1 | 22.5 | 80.0 | 225,000 | |
| 2 | a | 8.2 | 70.1 | 14.3 | 66.5 | 375,000 |
| b | 7.2 | 72.1 | 25.9 | 66.7 | 415,000 | |
| 3 | a | 6.3 | 67.8 | 13.5 | 82.5 | 250,000 |
| b | 5.5 | 81.3 | 18.0 | 61.6 | 300,000 | |
| Mean | 7.4 | 77.5 | 18.1 | 72.0 | 313,333 | |
| SD | 1.3 | 8.8 | 5.1 | 8.3 | 72,296 | |
Abbreviation: CFE, colony forming efficiency (measures the number of epithelial colonies that originate following culture).
A second round of validation studies demonstrated that the key quality attributes of the product were maintained when process refinements were introduced (such as the use of the 3T3‐J2 working cell bank, and the elimination of antibiotics from the second stage of cell culturing), and also encompassed stability studies for the final product and key process intermediates. Stability of the frozen ICB is significant as the amount of intermediate generated can be sufficient to enable more than one Holoclar graft to be produced from a single biopsy starting material. The opportunity to manufacture a second graft from a stored ICB can provide the patient with additional options in case of failure of the first graft or clinical need for a further graft. The recognition of a clinical need for a second engraftment is likely to be considered 6–12 months after the first Holoclar graft. Therefore a corresponding shelf life for the ICB would be of significant benefit for the patient to avoid the need for a new biopsy to generate more starting material. Based on the data shown in Table 3, a shelf life of 366 days for the ICB was accepted at the time of marketing authorization approval.
Table 3.
Percentage of p63bright and other cell populations during storage of the intermediate cell bank
| Batch | Marker | Time point 1 | Time point 2 | Time point 3 | Time point 4 | Time point 5 |
|---|---|---|---|---|---|---|
| 0116561 | Time | 14 days | 31 days | 164 days | 184 days | 366 days |
| p63bright (%) | 3.9 | 3.3 | 3.2 | 3.0 | 2.5 | |
| K3+ (%) | 70 | 76 | 64 | 57 | 53 | |
| Viability (%) | 71 | 65 | 64 | 61 | 65 | |
| 0117082 | Time | 12 days | 33 days | 160 days | 187 days | 368 days |
| p63bright (%) | 3.6 | 4.3 | 4.2 | 3.1 | 2.7 | |
| K3+ (%) | 69 | 65 | 70 | 67 | 67 | |
| Viability (%) | 72 | 81 | 70 | 81 | 81 | |
| 0117243 | Time | 9 days | 30 days | 156 days | 176 days | 366 days |
| p63bright (%) | 4.3 | 3.2 | 3.9 | 3.5 | 3.0 | |
| K3+ (%) | 71 | 71 | 64 | 77 | 73 | |
| Viability (%) | 84 | 84 | 75 | 80 | 71 |
As part of the study, data to support the assigned shelf life of 36 hours of drug product (between manufacture and use) were obtained from 31 lots of Holoclar manufactured from six cadaveric corneas and held for 40 hours, including vibration to simulate transport conditions and excursion temperature times. Material from these studies was also used to establish that the product maintains its properties when used, according to its instructions, within 15 minutes of opening of its primary container under ambient conditions.
Together, manufacturing process validation and sound product characterization (with validated analytical techniques) have given Holoclar the assurance that it can be produced and used under real life conditions with retention of its critical quality attributes.
Conclusion
The decision made by Chiesi together with Holostem and Academia to seek marketing authorization for Holoclar was not taken lightly as significant investments from all partners were required in order to bring the product production up to the required standard and to assemble the clinical and non‐clinical data for the filing. Resources were also required to progress through the regulatory procedure, including preparatory steps such as building and certifying a new facility, regulatory scientific advice procedures, application for orphan drug status, approval of a paediatric investigation plan, an ATMP classification procedure, and finally the marketing authorization application itself. This article has described the manufacturing upgrades that were undertaken for the application, and the benefits that these have brought to the patients that receive the product.
A critical element of manufacturing for an authorized product is validation. This ensures that a product manufactured under that authorization is consistent with that for which the positive benefit‐risk ratio (between efficacy and safety) has been established from clinical experience. For Holoclar, the scientific understanding that efficacy depends on the presence of LSCs (holoclones), and the knowledge that the content of these cells can be determined from immunostaining for the p63 marker, resulted in the correlation of this measurable product characteristic with positive clinical outcome. As a consequence, the manufacturing process has been optimized to maintain sufficient content of these cells, thus minimizing the chance of patients being administered a product with low efficacy. A number of measures were implemented that improved the safety of the product. Notably these included a comprehensive strategy toward elimination of microbial contamination that is inherently possible in the source tissue. This strategy had to be sufficiently robust to allow antibiotic‐free culture of cells in the later stages of the process, and was achieved by incorporating controls to minimize risks of any subsequent contamination. Another potential safety risk arose from the xenogeneic feeder cell layer needed for the cultivation of LSCs. While a number of groups use classic laboratory techniques utilizing primary human tissue, such as amniotic membrane as a support matrix (with or without heterologous donor cells present), such a source is less than ideal for the consistency and high safety standards expected of an authorized medicinal product. The 3T3‐J2 mouse cell line used for the feeder layer in Holoclar production is derived from established master and working cell banks providing (a) consistency of material, (b) security of supply, and (c) safety through thorough extensive testing for virological risks. In addition the cells are irradiated using a protocol that has been validated to demonstrate absence of residual cells with any ability to continue replication in a recipient. Studies have also been carried out to show that neither the autologous nor the feeder cells exhibit tumorigenic characteristics (Table 4).
Table 4.
Distinctive characteristics of Holoclar manufacturing and their implications for clinical use
| Manufacturing process | Rationale/implications for clinical use | |
|---|---|---|
| Raw materials | Use of compendial materials/highest possible standards | Minimization of contamination potential |
| Irradiation of bovine serum | Inactivation of potential viral contamination | |
| Consistency of production | Analysis of multiple batches from the same source material | Consistency of expected product characteristics |
| Source material (Biopsy) |
• Bactericidal antibiotics used in the primary culture • GMP facilities with rigorous control procedures • Sensitive microbiological testing |
Minimization of contamination potential |
| Serological examination of the patient and additional isolation of infected material | Possibility of treating also seropositive patients | |
| Feeder layer | Greater clonogenicity and LSC content of keratinocytes cultured on 3T3‐J2 mouse cells than on human fibroblasts | Maximization of clinical performance of the product |
| GMP compliant master cell bank fully tested for viral and other infectious agents | Minimization of contamination potential | |
| Validated irreversible lethal irradiation protocol | Absence of 3T3‐J2 cells in the recipient cornea | |
| Frozen intermediate cell bank | Validation of 366 days of shelf‐life | Potential opportunity to manufacture a second graft without the need of a new biopsy |
| Analysis of cultured autologous cells |
• Karyotype • Growth factor and feeder layer dependence • Soft‐agar assay • Long‐term replicative senescence, absence of immortalization |
Absence of tumorigenic potential |
| LSC content | Holoclar is released only if p63bright cells in the Drug Substance are in 2.5 ÷ 16% range | Association to higher clinical success rate |
| Drug product stability | Validation of 36 hours of total shelf‐life and 15 minutes from opening of primary container | Possibility to use under real life conditions |
Abbreviations: GMP, good manufacturing practice; LSC, limbal stem cell.
The efficacy and safety factors described above can also be seen as part of the overall quality package that is needed to achieve marketing authorization. The extensive characterization and demonstration of consistency (albeit within the context of the high intrinsic donor‐to‐donor variability) provided a basis on which the manufacturing process could be refined with confidence that the safety and efficacy of the product will not be compromised. Another important feature is validation of the storage times, transportation and in‐use procedures to provide assurance that the product retains its required properties from manufacture right up to the moment of administration to a patient.
The product upgrades required to achieve marketing authorization do require significant expenditure. As well as the costs for clinical studies, upgrading the product quality to meet EMA requirements will typically require investment in the manufacturing, including in facilities and their maintenance, additional analytical tests, raw material controls, and validation of the manufacturing process and analytical assays. Other costs will include those for regulatory filings, post‐approval variations, process revalidation, and pharmacovigilance.
While the theme of this paper has been the improvements made in Holoclar in order to obtain EU marketing authorization, the context in which non‐commercially authorized therapies may be used should not be forgotten. Clinical trials are essential for the development of new therapies, and provide the dosing, safety and efficacy data upon which a future marketing authorization can be based. At the clinical trial stage there are evidently risks inherent in the use of an experimental medicine that has had limited controlled assessment in humans, and developers must design their studies to minimize these risks. The manufacturer may not know precisely which product attributes are critical for clinical performance but should manufacture the product to GMP standards to avoid the introduction of additional risks. Treatments performed under compassionate use schemes and, even more so, hospital exemption provisions for ATMPs, also involve use without the support of approved clinical, non‐clinical and manufacturing data sets that can enable authorization. In the case of compassionate use, this may only be a temporary arrangement while an application for marketing authorization is ongoing. However, hospital exemption is designed for customized patient‐specific products for which a marketing authorization may never be feasible. In some such cases a therapy that is patient‐specific, and therefore apparently so non‐routine as to be unsuitable for the marketing authorization route, may subsequently find itself alongside a similar therapy that has actually achieved authorization. This may well have been the case with LSC products, like Holoclar, which some may have thought, as a complex autologous therapies, would not be amenable to marketing authorization. However, Chiesi and Holostem are pleased to have been able to demonstrate that this was possible for Holoclar, and the manufacture of such a therapy could be developed to the validated quality required. As a result, patients can have access to a therapy that is manufactured to high commercial standards and consistency. Holoclar is supported by clinical safety and efficacy data that have been subjected to intense regulatory review and by continuing obligations according to the risk management plan and other conditions of approval.
Author Contributions
G.P.: conception and design, provision of study materials or patients, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; D.A. and G.M.: collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; G.I. and D.P.: data analysis and interpretation, manuscript writing, final approval of manuscript; P.G. and M.D.L.: collection and/or assembly of data, data analysis and interpretation, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
G.P. is a member of the Board of Directors and R&D Director of Holostem Terapie Avanzate, and a consultant of J‐TEC (Japan). D.A., G.M., G.I., and D.P. is an employee of Chiesi Farmaceutici S.p.A. P.G. is an employee of Holostem Terapie Avanzate. M.D.L. is a member of the Board of Directors and Scientific Director of Holostem Terapie Avanzate, and a consultant of J‐TEC (Japan).
References
- 1. Dua HS, Azuara‐Blanco A. Limbal stem cells of the corneal epithelium. Surv Ophthalmol 2000;44:415–425. [DOI] [PubMed] [Google Scholar]
- 2. Barrandon Y, Green H. Three clonal types of keratinocyte with different capacities for multiplication. Proc Natl Acad Sci USA 1987;84:2302–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pellegrini G, Golisano O, Paterna P. Location and clonal analysis of stem cells and their differentiated progeny in the human ocular surface. J Cell Biol 1999;145:769–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lajtha LG. Stem cell concepts. Differentiation 1979;14:23–34. [DOI] [PubMed] [Google Scholar]
- 5. Lavker RM, Miller S, Wilson C et al. Hair follicle stem cells: Their location, role in hair cycle, and involvement in skin tumor formation. J Invest Dermatol 1993;101:16S–26S. [DOI] [PubMed] [Google Scholar]
- 6. Lehrer MS, Sun TT, Lavker RM. Strategies of epithelial repair: Modulation of stem cell and transit amplifying cell proliferation. J Cell Sci 1998;111:2867–2875. [DOI] [PubMed] [Google Scholar]
- 7. Barrandon Y. The epidermal stem cell: An overview. Dev Biol 1993;4:209–215. [Google Scholar]
- 8. Rama P, Matuska S, Paganoni G et al. Limbal stem‐cell therapy and long‐term corneal regeneration. N Engl J Med 2010;363:147–155. [DOI] [PubMed] [Google Scholar]
- 9. Shortt AJ, Secker GA, Notara MD et al. Transplantation of ex vivo cultured limbal epithelial stem cells: A review of techniques and clinical results. Surv Ophthalmol 2007;52:483–502. [DOI] [PubMed] [Google Scholar]
- 10. Baylis O, Figueiredo F, Henein C et al. 13 years of cultured limbal epithelial cell therapy: A review of the outcomes. J Cell Biochem 2011;112:993–1002. [DOI] [PubMed] [Google Scholar]
- 11. Pellegrini G, Rama P, Matuska S et al. Biological parameters determining the clinical outcome of autologous cultures of limbal stem cells. Regen Med 2013;8:553–567. [DOI] [PubMed] [Google Scholar]
- 12. Pellegrini G, Dellambra E, Golisano O et al. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci USA 2001;98:3156–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Di Iorio E, Barbaro V, Ruzza A et al. Isoforms of DeltaNp63 and the migration of ocular limbal cells in human corneal regeneration. Proc Natl Acad Sci USA 2005;102:9523–9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parsa R, Yang A, McKeon F et al. Association of p63 with proliferative potential in normal and neoplastic human keratinocytes. J Invest Dermatol 1999;113:1099–1105. [DOI] [PubMed] [Google Scholar]
- 15. Koster MI, Kim S, Mills AA et al. p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev 2004;18:126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nguyen BC, Lefort K, Mandinova A et al. Cross‐regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev 2006;20:1028–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Armstrong RA. The microbiology of the eye. Ophthalmic Physiol Opt 2000;20:429–441. [PubMed] [Google Scholar]
- 18. Donahue SP, Khoury JM, Kowalski RP. Common ocular infections. A prescriber's guide. Drugs 1996;52:526–540. [DOI] [PubMed] [Google Scholar]
- 19. Discher DE, Mooney DJ, Zandstra PW. Growth factors, matrices, and forces combine and control stem cells. Science 2009;324:1673–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol 1963;17:299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rheinwatd JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: The formation of keratinizing colonies from single cells. Cell 1975;6:331–343. [DOI] [PubMed] [Google Scholar]
- 22. Grueterich M, Espana EM, Tseng SC. Ex vivo expansion of limbal epithelial stem cells: Amniotic membrane serving as a stem cell niche. Surv Ophthalmol 2003;48:631–646. [DOI] [PubMed] [Google Scholar]
- 23. Kim JC, Tseng SC. Transplantation of preserved human amniotic membrane for surface reconstruction in severely damaged rabbit corneas. Cornea 1995;14:473–484. [PubMed] [Google Scholar]