

Abstract
γδT cells constitute a small proportion of lymphocytes in peripheral blood. Unlike αβT cells, the anti‐tumor activities are exerted through several different pathways in a MHC‐unrestricted manner. Thus, immunotherapy using γδT cells is considered to be effective for various types of cancer. Occasionally, however, ex vivo expanded cells are not as effective as expected due to cell exhaustion. To overcome the issue of T‐cell exhaustion, researchers have generated induced pluripotent stem cells (iPSCs) that harbor the same T‐cell receptor (TCR) genes as their original T‐cells, which provide nearly limitless sources for antigen‐specific cytotoxic T lymphocytes (CTLs). However, these technologies have focused on αβT cells and require a population of antigen‐specific CTLs, which are purified by cell sorting with HLA‐peptide multimer, as the origin of iPS cells. In the present study, we aimed to develop an efficient and convenient system for generating iPSCs that harbor rearrangements of the TCRG and TCRD gene regions (γδT‐iPSCs) without cell‐sorting. We stimulated human whole peripheral blood mononuclear cell (PBMC) culture using Interleukin‐2 and Zoledronate to activate γδT cells. Gene transfer into those cells with the Sendai virus vector resulted in γδT cell‐dominant expression of exogenous genes. The introduction of reprogramming factors into the stimulated PBMC culture allowed us to establish iPSC lines. Around 70% of the established lines carried rearrangements at the TCRG and TCRD gene locus. The γδT‐iPSCs could differentiate into hematopoietic progenitors. Our technology will pave the way for new avenues toward novel immunotherapy that can be applied for various types of cancer. stem cells translational medicine 2018;7:34–44
Keywords: Induced pluripotent stem cells, T‐lymphocytes, gamma‐delta TCR, immunotherapy
Significance Statement.
γδT cells constitute a small proportion of lymphocytes in peripheral blood, and immunotherapy using γδT cells is considered to be effective for various types of cancer. Occasionally, however, ex vivo expanded cells are not as effective as expected due to cell exhaustion. Induced pluripotent stem cells (iPSCs) have the potential to overcome this issue because they are capable of unlimited proliferation and multidirectional differentiation. In the present study, the authors successfully generated iPSCs from human γδT cells (γδT‐iPSCs) with a simple and clinically applicable method. The γδT‐iPSCs may provide an unprecedented source for cancer therapy.
Introduction
γδT cells are a small subset of T lymphocytes that express T‐cell receptors (TCRs) that are distinct from those expressed on the surface of αβT cells, a major subset of T lymphocytes 1, 2, 3. Among them, Vγ9Vδ2 T cells represent the major γδT cell subtype in human peripheral blood 4. There is substantial evidence to suggest that they represent an important player in the immune system's arsenal of effector cells and, that they have anti‐tumor activity 5. Unlike αβT cells, Vγ9Vδ2 T cells exhibit MHC‐unrestricted lytic activity against a wide variety of tumor cells 6, 7, because they recognize tumor ligands outside of MHC restriction. Thus, Vγ9Vδ2 T cells can exert potent cytotoxic effects against cancer with the reduced expression—or even in the absence—of human leukocyte antigen (HLA) 8. Additionally, the activation of γδ‐TCRs promotes γδT cell‐cytotoxicity through several different pathways, depending on granule exocytosis, the death receptor pathway and the secretion of cytokines 9, 10, 11, 12. For these reasons, immune therapy with Vγ9Vδ2 T cells is considered to be effective in the clinical setting.
There have been several clinical trials on the adoptive transfer of ex vivo expanded Vγ9Vδ2T cells into cancer patients 13, 14, 15, 16, 17, 18. The results of these trials revealed that adoptively transferred γδT cells are well tolerated by patients and that they can be safely used as immunotherapy. However, ex vivo expanded cells are occasionally not as effective as expected because long‐term stimulation drives tumor‐specific cytotoxic T lymphocytes (CTLs) toward a state of terminal differentiation and exhaustion 19, 20.
This limitation can be overcome by the use of induced pluripotent stem cells (iPSCs) 21, 22, 23. iPSCs possess the property of unlimited self‐renewal and multi‐lineage differentiation potential 24. Thus, T cells differentiated from iPSCs could become a source of near limitless and rejuvenated immune cells. In fact, recent studies have revealed that mature human T cells can be reprogrammed into iPSCs that can redifferentiate in vitro into functional T lymphocytes that express the same antigen‐specific TCR as their original cells 21, 22, 23. However, these previous technologies focused on αβT cells and required a population of CTLs that express a certain antigen‐specific TCR as the origin of the iPSCs and it was necessary to purify the cells by HLA‐peptide multimer selection.
In the present study, we established a simple and efficient method of generating iPSCs from human Vγ9Vδ2T cells without the use of HLA‐peptide multimer or antibodies and confirmed that the iPSC line could redifferentiate into the hematopoietic lineage. This technology may provide a therapeutic cell source for novel adoptive cell therapies.
Materials and Methods
γδT Cell‐Stimulating Culture of Whole PBMCs
Human γδT cells in whole peripheral blood mononuclear cell (PBMC) culture were activated according to a previously reported protocol, with slight modifications 9. Firstly, PBMCs were separated from whole blood samples using BD vacutainer blood collection tubes with sodium heparin (BD, Tokyo, Japan). Informed consent was obtained for the collection of whole blood from healthy volunteers. Subsequently, the PBMC cultures were stimulated with 5 µM zoledronic acid (Novartis, Basel, Switzerland) in RPMI 1640 medium (Life Technologies, Waltham, MA) supplemented with 10% fetal bovine serum (Life Technologies), 1.0 × 10−5 M 2Me (Nacalai Tesque, Kyoto, Japan), 100 U/ml penicillin and 100 µg/ml streptomycin (Life Technologies) at the beginning of culturing. One milliliter of the cell suspension was seeded in the wells of 24‐well multi‐culture dishes (Nunc, Waltham, MA) at a density of 1 × 106/ml and cultivated at 37°C in a humidified atmosphere containing 5% CO2.
From day 1, 100 IU/ml human recombinant IL‐2 (Shionogi Pharmaceuticals, Osaka, Japan) was added every day. Thereafter, the cultured cells were passed at a ratio of 1:2 into new wells every 2–3 days according to the degree of cellular proliferation. The cultured cells were analyzed at the indicated time points by flow cytometry.
Flow Cytometry
The cells were stained with monoclonal antibodies (mAb) and analyzed using a FACS Verse (Becton Dickinson, Franklin Lakes, NJ). The following mAbs were used for the phenotypic analysis: eFluor450‐labeled anti‐CD3 (eBiosciences, San Diego, CA, 48‐0037‐41), PE‐Cy7‐labeled anti‐CD3 (BD PharMingen, San Diego, CA, 563423), FITC‐labeled anti‐TCR Vγ9 (Beckman Coulter, Brea, CA, IM1463), Human APJ APC‐conjugated antibody (R&D systems, Minneapolis, MN, FAB856A), APC anti‐human CD34 (Biolegend, San Diego, CA, 343608) and Anti‐Human CD43 PE (eBioscience, 12‐0439).
On days 4 and 12 in hematopoietic differentiation, the cells were treated with 500 µl of Accutase at 37°C for 15 minutes and washed with 1% BSA in PBS after centrifuging, followed by reactions with antibodies. On day 12, the dissociated cells were resuspended in StemPro34 medium and plated onto plastic at 37°C for 1 hour, and the floating cells were collected to remove any adherent cells. Dead cells were excluded by 7‐AAD staining.
Generation of iPSCs from Human γδT Cell Culture
Stimulated PBMCs (1 × 104 cells) were suspended in 100 μl of γδT cell medium with CytoTune‐iPS 2.0 (DNAVEC, Tokyo, Japan, DV‐0304‐3) containing 2 type of Sendai viruses each of which individually encoded KLF4 and c‐MYC, and 1 type of Sendai virus that polycistronically encoded OCT3/4, KLF4, and SOX2 at an MOI of 10–30 and cultivated in a 96‐well plate. Twenty‐four hours later, the cells were plated on a 6‐well plate precoated with the recombinant laminin‐511 E8 fragments (iMatrix‐511, Nippi, Tokyo, Japan). On days 3, 5, and 7 after transduction, an additional 1 ml of hiPSC medium (Stem Fit, Ajinomoto, Tokyo, Japan) was added to the culture. From day 9, the hiPSC medium was changed every two days. From day 21, we started to pick up and expand colonies.
Human iPSCs Culture
We cultured the established iPSCs according to a previously described method 25. Briefly, the culture plates were precoated with recombinant laminin‐511 E8 fragments (iMatrix‐511, Nippi) (0.5mg/cm2), and the iPSCs were cultured in Stem Fit medium (Ajinomoto) at 37°C with 5% CO2. The medium was changed every other day and was passaged every 7–10 days using 0.5× TrypLE Select (1× TrypLE Select diluted 1:1 with 0.5 mM EDTA/PBS(‐), Life technologies) and Rock inhibitor (Y‐27632, WAKO, Osaka, Japan).
TCRG and TCRD Gene Rearrangement in iPSCs Derived from γδT Cell
To analyze the rearrangement of the TCRG and TCRD gene regions, cultured cells were lysed with a cell lysis solution (QIAGEN, Venlo, Netherlands) and treated with proteinase K. DNA was then extracted using a phenol–chloroform extraction‐based method followed by ethanol precipitation. A polymerase chain reaction (PCR) was performed according to the previous reports 26, 27 using the primers shown in Supporting Information Table S1. The amplified products were identified by 2% agarose gel electrophoresis and the dominant band within the expected size range was purified using a QIAquick gel‐extraction kit (QIAGEN). Direct sequencing was performed using a BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Waltham, MA) and an ABI 3130 genetic analyzer (Applied Biosystems) according to the manufacturer's instructions. Thereafter, these data were confirmed by capillary electrophoresis. Capillary electrophoresis was performed using the TCRB and TCRG T cell Clonality Assay Kit (InVivoScribe, San Diego, CA) according to the manufacturer's instructions. The PCR samples were electrophoresed on an ABI 3130 genetic analyzer and the results were analyzed using the GeneMapper software program (Applied Biosystems).
RT‐PCR and qRT‐PCR
To test the expression of pluripotent genes in γδT cell‐derived iPSC clones, total RNA was extracted using TRIzol reagent (Life technologies) and treated with a Turbo DNA‐free kit (Life technologies) to remove genomic DNA contamination. Five hundred nanograms of total RNA was reverse transcribed to single‐stranded cDNA using a PrimeScript II 1st Strand Synthesis Kit (Takara, Shiga, Japan) with oligo‐dT primers according to the manufacturer's instructions. A quantitative PCR was performed using SYBR Premix Ex Taq (TaKaRa) on a 7500 Real‐Time PCR System (Applied Biosystems). Each sample was analyzed in triplicate and the target genes were normalized to the GAPDH expression levels. The primer sequences of the RT‐PCR and qRT‐PCR are listed in Supporting Information Table S2.
Immunocytochemistry
Cells were fixed with PBS containing 4% paraformaldehyde for 10 minutes at room temperature. After washing with PBS, the cells were treated with PBS containing 5% donkey serum, 1% bovine serum albumin (BSA, WAKO) and 0.1% Triton X‐100 for 45 minutes at room temperature. The cells were incubated with primary antibodies 4°C overnight and then stained with secondary antibodies. The primary antibodies included Sendai virus (MBL, Nagoya, Japan, PD029), OCT3/4 (BD, 611202), NANOG (R&D Systems, AF1997), β‐III‐tubulin (Chemicon, Billerica, MA, MAB1637), α‐SMA (DAKO, Santa Clara, CA, M0851) and SOX17 (R&D systems, AF1924). All the secondary antibodies (Alexa Fluor 594‐conjugated anti‐rabbit, ‐mouse, ‐goat IgG and Alexa Fluor 488‐conjugated anti‐mouse, ‐goat IgG were obtained from Life technologies. Hoechst 33342 (WAKO) was used for nuclear staining.
Microarray Experiments and the PluriTest
Biotinylated cRNA was prepared from 100 ng of total RNA using a GeneChip 3'IVT PLUS Reagent kit (ThermoFisher Scientific, Waltham, MA) and was hybridized to the GeneChip PrimeView Human Gene Expression Array (ThermoFisher Scientific) for 16 hours at 45°C. Array slides were washed and stained in an Affymetrix Fluidics station 450, and scanned using a GeneChip scanner 3000 7G system. The data files were then uploaded to www.pluritest.org, scored for pluripotency, and deposited into the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database with accession no. GSE104605.
Karyotype Analyses
The G‐band karyotype analysis for γδT‐iPSC was performed at Chromocenter, Inc. (Yonago, Japan).
In Vitro Spontaneous Differentiation via Embryoid Body Formation
For embryoid body (EB) formation, undifferentiated iPSCs were dissociated into single cells, resuspended in Primate ES medium (Reprocell) containing 20 µM Rho‐associated kinase (Rock) inhibitor Y‐27632 (WAKO) and seeded on low‐cell‐adhesion 96‐well spindle‐bottom plates (PrimeSurface, Sumitomo Bakelite, Tokyo, Japan, MS‐9096M) at a density of 1 × 104 cells per well. After 7 days of culture, the EBs were transferred to gelatin‐coated 24‐well plates and were cultured in the same medium for another 8 days. The differentiated cells were immune‐stained with the indicated antibodies.
Differentiation of Human γδT‐iPSCs into Hematopoietic Progenitor Cells In Vitro
Prior to differentiation, single dissociated undifferentiated human γδT‐iPSCs were seeded onto a Laminin511‐E8‐coated 6‐well plate at a density of 2 × 103 cells and cultured in Stem Fit medium. When individual colonies grew to around 500 µm in diameter, the medium was replaced by Stem Fit medium supplemented with 4 µM of CHIR99021 (Tocris Bioscience, Bristol, UK), 80 ng/ml of rhBMP4 (R&D Systems) and 80 ng/ml of rhVEGF (R&D Systems). This day was defined as day 0 of our protocol. On day 2, the medium was refreshed. On day 4, the medium was replaced by Essential 6 medium (ThermoFisher Scientific) supplemented with 4 µM of SB43152 (WAKO), 80 ng/ml of rhVEGF, 50 ng/ml of rh‐bFGF (Reprocell) and 50 ng/ml of rhSCF (R&D Systems). On day 6, the medium was replaced by StemPro‐34SFM (Thermo Fisher Scientific) supplemented with 20 ng/ml of rhVEGF, 50 ng/ml of rhSCF, 50 ng/ml of rhIL‐3 (R&D Systems), 50 ng/ml of hIL‐6 (Roche, Basel, Switzerland), 50 ng/ml of rhFlt3 ligand (R&D Systems) and 10 IU/ml of recombinant human Erythropoietin α (EPO) (Kyowa Hakko Kirin, Tokyo, Japan). On day 8, the medium was replaced by StemPro‐34SFM supplemented with 50 ng/ml of rhSCF, 50 ng/ml of hIL‐6 (Roche) and 10 IU/ml of EPO. On day 10, the medium was refreshed. Cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Results
γδT Cell‐Dominant Gene Transduction in Whole PBMC Culture
γδT cells, which represent a small subset (1–5%) of the T cell population, have been reported to be activated with zoledronate (Zol) and interleukin (IL)‐2 9. To confirm this, we stimulated whole PBMC culture with Zol/IL‐2. We observed that cell aggregates formed and gradually grew, suggesting that some types of cells in culture were activated (Fig. 1A). We then examined whether γδT cells were dominantly activated by flow‐cytometry. CD3 and TCR Vγ9 co‐staining revealed that after 13 days of culture, a large portion—but not all—of the cultured cells were γδT cells with TCR Vγ9 expression (Fig. 1B); the percentages in three independent experiments were 81.6%, 79.8%, and 96.8%. These data indicated that we successfully activated γδT cells in whole PBMC culture.
Figure 1.
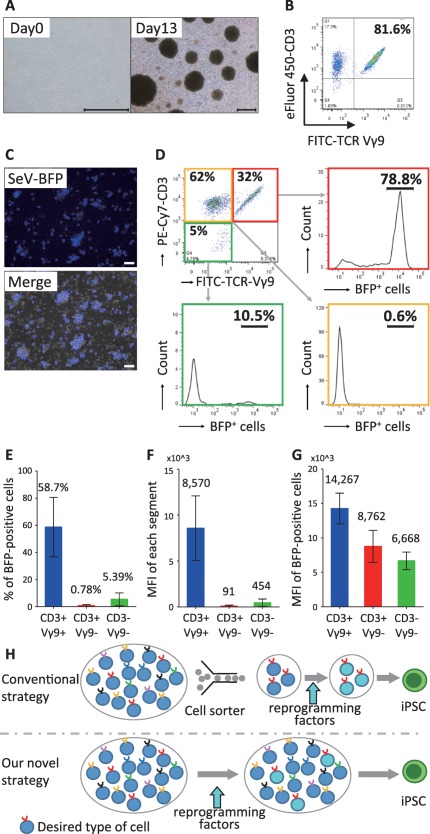
γδT cell‐dominant gene transfer in γδT cell‐stimulating culture of whole peripheral blood mononuclear cells (PBMCs). (A): A representative image of PBMCs just after isolation (Day 0) and after 13 days of cultures with stimulation by 5 µM zoledronate and 100 IU/ml IL‐2. The scale bar indicates 500 μm. (B): The flow cytometric analysis of CD3 and the TCR‐Vγ9 expression in γδT cell‐stimulating culture on day 13. (C): Two days after BFP gene transduction in the γδT cell‐stimulating culture. Upper panel: a fluorescence image, Lower panel: a merged image (fluorescence and phase contrast). The scale bars indicate 50 μm. (D): The flow cytometric analysis of the gene transduction efficiency of the SeV vector in CD3+/TCR Vγ9+ cells (γδT cells), CD3+/TCR Vγ9− cells (αβT cells) and CD3−/TCR Vγ9− cells (B cells and monocytes). (E–G): The proportions of BFP‐positive cells (E), the mean fluorescence intensity (MFI) of BFP (F) and the MFI of BFP in BFP‐positive cells (G) in each cell subsets. Data are the averages in three independent experiments. Each error bars indicate standard deviation. (H): The strategies for generating iPSCs from a specific desired type of cells. In the conventional strategy (upper panel), desired cells are purified with cell sorter and transduced with reprogramming factors to become iPSCs. In our novel strategy (lower panel), desired cells in a cell mixture are dominantly transduced with reprogramming factors. Abbreviations: TCR, T‐cell receptor; MFI, mean fluorescence intensity; BFP, blue fluorescent protein; iPSCs, induced pluripotent stem cells.
We next examined whether Sendai virus‐mediated gene transfection into whole PBMC culture stimulated with Zol/IL‐2 resulted in the transgene expression in a γδT cell‐dominant manner, because the Sendai virus was previously reported to specifically and efficiently transduce a foreign gene in activated murine and human T cells, but not in naive T cells 28. We infected Zol/IL‐2‐stimulated whole PBMC culture with SeV vector expressing the blue fluorescent protein (BFP) gene on day 4 and analyzed the cells on day 6. We observed the expression of BFP under fluorescence microscopy (Fig. 1C). In a representative experiment, the proportions of CD3+/TCR Vγ9+ cells (γδT cells), CD3+/TCR Vγ9− cells (αβT cells) and CD3‐/TCR Vγ9− cells (B cells and monocytes) were 32%, 63%, and 5%, respectively, and the proportions of BFP‐positive cells in the CD3+/TCR Vγ9+ cells, CD3+/TCR Vγ9 cells and CD3‐/TCR Vγ9− cells were 78.8%, 0.6% and 10.5%, respectively (Fig. 1D). The data of three independent experiments are shown in the Fig. 1E‐G. The proportion of BFP‐positive cells (Fig. 1E) and the mean fluorescence intensity (MFI) of BFP (Fig. 1F) in the CD3+/TCR Vγ9+ cells were much higher in comparison to the CD3+/TCR Vγ9− cells and CD3−/TCR Vγ9− cells. Furthermore, even among BFP‐positive cells, the MFI of BFP in the CD3+/TCR Vγ9+ cells was higher than that in the other subsets (Fig. 1G).
Taken together, we successfully demonstrated that a combination of Zol/IL‐2‐stimulation and Sendai virus vectors allowed for γδT cell‐dominant gene transduction in whole PBMC culture without cell sorting. Based on this finding, we conceived a novel strategy for generating iPSCs (Fig. 1H), in which the dominancy of gene transfer into desired cells, but not purity of the desired cells, allows for the generation of iPSCs with certain antigen‐specific TCR genes.
Generation of Human γδT Cell‐Derived iPSCs from Whole PBMC Culture
To generate iPSC lines from γδT cells, we transfected SeV vector encoding the OCT3/4, SOX2, KLF4, and c‐MYC reprogramming factors into whole PBMC culture stimulated with Zol/IL‐2 and subsequently cultivated the cells under human ESC conditions (Fig. 2A). After 3–4 weeks, we observed the emergence of human ESC‐like and non‐ESC‐like colonies (Fig. 2B, Supporting Information Fig. 1A). The numbers of hES‐like colonies/total colonies at 21 days post SeV‐infection were 0/0, 51/59, 40/49, 36/48 in dishes at MOIs of 0, 10, 20, 30, respectively. We picked up several human ESC‐like colonies and expanded the cells, thereby establishing 19 iPSC lines in an experiment.
Figure 2.
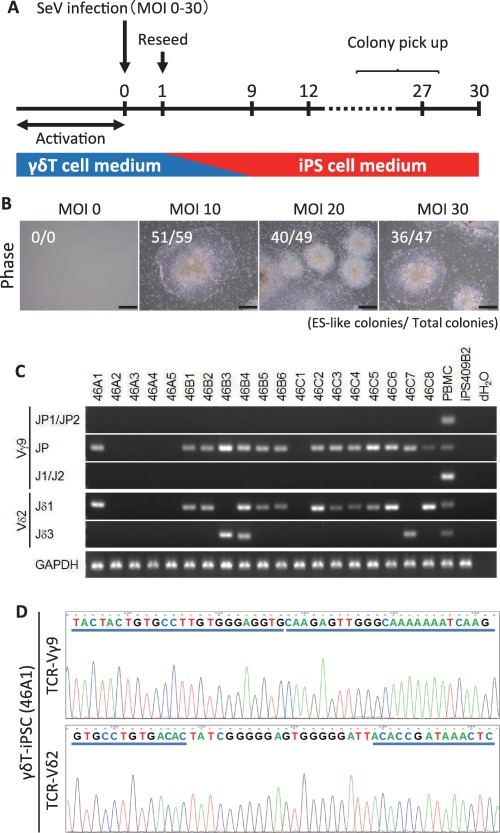
The generation of human γδT cell‐derived iPSCs from whole PBMC culture. (A): A schematic diagram of the reprogramming protocols used to generate iPSCs from γδT cells. Whole PBMC culture was stimulated with zoledronate and IL‐2 to activate γδT cells, and transduced with Sendai virus encoding OCT3/4, SOX2, KLF4, and c‐MYC. After 24 hours of cultivation in T cell medium, the transduced cells were reseeded on the Laminin511‐E8‐coated plate, and the medium was gradually changed into human iPSC medium. (B): Typical ESC‐like colonies on day 21 after gene transduction. The scale bars indicate 500 μm. (C): The genome PCR for detecting TCRG and TCRD gene rearrangement (V‐J assemblies). (D): Direct sequencing by amplifying the products of the Vγ9‐Jγ and Vδ2‐Jδ regions. The representative data show the nucleotides and deduced amino acid sequences of the junctional regions aligned relative to the germline. Abbreviations: iPSCs, induced pluripotent stem cells; PBMC, peripheral blood mononuclear cells; TCRD, T‐cell receptor delta; TCRG, T‐cell receptor gamma.
To examine the rearrangement at the TCRG and TCRD gene locus in these cell lines, we performed a genomic PCR. In 13 of the 19 iPSC lines, rearrangements of the TCRG and TCRD genes were identified as single bands representing Vγ9‐to‐JP and Vδ2‐to‐Jδ1 or ‐Jδ3 recombination, which indicated that these cell lines carried Vγ9Vδ2‐TCR genes (Fig. 2C). In one line (46B4) we detected both bands, which represented Vδ2‐to‐Jδ1 and Vδ2‐to‐Jδ3 recombination. The analysis of single‐cell sub‐clones derived from the 46B4 showed that the line was a mixture of clones harboring Vδ2‐to‐Jδ1 and Vδ2‐to‐Jδ3 recombination (data not shown). In addition, we also established iPSC lines from two other healthy donors using the same procedure, and reproducibly obtained iPSC lines carrying Vγ9Vδ2‐TCR genes (Supporting Information Fig. 1B, 1C); these were named the γδT‐iPSC lines.
Moreover, we determined the sequence of complementarity determining regions 3 (CDR3) of the TCRG and TCRD gene regions in the 14 established γδT‐iPSC lines, and identified a set of productive gene rearrangement in those cell lines (Fig. 2D; Table 1).
Table 1.
The sequence analysis of complementarity determining regions 3 (CDR3) of the TCRG and TCRD gene regions
| Clone ID | Rearrange | Nucleotide sequence | ||
|---|---|---|---|---|
| V segment | Junctional region | J segment | ||
| Germline | Vγ9‐JP | GCC TTG TGG GAG GTG | • • • | T GGG CAA GAG TTG |
| Vδ2‐Jδ1 | TGT GCC TGT GAC ACC | • • • | AC ACC GAT AAA CTC | |
| Vδ2‐Jδ3 | TGT GCC TGT GAC ACC | • • • | C TCC TGG GAC ACC | |
| 46A1 | Vγ9‐JP | GCC TTG TGG GAG GTG | — | CAA GAG TTG |
| Vδ2‐Jδ1 | TGT GCC TGT GAC AC | T ATC GGG GGA GTG GGG GAT T | AC ACC GAT AAA CTC | |
| 46B1 | Vγ9‐JP | GCC TTG TGG GAG G | GC TCT | CAA GAG TTG |
| Vδ2‐Jδ1 | TGT GCC TGT GAC | GCC CTC ATG GGG GGG ACA CC | C ACC GAT AAA CTC | |
| 46B2 | Vγ9‐JP | GCC TTG TGG GAG GTG | TGG | GAG TTG |
| Vδ2‐Jδ1 | TGT GCC TGT GAC AC | T TTG GGG GAT AGG AAA C | CC GAT AAA CTC | |
| 46B3 | Vγ9‐JP | GCC TTG TGG GAG GT | C TTA | CAA GAG TTG |
| Vδ2‐Jδ3 | TGT GCC TGT GAC | CGG CTA CTG GGG GAT ACT A | CC TGG GAC ACC | |
| 46B4 | Vγ9‐JP | GCC TTG TGG GAG G | GC TCT | CAA GAG TTG |
| Vδ2‐Jδ1 | TGT GCC TGT GAC | GCC CTC ATG GGG GGG ACA CC | C ACC GAT AAA CTC | |
| Vδ2‐Jδ3 | TGT GCC TGT GAC | GCT ACT GGG GGA CG | C TCC TGG GAC ACC | |
| 46B5 | Vγ9‐JP | GCC TTG TGG GAG GT | C | CAA GAG TTG |
| Vδ2‐Jδ1 | TGT GCC TGT GAC ACC | CTG GGG GTC G | AC ACC GAT AAA CTC | |
| 46B6 | Vγ9‐JP | GCC TTG TGG GAG | AAA | CAA GAG TTG |
| Vδ2‐Jδ1 | TGT GCC TGT GAC AC | T CTT GGG GGA T | CC GAT AAA CTC | |
| 46C2 | Vg9‐JP | GCC TTG TGG GAG G | CA CC | G CAA GAG TTG |
| Vd2‐Jd1 | TGT GCC TGT GAC AC | T CTT GGG GGA T | CC GAT AAA CTC | |
| 46C3 | Vγ9‐JP | GCC TTG TGG GAG G | CA CC | G CAA GAG TTG |
| Vδ2‐Jδ1 | TGT GCC TGT GAC AC | T CTT GGG GGA T | CC GAT AAA CTC | |
| 46C4 | Vγ9‐JP | GCC TTG TGG GAG GT | A ACA | CAA GAG TTG |
| Vδ2‐Jδ1 | TGT GCC TGT GAC ACC | GTA GGA CTA CTG GGG GAT ACG GG | T AAA CTC | |
| 46C5 | Vγ9‐JP | GCC TTG TGG GAG | CCC GAG GGT CAT TCC | TTG |
| Vδ2‐Jδ1 | TGT GCC TGT GAC ACC | CTG GGG GTC G | AC ACC GAT AAA CTC | |
| 46C6 | Vγ9‐JP | GCC TTG TGG GAG | CCC GAG GGT CAT TCC | TTG |
| Vδ2‐Jδ1 | TGT GCC TGT GAC ACC | GTA GGA CTA CTG GGG GAT ACG GG | T AAA CTC | |
| 46C7 | Vγ9‐JP | GCC TTG TGG GAG GT | T | GAG TTG |
| Vδ2‐Jδ3 | TGT GCC TGT GAC ACC | GTA AAA TTG GGC AG | C TCC TGG GAC ACC | |
| 46C8 | Vγ9‐JP | GCC TTG TGG GAG G | CA CC | G CAA GAG TTG |
| Vδ2‐Jδ1 | TGT GCC TGT GAC ACC | CTG GGG GTC G | AC ACC GAT AAA CTC | |
Characterization of γδT Cell‐Derived hiPSCs
The three established γδT‐iPSC lines (46A1, 46B1, 46C2) showed a human ESC‐like morphology (Supporting Information Fig. 2A). We first examined whether SeV remained in the lines by RT‐PCR and immune‐staining with anti‐SeV antibodies. At passage 5, almost all of the colonies in the three lines were positive for SeV vector (Fig. 3A, Supporting Information Fig. 2B). We then cultured these lines at 39°C for 5 days, since the SeV vector carries temperature‐sensitive mutations that enable the removal of the residual vector by shifting to a non‐permissive temperature (38°C–39°C). Consequently, there was an obvious decrease in the proportion of SeV‐positive colonies; however, it did not disappear (data not shown). We therefore performed sub‐cloning and expanded them to obtain SeV‐free iPSC lines. An RT‐PCR and immunostaining revealed that we could obtain SeV‐free subclones (46A1‐s3, 46B1‐s1, and 46C2‐s4) from the SeV‐positive parental lines (Fig. 3A, Supporting Information Fig. 2B). These subclones were used for the following assays.
Figure 3.
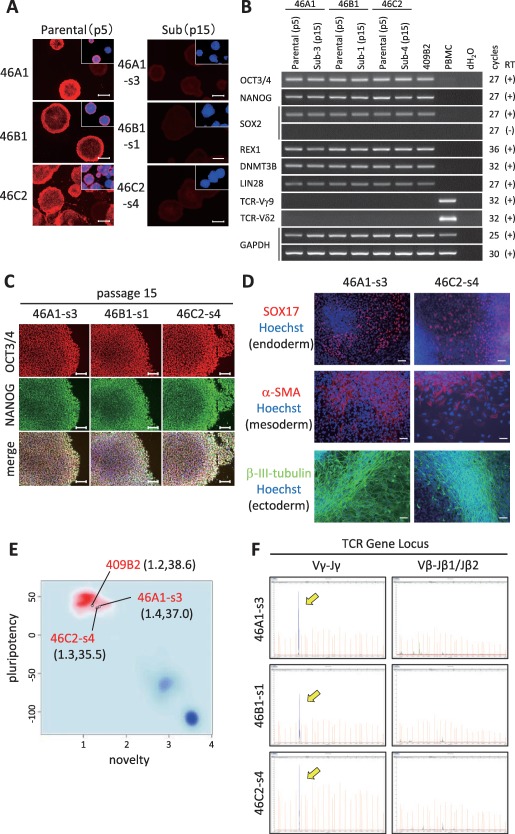
The characterization of γδT‐iPSCs. (A): An immunofluorescence analysis for the residual Sendai virus vectors in parental γδT‐iPSC lines (passage number 5, upper panels) and their subclones (passage number 15, lower panels). The boxes in each panel represent merged images with anti‐Sev in red and Hoechst33432 in blue. The scale bar indicates 500 μm. (B): The RT‐PCR for ES cell‐markers and TCR genes. The primers used for Oct3/4 and Sox2 detected the transcripts from the endogenous genes, but not from the Sendai viral transgenes. (C): An immunofluorescence analysis showing the expression of ES cell markers. The scale bars indicate 100 μm. (D): An in vitro differentiation via embryoid body formation. The images show immunofluorescence staining for SOX17 (endodermal marker), α‐smooth muscle actin (mesodermal marker), and βIII‐tubulin (ectodermal marker) in each iPSCs‐derived differentiated cells. The nuclei were stained with Hoechst 33342 (blue). The scale bar indicates 50 μm. (E): The PluriTest results of two γδT‐iPSC lines (46A1‐s3 and 46C2‐s4) and a conventional iPSC line (409B2). The background shows the density distribution of the Stem Cell Matrix samples 30, with the red and blue clouds representing the empirical density distributions for validated hPSCs (223 hESCs and 41 hiPSCs) and differentiated cells, respectively. The Novelty Score and Pluripotency Score of each of the clones are indicated in parentheses. (F): DNA fragment analysis by the capillary electrophoresis of the PCR products for the TCRG and TCRB gene regions. The yellow arrows indicated the rearrangement of the TCRG gene region in γδT‐iPSC lines. Abbreviations: TCR, T‐cell receptor; TCRG, T‐cell receptor gamma; TCRB, T‐cell receptor beta; PCR, polymerase chain reaction.
We next evaluated the stem cell marker expression of the γδT cell‐derived lines by an RT‐PCR and immunostaining. The RT‐PCR showed that these lines, like a conventional iPSC line (409B2) 29, expressed the mRNAs of typical ES cell markers such as OCT3/4, NANOG, SOX2, REX1, DNMT3B, and LIN28 (Fig. 3B, Supporting Information Fig. 2C). Immunostaining revealed that these lines expressed OCT3/4 and NANOG at the protein level (Fig. 3C). The 46A‐1‐s3 line was confirmed to have a normal 46XX karyotype by the G‐band technique (Supporting Information Fig. 2D).
To examine the differentiation potential of γδT‐iPSCs in vitro, we performed a differentiation assay via EB formation. The γδT‐iPSCs (46A1‐s3 and 46C2‐s4) were able to form EBs during suspension culture (data not shown). After subsequent attached culture for 8 days, we found cells that were positive for β III‐tubulin (ectoderm), α‐smooth muscle actin (α‐SMA, mesoderm) and SOX17(endoderm) (Fig. 3D), indicating that the γδT‐iPSCs could differentiate into three germ layers in vitro. Additionally, we performed a PluriTest, which is a robust open‐access bioinformatics assay to investigate pluripotency based on gene expression profiles 30. The analysis of these two γδT‐iPSCs and a conventional iPSC line (409B2) 29 by the PluriTest algorithm revealed that all the three lines were pluripotent and were similar to validated normal hPSCs; this was indicated by a Pluritency Score of >20 (y‐axis) and a Novelty Score of <1.67 (x‐axis) (Fig. 3E).
Furthermore, to reconfirm that the iPSC lines were derived from γδT cells and not αβT cells, we performed a DNA fragment analysis by the capillary electrophoresis of the PCR products for the TCRG and TCRB gene regions. The lines showed a specific peak for Vγ‐to‐Jγ recombination, but not for Vβ‐to‐Jβ1/Jβ2 recombination (Fig. 3F). These data demonstrated that the γδT‐iPSCs were absolutely derived from γδT cells and retained the same TCR genes as the original cells.
Redifferentiation of γδT‐iPSCs into the Hematopoietic Lineage
To test the hematopoietic differentiation potential of the γδT‐iPSCs, we examined whether or not γδT‐iPSCs could generate CD34+/CD43+ hematopoietic progenitor cells. We referred to and modified some previously reported methods 31, 32, 33 and established a hematopoietic differentiation protocol starting from iPS cell culture in Stem Fit medium on lamin‐511 E8 fragment (Fig. 4A). Two γδT‐iPSC lines (46A1‐s3 and 46C2‐s4) underwent differentiation with the protocol. On day 4, around two‐thirds of cells expressed APJ, an early hematovascular precursor marker 34, 35, 36, also known as APLNR (Fig. 4B). Flow cytometry on day 12 revealed that the γδT‐iPSC lines were able to generate CD34+/CD43+ hematopoietic progenitor cells efficiently; CD34/CD43 double‐positive cells accounted for more than 70% and 40% of the γδT‐iPSC‐46A1‐s3‐ and γδT‐iPSC‐46C2‐s4‐derived cells, respectively (Fig. 4C).
Figure 4.
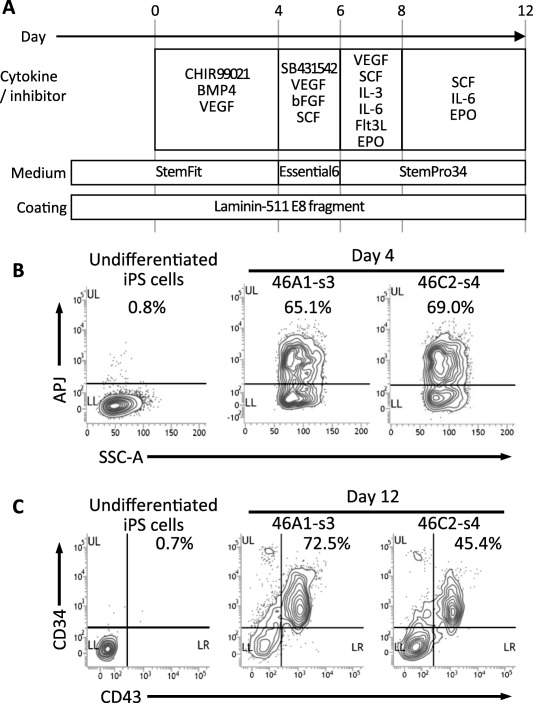
The redifferentiation of γδT‐iPSCs into hematopoietic lineage. (A): A schematic diagram showing the redifferentation protocol for deriving hematopoietic progenitor cells from γδT‐iPSCs. (B): The flow cytometric analysis for a hematovascular precursor marker (APJ) in γδT‐iPSC‐derived cells. The percentages of APJ‐positive cells are shown. (C): The flow cytometric analysis for hematopoietic lineage markers (CD34 and CD43) in γδT‐iPSC‐derived cells. The percentages of CD34/CD43 double‐positive cells are shown. Abbreviations: iPSCs, induced pluripotent stem cells.
Discussion
In the present study, we generated γδT cell‐derived iPSCs. Several previous studies have reported the generation of iPSCs from αβT cells (αβT‐iPSCs), raising the expectation that it will be possible to develop novel iPSC‐based cancer immune‐therapies 21, 22. However, a αβT‐iPSC line can only be used for a limited number of cancer patients due to the MHC‐restriction of αβT cell clones. In contrast, γδT cells exhibit MHC‐unrestricted lytic activity against a wide variety of tumor cells 6, 7. Thus, one γδT cell‐derived iPSC line can be used for a large number of patients. From the point of view of commercialization, which is one of the essential factors for the practical application of iPSC‐based therapy, one γδT‐iPSC‐based product could have a much larger market than one αβT‐iPSC‐based product. One of the obstacles to the development of iPSC‐based therapies is the high development and production costs 37. Thus, γδT‐iPSC‐based immunotherapy might be one of the most promising projects in terms of its clinical translation.
We generated iPSCs from the desired type of cells, or γδT cells without cell collection using a cell sorter. In contrast, conventional technologies for generating iPSCs from a particular cell types requires cell sorting with HLA‐peptide multimer or antibodies 21, 22. With reference to clinical applications, we should be concerned about the potential unknown effects of unintended residual materials that are used in the production process—including antibodies or HLA‐peptide multimer, which are used for the selection of the desired cells. Our novel technology could avoid this concern because both IL‐2 and Zoledronate have already been used in clinical practice.
Our novel strategy, in which the dominancy of gene transfer into the desired cells, but not the purity of the desired cells, allowed us to obtain iPSCs that were derived from particular cell type. It might be possible to apply this technology to generate iPSCs from various types of cells. Some previous reports have suggested that the differentiation propensity of iPSCs was influenced by their origin 38, 39, 40. This phenomenon is understood to be due, at least in part, to the epigenetic memory of the original somatic cells in iPS cells 41; thus, researchers might demand simple and clinically applicable technologies for generating iPSCs that use various cells (including T cells) according to the target cell type of the iPSC derivatives. In the current study, we used a combination of γδT cell‐dominant activation using with Zol/IL‐2 and activated T cell‐dominant gene transfer using a SeV vector to generate the γδT‐iPSCs. In the future, other methods should be developed according to the types of cells that are desired.
In the present study, we determined the CDR3 sequences of the rearranged TCRG and TCRD gene regions and confirmed that iPSC lines were successfully generated from several different γδT cells. The recognition of antigens by γδ‐TCRs is known to depend on the sequence of its peptide, especially that of CDR3 42. In addition, a recent report revealed that γδ‐TCRs recognized tumor antigens via the CDR3δ region 43 and CDR3δ‐grafted Vγ9Vδ2 T cells (peripheral blood lymphocytes expressing the γ9δ2‐TCR with a tumor antigen‐specific CDR3δ region) exhibited cytotoxicity against tumor cells 44. However, it has been difficult to determine the precise sequence of the CDR3δ region that exhibits the most effective antitumor activity. By assessing the antitumor activity of various γδT cell clones derived from a range of γδT‐iPSC lines with various CDR3 regions, we might be able to determine the γδT cells that have the most effective antitumor activity. Thus, in comparison to conventional adoptive immunotherapy, γδT‐iPSCs could provide both a source of near limitless and rejuvenated cells and more effective source of cells for cancer immunotherapy.
Conclusion
In summary, the results of the present study provide an efficient and convenient method for generating γδT‐iPSCs with the potential to differentiate into hematopoietic progenitor cells. Our technology will pave the way for new avenues for developing novel immune therapies that are applicable for various types of cancer.
Author Contributions
D.W.: collection and assembly of data, data analysis and interpretation, manuscript writing; M.K.‐A.: collection and assembly of data, conception and design, data analysis and interpretation, manuscript writing; M.T.‐I.: collection and assembly of data; Y.Y.: collection and assembly of data; T. Azuma: data analysis and interpretation; T. Aoi: collection and assembly of data, conception and design, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Note Added in Proof
This article was published online on 21 November 2018. Minor edits have been made that do not affect data. This notice is included in the online and print versions to indicate that both have been corrected 28 November 2018.
Supporting information
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Table
Acknowledgments
We wish to express our gratitude to Drs. Hiroshi Kawamoto, Kyoko Masuda, Takuya Maeda and Miho K. Furue for their helpful advice and to Dr. Shuji Terao for collaboration on the early stages of this work. We thank Drs. Sayumi Shimode, Ryusaku Matsumoto, Daisuke Yamamiya, and other members of our laboratory for scientific comment and valuable discussion, as well as Yukari Takatani for administrative support. This work was supported by Grant‐in‐Aid for JSPS Research Fellow (15J00925) to D.W. and a grant for Research Center Network for Realization of Regenerative Medicine (16817073) to T.Aoi from Japan Agency for Medical Research and Development, AMED.
References
- 1. Caccamo N, Dieli F, Wesch D et al. Sex‐specific phenotypical and functional differences in peripheral human Vgamma9/Vdelta2 T cells. J Leukoc Biol 2006;79:663–666. [DOI] [PubMed] [Google Scholar]
- 2. Carding SR, Egan PJ. Gammadelta T cells: Functional plasticity and heterogeneity. Nat Rev Immunol 2002;2:336–345. [DOI] [PubMed] [Google Scholar]
- 3. Kalyan S, Kabelitz D. Defining the nature of human gammadelta T cells: A biographical sketch of the highly empathetic. Cell Mol Immunol 2013;10:21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tyler CJ, Doherty DG, Moser B et al. Human Vgamma9/Vdelta2 T cells: Innate adaptors of the immune system. Cell Immunol 2015;296:10–21. [DOI] [PubMed] [Google Scholar]
- 5. Kabelitz D, Wesch D, Pitters E et al. Potential of human gammadelta T lymphocytes for immunotherapy of cancer. Int J Cancer 2004;112:727–732. [DOI] [PubMed] [Google Scholar]
- 6. Wrobel P, Shojaei H, Schittek B et al. Lysis of a broad range of epithelial tumour cells by human gamma delta T cells: Involvement of NKG2D ligands and T‐cell receptor‐ versus NKG2D‐dependent recognition. Scand J Immunol 2007;66:320–328. [DOI] [PubMed] [Google Scholar]
- 7. Silva‐Santos B, Serre K, Norell H. gammadelta T cells in cancer. Nat Rev Immunol 2015;15:683–691. [DOI] [PubMed] [Google Scholar]
- 8. Yoshida Y, Nakajima J, Wada H et al. gammadelta T‐cell immunotherapy for lung cancer. Surg Today 2011;41:606–611. [DOI] [PubMed] [Google Scholar]
- 9. Kondo M, Sakuta K, Noguchi A et al. Zoledronate facilitates large‐scale ex vivo expansion of functional gammadelta T cells from cancer patients for use in adoptive immunotherapy. Cytotherapy 2008;10:842–856. [DOI] [PubMed] [Google Scholar]
- 10. Kang N, Zhou J, Zhang T et al. Adoptive immunotherapy of lung cancer with immobilized anti‐TCRgammadelta antibody‐expanded human gammadelta T‐cells in peripheral blood. Cancer Biology & Therapy 2009;8:1540–1549. [DOI] [PubMed] [Google Scholar]
- 11. D'Asaro M, La Mendola C, Di Liberto D et al. V gamma 9V delta 2 T lymphocytes efficiently recognize and kill zoledronate‐sensitized, imatinib‐sensitive, and imatinib‐resistant chronic myelogenous leukemia cells. J Immunol 2010;184:3260–3268. [DOI] [PubMed] [Google Scholar]
- 12. Bonneville M, Scotet E. Human Vgamma9Vdelta2 T cells: Promising new leads for immunotherapy of infections and tumors. Curr Opin Immunol 2006;18:539–546. [DOI] [PubMed] [Google Scholar]
- 13. Abe Y, Muto M, Nieda M et al. Clinical and immunological evaluation of zoledronate‐activated Vgamma9gammadelta T‐cell‐based immunotherapy for patients with multiple myeloma. Exp Hematol 2009;37:956–968. [DOI] [PubMed] [Google Scholar]
- 14. Kobayashi H, Tanaka Y, Yagi J et al. Safety profile and anti‐tumor effects of adoptive immunotherapy using gamma‐delta T cells against advanced renal cell carcinoma: a pilot study. Cancer Immunol Immunother 2007;56:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bennouna J, Bompas E, Neidhardt EM et al. Phase‐I study of Innacell gammadelta, an autologous cell‐therapy product highly enriched in gamma9delta2 T lymphocytes, in combination with IL‐2, in patients with metastatic renal cell carcinoma. Cancer Immunol Immunother 2008;57:1599–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kobayashi H, Tanaka Y, Yagi J et al. Phase I/II study of adoptive transfer of gammadelta T cells in combination with zoledronic acid and IL‐2 to patients with advanced renal cell carcinoma. Cancer Immunol Immunother 2011;60:1075–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakajima J, Murakawa T, Fukami T et al. A phase I study of adoptive immunotherapy for recurrent non‐small‐cell lung cancer patients with autologous gammadelta T cells. Eur J Cardiothorac Surg 2010;37:1191–1197. [DOI] [PubMed] [Google Scholar]
- 18. Izumi T, Kondo M, Takahashi T et al. Ex vivo characterization of γδ T‐cell repertoire in patients after adoptive transfer of Vγ9Vδ2 T cells expressing the interleukin‐2 receptor β‐chain and the common γ‐chain. Cytotherapy 2013;15:481–491. [DOI] [PubMed] [Google Scholar]
- 19. Schietinger A Greenberg PD. Tolerance and exhaustion: Defining mechanisms of T cell dysfunction. Trends Immunol 2014;35:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karagiannis P, Iriguchi S, Kaneko S. Reprogramming away from the exhausted T cell state. Semin Immunol 2016;28:35–44. [DOI] [PubMed] [Google Scholar]
- 21. Nishimura T, Kaneko S, Kawana‐Tachikawa A et al. Generation of rejuvenated antigen‐specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013;12:114–126. [DOI] [PubMed] [Google Scholar]
- 22. Vizcardo R, Masuda K, Yamada D et al. Regeneration of human tumor antigen‐specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell 2013;12:31–36. [DOI] [PubMed] [Google Scholar]
- 23. Wakao H, Yoshikiyo K, Koshimizu U et al. Expansion of functional human mucosal‐associated invariant T cells via reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013;12:546–558. [DOI] [PubMed] [Google Scholar]
- 24. Takahashi K, Tanabe K, Ohnuki M et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 25. Nakagawa M, Taniguchi Y, Senda S et al. A novel efficient feeder‐free culture system for the derivation of human induced pluripotent stem cells. Sci Rep 2015;4:3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Dongen JJ, Langerak AW, Bruggemann M et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T‐cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED‐2 Concerted Action BMH4‐CT98–3936. Leukemia 2003;17:2257–2317. [DOI] [PubMed] [Google Scholar]
- 27. Patel KP, Pan Q, Wang Y et al. Comparison of BIOMED‐2 versus laboratory‐developed polymerase chain reaction assays for detecting T‐cell receptor‐gamma gene rearrangements. J Mol Diagn 2010;12:226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Okano S, Yonemitsu Y, Nagata S et al. Recombinant Sendai virus vectors for activated T lymphocytes. Gene Ther 2003;10:1381–1391. [DOI] [PubMed] [Google Scholar]
- 29. Okita K, Matsumura Y, Sato Y et al. A more efficient method to generate integration‐free human iPS cells. Nat Methods 2011;8:409–412. [DOI] [PubMed] [Google Scholar]
- 30. Muller FJ, Schuldt BM, Williams R et al. A bioinformatic assay for pluripotency in human cells. Nat Methods 2011;8:315–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohta R, Niwa A, Taniguchi Y et al. Laminin‐guided highly efficient endothelial commitment from human pluripotent stem cells. Sci Rep 2016;6:35680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takada S, Kambe N, Kawasaki Y et al. Pluripotent stem cell models of Blau syndrome reveal an IFN‐gamma‐dependent inflammatory response in macrophages. J Allergy Clin Immunol 2017. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 33. Kennedy M, Awong G, Sturgeon CM et al. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep 2012;2:1722–1735. [DOI] [PubMed] [Google Scholar]
- 34. Choi KD, Vodyanik MA, Togarrati PP et al. Identification of the hemogenic endothelial progenitor and its direct precursor in human pluripotent stem cell differentiation cultures. Cell Rep 2012;2:553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu QC, Hirst CE, Costa M et al. APELIN promotes hematopoiesis from human embryonic stem cells. Blood 2012;119:6243–6254. [DOI] [PubMed] [Google Scholar]
- 36. Slukvin II. Generating human hematopoietic stem cells in vitro ‐exploring endothelial to hematopoietic transition as a portal for stemness acquisition. FEBS Lett 2016;590:4126–4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Neofytou E, O'Brien CG, Couture LA et al. Hurdles to clinical translation of human induced pluripotent stem cells. J Clin Investig 2015;125:2551–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim K, Doi A, Wen B et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010;467:285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Polo JM, Liu S, Figueroa ME et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol 2010;28:848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aoi T. 10th anniversary of iPS cells: The challenges that lie ahead. J Biochem 2016;160:121–129. [DOI] [PubMed] [Google Scholar]
- 41. Kim M Costello J. DNA methylation: an epigenetic mark of cellular memory. Exp Mol Med 2017;49:e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rock EP, Sibbald PR, Davis MM et al. CDR3 length in antigen‐specific immune receptors. J Exp Med 1994;179:323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xu C, Zhang H, Hu H et al. Gammadelta T cells recognize tumor cells via CDR3delta region. Mol Immunol 2007;44:302–310. [DOI] [PubMed] [Google Scholar]
- 44. Zhao H, Xi X, Cui L et al. CDR3delta‐grafted gamma9delta2T cells mediate effective antitumor reactivity. Cell Mol Immunol 2012;9:147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Table