

INTRODUCTION
Crouzon syndrome is an autosomal dominant congenital disease due to a single activating point mutation in the fibroblast growth factor receptor 2 (FGFR-2) which causes a variety of craniofacial defects including craniosynostosis, maxillary hypoplasia, ocular proptosis, hypertelorism, and mandibular prognathism[1]. To better understand the role of FGFR2 and the consequences of gain-of-function mutations like those seen in Crouzon syndrome, the same mutation has been introduced in a murine model, resulting in a phenotype similar to that found in humans including hypertelorism, proptosis, and premature fusion of coronal and/or lambdoid sutures[2]. Previous investigations employing this model have shown human and murine adipose derived stem cells (ADSCs) to promote bone regeneration in critical size defects[3–5].
Furthermore, identifying effective biologic agents to incorporate into craniofacial reconstructions is of great clinical importance, reducing morbidity by obviating the need for additional autogenous bone grafting. A recent study by Laino et al. demonstrated equivalent percentage of bone formation in patients treated with autologous bone block from chin area as well as those treated with bone block allograft Puros[6]. Additionally, recombinant human bone morphogenetic protein-2 (rhBMP-2) has been shown to produce excellent results in both the restoration of continuity critical-sized defects of the mandible as well as an adjunct for sandwich osteotomies in immediate distraction osteogenesis[7, 8]. Finally, Petrauskaite et al. demonstrated successful osteoblast adhesion and proliferation on a mineralized cellulose matrix[9].
The purpose of this study is to evaluate the wound healing potential of osteoblasts harboring FGFR2 mutations by creating critical sized defects in the parietal bones of wild type CD-1 and FGFR2C342Y/+ Crouzon mice. We also evaluate the osteogenic potential of ADSCs harvested from fat pads of wild type and Crouzon animals. Finally, we investigate the role of the periosteum in calvarial bone regeneration in Crouzon mice. Due to the nature of the FGFR2 mutation we expect to observe increased bone regeneration in all mice treated with Crouzon osteoblasts when compared to WT osteoblast grafts.
MATERIALS AND METHODS
Wild type (WT) and Crouzon (CRZ) mice were acquired. All Crouzon FGFR2C342Y/+ mice on a CD-1 genetic background were graciously provided by Dr. V.P. Eswarakumar (Yale University School of Medicine, New Haven, CT). The animals were housed per IACUC guidelines, NIH Guide standards, the Animal Welfare Act, and ILAR guide. All animals were given water and food ad libitum. All protocols were approved by the Yale IACUC review board (protocol #2014-11460).
Next, ADSCs were harvested from the fat pads of both WT and CRZ mice. Adipose tissue dissected and excised from wild type CD-1 mice and Crouzon mice was minced into small pieces and digested in an enzymatic solution consisting of 0.15% collagenase Type I (Gibco, Grand Island, NY) in high-glucose Dulbecco’s modified Eagle medium (DMEM, Gibco) for 60 min in a shaking water bath at 37C. The tissue samples were triturated every 15 min. The digestion was stopped by adding fresh culture medium consisting of low-glucose DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS; Lonza, Walkersville, MD), 100 U/mL penicillin (Gibco), and 100 ug/mL streptomycin (Gibc0). Following centrifugation at 1200 rpm for 5 min, the resulting cell pellet was re-suspended in culture medium and filtered. Cells were then seeded in T-75 culture flasks with culture medium changed every 2–3 days. Cells were passaged as necessary when 80–90% confluent.
Critical sized skull defects were created in the parietal bones of all mice. Mice were anesthetized with 1–2% isofluorane and prepped by clipping the fur over the skull with scissors and applying Nair for 1–2 minutes to thoroughly remove all fur over the cranium. The surgical site was prepped with three rounds of ethanol followed by povidone-iodine. A longitudinal incision was made from the posterior aspect of the occiput to a point between the eyes to adequately expose the parietal bones of the skull. The periosteum was dissected off the cranium and pushed aside prior to creating the parietal bone defect. A Synthes Electric Pen Drive System fitted with a 3.75mm diamond-coated trephine drill bit was used to create a defect 3.75mm in diameter. The dura beneath was left intact as evidenced by superficial blood vessels and the bone fragment was gently lifted off the calvarium with jeweler’s forceps. The periosteum was put back in place and the incision closed with 5-0 Vicryl suture. For some experimental animals, the periosteum was removed from the right parietal bone prior to creating the defect.
Mice were either left untreated following surgery, transplanted with WT or CRZ ADSCs with fibrin scaffolds, treated with fibrin glue alone, or underwent removal of the periosteum. For implantation of ADSCs and fibrin glue, 1×106 cells in 10 μL of culture media was added to 10 μL of Evicel fibrin gel (Ethicon, Somerville, NJ) and the mixture was carefully pipetted into the defect. The periosteum was put back into place and the incision closed as outlined above.
Skulls were harvested at 8 and 16 weeks with the skin, muscles, and mandible completely removed. The skulls were preserved in 70% ethanol and imaged using Scanco Medical μCT 40 at -55 V and 145 μA at 16 μm resolution. A blinded investigator analyzed the resulting images using Osirix (64-bit).
Statistical analysis was conducted using SAS 9.3 statistical analysis software (SAS Institute Incorp., Cary, North Carolina, USA). The Analysis of Variance (ANOVA) F-test was used to compare the mean areas of each defect for each group.
RESULTS
Parietal Bone Critical-Sized Defects
Critical-sized defects 3.75 mm in diameter (area of 11.04 mm2) were created in the right parietal bones of CD-1 wild type and FGFR2C342Y/+ Crouzon mice to assess bone regeneration over time. To leave the dura intact, the calvarium was quickly scored with the trephine drill and a pair of jeweler’s forceps was used to fracture the posterior table and remove the bone fragment from the calvarium. The skulls were excised, cleaned, and evaluated with high-resolution computed tomography after 8 and 16 post-operative weeks. The average area of the defect after 8 and 16 weeks in Crouzon mice was 15.37±1.08 mm2 and 16.69±1.51 mm2, respectively (Table 1). Defects created in the skulls of wild type CD-1 mice averaged 14.17±1.88 mm2 and 14.96±2.26 mm2, respectively (Table 1). No statistically significant difference was detected between any of these groups (p=0.451). Interestingly, both groups experienced an increase in the size of the defect at 8 and 16 weeks.
Table 1.
Mean size defects listed by study group.
| Mean Size of Defect | ||
|---|---|---|
| WT | CRZ | |
| Untreated, 8 weeks | 14.17 cm2 | 15.37 cm2 |
| Untreated, 16 weeks | 14.96 cm2 | 16.69 cm2 |
| WT ADSC, 16 weeks | 15.35 cm2 | 12.98 cm2 |
| CRZ ADSC, 16 weeks | 15.47 cm2 | 14.22 cm2 |
| Fibrin glue only, 16 weeks | 14.92 cm2 | 14.43 cm2 |
| Removal of periosteum, 16 weeks | 13.23 cm2 | 13.54 cm2 |
Hematoxylin and eosin staining was used to evaluate the overall tissue composition around the defect while stains with Toluidine Blue and Von Kossa was used to assess bone mineralization along the defect edges (Figure 1). Additionally, immunohistochemistry was used to stain alkaline phosphatase, an osteoblast-specific enzyme, to evaluate osteoblast activity around the defect. After 8 and 16 post-operative weeks, no significant qualitative differences in architecture, bone mineralization or osteoblast activity could be seen between wild type and Crouzon animals. Small nodes of calcification can be seen in the center of the defects of both wild type and Crouzon mice; however, no differences in structure were detected between the groups of mice.
Figure 1.
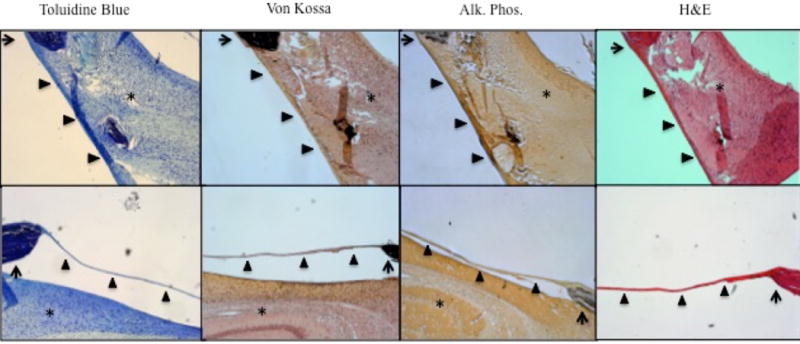
The edge of the defects (arrow) are imaged here. The arrowhead (▲) indicates the periosteum covering the defect, and the asterisks (*) indicate the underlying cerebral cortex. No significant differences in calcification, collagen deposition, or overall bony architecture were observed between wild type CD-1 mice and Crouzon mice.
Crouzon and wild type ADSCs do not promote reossification
Human adipose-derived stem cells (ADSC’s) have been shown to facilitate bone regeneration in murine skull critical-size defects[4]. Therefore, we sought to identify any osteogenic differences between wild type and Crouzon ADSC’s. Murine ADSC’s from wild type and Crouzon mice were harvested and expanded in vitro. These were then mixed with injectable fibrin glue just prior to implantation to act as a scaffold to anchor the stem cells to the defect. The fibrin-stem cell mixture was then gently pipetted into the defects of wild type and Crouzon mice and the defects were allowed to heal for 16 weeks.
Wild-type ADSCs implanted in wild type mice did not facilitate any significant closure of the defect. Instead, we found an average defect size of 15.35±1.34 mm2 after 16 weeks while wild-type ADSCs transplanted in Crouzon mice healed to an average size of 12.98±1.89 mm2 (Table 1). Alternatively, Crouzon ADSCs transplanted into the defects of wild type mice yielded an average area of 15.47±1.29 mm2 while Crouzon ADSCs added to defects created in Crouzon mice healed to an area of 14.22±3.32 mm2 (Table 1). Fibrin glue alone yielded an average defect area of 14.43±4.33 mm2 in Crouzon mice and 14.92±2.32 mm2 in wild type mice (Table 1). As mentioned previously, untreated defects in Crouzon mice averaged 16.69±1.51 mm2 while those in wild type mice averaged 14.96±2.26 mm2 after 16 weeks (Table 1). Statistical analysis with an ANOVA F test did not reveal a statistically significant difference between any of these groups (p=0.415).
The skulls were also evaluated histologically using the same stains described above. Fibrous connective tissue was found spanning the defect; otherwise, no significant histological differences in osteoblast activity, bone organization, or collagen deposition were observed. Moreover, islands of calcification nearly identical to those found in the untreated Crouzon and wild type mice at 8 and 16 post-operative weeks were observed histologically in mice treated with fibrin alone or ADSCs.
The Effects of the Periosteum on Bone Healing
Critical size defects were made in WT and Crouzon mice as previously described; however, the periosteum was completely removed to investigate whether the periosteum of the overlying scalp in Crouzon mice behaves differently than the periosteum of wild type mice. After 16 weeks, Crouzon mice were found to have defects that were 13.54±0.94 mm2 in size while wild type mice displayed defects that were 13.23±1.62 mm2 in size (Table 1). Again, statistical analysis with an ANOVA test did not reveal a statistically significant difference between these groups (p=.0415). Histological evaluation did not reveal a difference in bone formation or architecture between WT and CRZ mice.
DISCUSSION
Crouzon syndrome is caused by an activating mutation to the extracellular ligand-binding domain of FGFR2, resulting in constitutive activation of the receptor[10]. The purpose of this study was to evaluate the healing potential of this mutation applied in a murine model. The identification of sources both capable and incapable of generating accelerated healing rates may guide future efforts toward the development of novel therapeutics. Here we report that a heterozygous C342Y mutation in the FGFR2 protein does not confer any statistical advantage in either mature wild type or Crouzon mice to heal critical size defects made in the right parietal bone. Initial characterization of Crouzon mice noted that homozygous C342Y mutations resulted in ectopic ossification of the vertebra, pelvis, and joints in addition to craniosynostosis and many of the other hallmark features of Crouzon syndrome[11](Figure 2). Heterozygous mice also present with decreased frontal bone and tibial bone cortical density, suggesting ectopic, rather than excess bone formation as one of the primary abnormalities afflicting mice with Crouzon syndrome[11, 12]. Our findings are consistent with this hypothesis. Mice harboring the heterozygous Crouzon mutation did not exhibit excessive bone formation that would enable reossification of critical size defects in the right parietal bone.
Figure 2.
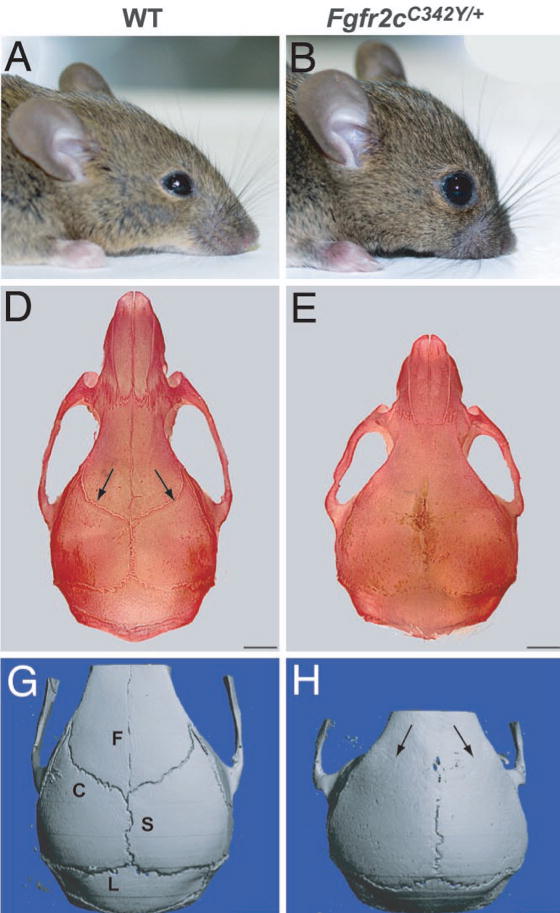
Panel A shows normal wild type CD-1 mouse. Panel B shows Crouzon-mutated mouse with fgfr2C342Y/+ mutation. Panel D indicates presence of coronal sutures in wild type mice with arrows. Panel E shows absence of coronal sutures in a Crouzon-mutated mouse due to premature fusion. Panel G is a 3D model of wild type cranium. Panel H is a 3D model of the cranium in a Crouzon-mutated mouse.
Our results show that not only is there no significant closure of the defects, but that the size of the defects created in the WT group were found to be larger when ADSC transplants were performed than when allowed to heal naturally at 16 weeks. It is possible this phenomenon is due to unintended damage to the underlying dura during trephination in some of the mice, however this is unlikely due to the presence of superficial blood vessels observed during the procedure. It is also possible that lack of irrigation while performing the craniotomies could have caused thermal damage to the surrounding tissue resulting in bone resorption due to necrosis. Lastly, it is important to note that although we appreciated enlargement of the defects in WT mice transplanted with ADSCs, no statistically significant difference in defect size was found. In a study by Cowan et al, ADSCs were shown to have contributed 84–99% of new bone formation by chromosomal detection, resulting in complete bony bridging by 12 weeks. Although we did not observe complete bony bridging in this study, it is unclear how the transplanted ADSCs contributed to the calvarial defect healing process since we did not perform chromosomal detection.
Critical size defects are defined as the smallest sized defect that cannot heal on its own[13]. It is not entirely known how new bone is regenerated to heal such defects in the cranium, but evidence strongly suggests that it likely takes place through interactions between the dura mater and the leading edge of the calvarial bone[14–18]. If these processes are not sufficient to heal the defect, fibrous connective tissue fills the defect permanently, which was found spanning the defects in wild type and Crouzon mice after 8 and 16 weeks[19, 20].
Additionally, small, punctate nodules or islands of mineralized bone were found within this fibrous tissue, suggesting osteoprogenitor cells from the periosteum or the dura mater deposited mineralized bone within the center of the defect (Figure 3). Notably, these nodules did not contribute at all to the closure of the defect. Although the periosteum is necessary for callus formation after long bone fractures, other studies evaluating critical size defects have noted that the periosteum does not produce a robust osteogenic response, suggesting that these nodules are more likely to originate from the dura mater[15, 21–23]. Moreover, the same punctate nodules were still present when the periosteum over the right parietal bone was removed surgically.
Figure 3.

After 16 weeks, fusion of multiple cranial sutures can be seen in 3D reconstructions of the Crouzon skulls. Note the small islands of bone growth in the center of the cranial defects.
In contrast, the dura mater has been shown to play a critical role during reossification of the calvarium[14–18]. This was first noted when craniotomies created in immature rabbits failed to heal when the dura mater below was disturbed[24, 25]. In vitro molecular studies reveal that the immature dura mater expresses osteogenic cytokines such as FGF2 and TGF-β that are necessary for orchestrating successful reossification of calvarial defects[16, 18, 26]. It is known that during craniofacial development, FGF signaling localizes to the dura mater lying below cranial sutures where it serves to regulate osteoblast differentiation and function[27–29]. Therefore, it is no surprise that similar patterns could govern the reossification process in the setting of critical size defects. In vitro co-culture experiments using juvenile dura mater cells and osteoblasts show a clear communication between these two cell populations where the dura mater promotes osteoblast differentiation[18, 30].
In addition to paracrine and/or autocrine signaling mechanisms, the dura mater also appears to contribute cellular elements to the reossification process[17, 31]. Wang et. al. created cranial defects in the parietal bones of rats and separated the dura from the leading bone edge with an impermeable cellulose acetate membrane[17]. They found that new bone was deposited from below, adjacent to the dura, rather than along the leading edge of calvarium, suggesting that the dura mater possesses or recruits cellular components capable of forming bone matrix[17]. Heterotopic transplantation of juvenile dura mater within mesenchymal pockets produces ectopic ossification, while exposure to recombinant human FGF2 in vitro induces immature dural cells to assume a more osteoblast-like phenotype[26, 31]. Thus, significant evidence points to a cellular component either recruited to the dura or emanating from the dura that contributes to reossification. None of the mice in our study displayed any significant degree of reossification; however, the bone nodules within the center of the defect suggest an equally mild, but insufficient response from the dura of both Crouzon and wild type mice. Over the course of 8 weeks, this response eventually ceased resulting in a permanent fibrous non-union, consistent with prior descriptions of unhealed critical size defects.
The inability of the dura to generate a sufficient wound healing response observed in this study is likely due to age-related differences between mature and juvenile dura mater. It is well known that infants before the age of 2 are capable of healing large cranial defects while older children and adults cannot, a pattern that is also found in animal models[26]. Fong et. al. calculated the tangential stretching forces on the dura mater that results as the brain grows and develops and found that the dura mater experiences significant strain (approximately 9.7%) until about 2 years of age[16]. In contrast, after 2 years of age when the brain has completed most of its growth, the dura mater experiences almost no strain (approximately 0.1%) according to their calculations. Thus, an important correlation between strain, age, and osteogenic capacity is clearly evident. Additionally, when immature dural cells were subjected to similar strain in vitro, they exhibited a significant up-regulation of genes involved in osteoblast differentiation and function, which is also consistent with the hypothesis that the dura mater possesses cellular elements that are capable of assuming an osteoblast-like phenotype[16]. Furthermore, molecular studies reveal that immature dural cells proliferate faster than adult dural cells and express significantly more osteogenic cytokines and growth factors such as FGF, TBGβ, osteocalcin, collagen type I, and alkaline phosphatase[18, 32]. Interestingly, adult dura mater transplanted under critical size defects made in immature animals was insufficient to stimulate reossification of the defect[15]. In contrast, juvenile dura mater transplanted in adult critical size defects was sufficient to elicit complete reossification, suggesting that mechanical strain may not be the only factor influencing osteogenesis[33].
These age-related differences in dural cell function could explain the inability of either group to reossify critical size defects in the right parietal bone. The C342Y mutation did not impart any significant improvement in even partial reossification of critical size defects; however, similar studies in immature mice might yield different results. Moreover, the fact that both animals were unable to heal critical size defects may mask any deficiencies in reossification that is associated with the Crouzon mutation. A different model that uses complete bone healing as a control rather than permanent non-union might be more effective. An additional limitation of this study was the lack of irrigation during the craniotomy procedure. Previous authors note the importance of constant saline irrigation to minimize thermal damage to surrounding tissue, including the dura. Due to the small size of the surgical field and the instruments at hand, however, we were unable to utilize this technique. Most groups in our study experienced bone resorption after 8 and 16 weeks, which could represent the area of necrosis that occurred secondary to thermal damage.
Only one study has examined the effects of FGFR-2 activating mutations on dural cells and its effects on paracrine signaling with osteoblasts. Ang et. al. harvested immature dural cells and transfected them with a constitutively active form of FGFR2, the same form found in humans with Crouzon syndrome[34]. These transfected dural cells were co-cultured with normal immature osteoblasts and found to increase osteoblast proliferation and drive the expression of genes involved osteoblast differentiation[34]. This coincides with studies of syndromic craniosynostosis, but not with the results we present here. Again, age-related differences could mask this effect since the dural cells harvested by Ang et. al. were taken from immature rats. Alternatively, dural cells from Crouzon animals may accelerate osteoblast differentiation, but not to a level sufficient to heal a large cranial defect. Unfortunately, there are no molecular and in vitro studies assessing dural cell biology of Crouzon mice.
The effects of periosteum on healing rates of critical sized defects were also evaluated. A previous study by Ozerdem et. al demonstrated periosteal contribution to cranial bone healing in rats[23]. Additionally, Uddstromer demonstrated modest bone formation from isolated periosteum in a rabbit model[22]. Our study does not demonstrate any significant changes in healing rates between groups with or without calvarial periosteum. In the study by Orzedem et. al, the contribution of the periosteum was noted to be synergistic with that of the dura, therefore it is likely that the same factors which contributed to lack of dural significance in this study are the cause of periosteal insignificance as well.
Human ADSCs represent a novel regenerative therapy paradigm applied to a wide range of diseases such as myocardial infarction, osteoarthritis, and diabetes mellitus to name just a few[35]. Therefore, we investigated whether ADSCs harboring the Crouzon syndrome mutation exhibited the same osteogenic potential as ADSCs harvested from wild type mice when implanted within a critical size defect. Previous studies using wild type and normal human ADSCs showed a robust osteogenic response sufficient to completely close critical size defects created in wild type and athymic nude mice after 8 weeks of healing[4, 36]. Other studies have also demonstrated improved osteogenesis with the use of rhBMP-2 in various clinical scenarios including giant cell tumor resection and osteonecrosis of the mandible secondary to bisphosphonate use[37, 38]. Lastly, in a study by Herford et al., rhBMP-2 was shown to induce increased cortical bone formation in primate mandibles[39].
In contrast, wild type ADSCs used in this study were unable to promote any significant degree of reossification in mature wild type or Crouzon mice. Previous studies implanted ADSCs on a poly(lactic-co-glycolic acid) PLGA scaffold[4, 36]. Although the scaffold alone was insufficient to heal critical size defects, it may serve as a better anchor for ADSCs than the fibrin scaffold used here. Scaffolds can vary significantly in their characteristics, which have a wide range of effects on cell behavior. Indeed, material science is a broad field within the realm of tissue engineering, a topic well beyond the scope of this discussion. Prior studies also use in-bred syngeneic mice or athymic nude mice to minimize any host immunity[4, 36]. Instead, CD-1 and Crouzon mice strains used here are not syngeneic; therefore, it is probable that a host immune response was elicited after implanting allogeneic stem cells within the defect. Inflammation is known to interfere with reossification and could have eliminated the stem cells before any benefit could be derived. Creating a line of in-bred Crouzon mice in the future will enable more thorough in vivo studies of the potential and behavior of ADSCs with the Crouzon mutation. Although this study demonstrates a lack of improved osteogenic potential of ADSCs and osteoblasts in CRZ mutated mice compared to WT mice it is limited by the use of a murine model, lack of irrigation during trephination as well as the fact that the mice used for both harvest of ADSCs and observation of defect healing were mature and non-syngeneic.
In conclusion, this study demonstrates that the activating FGFR2 mutation found in Crouzon syndrome does not promote reossification of critical size defects in mature wild type and Crouzon mice. Furthermore, ADSCs harvested from Crouzon mice do not possess any osteogenic advantage over wild type ADSCs. Future studies that use immature, in-bred strains of mice with PLGA scaffolds may prove more fruitful. Other bone healing models may also further elucidate the effects of activating FGFR2 mutations on reossification.
Contributor Information
Andre Alcon, Yale School of Medicine
Philipp Metzler, Yale School of Medicine
Jacob Eswarakumar, Yale School of Medicine
Alexander T. Wilson, Yale School of Medicine
Derek M. Steinbacher, Yale School of Medicine
References
- 1.Carinci F, Avantaggiato A, Curioni C. Crouzon syndrome: cephalometric analysis and evaluation of pathogenesis. Cleft Palate Craniofac J. 1994;31(3):201–9. doi: 10.1597/1545-1569_1994_031_0201_cscaae_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 2.Eswarakumar VP, Horowitz MC, Locklin R, et al. A gain-of-function mutation of Fgfr2c demonstrates the roles of this receptor variant in osteogenesis. Proc Natl Acad Sci U S A. 2004;101(34):12555–60. doi: 10.1073/pnas.0405031101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levi B, James AW, Nelson ER, et al. Acute skeletal injury is necessary for human adipose-derived stromal cell-mediated calvarial regeneration. Plast Reconstr Surg. 2011;127(3):1118–29. doi: 10.1097/PRS.0b013e318205f274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levi B, James AW, Nelson ER, et al. Human adipose derived stromal cells heal critical size mouse calvarial defects. PLoS One. 2010;5(6):e11177. doi: 10.1371/journal.pone.0011177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith DM, Cooper GM, Afifi AM, et al. Regenerative surgery in cranioplasty revisited: the role of adipose-derived stem cells and BMP-2. Plast Reconstr Surg. 2011;128(5):1053–60. doi: 10.1097/PRS.0b013e31822b65e4. [DOI] [PubMed] [Google Scholar]
- 6.Laino L, Iezzi G, Piattelli A, et al. Vertical ridge augmentation of the atrophic posterior mandible with sandwich technique: bone block from the chin area versus corticocancellous bone block allograft–clinical and histological prospective randomized controlled study. Biomed Res Int. 2014;2014:982104. doi: 10.1155/2014/982104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herford AS, Tandon R, Stevens TW, et al. Immediate distraction osteogenesis: the sandwich technique in combination with rhBMP-2 for anterior maxillary and mandibular defects. J Craniofac Surg. 2013;24(4):1383–7. doi: 10.1097/SCS.0b013e318292c2ce. [DOI] [PubMed] [Google Scholar]
- 8.Cicciu M, Herford AS, Cicciu D, et al. Recombinant human bone morphogenetic protein-2 promote and stabilize hard and soft tissue healing for large mandibular new bone reconstruction defects. J Craniofac Surg. 2014;25(3):860–2. doi: 10.1097/SCS.0000000000000830. [DOI] [PubMed] [Google Scholar]
- 9.Petrauskaite O, Gomes Pde S, Fernandes MH, et al. Biomimetic mineralization on a macroporous cellulose-based matrix for bone regeneration. Biomed Res Int. 2013;2013:452750. doi: 10.1155/2013/452750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mangasarian K, Li Y, Mansukhani A, et al. Mutation associated with Crouzon syndrome causes ligand-independent dimerization and activation of FGF receptor-2. J Cell Physiol. 1997;172(1):117–25. doi: 10.1002/(SICI)1097-4652(199707)172:1<117::AID-JCP13>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Nam HK, Wang E, et al. Further analysis of the Crouzon mouse: effects of the FGFR2(C342Y) mutation are cranial bone-dependent. Calcif Tissue Int. 2013;92(5):451–66. doi: 10.1007/s00223-013-9701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Kwon TG, Nam HK, et al. Craniosynostosis-associated Fgfr2(C342Y) mutant bone marrow stromal cells exhibit cell autonomous abnormalities in osteoblast differentiation and bone formation. Biomed Res Int. 2013;2013:292506. doi: 10.1155/2013/292506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hollinger JO, Kleinschmidt JC. The critical size defect as an experimental model to test bone repair materials. J Craniofac Surg. 1990;1(1):60–8. doi: 10.1097/00001665-199001000-00011. [DOI] [PubMed] [Google Scholar]
- 14.Gosain AK, Santoro TD, Song LS, et al. Osteogenesis in calvarial defects: contribution of the dura, the pericranium, and the surrounding bone in adult versus infant animals. Plast Reconstr Surg. 2003;112(2):515–27. doi: 10.1097/01.PRS.0000070728.56716.51. [DOI] [PubMed] [Google Scholar]
- 15.Hobar PC, Schreiber JS, McCarthy JG, et al. The role of the dura in cranial bone regeneration in the immature animal. Plast Reconstr Surg. 1993;92(3):405–10. doi: 10.1097/00006534-199309000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Fong KD, Warren SM, Loboa EG, et al. Mechanical strain affects dura mater biological processes: implications for immature calvarial healing. Plast Reconstr Surg. 2003;112(5):1312–27. doi: 10.1097/01.PRS.0000079860.14734.D6. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Glimcher MJ. Characterization of matrix-induced osteogenesis in rat calvarial bone defects: II. Origins of bone-forming cells. Calcif Tissue Int. 1999;65(6):486–93. doi: 10.1007/s002239900737. [DOI] [PubMed] [Google Scholar]
- 18.Greenwald JA, Mehrara BJ, Spector JA, et al. Immature versus mature dura mater: II. Differential expression of genes important to calvarial reossification. Plast Reconstr Surg. 2000;106(3):630–8. discussion 639. [PubMed] [Google Scholar]
- 19.Schmitz JP, Hollinger JO. The critical size defect as an experimental model for craniomandibulofacial nonunions. Clin Orthop Relat Res. 1986;(205):299–308. [PubMed] [Google Scholar]
- 20.Szpalski C, Barr J, Wetterau M, et al. Cranial bone defects: current and future strategies. Neurosurg Focus. 2010;29(6):E8. doi: 10.3171/2010.9.FOCUS10201. [DOI] [PubMed] [Google Scholar]
- 21.Honma T, Itagaki T, Nakamura M, et al. Bone formation in rat calvaria ceases within a limited period regardless of completion of defect repair. Oral Dis. 2008;14(5):457–64. doi: 10.1111/j.1601-0825.2007.01401.x. [DOI] [PubMed] [Google Scholar]
- 22.Uddstromer L. The osteogenic capacity of tubular and membranous bone periosteum. A qualitative and quantitative experimental study in growing rabbits. Scand J Plast Reconstr Surg. 1978;12(3):195–205. doi: 10.3109/02844317809012995. [DOI] [PubMed] [Google Scholar]
- 23.Ozerdem OR, Anlatici R, Bahar T, et al. Roles of periosteum, dura, and adjacent bone on healing of cranial osteonecrosis. J Craniofac Surg. 2003;14(3):371–9. doi: 10.1097/00001665-200305000-00016. discussion 380–2. [DOI] [PubMed] [Google Scholar]
- 24.Mossaz CF, Kokich VG. Redevelopment of the calvaria after partial craniectomy in growing rabbits: the effect of altering dural continuity. Acta Anat (Basel) 1981;109(4):321–31. doi: 10.1159/000145398. [DOI] [PubMed] [Google Scholar]
- 25.Reid CA, McCarthy JG, Kolber AB. A study of regeneration in parietal bone defects in rabbits. Plast Reconstr Surg. 1981;67(5):591–6. doi: 10.1097/00006534-198105000-00002. [DOI] [PubMed] [Google Scholar]
- 26.Spector JA, Greenwald JA, Warren SM, et al. Dura mater biology: autocrine and paracrine effects of fibroblast growth factor 2. Plast Reconstr Surg. 2002;109(2):645–54. doi: 10.1097/00006534-200202000-00035. [DOI] [PubMed] [Google Scholar]
- 27.Marie PJ. Fibroblast growth factor signaling controlling osteoblast differentiation. Gene. 2003;316:23–32. doi: 10.1016/s0378-1119(03)00748-0. [DOI] [PubMed] [Google Scholar]
- 28.Rice DP, Aberg T, Chan Y, et al. Integration of FGF and TWIST in calvarial bone and suture development. Development. 2000;127(9):1845–55. doi: 10.1242/dev.127.9.1845. [DOI] [PubMed] [Google Scholar]
- 29.Warren SM, Greenwald JA, Nacamuli RP, et al. Regional dura mater differentially regulates osteoblast gene expression. J Craniofac Surg. 2003;14(3):363–70. doi: 10.1097/00001665-200305000-00015. [DOI] [PubMed] [Google Scholar]
- 30.Spector JA, Greenwald JA, Warren SM, et al. Co-culture of osteoblasts with immature dural cells causes an increased rate and degree of osteoblast differentiation. Plast Reconstr Surg. 2002;109(2):631–42. doi: 10.1097/00006534-200202000-00033. discussion 643–4. [DOI] [PubMed] [Google Scholar]
- 31.Yu JC, McClintock JS, Gannon F, et al. Regional differences of dura osteoinduction: squamous dura induces osteogenesis, sutural dura induces chondrogenesis and osteogenesis. Plast Reconstr Surg. 1997;100(1):23–31. doi: 10.1097/00006534-199707000-00005. [DOI] [PubMed] [Google Scholar]
- 32.Greenwald JA, Mehrara BJ, Spector JA, et al. Biomolecular mechanisms of calvarial bone induction: immature versus mature dura mater. Plast Reconstr Surg. 2000;105(4):1382–92. doi: 10.1097/00006534-200004040-00018. [DOI] [PubMed] [Google Scholar]
- 33.Guzel MZ, Yildirim AM, Yucel A, et al. Osteogenic potential of infant dural grafts in different recipient beds. J Craniofac Surg. 1995;6(6):489–93. doi: 10.1097/00001665-199511000-00015. [DOI] [PubMed] [Google Scholar]
- 34.Ang BU, Spivak RM, Nah HD, et al. Dura in the pathogenesis of syndromic craniosynostosis: fibroblast growth factor receptor 2 mutations in dural cells promote osteogenic proliferation and differentiation of osteoblasts. J Craniofac Surg. 2010;21(2):462–7. doi: 10.1097/SCS.0b013e3181cfe9a0. [DOI] [PubMed] [Google Scholar]
- 35.Minteer D, Marra KG, Rubin JP. Adipose-derived mesenchymal stem cells: biology and potential applications. Adv Biochem Eng Biotechnol. 2013;129:59–71. doi: 10.1007/10_2012_146. [DOI] [PubMed] [Google Scholar]
- 36.Cowan CM, Shi YY, Aalami OO, et al. Adipose-derived adult stromal cells heal critical-size mouse calvarial defects. Nat Biotechnol. 2004;22(5):560–7. doi: 10.1038/nbt958. [DOI] [PubMed] [Google Scholar]
- 37.Cicciu M, Herford AS, Juodzbalys G, et al. Recombinant human bone morphogenetic protein type 2 application for a possible treatment of bisphosphonates-related osteonecrosis of the jaw. J Craniofac Surg. 2012;23(3):784–8. doi: 10.1097/SCS.0b013e31824dbdd4. [DOI] [PubMed] [Google Scholar]
- 38.Herford AS, Cicciu M. Recombinant human bone morphogenetic protein type 2 jaw reconstruction in patients affected by giant cell tumor. J Craniofac Surg. 2010;21(6):1970–5. doi: 10.1097/SCS.0b013e3181f502fa. [DOI] [PubMed] [Google Scholar]
- 39.Herford AS, Cicciù M, Eftimie LF, et al. rhBMP-2 applied as support of distraction osteogenesis: a split-mouth histological study over nonhuman primates mandibles. Int J Clin Exp Med. 2016;9(9):17187–17194. [Google Scholar]