

Abstract
Mesenchymal stem/stromal cells (MSCs) present a promising tool in cell‐based therapy for treatment of various diseases. Currently, optimization of treatment protocols in clinical studies is complicated by the variations in cell dosing, diverse methods used to deliver MSCs, and the variety of methods used for tracking MSCs in vivo. Most studies use a dose escalation approach, and attempt to correlate efficacy with total cell dose. Optimization could be accelerated through specific understanding of MSC distribution in vivo, long‐term viability, as well as their biological fate. While it is not possible to quantitatively detect MSCs in most targeted organs over long time periods after systemic administration in clinical trials, it is increasingly possible to apply pharmacokinetic modeling to predict their distribution and persistence. This Review outlines current understanding of the in vivo kinetics of exogenously administered MSCs, provides a critical analysis of the methods used for quantitative MSC detection in these studies, and discusses the application of pharmacokinetic modeling to these data. Finally, we provide insights on and perspectives for future development of effective therapeutic strategies using pharmacokinetic modeling to maximize MSC therapy and minimize potential side effects. Stem Cells Translational Medicine 2018;7:78–86
Keywords: Stem cells, Quantitative detection, Modeling, Kinetics
Significance Statement.
The establishment of optimal dosage and route of administration of mesenchymal stem/stromal cells (MSCs) requires the ability to quantitatively determine their in vivo distribution, long‐term viability as well as their biological fate. In this Concise Review, studies elucidating the in vivo kinetics of MSCs are summarized. This article highlights the use of modeling approaches for precise characterization and prediction for future development of efficacious MSC‐based therapies.
Introduction
While advancements in pharmaceuticals have significantly improved the quality of life and lifespans of millions, there remain a number of pathologies that are challenging to treat with drugs alone. Cell‐based therapies have emerged as the next generation major approach to treat a range of complex pathologies. The first clinical trial using mesenchymal stem/stromal cells (MSCs) was described in 1999, and it focused on the treatment of osteogenesis imperfecta, a rare skeletal condition 1. Since then, efforts have boomed to exploit MSCs as emerging therapeutics to regenerate damaged tissue and treatment of pathologies such as cardiovascular disease, liver cirrhosis, brain spinal cord injury, cartilage and bone injury, Crohn's disease, neurodegenerative disease, ischemic disease, and graft‐versus‐host disease 2. In 2015, there were 493 registered MSC clinical trials, and 2 years later in 2017 this number has increased to 743 3. Currently, MSCs are being exploited for both their unique multi‐lineage differentiation potential and immunomodulatory properties, which enable a range of potential applications. While increasingly mainstream, MSCs are being viewed from a traditional cell‐therapy perspective. We contend that the field would benefit from viewing MSCs as a pharmaceutical, and taking advantage of well‐established pharmaceutical approaches to modeling and predicting efficacy 4.
The therapeutic mode of action of transplanted MSCs may be different for each condition, with MSCs either directly contributing to new tissue formation through terminal differentiation, or modulation of endogenous repair processes and/or of inflammatory responses via paracrine signaling 5. Based on the assumed mode of action, the 743 registered trials declared in 2017 will use a range of total number of MSCs and involve different local or systemic delivery strategies. Accumulated data generated from these clinical trials, and experimental animal models, could be used to inform pharmacokinetic models. If robust models were generated, they could form the foundation for future predictions of efficacy, accelerating clinical research outcomes by reducing the number of conditions required in dose escalation studies and informing how to best deliver the cells. Such model development is normally regarded as a critical step in the development of any new therapeutic agent to establish the optimal dosage, route of administration, duration of treatment and targeting strategies to achieve the maximum effectiveness with lowest risk 6. MSC pharmacokinetic modeling would require data for MSC survival in vivo, percentage of cells that actually home to the diseased/damaged tissue, relative cytokine secretion and treatment outcome. It is possible and important to collect this data using effective MSC detecting and tracking technology for both the development of models and for the elucidation of biological mechanisms that underpin efficacy. MSC homing, distribution, and detection methods have been discussed in several review articles. These previous publications described specific aspects of MSC transplantation including tumor‐homing 7, cell‐tracking methods 8, study of dosing regimens 9, and general description of MSC biodistribution 10, 11. Similarly, there are an increasing number of articles describing techniques to quantify MSC in tissues, but few provide actual detailed descriptions of MSC being tracked using these methods, and importantly their limitations. In this Review, we pull these pieces together, critically assess technologies, and describe how they could be used to contribute to the development of pharmacokinetic modeling of MSC‐based therapies.
Methods for Quantitative MSC Detection
A critical step in generating MSC pharmacokinetic models is tracking the fate of cells following transplantation. An ideal quantification technique should have the following features: high sensitivity and specificity; long‐term detection and monitoring; and high spatial‐temporal resolution. The advantages and disadvantages of current available methods for quantitative MSC detection are summarized in Table 1. Without previous labeling, administered MSC numbers in tissue can be estimated using polymerase chain reaction (PCR) to amplify MSC‐specific DNA sequences (assuming that the MSC are labeled or allogeneic, providing a different DNA sequence than host cells) 12 or by labeling with MSC‐specific antigens in tissue biopsies on histology slices 13. Because of high repetition and species specificity, human Alu sequences are an endogenous marker of choice to detect administered human MSCs in animal organs in preclinical studies 5, 17. The assay of other sequences had similar sensitivity as the Alu assay but required additional manipulation of samples 5. The lower detection limit of quantitative PCR enabled detection of 100 MSCs per gram of organ tissue 5, 18, which makes it feasible to detect MSCs in patients using biopsies 12. Measuring HLA alleles and expression of SH3 in biopsies or postmortem samples has also been successfully used in clinical trials for detection of transplanted MSCs in tissues 13.
Table 1.
Comparison of methods used for quantitative mesenchymal stem/stromal cells (MSC) detection
| Technique | Detection | Detection limit (cells) | Advantages | Disadvantages | Reference |
|---|---|---|---|---|---|
| PCR/histology | MSC specific DNA sequences or antigens | 102 | High sensitivity, no need to label the cells | Need animal sacrifice, biopsy, or postmortem samples from patients | 12, 13 |
| Flow cytometry | Fluorescent dyes/proteins | 103 | High specificity, quantification of live cells | Preclinical study only | 14 |
| Optical imaging | Fluorescent dyes/proteins | 103 | High throughput, excellent for longitudinal studies | Small animals only, low resolution, low sensitivity | 8 |
| MRI | Contrast agents | 104 | High spatial resolution, whole‐body scanning, clinically useful, excellent for longitudinal studies | Quantification can be difficult, cytotoxicity of certain labeling agents | 15 |
| Radionuclear | Radioisotope labels | 104 | Quantification feasible using SPECT, whole‐body scanning; high sensitivity, | Limited spatial resolution; ionizing radiation | 16 |
Abbreviations: MRI, magnetic resonance imaging; PCR, polymerase chain reaction.
Both flow cytometry and optical imaging require labeling MSCs with fluorescent dyes/proteins. Flow cytometry enables estimation of the number of live MSCs per weight unit of tissue 14, but this requires that it be possible to harvest a portion of tissue for analysis. Flow cytometry is feasible in animal studies, but may not be feasible in clinical settings. Optical imaging uses a variety of dyes, such as 4′,6‐diamidino‐2‐phenylindole (DAPI) and bromodeoxyuridine, that can bind reversibly or irreversibly to the MSCs, and some studies suggest that these fluorescent dyes have no impact on cell morphology or function 18. However, DNA binding dyes are generally regarded as carcinogens 19 and, although their use in animal models is feasible, their use in humans would likely not be considered acceptable.
MSCs transduced with a fluorescent or luciferase reporter gene using lentiviral or retroviral vectors have been used extensively for tracking MSC in vivo within animal models 20. Quantification of luciferase labeled MSCs is less accurate than flow cytometry due to limitations in the quality of data that can be collected using live animal in vivo imaging. However, this approach does enable rapid and full body characterization that is not possible with flow cytometry. Luciferase and/or fluorescent cell labeling each have weaknesses, but in combination they can provide high‐resolution spatial and quantitative data. To date, luciferase and/or fluorescent cell labeling has only been used in preclinical studies. It is not likely that these methods will be extended to clinical quantification of MSC biodistribution in the near‐term due to risk aversion to genetic modification, with the only benefit being visual tracking of the transplanted cell population. Specifically, there is concern that viral vector‐mediated transduction of MSCs to produce fluorescence or bioluminescence may cause immunogenicity and/or insertional mutagenesis 21. We note that there is an increasing enthusiasm for gene therapy approaches, and many such approaches are increasingly regarded as safe 22, 23. A growing track record of safety in this space may influence the viability of genetic labeling approaches in the future.
In animal models, luciferase and/or fluorescent cell labeling has merit and is increasingly used. Even when cells are labeled, analysis can present challenges. Tissue harvest can be invasive, and once tissue is harvested, flow cytometry analysis or histology is required to quantify cell number. While tedious, this is often necessary as the quality of whole animal imaging can be limited by quality of signal that can be derived from cells that are deeper in the tissue or farther from the surface of the animal. Extrapolation of studies to humans would require amplification as many MSC‐targeted tissues and organs in humans are located much deeper than in the small animal models used in preclinical studies. Fluorescent dyes or even bioluminescence would not likely enable effective cell tracking in many human tissue repair applications.
In clinical settings, noninvasive methods for MSC tracking and quantification are required to better understand the cell viability, biodistribution, differentiation, and long‐term fate following engraftment, and thus optimizing MSC‐based therapies. Magnetic resonance imaging (MRI), positron emission tomography (PET), and single‐photon emission computed tomography (SPECT) are currently used modalities to image, track and quantify MSCs in patients 16. MRI has the advantages of a safe profile and 3‐dimensional capacity. Contrast agents bind to or are internalized by target MSCs ex vivo or in vivo, and high spatial resolution makes it ideal for tracking MSCs and their homing to organs 15. However, an important limitation of MRI is that it can be challenging to distinguish contrast agent‐labeled cells from free contrast agent or dead cells 8. Transfer of contrast agent from originally labeled MSCs to macrophages can also lead to false interpretation of MRI data, which imposes an additional challenge for in vivo tracking of therapeutic MSCs using MRI 7. Recently, a number of caspase‐3/7 targeted imaging probes have been developed to differentiate live and apoptotic MSCs in vivo with high specificity 24, 25, 26. Thus, it is conceivable that future agents will use such biomarkers for quantitative detection of MSCs. PET has the advantage of the higher sensitivity than SPECT and MRI, which allows more accurate quantification of cell number. PET is already capable of detecting administered MSCs over long time periods and of assessing their viability without the challenges of differentiating endogenous iron from labeled cells that confound MRI studies. However, PET suffers from limited spatial resolution, and leakage of radioisotopes from labeled MSCs. The labeling agents are excreted through renal and hepatobiliary pathways, creating the illusion that MSCs have homed to these tissues when in fact they might not have. This artifact can confound studies involving tracking of MSC to tissues of the liver, bladder, and intestine in patients 27. Since each imaging modality has its own strengths and limitations, novel contrast agents for sole imaging modality have been developed 28, 29, and multimodal imaging is expected to achieve more accurate information of the transplanted MSCs. For example, MRI and PET are two highly complementary methods for quantitative MSC detection in vivo, and the contrast agents for cell tracking with MRI‐PET have been developed to use MRI to localize and PET to measure the MSC number, viability, and other functional parameters following administration 30. Recently, a MRI/SPECT/fluorescent tri‐modal probe was synthesized and used to label MSCs preclinically 31. Multimodal imaging will provide a powerful approach for combined imaging of MSC distribution, target delivery, viability, and therapeutic efficacy in humans in the future.
In Vivo Kinetics of Systemically Applied MSCs
Akin to the use of pharmacokinetics for drug development, the overall goal of quantitative MSC detection is to elucidate their in vivo kinetics for enhancing therapeutic efficacy and to decrease toxicity. In some early qualitative studies, engraftment of MSCs was found to be saturated after increasing the dose of intravenous injected cells, suggesting that increasing cell dose may not improve outcomes 32, 33. This is an important consideration, not only from a risk mitigation perspective, but from a cost management perspective. If a lower cell dose can achieve the same outcomes, then this understanding may increase the economic feasibility of the therapy. These studies also suggested that transplanted MSCs exhibited limited proliferation and self‐renewal capacity following engraftment 32, 34. The kinetics of MSCs have been found to be significantly different between cell sources, disease models, methods used to derive MSCs, and delivery routes. Bone marrow‐derived MSCs are the most frequently investigated cell type and often designated as the gold standard, while MSCs originating from adipose tissue, peripheral blood, the lung, or the heart are also widely used in preclinical and clinical trials 35. However, MSCs from different tissues are not equivalent in phenotype, function, and biodistribution 36. For example, less amniotic fluid derived MSCs migrated into cryoinjured hearts than that of bone marrow derived MSCs in the short term (24 hours), but the long‐term (30 days) cell survival rates were similar between the two types of MSCs 37. Prolonged time in culture expansion can also induce a defect in MSCs that affects their engraftment into target organs 38. Various administration routes have been proposed for MSC transplantation ranging from classic intravenous infusion or subcutaneous injection to intra‐arterial, intra‐articular, intra‐portal, and intraperitoneal injections 11. Intravenous injection is still the main administration route in preclinical and clinical settings 39. Most of intravenously injected MSCs (>80%) are rapidly trapped in lungs, followed by rapid distribution of a portion of the injected MSCs to other tissues including liver, spleen, and inflammatory or injured sites. The accumulation of MSCs in the lungs is a key determining factor for their biodistribution, and the time duration of MSCs remaining in lungs is reported to vary from 7 days to 3 months depending on different detection methods and models applied 11. Using a different MSC delivery route has important effects on their distribution. Intra‐arterial injection provides direct delivery of MSCs to the target organs, as it bypasses the lung entrapment 17. Several studies revealed that the intra‐arterial injection could significantly decrease the number of MSCs that become trapped within the lungs and increased uptake in other organs, especially in the liver 40, 41, 42. Despite the benefits of delivery of MSCs to the target organs, there are increased risks of organ vascular occlusion which may lead to the development of ischemic lesions and cause mortality 42, and also one study has revealed the potential that the direct delivery method may influence MSC differentiation into an unintended lineage 43.
Table 2 summarized the in vivo distribution of therapeutic regenerative cells in clinical studies 12, 13, 27, 41, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54. Almost half of the fifteen clinical trials (7 in 15) used unselected bone marrow mononuclear cells, which represent a heterogenous population of various stem and progenitor cells, stromal cells, and hematopoietic cells at various stages of maturation. Some others used progenitor/stem cells from bone marrow or peripheral blood. Only three trials reported the biodistribution of culture‐expanded and enriched MSCs. The in vivo kinetics of MSCs are similar to that found in other therapeutic regenerative cells in terms of lung entrapment following intravenous infusion, with differences in recirculation and migration that could be related to the disease. Tissue‐specific homing has been demonstrated, indicating a response of all the administered stem cells to injured/diseased tissue. Compared with unselected bone marrow cells, more bone marrow stem cells, including MSCs, homed to infarcted human myocardium 45. After systemic administration, the concentration of stem/progenitor cells was greatest among tissues with acute injury, and progressively decreased in tissues in an intermediate phase or a chronic stage 54. However, variably low long‐term engraftment of MSCs was found in the target organs after systemic administration. For the treatment of liver cirrhosis, there were only 13.0%—17.4% of MSCs in the liver 10 days after intravenous infusion 41. Even after intracoronary injection, only 10% of the administered bone marrow stem cells were retained within the myocardium within 24 hours in patients with acute myocardial infarction, while the majority of MSC were found in the spleen and liver 44. Overall, these recent clinical trials suggest that only a small percentage of the original systemically administered therapeutic regenerative cells are capable of engrafting even under the best conditions, and of those that do engraft, only a small percentage have been shown to convey potent therapeutic responses.
Table 2.
Summary of regenerative cell in vivo distribution in clinical studies
| First Author/Year | Disease | Cell type | Cell dose | % stem cells | Delivery method | Quantification modality | In vivo distribution |
|---|---|---|---|---|---|---|---|
| Gholamrezanezhad/2011 [41] | Liver cirrhosis | MSC | 2.5–4 × 108 | >90 | Intravenous infusion | Planar whole‐body acquisitions/SPECT | MSCs accumulated in the lung first, MSCs in the liver increased from 0.0%–2.8% to 13.0%–17.4% in 10 days |
| Ringden/2006 [12] | Graft‐versus‐host disease | MSC | 0.7–9 × 106/kg | >90 | Intravenous infusion | PCR | MSC DNA was detected in lymph nodes and the gastrointestinal tract, but not in liver, spleen, and lung on day 96 |
| Koc/2000 [13] |
Breast cancer |
MSC | 1–2.2 × 106 | >95 | Intravenous injection | Histology | Circulating MSC detection rates were 43% at 15 minutes, and 14% at 1 hour |
| Kurpisz/2007 [44] | Acute myocardial infarction | Bone marrow stem cell | 2–4 × 106 | — | Intracoronary injection | SPECT | 10% of the cells were retained within the myocardium in 24 hours after infusion while their majority migrated to the spleen and liver |
| Hofmann/2005 [45] | Acute myocardial infarction | Bone marrow cell | 2.5 × 109 | 0.5 | Intracoronary or intravenous injection | PET | 1.3%–2.6% of Bone marrow cells and 14%–39% of bone marrow stem cells were detected in the infarcted myocardium 60 minutes after intracoronary injection, the remaining cells were found primarily in liver and spleen, after intravenous transfer, no cell was detected in the infarcted myocardium |
| Bone marrow stem cell | 1.7 × 109 | 66.6 | |||||
| Kang/2006 [46] | Chronic myocardial infarction | Peripheral blood stem cell | 4.5 × 108 | 8.3 | Intracoronary or intravenous injection | PET/CT | 1.5% of cells accumulated at the infarcted myocardium at 2 hours after intracoronary injection, intravenous injection showed a high initial lung uptake with no myocardial activity |
| Caveliers/2007 [47] | Chronic ischemic cardiomyopathy | Peripheral blood stem cell | 3.4 × 107 | 89 | Intracoronary injection | Planar whole‐body acquisitions/SPECT | 6.9%, 23.1%, and 3.1% of cells were in the heart, liver, and spleen in 1 hour after injection, and 2.3%, 23.8%, and 3.5% after 12 hours |
| Schächinger/2008 [48] | Acute myocardial infarction | Peripheral blood progenitor cell | 1.5 × 107 | — | Intracoronary injection | Planar whole‐body acquisitions/SPECT | 6.9% of cells were detected in the heart and declined to 2% after 3 to 4 days |
| Goussetis/2006 [27] | Chronic ischemic cardiomyopathy | Bone marrow progenitor cell | 1.6 × 107 | 74—92 | Intracoronary injection | Planar whole‐body acquisitions/SPECT | 9.2% and 6.8% of cells were localized in the infarcted area of the heart in 1 and 24 hours after injection, the remaining was distributed mainly to liver and spleen |
| Couto/2010 [49] | Liver cirrhosis | BMMC | 2–15 × 106 | 0.014 | Hepatic arterial infusion | Planar whole‐body acquisitions | Hepatic BMMC retentions of 41 and 32% in 3 and 24 hours after injection |
| Correa/2007 [50] | Acute ischemic stroke | BMMC | 3 × 107 | — | Cerebral arterial infusion | SPECT | BMMCs accumulated in the ipsilateral hemisphere, there was liver and spleen uptake, but no lung uptake 8 hours after infusion |
| Karpov/2005 [51] | Acute myocardial infarction | BMMC | 2–4 × 106 | — | Intracoronary injection | SPECT | BMMC in the myocardium were 7.8%, 6.8%, and 3.2% at 30 minutes, 2.5 hours, and 24 hours after injection, there was liver and spleen uptake |
| Barbosa da Fonseca/2011 [52] | Chronic chagasic cardiomyopathy | BMMC | 1–9.6 × 108 | 1.6 | Intracoronary injection | Planar whole‐body acquisitions/SPECT | 5.4%, 4.3%, and 2.3% of BMMCs in the heart after 1, 3, and 24 hours, The remaining cells was distributed to the liver, spleen, kidneys, and bladder. Intestinal uptake was observed after 24 hours |
| Barbosa da Fonseca/2009 [53] | Chronic ischemic stroke | BMMC | 1.25–5 × 106 | 1.6 | Cerebral arterial infusion | Planar whole‐body acquisitions/SPECT | 1.68% of cells were in the brain at 2 hours, while 43.56% distributed to liver, 7.20% to lungs, 3.98 to spleen, 4.35% to kidneys, and 9.01% to bladder, BMMCs were distributed mainly to the liver and spleen at 24 hours after injection |
| Penicka/2007 [54] | Acute and chronic myocardial infarction | BMMC | 2.5 × 109 | 0.6 | Intracoronary injection | Planar whole‐body acquisitions/SPECT | 1.31%–5.10% of cells were in the myocardium at 2 hours, BMMCs occupied the whole coronary artery territory at 2 hours, engraftment was confined to more distal parts of the infarction territory at 20 hours |
Abbreviations: BMMC, bone marrow mononuclear cell; MSCs, mesenchymal stem/stromal cells; PCR, polymerase chain reaction; PET, positron emission tomography; SPECT, single‐photon emission computed tomography; —, not reported.
Modeling the In Vivo Kinetics of Therapeutic MSCs
During the past 30 years, pharmacokinetic modeling approaches have been successfully applied to systematically analyze and predict the safety and efficacy of therapeutic agents including small molecules and biological products such as peptides or antibodies. Just as these models have been used for pharmaceuticals, it should be possible to adapt these models to facilitate understanding and prediction of the effects of MSCs. However, the use of living cells as therapeutic agents differs in many important ways from traditional pharmacology. It can be particularly challenging for MSC‐based therapy because of poorly understood pharmacokinetics and complex pharmacodynamics. MSCs convey therapeutic benefits by both engrafting and differentiating for regenerative applications, and relinquishing their molecular contents without directly contributing to new tissue formation. To simplify the explanation of their in vivo kinetics, the dynamics of systematically administered MSCs have previously been considered similar to that of inert micrometer‐scale particles injected into the bloodstream of animals 55. Shim et al. calculated the noncompartmental pharmacokinetic parameters (maximum serum concentration, area under curve and mean residence time) of MSCs as inert particles after intravenous and intra‐articular injection, and concluded that mean residence time values for intra‐articularly transplanted MSCs were longer than those for intravenously administered MSCs 17. In the view of MSC imparting therapeutic benefits by secreting soluble factors in vivo, MSCs may be better considered as drug delivery particles. Elman et al. reported the time to reach maximum serum concentration, the half‐life, and the elimination constant of MSCs and their secreted factors (monocyte chemoattractant protein‐1, interleukin‐6, and interleukin‐8) 56. This is the only study of pharmacokinetic analysis of MSC secretome in vivo. However, no pharmacokinetic model was developed in this study. Administered cells are not subject to conventional chemical analyses, making it unsuitable to apply standard pharmacokinetic modeling techniques and profiles. A multidisciplinary collaboration between experimental, modeling and clinical areas will be required to integrate the concepts of modern pharmacokinetic modeling in cell therapy, given the complexity of techniques required for experiments and the expertise needed for imaging, modeling, and clinical translation. Only recently, the two‐compartment pharmacokinetic model of therapeutic MSCs was developed to propose designs of dosing regimens 4. As shown in Figure 1A, this model incorporated four parameters: R i, injection rate; R c, clearance rate; K 1, rate of extravasation; and K 2, rate of intravasation. The model analysis suggests that the administered MSCs are only therapeutically active for a short period of time (less than 24 hours). The model predicted data are consistent with the cytokine response associated with MSC transplantation or MSC‐derived molecules (secretome) when the latter were administered to animals with systemic inflammation 5, 57. This study revealed that successive doses of MSCs within a shorter treatment period may allow for the maintenance of MSC‐based therapy within a therapeutic window that can sustain a long‐term biological response ultimately. Thus, MSCs may need to be administered at a greater magnitude and/or frequency to sustain a long‐term biological response. Both clinical and preclinical studies have demonstrated the safety and efficacy of multiple deliveries of MSCs 58, 59. In the treatment of graft‐versus‐host disease, smaller MSC doses with multiple infusions appear to be more promising compared with single bolus administration 60, 61. The question of frequency and timing rather than just dose as a factor in response for MSC‐based therapies remains to be explored in future studies.
Figure 1.
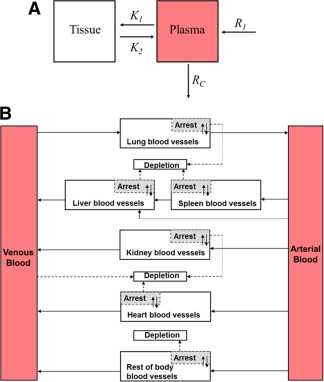
Modeling the in vivo kinetics of therapeutic mesenchymal stem/stromal cells (MSCs). (A): Schematic diagram of two‐compartment pharmacokinetic model incorporating four parameters: R i, injection rate; R c, clearance rate; K 1, rate of extravasation; and K 2, rate of intravasation. (B): Schematic diagram of the PBPK model. The whole body was separated into eight compartments: arterial blood, venous blood, lungs, spleen, liver, kidneys, heart, and the rest of body. Solid arrows indicate blood flow, dashed gray arrows indicate the depletion of MSCs, and gray boxes indicate the arrested MSCs isolated from blood circulation as in the extravascular space of organ.
Different to empirical pharmacokinetic models, physiologically based pharmacokinetic (PBPK) model is based on the anatomical structure of the living systems, with each important organ regarded as an individual compartment. All compartments are connected by blood flow. The complex, yet regulated, in vivo kinetics of administered therapeutic cells are amenable to PBPK model building and analysis. The applications of PBPK modeling for therapeutic cells have appeared as early as 1996 62. More efforts are being made to use PBPK models for the advancement of cell therapy, including T lymphocytes, tumor‐infiltrating lymphocytes, and natural killer cells. PBPK modeling has just been used in MSC‐based therapies recently and the first PBPK model for MSCs was published in 2016 14. In this study, the model development invoked assumptions based on direct visualization of MSC spatiotemporal disposition by intravital microscopy and assessment of cell quantity using flow cytometry. As shown in Figure 1B, the whole body was separated into eight compartments: arterial blood, venous blood, lungs, spleen, liver, kidneys, heart, and the rest of body. All compartments were interconnected via the systemic blood circulation. Key components included in the model were species‐specific physiological parameters (body weight, organ volume, and blood flow) and MSC‐specific parameters (partition coefficient, arrest rate constant, release rate constant, and depletion rate constant). The sensitivity analysis of this PBPK model suggest that the targeting efficiency of MSCs is determined by the lung retention and interaction between MSCs and target organs, including cell arrest, depletion and release. The clinical utility of the model was also tested with data obtained from stem cell‐based therapies to patients with liver cirrhosis 41, 49. This study suggests that instead of increasing the dose, possible strategies to further improve the target efficiency of MSC‐based therapies would be bypassing the initial lung entrapment and enhancing organ‐specific capture by modulating cell surface properties. Pharmacokinetic model studies have the potential for interspecies scaling, which allows prediction of the in vivo kinetics of therapeutic cells in humans using animal data. While for the other types of preclinical studies on MSC kinetics, it is not clear to what degree the findings in animals are quantitatively transferable to humans. By systematically examining the effects of changing individual model parameters, models can also identify key parameters and their values, and suggest possible strategies for improvements in biodistribution. Thus, we anticipate that after this theoretical framework of PBPK model for MSCs, more second‐generation pharmacokinetic models will be developed based on a more comprehensive view of MSCs and their secretome to reduce experimental costs due to a systematic minimization of required testing, increase throughput of discovery, and ultimately lead to more efficacious treatment regimens.
Conclusion
From both the completed and ongoing clinical trials, MSC‐based therapies represent an exciting approach that could potentially treat various diseases and maintain the promise of safety. However, much work remains to be done before MSCs can pass from clinical trials to the standard treatment protocols; the dosing regimen especially can be considered the Achilles’ heel of MSC‐based therapies. With the current dosing regimen, the majority of therapeutic regenerative cells cannot be located in target organs by sensitive quantitative detection techniques over long time periods after systemic administration. By using the pharmacokinetic modeling approach, researchers and clinicians have attempted to characterize and predict the in vivo kinetics of systemically applied MSCs in order to maximize their therapeutic activity while minimizing potential side effects. Recently published pharmacokinetic models for MSCs suggest that MSCs may need to be administered at a greater magnitude and/or frequency to sustain a long‐term biological response. Other possible strategies to further improve the target efficiency of MSC‐based therapies could be bypassing the initial lung entrapment and enhancing organ‐specific capture by modulating cell surface properties. More second‐generation pharmacokinetic models will be developed to facilitate design of new formulations and dosing regimens for cell therapies.
Author Contributions
A.B., K.F., and X. Liang: literature review and analysis, manuscript writing; X.H.: literature review and analysis; X. Liu., D.H.G.C., M.R.D., and M.S.R.: manuscript writing, financial support; H.W.: conception and design, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
Dr. Doran holds a Fellowship with the National Health and Medical Research Council of Australia. Dr. Roberts provided expert independent advice to the U.S. Federal Trade Commission and others including various paten cases. He is an Australian National Health & Medical Research Council project grant holder and Fellow, a member of a compulsory UniSuper superannuation action fund which manages various and Director of a family company which manages consulting disclosed above. The other authors indicated no potential conflicts of interest.
Note Added in Proof
This article was published online on 06 December 2017. Minor edits have been made that do not affect data. This notice is included in the online and print versions to indicate that both have been corrected 28 December 2017.
Acknowledgments
This work was supported by grants from National Health and Medical Research Council (APP1126091 and APP1055176).
Contributor Information
Michael S. Roberts, Email: m.roberts@uq.edu.au
Haolu Wang, Email: h.wang21@uq.edu.au.
References
- 1. Horwitz EM, Prockop DJ, Fitzpatrick LA et al. Transplantability and therapeutic effects of bone marrow‐derived mesenchymal cells in children with osteogenesis imperfecta. Nat Med 1999;5:309–313. [DOI] [PubMed] [Google Scholar]
- 2. Squillaro T, Peluso G, Galderisi U. Clinical trials with mesenchymal stem cells: An update. Cell Transplant 2016;25:829–848. [DOI] [PubMed] [Google Scholar]
- 3.Clinical Trials Available at https://clinicaltrials.gov/. Accessed July 21, 2017.
- 4. Parekkadan B, Milwid JM. Mesenchymal stem cells as therapeutics. Annu Rev Biomed Eng 2010;12:87–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee RH, Pulin AA, Seo MJ et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti‐inflammatory protein TSG‐6. Cell Stem Cell 2009;5:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhao P, Zhang L, Grillo JA et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin Pharmacol Ther 2011;89:259–267. [DOI] [PubMed] [Google Scholar]
- 7. Reagan MR, Kaplan DL. Concise review: Mesenchymal stem cell tumor‐homing: Detection methods in disease model systems. Stem Cells 2011;29:920–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kircher MF, Gambhir SS, Grimm J. Noninvasive cell‐tracking methods. Nat Rev Clin Oncol 2011;8:677–688. [DOI] [PubMed] [Google Scholar]
- 9. Golpanian S, Schulman IH, Ebert RF et al. Concise review: Review and perspective of cell dosage and routes of administration from preclinical and clinical studies of stem cell therapy for heart disease. Stem Cells Translational Medicine 2016;5:186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karp JM, Leng Teo GS. Mesenchymal stem cell homing: The devil is in the details. Cell Stem Cell 2009;4:206–216. [DOI] [PubMed] [Google Scholar]
- 11. Leibacher J, Henschler R. Biodistribution, migration and homing of systemically applied mesenchymal stem/stromal cells. Stem Cell Res Ther 2016;7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ringden O, Uzunel M, Rasmusson I et al. Mesenchymal stem cells for treatment of therapy‐resistant graft‐versus‐host disease. Transplantation 2006;81:1390–1397. [DOI] [PubMed] [Google Scholar]
- 13. Koc ON, Gerson SL, Cooper BW et al. Rapid hematopoietic recovery after coinfusion of autologous‐blood stem cells and culture‐expanded marrow mesenchymal stem cells in advanced breast cancer patients receiving high‐dose chemotherapy. J Clin Oncol 2000;18:307–316. [DOI] [PubMed] [Google Scholar]
- 14. Wang H, Liang X, Xu ZP et al. A physiologically based kinetic model for elucidating the in vivo distribution of administered mesenchymal stem cells. Sci Rep 2016;6:22293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bulte JW. In vivo MRI cell tracking: Clinical studies. AJR Am J Roentgenol 2009;193:314–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nguyen PK, Riegler J, Wu JC. Stem cell imaging: From bench to bedside. Cell Stem Cell 2014;14:431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shim G, Lee S, Han J et al. Pharmacokinetics and in vivo fate of intra‐articularly transplanted human bone marrow‐derived clonal mesenchymal stem cells. Stem Cells Dev 2015;24:1124–1132. [DOI] [PubMed] [Google Scholar]
- 18. Sensebe L, Fleury‐Cappellesso S. Biodistribution of mesenchymal stem/stromal cells in a preclinical setting. Stem Cells Int 2013;2013:678063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luch A. Nature and nurture ‐ lessons from chemical carcinogenesis. Nat Rev Cancer 2005;5:113–125. [DOI] [PubMed] [Google Scholar]
- 20. Swart JF, de Roock S, Hofhuis FM et al. Mesenchymal stem cell therapy in proteoglycan induced arthritis. Ann Rheum Dis 2015;74:769–777. [DOI] [PubMed] [Google Scholar]
- 21. Stender S, Murphy M, O'Brien T et al. Adeno‐associated viral vector transduction of human mesenchymal stem cells. Eur Cell Mater 2007;13:93–99; discussion 99. [DOI] [PubMed] [Google Scholar]
- 22. Ginn SL, Alexander IE, Edelstein ML et al. Gene therapy clinical trials worldwide to 2012 ‐ an update. J Gene Med 2013;15:65–77. [DOI] [PubMed] [Google Scholar]
- 23. Nathwani AC, Reiss UM, Tuddenham EG et al. Long‐term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 2014;371:1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nejadnik H, Ye D, Lenkov OD et al. Magnetic resonance imaging of stem cell apoptosis in arthritic joints with a caspase activatable contrast agent. ACS Nano 2015;9:1150–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ye D, Shuhendler AJ, Pandit P et al. Caspase‐responsive smart gadolinium‐based contrast agent for magnetic resonance imaging of drug‐induced apoptosis. Chem Sci 2014;4:3845–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ye D, Shuhendler AJ, Cui L et al. Bioorthogonal cyclization‐mediated in situ self‐assembly of small‐molecule probes for imaging caspase activity in vivo. Nat Chem 2014;6:519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goussetis E, Manginas A, Koutelou M et al. Intracoronary infusion of CD133+ and CD133‐CD34+ selected autologous bone marrow progenitor cells in patients with chronic ischemic cardiomyopathy: Cell isolation, adherence to the infarcted area, and body distribution. Stem Cells 2006;24:2279–2283. [DOI] [PubMed] [Google Scholar]
- 28. Kim T, Lee N, Arifin DR et al. In vivo micro‐CT imaging of human mesenchymal stem cells labeled with gold‐poly‐L‐lysine nanocomplexes. Adv Funct Mater 2017;27:1604213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang L, Wang Y, Tang Y et al. High MRI performance fluorescent mesoporous silica‐coated magnetic nanoparticles for tracking neural progenitor cells in an ischemic mouse model. Nanoscale 2013;5:4506–4516. [DOI] [PubMed] [Google Scholar]
- 30. Patel D, Kell A, Simard B et al. The cell labeling efficacy, cytotoxicity and relaxivity of copper‐activated MRI/PET imaging contrast agents. Biomaterials 2011;32:1167–1176. [DOI] [PubMed] [Google Scholar]
- 31. Tang Y, Zhang C, Wang J et al. MRI/SPECT/fluorescent tri‐modal probe for evaluating the homing and therapeutic efficacy of transplanted mesenchymal stem cells in a rat ischemic stroke model. Adv Funct Mater 2015;25:1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marino R, Martinez C, Boyd K et al. Transplantable marrow osteoprogenitors engraft in discrete saturable sites in the marrow microenvironment. Exp Hematol 2008;36:360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu J, Sun Z, Sun HS et al. Intravenously administered bone marrow cells migrate to damaged brain tissue and improve neural function in ischemic rats. Cell Transplant 2008;16:993–1005. [PubMed] [Google Scholar]
- 34. Dominici M, Marino R, Rasini V et al. Donor cell‐derived osteopoiesis originates from a self‐renewing stem cell with a limited regenerative contribution after transplantation. Blood 2008;111:4386–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hass R, Kasper C, Bohm S et al. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue‐derived MSC. Cell Commun Signal 2011;9:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Phinney DG, Sensebe L. Mesenchymal stromal cells: Misconceptions and evolving concepts. Cytotherapy 2013;15:140–145. [DOI] [PubMed] [Google Scholar]
- 37. Iop L, Chiavegato A, Callegari A et al. Different cardiovascular potential of adult‐ and fetal‐type mesenchymal stem cells in a rat model of heart cryoinjury. Cell Transplant 2008;17:679–694. [DOI] [PubMed] [Google Scholar]
- 38. Rombouts WJC, Ploemacher RE. Primary murine MSC show highly efficient homing to the bone marrow but lose homing ability following culture. Leukemia 2003;17:160–170. [DOI] [PubMed] [Google Scholar]
- 39. Zhang J, Huang X, Wang H et al. The challenges and promises of allogeneic mesenchymal stem cells for use as a cell‐based therapy. Stem Cell Res Ther 2015;6:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Makela T, Takalo R, Arvola O et al. Safety and biodistribution study of bone marrow‐derived mesenchymal stromal cells and mononuclear cells and the impact of the administration route in an intact porcine model. Cytotherapy 2015;17:392–402. [DOI] [PubMed] [Google Scholar]
- 41. Gholamrezanezhad A, Mirpour S, Bagheri M et al. In vivo tracking of 111In‐oxine labeled mesenchymal stem cells following infusion in patients with advanced cirrhosis. Nucl Med Biol 2011;38:961–967. [DOI] [PubMed] [Google Scholar]
- 42. Walczak P, Zhang J, Gilad AA et al. Dual‐modality monitoring of targeted intraarterial delivery of mesenchymal stem cells after transient ischemia. Stroke 2008;39:1569–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kunter U, Rong S, Boor P et al. Mesenchymal stem cells prevent progressive experimental renal failure but maldifferentiate into glomerular adipocytes. J Am Soc Nephrol 2007;18:1754–1764. [DOI] [PubMed] [Google Scholar]
- 44. Kurpisz M, Czepczynski R, Grygielska B et al. Bone marrow stem cell imaging after intracoronary administration. Int J Cardiol 2007;121:194–195. [DOI] [PubMed] [Google Scholar]
- 45. Hofmann M, Wollert KC, Meyer GP et al. Monitoring of bone marrow cell homing into the infarcted human myocardium. Circulation 2005;111:2198–2202. [DOI] [PubMed] [Google Scholar]
- 46. Kang WJ, Kang HJ, Kim HS et al. Tissue distribution of 18F‐FDG‐labeled peripheral hematopoietic stem cells after intracoronary administration in patients with myocardial infarction. J Nucl Med 2006;47:1295–1301. [PubMed] [Google Scholar]
- 47. Caveliers V, De Keulenaer G, Everaert H et al. In vivo visualization of 111In labeled CD133+ peripheral blood stem cells after intracoronary administration in patients with chronic ischemic heart disease. Q J Nucl Med Mol Imaging 2007;51:61–66. [PubMed] [Google Scholar]
- 48. Schachinger V, Aicher A, Dobert N et al. Pilot trial on determinants of progenitor cell recruitment to the infarcted human myocardium. Circulation 2008;118:1425–1432. [DOI] [PubMed] [Google Scholar]
- 49. Couto BG, Goldenberg RC, da Fonseca LM et al. Bone marrow mononuclear cell therapy for patients with cirrhosis: A Phase 1 study. Liver Int 2011;31:391–400. [DOI] [PubMed] [Google Scholar]
- 50. Correa PL, Mesquita CT, Felix RM et al. Assessment of intra‐arterial injected autologous bone marrow mononuclear cell distribution by radioactive labeling in acute ischemic stroke. Clin Nucl Med 2007;32:839–841. [DOI] [PubMed] [Google Scholar]
- 51. Karpov RS, Popov SV, Markov VA et al. Autologous mononuclear bone marrow cells during reparative regeneratrion after acute myocardial infarction. Bull Exp Biol Med 2005;140:640–643. [DOI] [PubMed] [Google Scholar]
- 52. Barbosa da Fonseca LM, Xavier SS, Rosado de Castro PH et al. Biodistribution of bone marrow mononuclear cells in chronic chagasic cardiomyopathy after intracoronary injection. Int J Cardiol 2011;149:310–314. [DOI] [PubMed] [Google Scholar]
- 53. Barbosa da Fonseca LM, Gutfilen B, Rosado de Castro PH et al. Migration and homing of bone‐marrow mononuclear cells in chronic ischemic stroke after intra‐arterial injection. Exp Neurol 2010;221:122–128. [DOI] [PubMed] [Google Scholar]
- 54. Penicka M, Lang O, Widimsky P et al. One‐day kinetics of myocardial engraftment after intracoronary injection of bone marrow mononuclear cells in patients with acute and chronic myocardial infarction. Heart 2007;93:837–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sjoholm I, Edman P. Acrylic microspheres in vivo. I. Distribution and elimination of polyacrylamide microparticles after intravenous and intraperitoneal injection in mouse and rat. J Pharmacol Exp Ther 1979;211:656–662. [PubMed] [Google Scholar]
- 56. Elman JS, Murray RC, Wang F et al. Pharmacokinetics of natural and engineered secreted factors delivered by mesenchymal stromal cells. PLoS One 2014;9:e89882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zangi L, Margalit R, Reich‐Zeliger S et al. Direct imaging of immune rejection and memory induction by allogeneic mesenchymal stromal cells. Stem Cells 2009;27:2865–2874. [DOI] [PubMed] [Google Scholar]
- 58. Ball LM, Bernardo ME, Roelofs H et al. Multiple infusions of mesenchymal stromal cells induce sustained remission in children with steroid‐refractory, grade III‐IV acute graft‐versus‐host disease. Br J Haematol 2013;163:501–509. [DOI] [PubMed] [Google Scholar]
- 59. Kol A, Wood JA, Carrade Holt DD et al. Multiple intravenous injections of allogeneic equine mesenchymal stem cells do not induce a systemic inflammatory response but do alter lymphocyte subsets in healthy horses. Stem Cell Res Ther 2015;6:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhou H, Guo M, Bian C et al. Efficacy of bone marrow‐derived mesenchymal stem cells in the treatment of sclerodermatous chronic graft‐versus‐host disease: Clinical report. Biol Blood Marrow Transplant 2010;16:403–412. [DOI] [PubMed] [Google Scholar]
- 61. Lin Y, Hogan WJ. Clinical application of mesenchymal stem cells in the treatment and prevention of graft‐versus‐host disease. Adv Hematol 2011;2011:427863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhu H, Melder RJ, Baxter LT et al. Physiologically based kinetic model of effector cell biodistribution in mammals: Implications for adoptive immunotherapy. Cancer Res 1996;56:3771–3781. [PubMed] [Google Scholar]