

Abstract
The exponential growth of genomic variants uncovered by next generation sequencing necessitates efficient and accurate computational analyses to predict their functional effects. A number of computational methods have been developed for the task, but few unbiased comparisons of their performance are available. To fill the gap, The Critical Assessment of Genome Interpretation (CAGI) comprehensively assesses phenotypic predictions on newly collected experimental datasets. Here, we present the results of the SUMO conjugase challenge where participants were predicting functional effects of missense mutations in human SUMO-conjugating enzyme UBE2I. The performance of the predictors is similar to each other and is far from perfection. Evolutionary information from sequence alignments dominates the success: deleterious mutations at conserved positions and benign mutations at variable positions are accurately predicted. Prediction accuracy of other mutations remains unsatisfactory, and this fast-growing field of research is yet to learn the use spatial structure information to improve the predictions significantly.
Keywords: CAGI, substitutions, predictors, performance, SUMO
Introduction
Ever-growing next generation sequencing efforts identify copious missense variants that lead to single amino acid substitutions in proteins. Phenotypic effects for most of these variants are unknown, and their comprehensive functional studies are not feasible considering the scale. A number of computational methods have been developed to predict the effects of missense mutations and prioritize them for experimental work. These methods can be divided into three main categories: single predictors based on evolutionary considerations from sequence, single predictors deriving from additional information such as 3D structure and physicochemical attributes of amino acids, and meta-predictors that integrate scores from several other predictors(Gnad, et al., 2013; Miosge, et al., 2015). Compared to the abundance of prediction methods(Adebali, et al., 2016; Adzhubei, et al., 2010; Bromberg and Rost, 2007; Choi and Chan, 2015; Katsonis and Lichtarge, 2014; Kircher, et al., 2014; Kumar, et al., 2009; Li, et al., 2009; Martelli, et al., 2016; Niroula, et al., 2015; Pejaver, et al., 2017; Thomas and Kejariwal, 2004; Yue, et al., 2006), independent studies assessing their performance are scarce(Martelotto, et al., 2014; Miosge, et al., 2015; Schiemann and Stowell, 2016). The reliability of previous assessments remains unclear because predictors perform inconsistently across benchmarks, which may indicate unintended overlap between training and testing data sets(Grimm, et al., 2015). Therefore, a community-wide experiment on de novo generated testing sets is required to: (1) objectively assess different predictors, (2) reveal the strengths and weaknesses of methods, (3) highlight the most promising trends in the field, and (4) provide guidance for people outside the field in choosing optimal methods.
Here we evaluate the SUMO conjugase challenge in the Critical Assessment of Genome Interpretation (CAGI), in which participants were asked to predict the fitness effects of missense mutations in the human SUMO-conjugating enzyme (UBC9, also known as UBE2I). UBE2I is the only known human SUMO ligase (E2 enzyme) that transfers SUMO from the E1 complex to downstream substrates through a conserved Cys residue. UBE2I recognizes substrates by their consensus motif and catalyzes the sumoylation reaction, which can be assisted by E3 ligases(Geiss-Friedlander and Melchior, 2007). The SUMO pathway affects multiple transcription factors and regulates diverse cellular processes including protein degradation, cell proliferation, signal transduction, nuclear transport, and chromosome segregation(Flotho and Melchior, 2013; Gareau and Lima, 2010). UBE2I regulates proliferation and transformation in different cancers and is targeted by multiple viruses, including HIV, EBV and HPV(Everett, et al., 2013; Jaber, et al., 2009; Li, et al., 2007; Qin, et al., 2011; Seeler and Dejean, 2017). A large number of novel missense mutations in UBE2I have been identified recently in cancer patients, but their functional impact remains unclear(Wu, et al., 2014). Furthermore, high-resolution spatial structures of different macromolecular complexes of UBE2I with its substrates and regulators are available, enabling predictors to use extensive structural information(Alontaga, et al., 2015; Bernier-Villamor, et al., 2002; Capili and Lima, 2007; Streich and Lima, 2016). Therefore, UBE2I is an excellent target for testing computational predictions of mutational effects.
The dataset used here to assess predictions was a collection of functional impact scores for human UBE2I mutants measured in a companion study (Weile et al., unpublished results). Briefly, effects of missense mutations in UBE2I were measured using a previously-described Saccharomyces cerevisiae (yeast) complementation assay, in which a yeast strain carrying a temperature-sensitive allele of the endogenous UBC9 gene exhibits loss of growth that can be rescued by expressing human UBE2I(Sun, et al., 2016). The impact of specific mutants or mutant combinations was assessed by the relative growth rate in a competitive growth assay. This high-throughput experiment measured over 5,000 UBE2I mutants (682 single mutations, and 4,427 distinct mutation combinations) for predictions.
We received 16 prediction datasets from 9 groups. The assessment shows that most predictors are capturing qualitative (e.g., deleterious vs. benign) effects of mutations on proteins. However, the quantitative agreement between predictions and experimental measurements remains modest. The accuracy of predictions varies greatly among mutations and correlates strongly with the evolutionary signal in sequence alignment. While deleterious mutations at conserved positions are predicted best, predictions are poor for deleterious mutations at non-conserved positions and benign or beneficial mutations at conserved sites. Thus, significant improvements are needed and may come from more rigorous integration of features, better treatment of 3D structural information, consideration of epistatic effects, and analysis of interacting partners.
Materials and Methods
Experimental procedures
Full experimental details can be found in the companion study that reports the primary experimental results (Weile et al, submitted), but we briefly summarize the methods here.
Library construction
A library of over 5000 UBE2I variants using codon-replacement mutagenesis [Weile et al, submitted] was constructed. For each UBE2I codon we designed an oligonucleotide targeting that codon. Each oligo was synthesized with an NNK degeneracy at the position, thus allowing it to encode any amino acid, but only one stop codon. The UBC9 ORF was amplified in the presence of dUTP to generate uracil-doped template for the mutagenesis reaction. Oligonucleotides were then pooled and hybridized to the template. Gaps between hybridizations were filled with a non-strand-displacing polymerase and sealed by ligation. The uracil-doped template was removed using Uracil-DNA-Glycosylase (UDG). The mutagenesis product was then amplified, adding flanking site-specific recombination sites to allow subcloning into Gateway Entry vectors. The resulting Entry pool was subcloned en masse into a library of barcoded Gateway Destination expression vectors. The Destination library was then transformed en masse into Escherichia coli. Over 10,000 individual colonies were picked and arrayed onto 384-well plates.
To establish the identity of each plasmid barcode and its associated set of mutations in the target ORF we used kiloSEQ (SeqWell Inc., Beverly, MA). Using the resulting sequence information, we determined the subset of clones that (i) contained a minimum of one missense mutation, (ii) contain no insertions or deletions, (iii) contained no mutations outside of the ORF, (iii) had unique barcodes, and (iv) had sufficient read coverage during kiloSEQ to allow for confident genotyping. This high-quality subset of clones was re-arrayed to form the final clone library.
Complementation assay
The library of mutant clones was pooled and transformed into a mutant yeast strain carrying a temperature-sensitive (ts) allele of UBC9. As positive and negative controls, sets of barcoded null-allele and wild type allele-bearing clones were also added. The pool was then split into six replicate plates; three replicates were grown at the permissive (non-selective) temperature (25°C), and three replicates were grown at the restrictive (selective) temperature (37°C). After 48 hours, the confluent plates were scraped and barcode loci amplified in preparation for next-generation sequencing. Barcode reads were then counted and used to calculate the relative abundance of each clone in the pool for each condition and replicate.
Competitive growth score calculation
For each clone in the assay, a log ratio was calculated of the average barcode read count at the restrictive temperature to the average count at the permissive temperature. These log ratios were then normalized to the log ratios observed for null and wild type controls, respectively, such that the resulting score will be zero if a clone's log ratio matches that of the null control, and it will be 1 if it matches the wild-type UBE2I controls.
To determine whether deviations from the wild type and null controls were significant, a Student’s t-test was used. Benjamini-Hochberg corrected q-values were then derived from the t-test p-values and used to filter the results (q < 0.05). The test revealed that clones that received negative scores due to their apparent growth being weaker than that of the null controls, did not significantly differ from the controls and can thus simply be considered as complete loss of function variants. On the other hand, a number of clones were found to grow significantly faster than the wild type controls.
Clones for which replicate experiments at the permissive temperature yielded fewer than ten barcoded counts were poorly measured, and excluded from the downstream analysis. Because empirical standard deviations calculated for each clone or mutation based on a small number of replicates are expected to be imprecise, we used regularized error estimation(Baldi and Long, 2001).
Three subsets of data were provided in the challenge. Subset 1 is the most accurate and consists of 219 single amino acid mutations for which at least three independent barcoded clones are represented, providing internal replicates of the experiment. Subset 2 contains another 463 single variants, while Subset 3 contains 4,427 mutants with two or more substitutions. To help participants calibrate numeric values, the distribution of experimental growth scores was provided in the challenge.
Positive control and the baseline predictor
To provide a reference for predictions, we defined a positive and a baseline predictor control. The positive control was the “perfect” prediction one would expect when experimental errors were considered. A prediction for each variant in the positive control was a randomly selected value from a Gaussian distribution with the given competitive growth score as mean and the experimental standard error as the standard deviation. The baseline predictor was based on the frequency of amino acids at each position in a UBC9 family multiple sequence alignment (MSA). The MSA was constructed using Promals3D(Pei and Grishin, 2014) from UBC9 and its 228 orthologs/inparalogs from the InPararoid(Sonnhammer and Ostlund, 2015) database (sequences and alignment are included in Supp. Material). For Subsets 1 and 2, predictions were calculated using the following formula:
where Qm and Qw are the estimated probabilities of mutated and wild-type amino acids at a mutated position in the alignment as defined in, and Pm and Pw are Robinson-Robinson background frequencies(Robinson and Robinson, 1991) of the mutated and wild type amino acids. For Subset 3 with multiple mutations, we used the sum of the predictions for each single mutation.
Quantile transformation of original predictions
Most participants ignored calibrating their predictions using the distribution of experimental growth score given to them. Thus, rescaling of predictions was required to make predictors comparable in their scale, which is especially important for numeric comparison. We performed quantile transformation of the original predictions from participants and of our baseline predictor. Because predictors were not allowed to predict negative values and the negative competitive growth scores obtained in experiments did not show statistically significant difference from 0, all negative competitive growth scores were shifted to 0 before transformation. The mutations were ranked by the predicted values, and each mutation was assigned the experimental score with the same rank. The assigned experimental scores for mutants that are predicted to be ties are further averaged to obtain the final transformed predictions.
Scores for prediction assessment
Each method was evaluated by their ability to: (1) to classify mutations into categories such as deleterious and non-deleterious mutations (classification), (2) to rank mutations by their impacts on the protein function (ordinal association) and (3) to predict experimental competitive growth scores (numeric comparison). For the assessment, mutations were assigned by the growth score to the following categories: lower or equal to 0.3 for deleterious, between 0.3 and 0.7 for intermediate, from 0.7 to 1.3 for wild-type, and greater than 1.3 for advantageous. Table 1 summarizes scores for each aspect. Four out of these scores, i.e., Area Under ROC (AUC) for classification of deleterious mutations and the three ordinal association scores, rely on the rank of experimental scores and predictions, both of which contain ties and requires special treatment as noted in Table 1.
Table 1.
Summary of Measurements in Assessments
| Classification | |
|---|---|
| Area Under ROC |
P: number of true deleterious mutations; N: number of true non-deleterious mutations. Mutations are ranked by the predicted growth score. is the count of true deleterious mutations that are ranked no worse than the true non-deleterious mutation. Each true deleterious mutation ranked the same as the true non-deleterious mutation is counted as 0.5. |
| MCC |
i ∊ (deleterious, intermediate, benign and advantageous); TP: true positive; TN: true negative; FP: false positive; FN: false negative. |
| F1 |
, ; TP: true positive; TN: true negative; FP: false positive; FN: false negative. |
| Ordinal association | |
| Kendall tau-b rank correlation | ; nc the number of concordant pairs; , the number of discordant pairs; n, the total number of pairs; , number of values in the group of ties by predictions; , number of values in the group of ties by experimental scores. |
| Spearman’s rank correlation |
, covariance between predicted and experimental ranks of mutants; and , standard deviations of predicted and experimental ranks, respectively. Ties were randomly assigned distinct ranks first and then the average of these ranks were assigned to each of them. |
| Rank agreement test |
is the number of mutants with the difference between the predicted and experimental ranks below a certain cutoff i, 0≤i≤n−1, where n is the total number of mutations in a data set. Ties were randomly assigned distinct ranks. This random assignment was performed 50 times and the resulting scores were averaged. |
| Numeric comparison | |
| Pearson’s correlation | , covariance between predictions and experimental scores; , standard deviation of predictions; , standard deviation of experimental scores |
| RMSD |
N, the size of a dataset; , predictions; , experimental scores |
| Value agreement test |
is the number of mutants with the difference between the predicted and experimental growth scores below a certain cutoff i. The cutoffs are taken from 0 to the number larger than maximal difference between experimental and predicted growth scores in the dataset, with an incremental of 0.01. The normalized area was used as the measurement. |
Evaluation of overall performance and its statistical significance
Four (three scores for ordinal association and Area Under ROC) of the measurements listed in Table 1 were purely based on rank and were not sensitive to the distribution of numeric values. Five others depended on the distribution of numeric values and thus were calculated with both original and quantile-transformed predictions. For each measurement, we transformed the original scores to Z-scores, and positive control and baseline predictor were excluded from the calculation of mean and standard deviation of original scores to avoid their influence on the score distribution. The average Z-scores of the rank-based, original-value-based, and transformed-value-based measurements were computed and summed up to be the final score to assess the performance on each subset. The final assessment score was a weighted sum of the scores from three subsets. Because the experimental competitive scores in Subset 1 were more accurate (with replicates) than those in Subset 2 and 3, Subset 1 was weighted twice as much as the other two subsets.
To take experimental errors into consideration, we assumed that the growth score for each mutant in a dataset (Subset 1, 2, and 3) can be randomly drawn from a Gaussian distribution defined by the reported growth score and the standard error. We repeated this procedure 1000 times to generate 1000 derived datasets from Subset 1. Then, we performed bootstrap resampling on each derived dataset 40 times, and thus generated 40,000 samples from Subset 1. Similarly, we obtained 40,000 samples from Subset 2 but just 200 samples (40 derived datasets each resampled 5 times with bootstrap) for Subset 3 due to time constraints required by the large number of mutants in it. We randomly chose three simulated samples from Subsets 1, 2 and 3 to form a new test set. A total of 40,000 new test sets were generated and used to assess the predictors using the same procedure as described above. We obtained the distribution of ranks for each group on these test sets. In addition, for each pair of groups, we compared their performance on each of the new test sets and counted their number of wins (head-to-head test).
Identification and characterization of well predicted and poorly predicted variants
The absolute difference between the experimental score and transformed prediction was used to assess the prediction quality for each mutation by each group. A heat map was plotted and visualized by ClustVis(Metsalu and Vilo, 2015) to illustrate the prediction quality of each mutation from every predictor and the baseline prediction control. To find common properties shared by well and poorly predicted variants, we calculated conservation by AL2CO(Pei and Grishin, 2001) and relative solvent accessibility of residues by DSSP(Kabsch and Sander, 1983).
Results
UBE2I mutation bias towards being deleterious to competitive growth
The effect of variants in the single-mutation high-accuracy Subset 1 on competitive growth is illustrated in Figure 1A. Out of 219 variants, six without a given competitive growth score or standard error were excluded from the analysis. The competitive growth scores were scaled so that mutant clones with growth identical to a null control were 0 and those with growth identical to a wild-type control were 1. 41% of all high-accuracy Subset 1 mutations were deleterious (Figure 1A).
Figure 1. UBE2I Competitive Growth Score Distribution of Single Mutations.
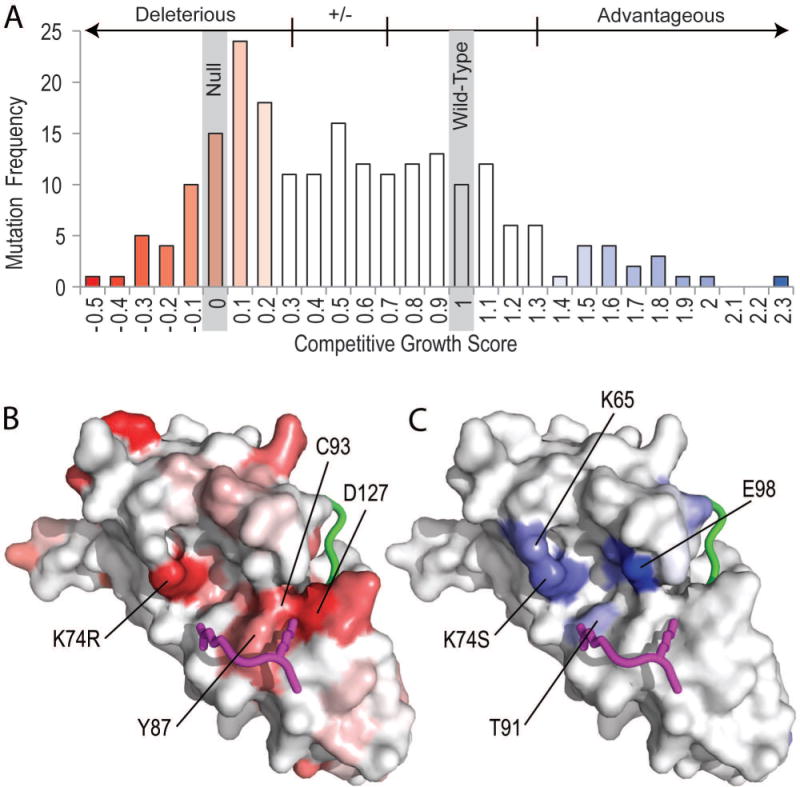
A) A histogram depicts the frequency of competitive growth scores for UBE2I mutations in the high-accuracy Subset1 denoting 213 single amino acid variants for which at least three independent barcoded clones are represented. Four growth response categories grouping mutations are labeled above the graph with their boundaries marked by vertical lines. Bars are colored in gradient from red (growth slower than the null control) to white (+/− growth) for deleterious mutations (−0.5 to 0.3) and from white (wild type) to blue (more growth than wild type) for advantageous mutations (1.3 to 2.3). The same competitive growth score gradient scales are applied to B-factors of corresponding residue positions in surface representations of the UBE2I structure (PDB: 1a3s) for B) deleterious competitive growth score mutations and C) advantageous competitive growth score mutations. The consensus substrate tetrapeptide PsiKxE (magenta, with K and E in stick) and the C-terminal sumo peptide (green) from PDB: 1z5s superimposed with UBE2I highlights the active site. Select residues near the active site are labeled.
The remaining single amino acid variants (Subset 2, 410 informative mutations) without replicates followed a similar distribution (Supp. Figure S1A), with 48% of Subset 2 mutations falling in the deleterious category. For those remaining variants with multiple amino acid substitutions (Subset 3, 3,872 clones with experimental measurements, Supp. Figure S1A), the distribution shifts further towards null (67% of Subset 3 mutations). Thus, almost half of the UBE2I single amino acid mutations and a majority of multiple mutations were detrimental to growth. On the other hand, relatively few UBE2I amino acid variants (42 or 6.7% of single-mutant variants and 209 or 5.4% of multiple-mutant variants) were advantageous.
Deleterious and advantageous mutations mapped to the UBE2I structure suggest functional effects
High-accuracy Subset 1 deleterious mutations mapped to the UBE2I structure distribute across the surface (Figure 1B), with only 8% of the mutations being completely buried (7 out of 87 have solvent accessibility score 0). Many of the mutations cluster around the active site C93, with seven deleterious mutation positions being within 5 Å of the catalytic residue. For example, mutation of either of the two residues (D127 to V or G and Y87 to C or N) that surround the consensus substrate tetrapeptide Lysine residue that gets ligated to the C-terminus of Sumo results in null-level growth. A more conservative mutation of the adjacent Y87 to H results in an intermediate growth phenotype with a score (0.318) close to the deleterious boundary. The distribution of these mutations near the active site suggests that correct positioning of the substrate Lysine is required for wild-type UBE2I activity. As such, a distribution of all variants that include the active site C93 tends to surround the null score 0, with some of the multiple variants extending slightly towards wild-type growth scores (Supp. Figure S1B). The distribution of all variants that include the adjacent D127 is similar to that of the active site C93, except it includes a minor tail that extends towards higher competitive growth scores (Supp. Figure S1C).
A wealth of UBE2I structure information exists to aid in mutation prediction, including the UBE2I structure alone and numerous complex co-crystal structures. Inspection of the co-complexes superimposed using UBE2I highlights binding surfaces on the ligase that overlap with mapped mutations. Quaternary complex structures with RanGap1 substrate, E3 ligase RanBP2 fragments, and SUMO1/2 show subtle conformational changes that help the E3 ligase achieve SUMO specificity(Gareau, et al., 2012) (Figure 2). In this quaternary complex, the SUMO C-terminus is poised to modify the RanGap1 substrate lysine, and the RanBP2 E3 fragments adopt an extended conformation with the N-terminus wrapping around SUMO and C-terminus wrapping around UBE2I. In addition to the substrate lysine binding residues described above, several deleterious mutations map (within 4A) to the RanGap1 substrate binding surface (K74, A129, Q126, Y134, and T135), the SUMO binding surface (N85, S95, R104, I107, L114, G115, L119, and N124), and the E3 ligase binding surface (S2, I4, E12, R13, P28, K59, and S70). The overlap of deleterious mutations with the quaternary complex structure binding sites suggests a similar E3 ligation mode positioning SUMO for ligation to substrate is required for wild type yeast growth.
Figure 2. Advantageous Growth Mutations.
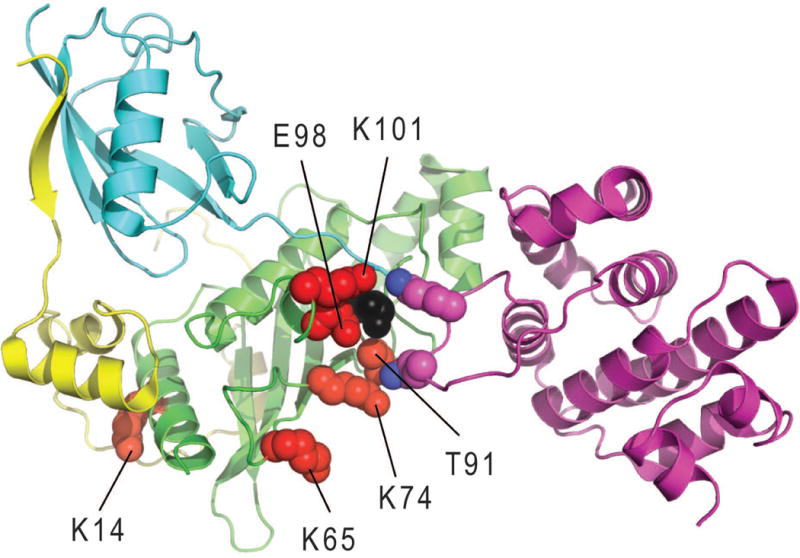
The Ubc9 (green cartoon) structure complex (PDB: 3uio) bound to RanGAP1 (magenta cartoon), SUMO2 (cyan cartoon) and the E3 ligase IR1 domain of RanBP2 (yellow cartoon) highlights positions of residues with advantageous growth mutations (red spheres, labeled according to WT position) with respect to the active site Cys (black sphere) and the consensus substrate tetrapeptide E and K residues (magenta spheres)
In addition to the various UBE2I target structure complexes, binary complex structures of UBE2I with other binding partners are known. A complex with the cys domain of SUMO E1 (SAE1, PDB: 2px9) reveals a common binding surface on UBE2I for this binding partner that overlaps with the SUMO1/2 domains of the quaternary complex competent for modifying substrate. The structure of UBE2I with SUMO-activating enzyme subunit 2 (SAE2, PDB: 2px9), the null growth D127 mutants cannot form hydrogen bonds to an interacting loop of SAE2 in a similar manner as the native residue. The UBE2I SAE2 interaction surface (residues in UBE2I within 4A of SAE2) includes the side chains of seven positions with deleterious mutations and three positions with intermediate mutations, with 5 of these forming side chain specific hydrogen bonds to SAE2 that would be lost in the mutant. The interface also includes five wild type positions having either relatively benign mutations (K48R, K49N, I96L, and K101E) or lacking mutations (P128). Two SAE2 interface residues in UBE2I have mutations (T91A and E98G) that confer advantageous growth, which perhaps suggests that SUMO transfer from E1 improves in these mutants.
Non-covalent (PDB: 2pe6 and 2uyz) and covalently modified (PDB: 2vrr) complex structures between UBE2I and SUMO1 reveal two alternate binding modes for SUMO1 that differ from the modification competent site found in the quaternary structures. Both alternate sites contain a number of additional deleterious mutations: R13, W16, and H20 in the non-covalent site and G3, R13, T35, and L38 in the covalent site. The role of these mutations is less clear, given the fact that each of these sites overlaps with other binding partners. The non-covalent site significantly overlaps with that of the bound E3 ligase RanBP2 C-terminus while the covalent site overlaps with that of a bound RWD domain from RWDD3 (PDB: 4y1l), the ubiquitin conjugating enzyme UBE2K (PDB: 2o25), as well as part of the C-terminal E3 ligase RanBP2 surface. Finally, a complex between UBE2I and importin highlights UBE2I binding components leading to nuclear import (PDB: 2xwu). These interface positions include 15 deleterious mutations (12 with relatively severe alterations from the native structure). The presence of numerous deleterious mutations in the importin interface suggests that USB9 localization to the nucleus is required for the growth phenotype.
The relatively smaller proportion of high-accuracy Subset 1 mutations that are advantageous for growth also distribute across the UBE2I surface, with two of the residues being near the active site (T91 and E98). A distribution of competitive growth scores for all variants that include E98 shifts towards higher scores, displaying a broad second peak in the advantageous growth category (Supp. Figure S1C). Interestingly, both T91 and E98 can provide substrate contacts. For example, T91 forms hydrogen bonds with the consensus substrate tetrapeptide E, while E98 approaches a K just N-terminal to the conserved consensus tetrapeptide in one available UBE2I structure bound to substrate (PDB: 5d2m). Two of the activating mutations, K74 and K65, belong to a basic patch that discriminates negatively charged amino acid-dependent sumoylation motif (NDSM) substrates(Yang, et al., 2006). These NDSM substrates include an additional defined motif PsiKxE(xxSP) whose conserved S gets phosphorylated to promote sumoylation of several substrates(Hietakangas, et al., 2006). Thus, activating mutations appear to discriminate between sumoylation substrates, potentially shifting activity away from phosphorylation-dependent substrates and towards those alternate substrates that contribute to competitive growth.
Negative growth scores and disparate distributions of predicted scores are a challenge for the assessment
Many clones grew slower than the null control, resulting in negative growth scores. FDR-corrected t-tests based on the regularized standard deviations showed that none of these clones' growth scores were significantly different from the null controls. Thus, predictors were not allowed to assign negative growth scores. Unfortunately, this rule required reassignment of all the experimental mutations with negative growth scores. In an attempt to best accommodate predictions provided by participating groups (Figure 3), negative growth scores were shifted to 0, since most of the groups (11 out of 16) submitted multiple 0's and none of them submitted negative predictions. Such a reassignment of experimental growth scores resulted in many ties and required special attention to ties in the assessment (see Methods).
Figure 3. Competitive Growth Score Prediction Distributions.
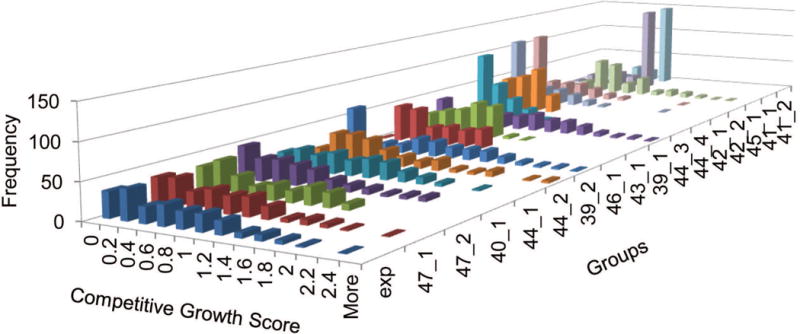
A three-dimensional plot depicting the frequencies (vertical axis) of experimental competitive growth scores (horizontal axis) for Subset1 (exp) alongside all group predictions for competitive growth scores (depth axis). Groups are ordered by their KS test statistic from low (closer to experimental distribution) to high (farther from experimental distribution).
Although the groups were given the distribution of growth scores for each of the subsets, their distributions tended to be significantly different from the experimental distribution, with most of the groups (13) having a Kolmogorov–Smirnov (KS) test P-value less than 0.1 (Supp. Table S1). Only three groups (47_1, 47_2, and 40_1) submitted scores with distributions similar to those provided. Some groups over-predicted null mutations (44_3, 42_1, and 42_2) while others over-predicted wild-type mutations (41_1 and 41_2). Many of the groups also tended to ignore the advantageous mutations (41_1, 41_2, 42_1, 42_2, 44_3, 44_4, 46_1, and 43_1). The difference in these score distributions does not affect assessment based on ranks, but causes problems in evaluation based on numeric values. For instance, we observed that re-scaling of the predicted growth scores to reduce the standard deviation could result in a considerable boost in the performance measured by RMSD between the predicted and experimental values. Therefore, we applied quantile transformation (see Methods) to the predictions to convert all the predictions to the same distribution as the experimental results, and the evaluation was done on both the original and the transformed predictions.
Assessment revealed modest performance comparable between the predictors
The predictors were evaluated by their ability to (1) classify mutations into fitness categories; (2) rank mutations by their effects on fitness (i.e., competitive growth rate of yeast); and (3) numerically predict competitive growth scores of mutants (Table 1). Table 2 summarizes the overall performance of the predictors on three subsets. All participants except group 45 show significantly better than random performance, with the best performing groups being able to rank about 67% (Kendall-tau rank correlation coefficient: 0.338) pairs of single mutants correctly (Supp. Table S2). The RMSD between transformed predictions and experimental scores is significantly better than random for all groups except group 45 (P > 0.05 for both original and transformed predictions). The results show a definite promise of predicting fitness effects of mutations. However, the current accuracy of the predictions is rather low, which is revealed by the large gap between the positive control (see Methods) and predictions. The performances on Subsets 1 and 2 (single mutation) are comparable, and are significantly better than that on Subset 3 (multiple substitutions), reflecting the difficulty in predicting effects of multiple mutations.
Table 2.
Summary of assessments on three subsets.
| CAGI Sumo-Ligase Challenge Performance | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| group | Subset 1 Z-Scores | Subset 2 Z-Scores | Subset 3 Z-Scores | Overall | |||||||||||||
| Rank Avg | Trans Avg | Orig Avg | Sum | Rank | Rank Avg | Trans Avg | Orig Avg | Sum | Rank | Rank Avg | Trans Avg | Orig Avg | Sum | Rank | Sum | Rank | |
| positive | 6.43 | 10.50 | 6.94 | 23.86 | na | 4.40 | 7.24 | 6.26 | 17.90 | na | 27.57 | 24.54 | 13.18 | 65.29 | na | 130.91 | na |
| 43-1 | 0.97 | 0.58 | 1.07 | 2.62 | 3 | 0.51 | 0.33 | 0.98 | 1.82 | 1 | 1.79 | 1.18 | 1.58 | 4.55 | 1 | 11.62 | 1 |
| conserv | 0.71 | 0.43 | 0.51 | 1.65 | 5 | 0.47 | 0.50 | 0.57 | 1.54 | 4 | 1.29 | 0.94 | 0.63 | 2.85 | 2 | 7.69 | 2 |
| 47-2 | 0.99 | 0.94 | 0.95 | 2.87 | 1 | 0.57 | 0.57 | 0.01 | 1.16 | 5 | −0.51 | −0.84 | −0.20 | −1.56 | 11 | 5.35 | 3 |
| 47-1 | 1.00 | 0.92 | 0.77 | 2.68 | 2 | 0.57 | 0.57 | 0.61 | 1.75 | 2 | −0.71 | −0.94 | −0.49 | −2.13 | 12 | 4.99 | 4 |
| 46-1 | 0.69 | 0.60 | 1.01 | 2.30 | 4 | 0.10 | 0.17 | −0.10 | 0.17 | 10 | −0.67 | 0.49 | −0.03 | −0.21 | 7 | 4.57 | 5 |
| 41-2 | 0.40 | 0.46 | 0.25 | 1.11 | 7 | 0.43 | 0.40 | −0.11 | 0.72 | 7 | 1.11 | 0.35 | −0.60 | 0.87 | 5 | 3.80 | 6 |
| 41-1 | 0.40 | 0.46 | 0.25 | 1.12 | 6 | 0.42 | 0.40 | −0.11 | 0.72 | 8 | 0.83 | 0.15 | −1.00 | −0.01 | 6 | 2.94 | 7 |
| 39-2 | 0.17 | 0.33 | −0.23 | 0.27 | 11 | −0.04 | 0.13 | −0.30 | −0.21 | 11 | −0.39 | 1.62 | 0.75 | 1.97 | 4 | 2.29 | 8 |
| 44-4 | −0.15 | 0.16 | 0.56 | 0.56 | 8 | 0.22 | 0.27 | 1.08 | 1.57 | 3 | −0.09 | −0.54 | −0.42 | −1.06 | 9 | 1.64 | 9 |
| 40-1 | 0.27 | 0.05 | 0.15 | 0.46 | 9 | na | na | na | na | na | na | na | na | na | na | 0.93 | 10 |
| 39-1 | 0.11 | 0.28 | −0.81 | −0.42 | 14 | 0.06 | 0.11 | −0.62 | −0.45 | 13 | 0.45 | 1.05 | 0.51 | 2.01 | 3 | 0.72 | 11 |
| 44-1 | −0.13 | 0.16 | −0.09 | −0.07 | 13 | 0.25 | 0.27 | 0.45 | 0.97 | 6 | −0.12 | −0.54 | −0.01 | −0.67 | 8 | 0.17 | 12 |
| 44-3 | 0.31 | −0.04 | 0.08 | 0.35 | 10 | 0.07 | −0.03 | 0.35 | 0.39 | 9 | −0.70 | −0.99 | 0.40 | −1.29 | 10 | −0.20 | 13 |
| 44-2 | 0.31 | −0.04 | −0.26 | 0.01 | 12 | 0.08 | −0.03 | −0.41 | −0.36 | 12 | −0.98 | −1.00 | −0.49 | −2.47 | 13 | −2.82 | 14 |
| 42-2 | −1.21 | −0.82 | −0.70 | −2.72 | 15 | na | na | na | na | na | na | na | na | na | na | −5.44 | 15 |
| 42-1 | −1.43 | −1.16 | −0.71 | −3.31 | 16 | na | na | na | na | na | na | na | na | na | na | −6.61 | 16 |
| 45-1 | −2.69 | −2.87 | −2.28 | −7.84 | 17 | −3.25 | −3.19 | −1.83 | −8.27 | 14 | na | na | na | na | na | −23.95 | 17 |
The scores were divided into three classes (see Methods). rank avg, the average Z scores of measurements including Kendall Tau-B, Spearman’s correlation, rank agreement test and ROC curve for deleterious mutations. Trans Avg, the average Z-score of F1, MCC, RMSD, value agreement test and Pearson’s correlation for transformed predictions, Orig Ave, the average of Z scores of the same measurements as Trans Ave but for original predictions. Overall, the weighted sum of final score from three subsets. 2*Zsubset1+Zsubset 2 +Zsubset3. positive, the positive control excluded in the rank; conserv, the baseline control included in the rank.
Group 43 consistently outperformed most other methods across the three subsets, ranking first in the final assessment. Group 47 was best-performing in Subset 1, but was relatively poor in predicting the effect of multiple mutations. Surprisingly, the performance of our baseline predictor that uses only amino acid frequencies from the multiple alignment was better than many of the more complex predictors on all subsets, ranking second in the overall assessment. To assess how the ranking of predictors is affected by experimental errors or by the set of mutants obtained in the experiment, we performed the same assessment on 40,000 additional simulated experimental datasets (contains Subset 1, 2, and 3, see Methods). The distribution of the ranks for predictors on these simulated datasets is shown in Figure 4A. Except group 43, all others revealed a wide distribution in ranks. Similarly, head-to-head test (Supp. Figure 2A) shows that group 43 ranks better than all other participants on over 99% of simulated datasets respectively, and it ranks better than our baseline control on 90% of the simulated datasets.
Figure 4. Assessments on Predictors.
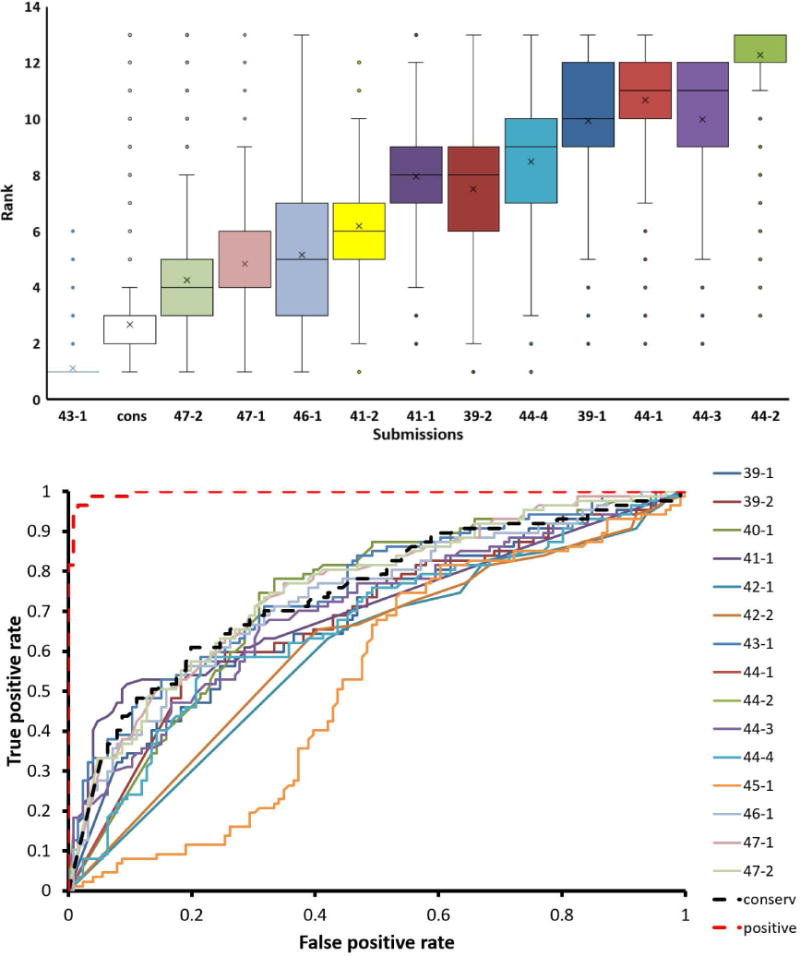
A) Testing sets (from Subset 1,2 and 3) simulated to reflect confidence were repeated to obtain a distribution showing the robustness of relative ranks. The bottom and top of the box represents the first and third quantile of the distributions, respectively. the mean of the distribution (X); the median of the distribution (line); and the outliers which are 1.5 times the length of the interquartile range (circle) are distinguished. The lower rank indicates better performance. B) ROC for deleterious mutations. conserv, the baseline control.
However, the superior performance of Group 43 to group 47 mainly results from its better ability in predicting the effect of multiple mutations (Subset 3). For Subset 3, Pearson’s or Spearman’s correlation coefficients are significantly different between Group 43 and other predictors (44, 46, 47, P < 0.05). In contrast, except Group 45 and 42, the ability for participants to predict the effects of single mutants (Subsets 1 and 2) is comparable (Table 2). Their Pearson or Spearman’s rank correlation coefficients between experimental scores and predictions do not show statistically significant differences (P > 0.05) in subset 1. Besides, Group 47 only marginally outperformed Group 43 in about 57% of 40,000 simulated datasets from Subsets 1 and 2 for single mutations. (Supp. Figure 2B)
Predictors are adequate at detecting deleterious mutations
One of the primary goals for prediction of mutation effects is to identify deleterious mutations. Thus, we specifically evaluated predictors’ ability in detecting mutations that result in a detrimental growth phenotype (growth score ≤ 0.3). The ROC curve of predicting deleterious mutations in Subset 1 by different predictors is shown in Figure 4B and Table S2. Except groups 45 and 42, other groups show comparable and adequate performance (AUC > 0.7 for top groups). In addition, we applied Matthews correlation coefficients (MCC) to evaluate the ability of predictors to partition mutations (Table S3) into the following categories: deleterious, intermediate, benign, and advantageous (see Method). MCC for discriminating deleterious mutations (most are above 0.3 for both original and transformed predictions) is consistently higher than the MCC for distinguishing mutations in other categories (from 0 to 0.2), indicating that the predictors are more reliable in detecting deleterious mutations. However, the ability for predictors to separate intermediate and advantageous mutations is not clearly better than random. This finding was further confirmed by the decrease in the Pearson correlation coefficient (Table S4) between predictions and experimental scores when deleterious mutations were excluded from Subset 1.
Identification and characterization of mutations predicted well and poorly
The heat map of prediction quality by each group on each mutant in Subset 1 is shown in Figure 5A. The mutants form three clusters on the heat map: variants that are well or poorly predicted by almost all predictors formed clusters 1 and 3, respectively (Supp. Figure S3); while cluster 2 contained mutations well or poorly predicted only by some predictors. In accordance with the performance on classification of mutations reflected by MCC, most poorly predicted variants were advantageous while most well predicted variants were deleterious.
Figure 5. Performance Evaluation on Single Mutations.
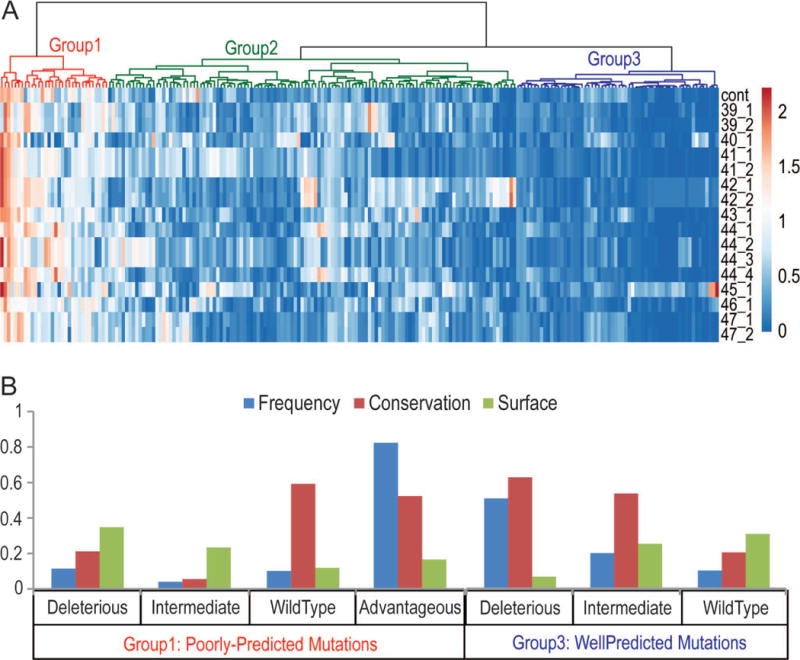
A) The absolute difference between experimental competitive growth scores and that predicted by each group method (using transformed data), as well as a baseline predictor (cont) based on residue frequency in multiple alignment, were calculated to reflect prediction quality for each mutation in Subset1. Difference data was uploaded to the ClustVis web tool to visualize the corresponding heatmap, with mutations (horizontal axis) colored from red (high difference) to blue (low difference). Mutations were clustered (depicted as a tree above the heatmap) using Euclidean distance with Ward minimum variance method linkage criterion. The three largest clusters correspond to overall poor prediction quality (red), intermediate prediction quality (green), and good prediction quality (blue). B) Group 1 poorly-predicted mutations (left, labeled below) and group 3 well predicted mutations (right, labeled below) were split into growth performance categories according to experimental growth score: deleterious, intermediate, wild type, and advantageous (none in well predicted mutations). Properties of the categorized mutations are illustrated in a bar chart: the frequency of mutations in each category with respect to the total in the group (blue bars, Fraction), the average conservation fraction (red bars, Conservation) measured by Al2Co, and the average solvent accessibility fraction (green bars, Surface) measured by DSSP.
Nevertheless, some deleterious mutations were poorly predicted. These mutations were at positions that are less conserved and more accessible to solvent. On the contrary, among wild-type mutations, poorly predicted ones were more conserved with lower relative solvent accessibility than the well predicted mutations (Figure 5B). In addition, among intermediate mutants, the poorly and well predicted ones showed similar solvent accessibility while well predicted intermediate variants were more conserved. Given the fact that solvent accessibility correlated with conservation (Pearson’s correlation=0.5), the sharp contrast between well and poorly predicted mutations in each category suggested that positional conservation in sequence alignment may dominate the predictions.
Discussion
To improve the value of assessment and thus have a positive influence on the development of better predictors, several challenges remain to be overcome. One major challenge is the generation of datasets for testing. Although disease-causing mutations may be most valuable for evaluating the performance of predictors, bias in their assessments may arise due to errors in public databases and possible inclusion of their data in sets against which the predictors are trained. To avoid these problems in the CAGI SUMO conjugase challenge, testing datasets were generated de novo by high-throughput yeast complementation assays. However, mutations may have different effects in yeast and human. Despite the large number of orthologues and a striking conservation of biological processes shared by yeast and human, unique properties exist in both biological systems. The sequence identity between yeast UBC9 and human UBE2I is only about 56%. In addition, orthologues of some interacting partners of UBE2I, such as RanBP2, is missing in yeast (Fauser, et al., 2001) while others like RanGAP1 share low sequence identity. And, interestingly, many substitutions of residues K65, K74 and K76 responsible for interactions between UBE2I with RanGAP1 showed beneficial effects in yeast complementation assays. In addition, these residues are also responsible for recognizing phosphorylated substrates(Gareau and Lima, 2010). Due to the absence of interacting partners in yeast, it is unclear whether these variants would have the same effects in human. For variants of the catalytic site position (C93), the yeast complementation assay showed functional disruptions similarly to human cells(Lin, et al., 2002). In Subset 1 and 2, all variants at the catalytic site were nearly null. The variants at R13 and R17, residues important for interaction with SUMO1 and nuclear import of UBE2I(Tatham, et al., 2003), were also deleterious in Subset 1 and 2. Thus, the yeast complementation assay can be used to identify functional effects of variants with certain limits. This uncertainty is expected to be resolved in future CAGI by using unpublished clinical data or data from human-derived cell lines.
Another problem arising in the CAGI assessment was the difference in numeric scale of predictions. Different numeric scales of predictions can skew the performance. For example, the shrinkage of numeric prediction scale may lead to better RMSD, which does not necessarily reflect better predictions. We tried several transformation methods, including normalization and standardization; however, they did not lead to consistent results in numeric comparison. Therefore, the assessment was affected by transformation and there were possible biases introduced. However, compared with other re-scaling methods, our choice of directly assigning experimental competitive scores to predictions by comparing ranks was expected to minimize the differences between prediction and experimental score distributions. To address possible biases of score transformations, we introduced binary classification and ordinal association scores, which were more tolerant to numeric differences. We also evaluated scores for both original and transformed predictions and included them in the assessment to offset the concerns triggered by transformation. All of these score choices were aimed at removing bias and resulted in reasonable assessments. To address the problem in the future CAGI, a standard and agreed upon re-scaling procedure for submitted predictions could be automatically applied when participants submit their results.
Participants of CAGI4 included five published predictors: SAVER(Adebali, et al., 2016) (group 40), SNAP(Bromberg and Rost, 2007) (group 41), INPS3D(Savojardo, et al., 2016) (group 42), evolutionary action method(Katsonis and Lichtarge, 2014) (group 43), MutPred(Li, et al., 2009) & MutPred2(Pejaver, et al., 2017) (group 44) and four newly developed predictors (groups 39, 45, 46 and 47). These predictors can be classified into three groups: purely sequence-based (groups 39, 40, 41 and 43), those that combine sequence and spatial structure (groups 42, 44 and 46), and meta-predictors that integrate various predictions (groups 45 and 47) (Supp. Table S5). Each predictor offers unique implementation and combination of features. While all predictors use sequence alignments, they differ in how alignments are constructed and how alignment information is used. For instance, groups 39 and 41 aligned all confident HHblits(Remmert, et al., 2011) and PSI-BLAST(Altschul, et al., 1997) hits as defined by E-value cutoff and length coverage, while group 40 differentiated orthologs and paralogs based on phylogenetic trees. Most groups used substitution frequencies to predict the effects of mutations, but group 41 predicted structural (solvent accessibility) and functional (annotation from UniProt) features from the sequences and integrated them with machine learning methods. Solvent accessibility is used by all predictors that incorporate structure information. Other features such as B-factor and secondary structure are also frequently used. In addition, group 46 analyzed biological assemblies and used different interaction interfaces present in all available structures of UBE2I. Most predictors use machine learning methods to integrate various features and predictions. For example, group 47 integrated 12 available predictors including two of the most popular methods SIFT(Kumar, et al., 2009) and POLYPHEN2 (Adzhubei, et al., 2010) using SVM with an RBF kernel.
Performance of most predictors was comparable on single mutations regardless of different factors and methods they used to make predictions. Most predictors could adequately predict deleterious mutations, especially those in conserved positions, and wild-type mutations in non-conserved positions. Similar performance of most predictors suggest predictions may heavily rely on sequence conservation. This idea was further strengthened by the surprising result that a simple conservation-based baseline predictor ranked among the top. This performance suggests that other attributes such as protein structures may not be fully utilized by current predictors. However, whether protein structures can improve predictions significantly is still a matter of debate(Capriotti and Altman, 2011; Kumar, et al., 2009; Saunders and Baker, 2002; Schaefer and Rost, 2012). While some previous studies implied that addition of protein structure considerations to predictions only marginally elevates the performance(Kumar, et al., 2009; Saunders and Baker, 2002), others claimed the introduction of protein structures led to 6% improvements(Capriotti and Altman, 2011). It is possible that the usefulness of protein structures for predictions depends on the protein of interest, the positions of variants, or even on the quality of sequence conservation analysis performed by a predictor. More comprehensive studies are expected to clarify whether the introduction of structural features can contribute to better predictions.
For Subset 3, where each target is a combination of single variants, the performance of predictors decreased. Different summation schemes of predictions on single variants were used to assign the final prediction to the target. The best-performing Group 43 in Subset 3 assigned the sum of the predictions of single variants to the target, while Group 47, which scored best in Subset 1, assigned predictions of most deleterious variant to the target. The discrepancy in performance between these groups suggested that the effects of multiple mutations on fitness may be additive and should be taken into account. However, experiments with our baseline predictor did not support this explanation. We compared the performance of two baseline predictors: the one that assigns the sum of scores for all mutations with the one that assigns the minimal score (i.e., most deleterious mutation only). Performance of the two predictors did not differ significantly (Kendall tau-b values for correlation between predictions and experimental data were 0.17 and 0.18). In addition, the Pearson’s correlation between the two controls was 0.91, suggesting highly similar predictions. Therefore, it remains unclear whether the consideration on additive effects of variants was the major reason that Group 43 had the best performance in Subset 3.
Notably, many variants have high standard errors even in Subset 1 (with several replicates). The small number of replicates is a possible reason. However, the distributions of experimental competitive growth scores for both null mutants and wild-type clones with more than three replicates also showed a wide distribution, implying varying responses from individuals with the same mutation. Such dispersion may be reflective of the measurement precision one might expect in a growth-based assay.
In summary, the SUMO conjugase challenge highlights better performance of methods for predictions of deleterious mutations at conserved sites, the type of mutations that is highly likely to cause disease. It also reveals that substantial improvements of predictions are needed to predict deleterious variants at non-conserved sites and benign mutations at conserved sites.
Supplementary Material
Acknowledgments
Grant number: The CAGI experiment coordination is supported by NIH U41 HG007346 and the CAGI conference by NIH R13 HG006650
References
- Adebali O, Reznik AO, Ory DS, Zhulin IB. Establishing the precise evolutionary history of a gene improves prediction of disease-causing missense mutations. Genet Med. 2016;18(10):1029–36. doi: 10.1038/gim.2015.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alontaga AY, Ambaye ND, Li YJ, Vega R, Chen CH, Bzymek KP, Williams JC, Hu W, Chen Y. RWD Domain as an E2 (Ubc9)-Interaction Module. J Biol Chem. 2015;290(27):16550–9. doi: 10.1074/jbc.M115.644047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi P, Long AD. A Bayesian framework for the analysis of microarray expression data: regularized t -test and statistical inferences of gene changes. Bioinformatics. 2001;17(6):509–19. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell. 2002;108(3):345–56. doi: 10.1016/s0092-8674(02)00630-x. [DOI] [PubMed] [Google Scholar]
- Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35(11):3823–35. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capili AD, Lima CD. Structure and analysis of a complex between SUMO and Ubc9 illustrates features of a conserved E2-Ubl interaction. J Mol Biol. 2007;369(3):608–18. doi: 10.1016/j.jmb.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriotti E, Altman RB. Improving the prediction of disease-related variants using protein three-dimensional structure. BMC Bioinformatics 12 Suppl. 2011;4:S3. doi: 10.1186/1471-2105-12-S4-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745–7. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Boutell C, Hale BG. Interplay between viruses and host sumoylation pathways. Nat Rev Microbiol. 2013;11(6):400–11. doi: 10.1038/nrmicro3015. [DOI] [PubMed] [Google Scholar]
- Fauser S, Aslanukov A, Roepman R, Ferreira PA. Genomic organization, expression, and localization of murine Ran-binding protein 2 (RanBP2) gene. Mamm Genome. 2001;12(6):406–15. doi: 10.1007/s003350010291. [DOI] [PubMed] [Google Scholar]
- Flotho A, Melchior F. Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem. 2013;82:357–85. doi: 10.1146/annurev-biochem-061909-093311. [DOI] [PubMed] [Google Scholar]
- Gareau JR, Lima CD. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol. 2010;11(12):861–71. doi: 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareau JR, Reverter D, Lima CD. Determinants of small ubiquitin-like modifier 1 (SUMO1) protein specificity, E3 ligase, and SUMO-RanGAP1 binding activities of nucleoporin RanBP2. J Biol Chem. 2012;287(7):4740–51. doi: 10.1074/jbc.M111.321141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007;8(12):947–56. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- Gnad F, Baucom A, Mukhyala K, Manning G, Zhang Z. Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genomics. 2013;14(Suppl 3):S7. doi: 10.1186/1471-2164-14-S3-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm DG, Azencott CA, Aicheler F, Gieraths U, MacArthur DG, Samocha KE, Cooper DN, Stenson PD, Daly MJ, Smoller JW, et al. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum Mutat. 2015;36(5):513–23. doi: 10.1002/humu.22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietakangas V, Anckar J, Blomster HA, Fujimoto M, Palvimo JJ, Nakai A, Sistonen L. PDSM, a motif for phosphorylation-dependent SUMO modification. Proc Natl Acad Sci U S A. 2006;103(1):45–50. doi: 10.1073/pnas.0503698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber T, Bohl CR, Lewis GL, Wood C, West JT, Jr, Weldon RA., Jr Human Ubc9 contributes to production of fully infectious human immunodeficiency virus type 1 virions. J Virol. 2009;83(20):10448–59. doi: 10.1128/JVI.00237-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22(12):2577–637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Katsonis P, Lichtarge O. A formal perturbation equation between genotype and phenotype determines the Evolutionary Action of protein-coding variations on fitness. Genome Res. 2014;24(12):2050–8. doi: 10.1101/gr.176214.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, Mooney SD, Radivojac P. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25(21):2744–50. doi: 10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lu J, Prochownik EV. Dual role for SUMO E2 conjugase Ubc9 in modulating the transforming and growth-promoting properties of the HMGA1b architectural transcription factor. J Biol Chem. 2007;282(18):13363–71. doi: 10.1074/jbc.M610919200. [DOI] [PubMed] [Google Scholar]
- Lin D, Tatham MH, Yu B, Kim S, Hay RT, Chen Y. Identification of a substrate recognition site on Ubc9. J Biol Chem. 2002;277(24):21740–8. doi: 10.1074/jbc.M108418200. [DOI] [PubMed] [Google Scholar]
- Martelli PL, Fariselli P, Savojardo C, Babbi G, Aggazio F, Casadio R. Large scale analysis of protein stability in OMIM disease related human protein variants. BMC Genomics. 2016;17(Suppl 2):397. doi: 10.1186/s12864-016-2726-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelotto LG, Ng CK, De Filippo MR, Zhang Y, Piscuoglio S, Lim RS, Shen R, Norton L, Reis-Filho JS, Weigelt B. Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome Biol. 2014;15(10):484. doi: 10.1186/s13059-014-0484-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015;43(W1):W566–70. doi: 10.1093/nar/gkv468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miosge LA, Field MA, Sontani Y, Cho V, Johnson S, Palkova A, Balakishnan B, Liang R, Zhang Y, Lyon S, et al. Comparison of predicted and actual consequences of missense mutations. Proc Natl Acad Sci U S A. 2015;112(37):E5189–98. doi: 10.1073/pnas.1511585112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niroula A, Urolagin S, Vihinen M. PON-P2: prediction method for fast and reliable identification of harmful variants. PLoS One. 2015;10(2):e0117380. doi: 10.1371/journal.pone.0117380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J, Grishin NV. AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics. 2001;17(8):700–12. doi: 10.1093/bioinformatics/17.8.700. [DOI] [PubMed] [Google Scholar]
- Pei J, Grishin NV. PROMALS3D: multiple protein sequence alignment enhanced with evolutionary and three-dimensional structural information. Methods Mol Biol. 2014;1079:263–71. doi: 10.1007/978-1-62703-646-7_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pejaver V, Urresti J, Lugo-Martinez J, Pagel KA, Lin GN, Nam HJ, Mort M, Cooper DN, Sebat J, Iakoucheva LM, Mooney SD. MutPred2: inferring the molecular and phenotypic impact of amino acid variants. bioRxiv. 2017:134981. doi: 10.1038/s41467-020-19669-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Xu J, Aysola K, Begum N, Reddy V, Chai Y, Grizzle WE, Partridge EE, Reddy ES, Rao VN. Ubc9 mediates nuclear localization and growth suppression of BRCA1 and BRCA1a proteins. J Cell Physiol. 2011;226(12):3355–67. doi: 10.1002/jcp.22695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmert M, Biegert A, Hauser A, Soding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2011;9(2):173–5. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- Robinson AB, Robinson LR. Distribution of glutamine and asparagine residues and their near neighbors in peptides and proteins. Proc Natl Acad Sci U S A. 1991;88(20):8880–4. doi: 10.1073/pnas.88.20.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Baker D. Evaluation of structural and evolutionary contributions to deleterious mutation prediction. J Mol Biol. 2002;322(4):891–901. doi: 10.1016/s0022-2836(02)00813-6. [DOI] [PubMed] [Google Scholar]
- Savojardo C, Fariselli P, Martelli PL, Casadio R. INPS-MD: a web server to predict stability of protein variants from sequence and structure. Bioinformatics. 2016;32(16):2542–4. doi: 10.1093/bioinformatics/btw192. [DOI] [PubMed] [Google Scholar]
- Schaefer C, Rost B. Predict impact of single amino acid change upon protein structure. BMC Genomics. 2012;13(Suppl 4):S4. doi: 10.1186/1471-2164-13-S4-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemann AH, Stowell KM. Comparison of pathogenicity prediction tools on missense variants in RYR1 and CACNA1S associated with malignant hyperthermia. Br J Anaesth. 2016;117(1):124–8. doi: 10.1093/bja/aew065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeler JS, Dejean A. SUMO and the robustness of cancer. Nat Rev Cancer. 2017;17(3):184–197. doi: 10.1038/nrc.2016.143. [DOI] [PubMed] [Google Scholar]
- Sonnhammer EL, Ostlund G. InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 2015;43:D234–9. doi: 10.1093/nar/gku1203. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streich FC, Jr, Lima CD. Capturing a substrate in an activated RING E3/E2-SUMO complex. Nature. 2016;536(7616):304–8. doi: 10.1038/nature19071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Yang F, Tan G, Costanzo M, Oughtred R, Hirschman J, Theesfeld CL, Bansal P, Sahni N, Yi S, et al. An extended set of yeast-based functional assays accurately identifies human disease mutations. Genome Res. 2016;26(5):670–80. doi: 10.1101/gr.192526.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatham MH, Kim S, Yu B, Jaffray E, Song J, Zheng J, Rodriguez MS, Hay RT, Chen Y. Role of an N-terminal site of Ubc9 in SUMO-1, -2, and -3 binding and conjugation. Biochemistry. 2003;42(33):9959–69. doi: 10.1021/bi0345283. [DOI] [PubMed] [Google Scholar]
- Thomas PD, Kejariwal A. Coding single-nucleotide polymorphisms associated with complex vs. Mendelian disease: evolutionary evidence for differences in molecular effects. Proc Natl Acad Sci U S A. 2004;101(43):15398–403. doi: 10.1073/pnas.0404380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TJ, Shamsaddini A, Pan Y, Smith K, Crichton DJ, Simonyan V, Mazumder R. A framework for organizing cancer-related variations from existing databases, publications and NGS data using a High-performance Integrated Virtual Environment (HIVE) Database (Oxford) 2014;2014 doi: 10.1093/database/bau022. bau022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Galanis A, Witty J, Sharrocks AD. An extended consensus motif enhances the specificity of substrate modification by SUMO. EMBO J. 2006;25(21):5083–93. doi: 10.1038/sj.emboj.7601383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue P, Melamud E, Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.