

Abstract
Huntington disease (HD) is an inherited neurodegenerative disorder characterized by motor and cognitive dysfunction caused by expansion of polyglutamine (polyQ) repeat in exon 1 of huntingtin (HTT). In patients, the number of glutamine residues in polyQ tracts are over 35, and it is correlated with age of onset, severity, and disease progression. Expansion of polyQ increases the propensity for HTT protein aggregation, process known to be implicated in neurodegeneration. These pathological aggregates can be transmitted from neuron to another neuron, and this process may explain the pathological spreading of polyQ aggregates. Here, we developed an in vivo model for studying transmission of polyQ aggregates in a highly quantitative manner in real time. HTT exon 1 with expanded polyQ was fused with either N-terminal or C-terminal fragments of Venus fluorescence protein and expressed in pharyngeal muscles and associated neurons, respectively, of C. elegans. Transmission of polyQ proteins was detected using bimolecular fluorescence complementation (BiFC). Mutant polyQ (Q97) was transmitted much more efficiently than wild type polyQ (Q25) and forms numerous inclusion bodies as well. The transmission of Q97 was gradually increased with aging of animal. The animals with polyQ transmission exhibited degenerative phenotypes, such as nerve degeneration, impaired pharyngeal pumping behavior, and reduced life span. The C. elegans model presented here would be a useful in vivo model system for the study of polyQ aggregate propagation and might be applied to the screening of genetic and chemical modifiers of the propagation.
Keywords: Aging, Bimolecular Fluorescence Complementation, C. elegans, Huntington disease, Huntingtin, Protein aggregation
Graphical Abstract
INTRODUCTION
The abnormal aggregation and accumulation of specific proteins in the form of cytoplasmic inclusion is common pathological feature of most age-related neurodegenerative diseases, such as Alzheimer disease (AD), Parkinson disease (PD), Huntington disease (HD) and amyotrophic lateral sclerosis (ALS). In general, higher the age, greater the incidence of the disease onset. Protein aggregation is promoted with aging [1], probably due to increased oxidative stress [2,3] and the decline in protein degradation system [4,5].
HD is a representative autosomal dominant inherited disease. Clinically, HD manifests motor defects, including facial convulsions, tremors, and chorea, and non-motor symptoms, such as memory disorders, depression, and anxiety [6]. Classically, this neurological disorder has been associated with the progressive degeneration of the medium spiny neurons (MSNs) in the striatum [7], although evidence states that several brain areas are involved [8,9].
HD has a comprehensible cause, that is, an expansion of CAG repeat in the exon 1 of huntingtin gene, which translates to the expanded polyglutamine (polyQ) tract over pathogenic threshold (Q34) in huntingtin (HTT) protein [10]. Extension of polyQ causes aggregation of HTT protein. The huntingtin aggregates were initially found in the nucleus [11,12] and also discovered in the cytoplasm and the processes of neurons in the brains of HD patients [13]. Although the precise mechanism of neurodegeneration in HD has not been elucidated yet, misfolding and intracellular aggregation of HTT with expanded polyQ is clearly the key element of the pathogenesis and progression of the disease, leading to a toxic gain of function and neuronal cell death [14].
The pathological polyQ aggregates begin to appear in specifically in the putamen and caudate nuclei in the basal ganglia region, and subsequently spread to a wide area of the cerebral cortex, including motor cortex and frontal cortex [15]. It has been suggested that the aggregate spreading is mediated by the direct cell-to-cell transmission of polyQ aggregates, a process that involves seeded polymerization of the protein in a prion like-fashion [16,17,18].
Elucidating the mechanism of aggregation transmission is an essential step towards the understanding of the development and progression of brain diseases. Here, we generated an in vivo model for the cell-to-cell transmission of huntingtin aggregates in Caenorhabditis elegans (C. elegans) using bimolecular fluorescence complementation (BiFC) technique and investigated the effects of polyQ length and aging on huntingtin aggregate propagation and the associated degenerative phenotypes.
MATERIALS AND METHODS
Culturing of nematodes
All worms were maintained, grown, on nematode growth medium (NGM) plates fed with Escherischia coli (E. coli) strain OP50 at 20℃ and were handled by using standard procedures [19]. Wild-type Bristol N2 was obtained from the Caenorhabditis Genetics Center (CGC, University of Minnesota, St. Paul, MN).
Plasmids construction
Plasmids, including Pmyo-2::V1S, Pflp-21::SV2-ICR-DsRed and Pmyo-2::V1Q25 vector, created as previously described [20] were used.
To make vectors expressing the human huntingtin exon 1 with a polyglutamine stretch, a sense primer containing a SalI site, 5′-TAAGCAGTCGACCGCCATGGCGACCCTGGA-3′ and an anti-sense primer containing an XhoI site, 5′-TGCTTACTCGAGAGTGCTTACTCGAGAGGTCGGTGCAGAGGCTCCTC-3′ were used to amplify polyQ products obtained from pcDNA3.1 Myc-His polyQ25 or Q97 vector, which were generous gifts from Dr. George Lawless (UCLA HD group, CA, USA). The human SNCA fragment of Pflp-21::SV2-ICR-DsRed was replaced by the PCR-amplified polyQ fragment to construct Pflp-21::Q25V2-ICR-DsRed, Pflp-21::Q97V2-ICR-DsRed. In addition, to construct Pmyo-2::V1Q25 and Pmyo-2::V1Q97, a sense primer containing a ClaI site, 5′-TAAGCAATCGATATGGCGACCCTGGAAAAGCTG-3′ and an anti-sense primer containing a ClaI site, 5′-TGCTTAATCGATAGGTCGGTGCAGAGGCTCCTC-3′ were used. The plasmid Pmyo-2::V1Q25, Pmyo-2::V1Q97 were generated by replacing SNCA with the HTT-exon1 polyQ fragment in Pmyo-2::V1S.
Establishment of BiFC transgenic lines
The procedure for BiFC transgenic lines was performed as previously described [20]. To analyze the effects of expanded polyglutamine on aggregate transmission, Pmyo-2::V1Q97 and Pflp-21::Q97V2-ICR-DsRed plasmids were co-injected into the gonads of late L4-stage N2 worms with a selection marker, pRF4 which expresses a mutant collagen gene, rol-6(su1006) [21], to make a double transgenic line expressing the BiFC pair and huntingtin exon 1 with a polyglutamine stretch. Additionally, to analyze the effects of polyQ length on aggregates transmission, Pmyo-2::V1Q25 and Pflp-21::Q25V2-ICR-DsRed plasmids were co-injected into N2 worms with pRF4. All of these transgenic worms showed a roller phenotype and expression of DsRed fluorescence in the pharyngeal neurons.
Single-worm PCR (polymerase chain reaction)
Single-worm PCR analyses were performed as previously described [20]. The genomic DNA released from a gravid single worm in each line was mixed with Ex Taq™ polymerase (RR001A; Takara Shuzo Co. Ltd, Shiga, Japan) and then single-worm PCR for target genes was performed in Bio-Rad MyCycler PCR Thermal Cycler system (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Immunoblotting
The procedure for western blotting was performed as previously described [20]. Protein samples obtained from the worm pellets were loaded onto 12% SDS-PAGE gels. Monoclonal anti-polyglutamine primary antibody was used for western blotting (MAB1574; Millipore Corporation, Temecula, CA, USA). Chemiluminescence detection was performed with ECL™ prime solution (RPN2232; GE Healthcare Life Sciences, Marlborough, MA, USA), and images were obtained using the Amersham imager 600 (GE Healthcare Life Sciences, Marlborough, MA, USA) and quantified with Multi Gauge (v3.0) software (Fujifilm, Tokyo, Japan).
Fluorescence microscopy of live worms
Worms were collected, washed with M9 buffer (22 mM KH2PO4, 22 mM Na2HPO4, 85 mM NaCl, 1 mM MgSO4), and then immobilized with 10 mM sodium azide (S2002, Sigma-Aldrich) in M9 buffer. After removing the buffer, drop worms were placed on microscope cover glasses (HSU-0101242; Marienfeld Laboratory Glassware, Lauda-Königshofen, Germany), and covered with a coverslip. All images were obtained using Olympus FV1000 confocal laser scanning microscopy (Olympus, Tokyo, Japan).
Pharyngeal pumping analysis
Pharyngeal pumping rate of each line was monitored for 1 min at room temperature using Axio observer a1 inverted microscope (Carl Zeiss MicroImaging Inc., Göttingen, Germany). The pumping count was symbolized as PPM (Pumps Per Minute).
Life span assay
Life span analyses were performed as previously described [20]. Eggs produced from gravid worms were synchronously grown up to the larval 4 (L4)-stage on NGM plates seeded with E. coli OP50 at 20℃. The L4-stage worms were transferred to NGM plates containing 100 mM 5-fluoro-2′-deoxyuridine (FudR; F0503, Sigma-Aldrich). The number of worms that were alive or dead was recorded every 1~2 days. Worms that ruptured, burrowed, or crawled off the plates were censored but included in the life span analysis as censored animals. The survival data and the mean lifespan assays were analyzed by using online application for the survival analysis (OASIS; http://sbi.postech.ac.kr/oasis/surv/) [22].
Statistical analysis
All experiments were performed blind-coded and repeated at least three times. The graphs were drawn using Prism 5 software (Graphpad Software Inc., San Diego, CA, USA) and the values in the figures are represented as mean±s.e.m. All data were analyzed, compared for statistical significance by one-way ANOVA with Tukey's post-hoc test using Graphpad InStat version 3.05 software (Graphpad Software Inc., San Diego, CA, USA).
RESULTS
Generation of C. elegans model for cell-to-cell transmission of HTT protein
BiFC has been applied to visualization of dimerization and oligomerization of proteins in cells [23]. This technique has also been successfully used to investigate the cell-to-cell transmission of α-synuclein in mammalian cells [24] and C. elegans [20]. To investigate cell-to-cell transmission of HTT aggregates in vivo, we generated transgenic C. elegans lines in which the transmission can be detected real-time in a quantitative manner (Fig. 1A). For the generation of transgenic lines, we constructed a several vectors expressing polyQ proteins conjugated to either N-terminal or C-terminal fragment of Venus. The N-terminal part of Venus (V1) fused to the huntingtin exon 1 with a polyQ tract was expressed in pharyngeal muscle under the myo-2 promoter (Pmyo-2) [25]. And the C-terminal part of Venus (V2) linked to the huntingtin exon 1 with a polyQ stretch and DsRed, which used as a neuronal marker, were co-expressed using the flp-21 promoter (Pflp-21) in neurons that are connected with the pharyngeal muscle (Fig. 1B) [26]. The wild type (V1Q25 and Q25V2) and the mutant pair (V1Q97 and Q97V2) were injected into the L4-stage N2 worms, and several extrachromosomal lines were obtained for each model. The presence of transgenes in these lines was confirmed by single-worm PCR (Fig. 1C). Expression levels of polyQ proteins in pharyngeal muscles were measured by western blotting. We selected three lines that have the similar expression levels in each model for experimental analysis (Fig. 1D).
Fig. 1. Generation of C. elegans model for transmission of HTT aggregates. (A) Scheme of the trans-cellular polyQ transmission model in C. elegans. (B) Transgenes used for generating the BiFC model in C. elegans. (C) Single-worm PCR analysis. Presence of transgenes were validated. V1Q25: 937-bp, V1Q97: 1050-bp; Q97V2: 761-bp, Q25V2: 557-bp. (D) Expression levels of polyQ proteins were measured by immunoblotting in each line. Three lines that have the similar expression levels were selected for experimental analysis (red numbers).

Trans-cellular polyQ transmission in C. elegans
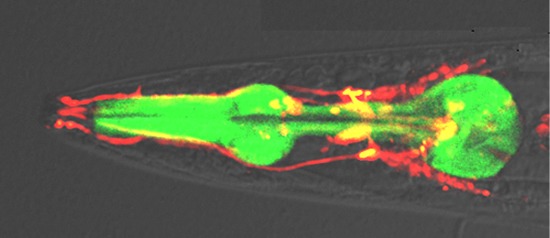
To determine the effects of the length of polyQ on aggregate transmission, we analyzed the BiFC signals in the pharynx using confocal microscopy. The BiFC fluorescence in our system solely depends on the extent of transmission rather than the aggregation propensity, because the cell-autonomous protein aggregation would not generate BiFC fluorescence. All lines we generated produced BiFC fluorescence in both the pharyngeal muscle and neighboring neurons (Fig. 2A). This suggests that the transmission does occur between these two cell types and in a bidirectional manner. However, the lines with long polyQ tracts (Q97) exhibited stronger BiFC fluorescence than the lines with short polyQ length (Fig. 2A and B).
Fig. 2. Cell-to-Cell transmission of the polyQ protein aggregates in C. elegans. (A) Alteration of BiFC fluorescence with aging in the pharynx. The red arrowheads point to BiFC-positive inclusions in the pharynx. Scale bars: 200 µm. (B) Quantification of BiFC fluorescence with aging. Twenty worms for each line were used, *p<0.05. (C-E) BiFC-positive inclusions. Percentage of worms that have BiFC-positive inclusions at different ages (C, D). Worms with BiFC-positive inclusions were quantified separately in individual lines (E). Twenty worms for each line were used, n=3, ***p<0.001.

The transmission of both wild type and mutant polyQ proteins was increased with age. At all ages, the Q97 lines showed higher BiFC signal than the Q25 lines. While transgenic animals with Q25 exhibited largely diffuse patterns in the pharynx until day 8 (Fig. 2A), some inclusions did appear at day 13 in these animals (Fig. 2B). On the other hand, the Q97 animals developed the inclusion bodies as early as two day after the L4-stage (Fig. 2C~E), and the numbers of BiFC-positive inclusion bodies were considerably higher in the Q97 lines compared to the Q25 lines at all ages (Fig. 2C and D). These data indicate that both transgenic animals showed an age-dependent increase in aggregate transmission with polyQ expansion accelerating the rate of aggregate transmission between muscles and adjacent neurons.
Degenerative phenotypes during the polyQ transmission in C. elegans
In order to investigate the effects of different polyQ lengths on the degenerative phenotypes in C. elegans, we first examined nerve degeneration of axonal processes from the URA motor neuron [26] that were labeled with DsRed. While the wild-type N2 (Non-tg) worms showed undamaged axonal processes at day 8 and 13, transgenic animals expressing Q25 protein exhibited a small degree of nerve degeneration, determined by neuritic bleb formation and fragmentation of axonal process, on both day 8 and day13 (Fig. 3A and B). The nerve degeneration was further worsened at all ages when pathogenic huntingtin mutant, Q97, was expressed in muscle and neurons (Fig. 3A and B), showing correlation between the extents of polyQ transmission and those of nerve degeneration.
Fig. 3. Phenotypic changes in C. elegans models during polyQ transmission. (A, B) Nerve degeneration. Axonal processes from the URA motor neurons with DsRed fluorescence were analyzed for nerve degeneration. Worms with nerve fragmentation (A) and axonal blebs (B) in the URA motor neurons were quantified at days 8 and 13. Twenty worms for each line were analyzed; n=3, *p<0.05, ns: not significant. (C) Pharyngeal pumping rates at days 8 and 13. Twenty worms for each line were analyzed; n=3, **p<0.01, ***p<0.001. (D) Life-span analyses. The survival rates of all the lines in respective transgenic groups are plotted. One hundred worms for each line were analyzed. (E) Mean life span. One hundred worms in each line were analyzed; n=3, **p<0.01, ***p<0.001.

To analyze behavioral changes in these animals, we measured the pharyngeal pumping rates. The pumping rates of transgenic worms containing Q25, compared to the wild-type N2, did not change significantly at day 8 (Fig. 3C). However, at day 13, the reduction of pumping rates in Q25 animals had become significant (Fig. 3C). The pumping rates of the Q97 animals were significantly decreased already at day 8 and exhibited more deficits at all ages than the Q25 animals (Fig. 3C).
In life span analyses, transgenic worms with Q25 exhibited a slightly, but not significantly, decreased life span compared to the N2 worms at day 8 (Fig. 3D), whereas the life span of Q97 animals were significantly decreased than the other lines (Fig. 3D). There is no significant difference of the survival rates among three independent lines in each transgenic model, validating consistency of the data. When average life span was concerned, the transgenic lines with Q25 exhibited shortened life span compare with N2 worms, while the Q97 worms showed further reduced mean lifespan (Fig. 3E).
DISCUSSION
Previous studies have shown that aggregates generated by HTT fragments with expanded polyQ propagated between cells in both cultured neurons and animal models [16,17,18,27,28]. This occurs transsynaptically [18] as well as between neurons and glia [17]. In humans, evidence suggested spreading of HTT aggregates, showing the presence of mutant HTT in neuronal grafts in the cortex [29]. In the current study, we developed an in vivo model in C. elegans, in which trans-cellular polyQ transmission can be analyzed in real time in a highly quantitative manner. In the C. elegans model, transmission of polyQ proteins increased with age, and the rate of transmission was higher with mutant polyQ than with wild type polyQ. The rate of transmission correlated with degenerative phenotypes, which include nerve degeneration, impaired pharyngeal pumping, and reduced life span. These findings demonstrate that the C. elegans model represents the trans-cellular aggregate transmission and the associated degenerative phenotypes in a quantitative manner, and thus can be used in screenings for genetic and chemical modifiers of transmission.
Here, we suggested that the rate of transmission correlated with degenerative phenotype. However, overexpression of mutant polyQ can be toxic cell-autonomously even without transmission. Therefore, the degenerative phenotypes we observed may not necessarily be due solely to the transmission of polyQ. In the current study, we cannot rule out the possibility of cell-autonomous cytotoxicity of polyQ proteins.
We chose to use the pharyngeal system to study the relationship between aggregate transmission and the associated phenotypes. The pharynx has its own autonomic nervous system, which are known as pharyngeal nervous system, and its neuronal processes are connected with adjacent pharyngeal muscles [30]. Because pharyngeal specific motor neurons regulate movement of the muscle, degeneration of axonal processes would lead to abnormal motor symptoms and affect the lifespan. One of the strengths of our C. elegans model is that the HTT transmission is associated with nerve degeneration and the behavioral phenotypes. This property of the model allows for the investigation into the relationship between aggregate transmission and neurodegeneration, which has been elusive due to the lack of consistent association between the two events in mouse models. One approach to this problem might be to search for the genetic modifiers that could either dissociate or strengthen the relationship between the HTT transmission and neurodegeneration.
Another strength of the model is the age-dependent manner of aggregate transmission, which would allow for the study of effects of aging, a biggest risk factor for many neurodegenerative diseases. In the previous study using C. elegans, we found that the agingdependent transmission of α-synuclein aggregates was due to the decline of lysosomal degradation activity with aging [20]. Many different neurodegenerative diseases, such as PD and HD, are considered to share the same pathogenic mechanism. In particular, impairment of proteostasis has been the prime suspect for causing aggregate formation and neuronal dysfunction/degeneration [31]. Additionally, intracellular protein aggregation has been shown to cause alteration of the proteostasis system including ubiquitinproteaosomal activity [32,33], dysfunction of autophagy and lysosomes [34], leading to impairment of synapse, axonal transport, and mitochondrial dysfunction [35,36]. Our C. elegans model might be useful in the study of this reciprocal relationship between protein aggregation/transmission and proteostasis impairment.
In conclusion, we have developed a novel in vivo model for the study of HTT aggregate transmission. Our model would be useful not only for identification of pharmacological and genetic modifiers of HTT transmission, but also for understanding the mechanism of HTT-mediated neurodegeneration.
ACKNOWLEDGEMENTS
This work was supported by Research Resettlement Fund for the new faculty of Seoul National University and by the National Research Foundation (NRF) grant funded by the Korean Government (MEST) (NRF-2015R1A2A1A10052540, NRF-2015R1A2A1A15053661).
References
- 1.Li XJ, Li S. Proteasomal dysfunction in aging and Huntington disease. Neurobiol Dis. 2011;43:4–8. doi: 10.1016/j.nbd.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perluigi M, Poon HF, Maragos W, Pierce WM, Klein JB, Calabrese V, Cini C, De Marco C, Butterfield DA. Proteomic analysis of protein expression and oxidative modification in r6/2 transgenic mice: a model of Huntington disease. Mol Cell Proteomics. 2005;4:1849–1861. doi: 10.1074/mcp.M500090-MCP200. [DOI] [PubMed] [Google Scholar]
- 3.Stoy N, Mackay GM, Forrest CM, Christofides J, Egerton M, Stone TW, Darlington LG. Tryptophan metabolism and oxidative stress in patients with Huntington's disease. J Neurochem. 2005;93:611–623. doi: 10.1111/j.1471-4159.2005.03070.x. [DOI] [PubMed] [Google Scholar]
- 4.Holmberg CI, Staniszewski KE, Mensah KN, Matouschek A, Morimoto RI. Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 2004;23:4307–4318. doi: 10.1038/sj.emboj.7600426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walker FO. Huntington's disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 7.Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cudkowicz M, Kowall NW. Degeneration of pyramidal projection neurons in Huntington's disease cortex. Ann Neurol. 1990;27:200–204. doi: 10.1002/ana.410270217. [DOI] [PubMed] [Google Scholar]
- 9.Rosas HD, Liu AK, Hersch S, Glessner M, Ferrante RJ, Salat DH, van der Kouwe A, Jenkins BG, Dale AM, Fischl B. Regional and progressive thinning of the cortical ribbon in Huntington's disease. Neurology. 2002;58:695–701. doi: 10.1212/wnl.58.5.695. [DOI] [PubMed] [Google Scholar]
- 10.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 11.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 12.Becher MW, Kotzuk JA, Sharp AH, Davies SW, Bates GP, Price DL, Ross CA. Intranuclear neuronal inclusions in Huntington's disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis. 1998;4:387–397. doi: 10.1006/nbdi.1998.0168. [DOI] [PubMed] [Google Scholar]
- 13.Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang W, Dunlap JR, Andrews RB, Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet. 2002;11:2905–2917. doi: 10.1093/hmg/11.23.2905. [DOI] [PubMed] [Google Scholar]
- 15.Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol. 2010;11:301–307. doi: 10.1038/nrm2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009;11:219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pearce MM, Spartz EJ, Hong W, Luo L, Kopito RR. Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat Commun. 2015;6:6768. doi: 10.1038/ncomms7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pecho-Vrieseling E, Rieker C, Fuchs S, Bleckmann D, Esposito MS, Botta P, Goldstein C, Bernhard M, Galimberti I, Müller M, Lüthi A, Arber S, Bouwmeester T, van der Putten H, Di Giorgio FP. Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons. Nat Neurosci. 2014;17:1064–1072. doi: 10.1038/nn.3761. [DOI] [PubMed] [Google Scholar]
- 19.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim DK, Lim HS, Kawasaki I, Shim YH, Vaikath NN, El-Agnaf OM, Lee HJ, Lee SJ. Anti-aging treatments slow propagation of synucleinopathy by restoring lysosomal function. Autophagy. 2016;12:1849–1863. doi: 10.1080/15548627.2016.1207014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang JS, Nam HJ, Seo M, Han SK, Choi Y, Nam HG, Lee SJ, Kim S. OASIS: online application for the survival analysis of lifespan assays performed in aging research. PLoS One. 2011;6:e23525. doi: 10.1371/journal.pone.0023525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kerppola TK. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu Rev Biophys. 2008;37:465–487. doi: 10.1146/annurev.biophys.37.032807.125842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bae EJ, Yang NY, Song M, Lee CS, Lee JS, Jung BC, Lee HJ, Kim S, Masliah E, Sardi SP, Lee SJ. Glucocerebrosidase depletion enhances cell-to-cell transmission of alpha-synuclein. Nat Commun. 2014;5:4755. doi: 10.1038/ncomms5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKay JP, Raizen DM, Gottschalk A, Schafer WR, Avery L. eat-2 and eat-18 are required for nicotinic neurotransmission in the Caenorhabditis elegans pharynx. Genetics. 2004;166:161–169. doi: 10.1534/genetics.166.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rogers C, Reale V, Kim K, Chatwin H, Li C, Evans P, de Bono M. Inhibition of Caenorhabditis elegans social feeding by FMRFamide-related peptide activation of NPR-1. Nat Neurosci. 2003;6:1178–1185. doi: 10.1038/nn1140. [DOI] [PubMed] [Google Scholar]
- 27.Costanzo M, Abounit S, Marzo L, Danckaert A, Chamoun Z, Roux P, Zurzolo C. Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J Cell Sci. 2013;126:3678–3685. doi: 10.1242/jcs.126086. [DOI] [PubMed] [Google Scholar]
- 28.Babcock DT, Ganetzky B. Transcellular spreading of huntingtin aggregates in the Drosophila brain. Proc Natl Acad Sci U S A. 2015;112:E5427–E5433. doi: 10.1073/pnas.1516217112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cicchetti F, Lacroix S, Cisbani G, Vallières N, Saint-Pierre M, St-Amour I, Tolouei R, Skepper JN, Hauser RA, Mantovani D, Barker RA, Freeman TB. Mutant huntingtin is present in neuronal grafts in Huntington disease patients. Ann Neurol. 2014;76:31–42. doi: 10.1002/ana.24174. [DOI] [PubMed] [Google Scholar]
- 30.White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- 31.Kim DK, Kim TH, Lee SJ. Mechanisms of aging-related proteinopathies in Caenorhabditis elegans. Exp Mol Med. 2016;48:e263. doi: 10.1038/emm.2016.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verhoef LG, Lindsten K, Masucci MG, Dantuma NP. Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum Mol Genet. 2002;11:2689–2700. doi: 10.1093/hmg/11.22.2689. [DOI] [PubMed] [Google Scholar]
- 33.Ortega Z, Lucas JJ. Ubiquitin-proteasome system involvement in Huntington's disease. Front Mol Neurosci. 2014;7:77. doi: 10.3389/fnmol.2014.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin DD, Ladha S, Ehrnhoefer DE, Hayden MR. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015;38:26–35. doi: 10.1016/j.tins.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 35.Reddy PH, Shirendeb UP. Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington's disease. Biochim Biophys Acta. 2012;1822:101–110. doi: 10.1016/j.bbadis.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]