

Abstract
Emerging evidence has suggested that the gut microbiota contribute to brain dysfunction, including pathological symptoms of Alzheimer disease (AD). Microbiota secrete membrane vesicles, also called extracellular vesicles (EVs), which contain bacterial genomic DNA fragments and other molecules and are distributed throughout the host body, including blood. In the present study, we investigated whether bacteria-derived EVs in blood are useful for metagenome analysis in an AD mouse model. Sequence readings of variable regions of 16S rRNA genes prepared from blood EVs in Tg-APP/PS1 mice allowed us to identify over 3,200 operational taxonomic units corresponding to gut microbiota reported in previous studies. Further analysis revealed a distinctive microbiota landscape in Tg-APP/PS1 mice, with a dramatic alteration in specific microbiota at all taxonomy levels examined. Specifically, at the phylum level, the occupancy of p_Firmicutes increased, while the occupancy of p_Proteobacteria and p_Bacteroidetes moderately decreased in Tg-APP/PS1 mice. At the genus level, the occupancy of g_Aerococcus, g_Jeotgalicoccus, g_Blautia, g_Pseudomonas and unclassified members of f_Clostridiale and f_Ruminococcaceae increased, while the occupancy of g_Lactobacillus, unclassified members of f_S24-7, and g_Corynebacterium decreased in Tg-APP/PS1 mice. A number of genus members were detected in Tg-APP/PS1 mice, but not in wild-type mice, while other genus members were detected in wild-type mice, but lost in Tg-APP/PS1 mice. The results of the present study suggest that the bodily microbiota profile is altered in Tg-APP/PS1 mice, and that blood EVs are useful for the metagenome analysis of bodily microbiota in AD.
Keywords: Alzheimer Disease, Microbiota, Metagenomics
Graphical Abstract
INTRODUCTION
Alzheimer disease (AD) is a neurodegenerative disease characterized by Aβ plaque deposition and cognitive impairment. Recent studies have indicated that the gut microbiota contribute to brain dysfunction in various brain disorders [1,2,3]. The recent progress in studies of microbiota composition changes in AD has been remarkable. An analysis of selected gut microbiota from the stool of AD patients revealed an increase of Escherichia/Shigella and a decrease of Eubacterium rectale [4]. DNA sequencing analysis of 16S ribosomal genes from post-mortem brain extracts of AD patients identified a number of signatures of gut microbiomes in the brain [5]. Sequence readings of 16S ribosomal RNA genes from fecal samples manifested a remarkable shift in gut microbiota in APP/PS1 mice, whereas cerebral Aβ amyloid pathology was reduced in APP/PS1 mice raised in the germ-free condition [6]. When 3xTg-AD mice were treated with a SLAB51 probiotic formulation, the compositions of their gut microbiota changed, Aβ aggregate accumulation was reduced, and cognitive decline was delayed [7]. Sequence readings of 16S ribosomal RNA genes from fecal samples indicated that the genera Odoribacter and Helicobacter increased in APP/PS1 mice, while the genus Prevotella decreased in APP/PS1 mice [8]. Sequence readings of 16S ribosomal RNA genes from selected phyla and species in fecal samples of 5xFAD mice indicated that the phylum Firmicutes increased, while the phylum Bacteroidetes decreased, and Clostridium leptum increased [9]. Overall, the available information suggests that microbiota compositions are altered in AD patients and AD animal models, although detailed microbiota profiles are complex and require further studies.
Metagenome analyses of bodily microbiota are carried out mostly with fecal samples or small-intestine fluid. Such approaches directly provide the landscape of gut microbiota profiles. Recent studies have suggested that blood serum contains bacterial genome DNA fragments. Microbiomes secrete membrane vesicles, also called extracellular vesicles (EVs) or shedding microvesicles [10,11]. Gram-negative bacterial EVs are small in size (10 to 300 nm in diameter) and have a lipopolysaccharide (LPS)-containing outer membrane and periplasmic constituents [11,12]. Gram-positive bacterial EVs are also small in size (50~150 nm in diameter) [13,14]. Bacterial EVs contain bacterial DNA fragments, RNAs, adhesins, toxins, lipoproteins, phospholipids, peptidoglycans and immunomodulatory compounds [10,13,14,15,16,17]. Due to their small size, bacterial membrane vesicles are permeable to the cellular membrane of the intestinal barrier, and are thereby distributed throughout the body including blood [18,19]. These results suggest that blood serum can be used as an alternative route for metagenome analysis of bodily microbiota.
In the present study, we investigated whether bacterial EVs in blood would be useful for the assessment of bodily microbiota in Tg-APP/PS1 mice.
MATERIALS AND METHODS
Animals
Tg-APPswe/PS1dE9 (Tg-APP/PS1 for short) mice [20] were obtained from the Jackson Laboratories (Bar Harbor, ME, USA). Tg-APP/PS1 mice were crossed with C57BL6 mice for more than 10 generations. For genotyping, the following primer sets were used; 5′-AGGACTGACCACTCGACCAG-3′ and 5′-CGGGGGTCTAGTTCTGCAT-3′ for APP; 5′-AATAGAGAACGGCAGGAGCA-3′ and 5′-GCCATGAGGGCACTAATCAT-3′ for PSEN. Two to three mice were housed per cage under a 12 h light/dark cycle in a humidity- and temperature-controlled room, and were allowed access to a diet of lab chow and water ad libitum. All animals were handled in accordance with the animal care guidelines of the Ewha Womans University (IACUC 2013-01-007).
Isolation of bacterial EVs and extraction of DNA from mouse serum samples
Bacterial EVs were isolated from the sera as described previously [21,22,23]. Male Tg-APPswe/PS1dE9 mice were sacrificed at the age of 8 months and blood was collected from the hearts of sacrificed mice. Collected blood was centrifuged at 1,500 × g for 15 min at 4℃, and sera were separated, frozen and stored at −70℃ until use.
The sera were diluted 1 in 3 with 1x PBS (pH 7.4; ML008-01, Welgene Inc., Gyeongsan, Korea) and centrifuged at 10,000 × g for 1 min at 4℃. Then, the supernatants were collected and filtered through a 0.22-µm filter to remove bacteria and foreign particles. The separated bacterial EVs were boiled at 100℃ for 40 min. They were then centrifuged at 13,000 rpm for 30 min at 4℃, and the supernatants were collected. Bacterial DNA was extracted from the boiled EVs with a PowerSoil DNA Isolation Kit (MO BIO Laboratories Inc., Carlsbad, CA USA) in accordance with the manufacturer's instructions. The DNA from the EVs in each sample was quantified with a QIAxpert system (QIAGEN, Hilden, Germany).
PCR amplification and sequencing of variable regions of the 16S rRNA gene
Bacterial DNA was extracted from isolated EVs with a genomic DNA extraction kit (Bioneer Inc., Korea) as described previously [21]. PCR amplification of bacterial 16S ribosomal RNA genes was carried out with the primer set of 16S_V3_F (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′) and 16S_V4_R (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′), which were specific for the V3-V4 hypervariable regions of 16S rDNA.
The libraries were prepared with PCR products in accordance with the MiSeq System guide (Illumina Inc., San Diego, CA, USA), and the prepared libraries were quantified with a QIAxpert (QIAGEN, Germany). The prepared libraries were pooled at equimolar ratios, and sequenced on a MiSeq (Illumina Inc.) in accordance with the manufacturer's recommendations.
Taxonomic assignments by sequence reads of 16S rRNA genes and following microbiota analysis
Taxonomic assignments were made by sequence reads of the 16S rRNA genes as described previously [21]. Briefly, the pyrosequencing reads obtained were filtered bythe barcode and primer sequences on a MiSeq (Illumina, USA). Taxonomic assignment was performed with the profiling program MDx-Pro ver.1 (MD Healthcare, Seoul, Korea). Briefly, the quality of sequence reads was controlled through the inclusion of sequences with read lengths longer than 300 bp and average PHRED scores higher than 20. Operational taxonomy units (OTUs) were clustered by the sequence clustering algorithm CD-HIT. Subsequently, taxonomy assignments were achieved by means of UCLUST and QIIME on the basis of sequence similarities with the 16S rDNA sequence database of GreenGenes 8.15.13. In the event that clusters could not be assigned at the genus level due to the lack of sequences in the database or redundant sequences, the taxon was assigned at a higher level and indicated in parentheses. Taxonomic assignments were achieved on the basis of similarities at the following levels: genus, >94% similarity; family, >90% similarity; order, >85% similarity; class, >80% similarity; and phylum, >75% similarity.
Statistical analysis
Data are presented as the mean percentage±SEM. The z-score was calculated as X − µ/σ, where µ=mean; X=individual score; σ=standard deviation (SD).
RESULTS
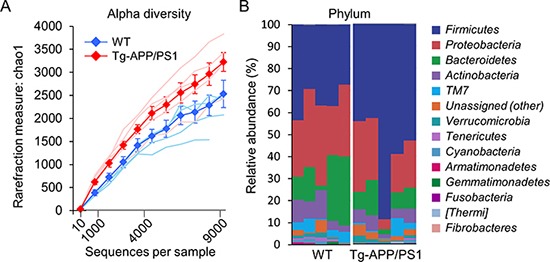
Tg-APP/PS1 mice show plaque deposition in the brain starting from 6.5 months of age and severe cognitive deficits at 7~7.5 months [24,25,26]. Bacterial EVs in blood were collected from 8-month-old Tg-APP/PS1 mice and their wild-type controls, and bacterial DNA was extracted from prepared EVs as described in the Materials and Methods. PCR amplification of variable regions of 16S ribosomal RNA (rRNA) genes and subsequent sequence readings led us to identify approximately 2,500 and 3,200 OTUs for wild-type and Tg-APP/PS1 mice, respectively. Although a slightly higher number of OTUs was identified for Tg-APP/PS1 mice than for controls, the difference was not statistically significant (Fig. 1A). Among the identified OTUs, 30 OTUs at the phylum level, 72 OTUs at the class level, 133 OTUs at the order level, 263 OTUs at the family level, and 571 OTUs at the genus level were assigned to either wild-type or Tg-APP/PS1 mice. Among the identified OTUs, in the present study we focused on the OTUs with occupancies ≥0.1% at each taxon level.
Fig. 1. The diversity and composition of microbiota at the phylum level in wild-type and Tg-APP/PS1 mice. (A) Rarefaction curves representing the number of OTUs for each sample (fine lines) and the mean OTUs (thick lines and filled circles) +/− SEM over the identified sequences in wild-type control (blue) and Tg-APP/PS1 mice (red) (n=5, each). (B) Clustered stacked columns representing the overall compositions of microbiomes at the phylum level in wild-type control and Tg-APP/PS1 mice. Those of occupancies 0.1% and higher in wild-type control and/or Tg-APP/PS1 mice are presented. (C) The percent compositions, fold-changes and z-scores of microbiomes at the phylum level in wild-type and Tg-APP/PS1 mice. Data are presented as the mean percentage +/− SEM (n=5, each). The z-score is X − µ/σ, where µ=mean; X=individual score; σ=standard deviation (SD). Increases and decreases of the mean occupancy by more than one z in Tg-APP/PS1 are marked by ↑ and ↓, respectively. « and » denote infinite increases and decreases, respectively.

Among the identified OTUs at the phylum level, 14 phylum members had occupancies ≥0.1% in either wild-type or Tg-APP/PS1 mice (Fig. 1B). The top 5 phylum members (p_Firmicutes, p_Proteobacteria, p_Bacteroidetes, p_Actinobacteria, and p_TM7[Saccharibacteria]) comprised nearly 95% of the identified OTUs in both wild-type and Tg-APP/PS1 mice, whereas their relative occupancies differed between the two groups. The following phylum members were notable because their occupancies in Tg-APP/PS1 mice differed by more than one standard deviation (SD; one z-score) from those in wild-type mice. The occupancy of p_Firmicutes increased from 34.7 to 57.5% in Tg-APP/PS1 mice, whereas the occupancy of p_Proteobacteria decreased from 30.5 to 20.7% (Fig. 1C). In addition, the occupancies of p_Fibrobacteres and p_[Thermi] (Deinococcus-Thermus) increased from 0 to 0.1% and 0.2%, respectively, in Tg-APP/PS1 mice, whereas the occupancy of p_Tenericutes decreased from 0.9 to 0.3% (Fig. 1C).
The microbiota for which occupancies in Tg-APP/PS1 mice were altered at the class, order, and family levels are further specified and summarized in Table 1. Among p_Firmicutes members, the changes in c_Bacilli is remarkable, particularly the increase of f_Aerococcaceae and f_Leuconostocaceae and the decrease of f_Lactobacillaceae in Tg-APP/PS1 mice. Among p_Proteobacteria, f_Sphingomonadaceae, f_Comamonadaceae and f_Rhodocyclaceae decreased in Tg-APP/PS1 mice, whereas f_Pseudomonadaceae and f_Caulobacteraceae increased. Among p_Bacteroidetes, f_S24-7 and f_Flavobacteriaceae decreased in Tg-APP/PS1 mice, whereas f_Cytophagaceae and f_Sphingobacteriaceae increased. Among p_Actinobacteria, f_Corynebacteriaceae notably decreased in Tg-APP/PS1 mice (Table 1).
Table 1. The percent compositions of microbiomes at the levels of class, order, and family in wild-type and Tg-APP/PS1 mice.
| Phylum | Clas | Order | Family | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Taxon | Occupancy (%; Mean) | z-score | Taxon | Occupancy (%; Mean) | z-score | Taxon | Occupancy (%; Mean) | z-score | Taxon | Occupancy (%; Mean) | z-score | ||||
| WT | Tg | WT | Tg | WT | Tg | WT | Tg | ||||||||
| Firmicutes | 34.7 | 57.5 | ↑ 3.4 | Clostridia | 18.1 | 25.9 | ↑ 1.4 | Clostridiales | 18.1 | 25.9 | ↑ 1.4 | *Unidentified | 4.7 | 7 | ↑ 1.1 |
| Lachnospiraceae | 3.1 | 5.9 | ↑ 1.5 | ||||||||||||
| [Mogibacteriaceae] | 0 | 0.1 | ↑ << | ||||||||||||
| Eubacteriaceae | 0 | 0.1 | ↑ << | ||||||||||||
| Bacilli | 16 | 30.3 | ↑ 5.4 | Lactobacillales | 8.3 | 17 | ↑ 3.2 | Lactobacillaceae | 5.7 | 3.1 | ↓ −1.7 | ||||
| Aerococcaceae | 0.6 | 11.1 | ↑ 10.6 | ||||||||||||
| Leuconostocaceae | 0 | 0.3 | ↑ 4.2 | ||||||||||||
| Bacillales | 7.3 | 12.9 | ↑ 1.1 | Staphylococcaceae | 6.4 | 12 | ↑ 1 | ||||||||
| Other | 0 | 0.3 | ↑ 27.8 | Other | 0 | 0.3 | ↑ 27.8 | ||||||||
| Proteobacteria | 30.5 | 20.7 | ↓ −1.6 | Gammaproteobacteria | 14.4 | 13 | Pseudomonadales | 9.1 | 8.8 | Pseudomonadaceae | 3.1 | 4.5 | ↑ 1.4 | ||
| Xanthomonadales | 0 | 0.1 | ↑ 5.6 | Xanthomonadaceae | 0 | 0.1 | ↑ 5.6 | ||||||||
| Alphaproteobacteria | 10.2 | 3.7 | Sphingomonadales | 3.8 | 0.7 | ↓ −1.1 | Sphingomonadaceae | 3.8 | 0.7 | ↓ −1.1 | |||||
| Rhizobiales | 1 | 1.4 | Bartonellaceae | 0 | 0.1 | ↑ << | |||||||||
| Caulobacterales | 0.2 | 0.9 | ↑ 2.1 | Caulobacteraceae | 0.2 | 0.9 | ↑ 2.1 | ||||||||
| Betaproteobacteria | 4.7 | 3.4 | Burkholderiales | 4.2 | 3.2 | Comamonadaceae | 2.5 | 1.1 | ↓ −1.1 | ||||||
| Rhodocyclales | 0.2 | 0 | ↓ >> | Rhodocyclaceae | 0.2 | 0 | ↓ >> | ||||||||
| Bacteroidetes | 17.8 | 8.7 | Bacteroidia | 17.5 | 7.9 | Bacteroidales | 17.5 | 7.9 | S24-7 | 6.3 | 1.8 | ↓ −1 | |||
| [Odoribacteraceae] | 0 | 0.1 | ↑ << | ||||||||||||
| Flavobacteriia | 0.3 | 0.3 | Flavobacteriales | 0.3 | 0.3 | Flavobacteriaceae | 0.2 | 0 | ↓ >> | ||||||
| Cytophagia | 0 | 0.2 | ↑ << | Cytophagales | 0 | 0.2 | ↑ << | Cytophagaceae | 0 | 0.2 | ↑ << | ||||
| Sphingobacteriia | 0 | 0.2 | ↑ 2.6 | Sphingobacteriales | 0 | 0.2 | ↑ 2.6 | Sphingobacteriaceae | 0 | 0.2 | ↑ 2.6 | ||||
| Actinobacteria | 7.7 | 5.2 | Actinobacteria | 6.9 | 4.3 | Actinomycetales | 6.1 | 3.3 | Corynebacteriaceae | 3.5 | 0.9 | ↓ −1.4 | |||
| Micrococcaceae | 0 | 0.1 | ↑ << | ||||||||||||
| Nocardioidaceae | 0 | 0.1 | ↑ 6.2 | ||||||||||||
| TM7 | 3.8 | 2.6 | TM7-3 | 3.8 | 2.6 | *Unidentified | 0 | 0.2 | ↑ << | *Unidentified | 0 | 0.2 | ↑ << | ||
| Tenericutes | 0.9 | 0.3 | ↓ −1.1 | Mollicutes | 0.9 | 0.3 | ↓ −1.1 | RF39 | 0.9 | 0.3 | ↓ −1.1 | *Unidentified | 0.9 | 0.3 | ↓ −1.1 |
| Cyanobacteria | 0.9 | 0.5 | 4C0d-2 | 0 | 0.2 | ↑ 1.4 | YS2 | 0 | 0.2 | ↑ 1.4 | *Unidentified | 0 | 0.2 | ↑ 1.4 | |
| Gemmatimonadetes | 0.2 | 0 | Gemmatimonadetes | 0.2 | 0 | ↓ >> | Gemmatimonadales | 0.1 | 0 | ↓ >> | *Unidentified | 0.1 | 0 | ↓ >> | |
| *Unidentified | 0.1 | 0 | ↓ >> | *Unidentified | 0.1 | 0 | ↓ >> | ||||||||
| [Thermi] | 0 | 0.2 | ↑ << | Deinococci | 0 | 0.2 | ↑ << | Deinococcales | 0 | 0.2 | ↑ << | Deinococcaceae | 0 | 0.2 | ↑ << |
| Fibrobacteres | 0 | 0.1 | ↑ << | TG3 | 0 | 0.1 | ↑ << | TG3-1 | 0 | 0.1 | ↑ << | TSCOR003-O20 | 0 | 0.1 | ↑ << |
| Summation | 96.5 | 95.6 | 93.1 | 92.6 | 77.3 | 83.9 | 41.5 | 51.8 | |||||||
The family members of microbiomes for which occupancy differed by more than one standard deviation (SD; one z) in Tg-APP/PS1 mice from that of wild-type mice are presented, along with the higher taxonomy levels. Those occupying 0.1% and higher in either wild-type or Tg-APP/PS1 mice were included.
Data are presented as the mean percentages and z-scores. The z-score is X − µ/σ, where µ=mean; X=individual score; σ=standard deviation (SD). Increases and decreases in the mean occupancy by more than one z in Tg-APP/PS1 are indicated by ↑ and ↓, respectively. « and » denote infinite increases and decreases, respectively.
1)APPswe/PSEN1dE9; 2)APPswe/PSEN1-L166P
We continued to analyze the occupancies of specific genus members in Tg-APP/PS1 mice relative to wild-type controls. Of the 130 genus members with relative occupancies ≥0.1% in either wild type or Tg-APP/PS1 mice, 39 members were upregulated, and 16 members were downregulated in Tg-APP/PS1 mice, by more than one SD relative to wild-type mice, while the remaining 70 members were unchanged in Tg-APP/PS1 mice. Of these altered genus members, 17 were newly detected in Tg-APP/PS1 mice, but not in wild-type mice, while 12 were detected in wild-type mice, but lost in Tg-APP/PS1 mice (Table 2). The heat-map analysis in Fig. 2A represents these changes pictorially (Fig. 2A). The total relative occupancy of these 55 (39+16) altered genus members was 29.9% in wild-type mice and 52.2% in Tg-APP/PS1 mice (Table 2). Of the 55 altered genus members, 9 had occupancies ≥0.1% in either wild-type or Tg-APP/PS1 mice. Specifically, g_Corynebacterium decreased from 3.50% in wildtype mice to 0.94% in Tg-APP/PS1 mice, an unclassified member of f_S24-7 decreased from 6.30 to 1.81%, and g_Lactobacillus decreased from 5.57 to 3.13% (Table 2 and Fig. 2B). On the other hand, g_Aerococcus increased from 0.02% in wildtype mice to 10.45% in Tg-APP/PS1 mice, g_Jeotgalicoccus increased from 0.83 to 8.50%, an unclassified member of o_Clostridiales increased from 4.66 to 7.03%, g_Blautia increased from 0.12 to 1.37%, an unclassified member of f_Ruminococcaceae increased from 2.44 to 6.09%, and g_Pseudomonas increased from 2.78 to 4.13% (Table 2 and Fig. 2B).
Table 2. The percent compositions of microbiomes at the genus level in wild-type and Tg-APP/PS1 mice.
| Taxon | Occupancy (%; Mean±SEM) | Fold change | z-score | |
|---|---|---|---|---|
| WT | Tg-APP/PS | |||
| p__Bacteroidetes;c__Bacteroidia;o__Bacteroidales;f__S24-7;g__ | 6.3±1.97 | 1.81±0.53 | ↓ −3.5 | −1 |
| p__Firmicutes;c__Bacilli;o__Lactobacillales;f__Lactobacillaceae;g__Lactobacillus | 5.57±0.58 | 3.13±0.85 | ↓ −1.8 | −1.9 |
| p__Firmicutes;c__Clostridia;o__Clostridiales;f__;g__ | 4.66±1 | 7.03±2.78 | ↑ 1.5 | 1.1 |
| p__Actinobacteria;c__Actinobacteria;o__Actinomycetales;f__Corynebacteriaceae;g__Corynebacterium | 3.5±0.79 | 0.94±0.39 | ↓ −3.7 | −1.4 |
| p__Proteobacteria;c__Gammaproteobacteria;o__Pseudomonadales;f__Pseudomonadaceae;g__Pseudomonas | 2.78±0.38 | 4.13±0.87 | ↑ 1.5 | 1.6 |
| p__Firmicutes;c__Clostridia;o__Clostridiales;f__Ruminococcaceae;g__ | 2.44±0.65 | 6.09±2.2 | ↑ 2.5 | 2.5 |
| p__Tenericutes;c__Mollicutes;o__RF39;f__;g__ | 0.92±0.26 | 0.26±0.15 | ↓ −3.6 | −1.1 |
| p__Firmicutes;c__Bacilli;o__Bacillales;f__Staphylococcaceae;g__Jeotgalicoccus | 0.83±0.62 | 8.5±5.49 | ↑ 10.2 | 5.5 |
| p__Proteobacteria;c__Alphaproteobacteria;o__Rhodobacterales;f__Rhodobacteraceae;g__Amaricoccus | 0.41±0.41 | 0±0 | ↓ >> | >> |
| p__Proteobacteria;c__Betaproteobacteria;o__Burkholderiales;f__Oxalobacteraceae;g__Ralstonia | 0.21±0.16 | 0±0 | ↓ >> | >> |
| p__Firmicutes;c__Erysipelotrichi;o__Erysipelotrichales;f__Erysipelotrichaceae;g__ | 0.19±0.08 | 0.47±0.19 | ↑ 2.5 | 1.5 |
| p__Fusobacteria;c__Fusobacteriia;o__Fusobacteriales;f__Leptotrichiaceae;g__Leptotrichia | 0.18±0.11 | 0±0 | ↓ >> | >> |
| p__Bacteroidetes;c__Flavobacteriia;o__Flavobacteriales;f__Flavobacteriaceae;g__Flavobacterium | 0.16±0.16 | 0±0 | ↓ >> | >> |
| p__Proteobacteria;c__Betaproteobacteria;o__Burkholderiales;f__Alcaligenaceae;g__Oligella | 0.15±0.15 | 0±0 | ↓ >> | >> |
| p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Lachnospira | 0.15±0.1 | 0±0 | ↓ >> | >> |
| p__Firmicutes;c__Bacilli;o__Bacillales;f__Planococcaceae;g__Sporosarcina | 0.14±0.14 | 0.49±0.49 | ↑ 3.6 | 1.2 |
| p__Actinobacteria;c__Actinobacteria;o__Bifidobacteriales;f__Bifidobacteriaceae;g__ | 0.14±0.08 | 0±0 | ↓ >> | >> |
| p__Proteobacteria;c__Betaproteobacteria;o__Rhodocyclales;f__Rhodocyclaceae;g__Methyloversatilis | 0.13±0.13 | 0±0 | ↓ >> | >> |
| p__Proteobacteria;c__Alphaproteobacteria;o__Sphingomonadales;f__Sphingomonadaceae;g__Sphingopyxis | 0.13±0.08 | 0±0 | ↓ >> | >> |
| p__Firmicutes;c__Bacilli;o__Lactobacillales;f__Aerococcaceae;g__Alloiococcus | 0.13±0.09 | 0±0 | ↓ >> | >> |
| p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Blautia | 0.12±0.11 | 1.37±0.95 | ↑ 11.1 | 5.3 |
| p__Gemmatimonadetes;c__Gemmatimonadetes;o__;f__;g__ | 0.11±0.11 | 0±0 | ↓ >> | >> |
| p__Gemmatimonadetes;c__Gemmatimonadetes;o__Gemmatimonadales;f__;g__ | 0.11±0.11 | 0±0 | ↓ >> | >> |
| p__Proteobacteria;c__Betaproteobacteria;o__Burkholderiales;f__Oxalobacteraceae;g__ | 0.08±0.05 | 0.28±0.21 | ↑ 3.4 | 1.6 |
| p__Proteobacteria;c__Alphaproteobacteria;o__Rhizobiales;f__Methylobacteriaceae;g__ | 0.06±0.05 | 0.34±0.2 | ↑ 5.8 | 2.3 |
| p__Bacteroidetes;c__Bacteroidia;o__Bacteroidales;f__Porphyromonadaceae;g__Parabacteroides | 0.06±0.06 | 0.28±0.19 | ↑ 5 | 1.8 |
| p__Cyanobacteria;c__4C0d-2;o__YS2;f__;g__ | 0.05±0.05 | 0.19±0.12 | ↑ 4 | 1.4 |
| p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;Other | 0.04±0.03 | 0.45±0.41 | ↑ 10.1 | 6.9 |
| p__Proteobacteria;c__Alphaproteobacteria;o__Caulobacterales;f__Caulobacteraceae;g__ | 0.04±0.01 | 0.81±0.51 | ↑ 18.7 | 29.6 |
| p__Proteobacteria;c__Gammaproteobacteria;o__Enterobacteriales;f__Enterobacteriaceae;Other | 0.04±0.02 | 0.42±0.19 | ↑ 10.4 | 7.7 |
| p__Proteobacteria;c__Betaproteobacteria;o__Neisseriales;f__Neisseriaceae;g__Neisseria | 0.03±0.03 | 0.2±0.14 | ↑ 7 | 2.7 |
| p__Proteobacteria;c__Gammaproteobacteria;o__Pasteurellales;f__Pasteurellaceae;g__Haemophilus | 0.02±0.02 | 0.14±0.1 | ↑ 6.2 | 2.3 |
| p__Firmicutes;c__Bacilli;o__Lactobacillales;f__Aerococcaceae;g__Aerococcus | 0.02±0.01 | 10.45±8.55 | ↑ 630 | 434.2 |
| p__Actinobacteria;c__Actinobacteria;o__Actinomycetales;f__Nocardioidaceae;g__ | 0.01±0.01 | 0.11±0.07 | ↑ 15 | 6.2 |
| p__Firmicutes;c__Bacilli;Other;Other;Other | 0.01±0.01 | 0.32±0.3 | ↑ 48.6 | 27.8 |
| p__Firmicutes;c__Clostridia;o__Clostridiales;f__Ruminococcaceae;Other | 0.01±0 | 0.24±0.23 | ↑ 40.1 | 23.4 |
| p__Firmicutes;c__Bacilli;o__Bacillales;f__Bacillaceae;g__Bacillus | 0±0 | 0.27±0.16 | ↑ 72.4 | 32 |
| p__Firmicutes;c__Bacilli;o__Lactobacillales;f__Leuconostocaceae;g__Weissella | 0±0 | 0.18±0.18 | ↑ 149.8 | 66.6 |
| p__Firmicutes;c__Clostridia;o__Clostridiales;f__[Mogibacteriaceae];g__ | 0±0 | 0.1±0.06 | ↑ << | << |
| p__Fibrobacteres;c__TG3;o__TG3-1;f__TSCOR003-O20;g__ | 0±0 | 0.11±0.11 | ↑ << | << |
| p__Bacteroidetes;c__Bacteroidia;o__Bacteroidales;f__[Odoribacteraceae];g__Odoribacter | 0±0 | 0.11±0.08 | ↑ << | << |
| p__Proteobacteria;c__Deltaproteobacteria;o__Desulfovibrionales;f__Desulfovibrionaceae;g__ | 0±0 | 0.11±0.1 | ↑ << | << |
| p__Bacteroidetes;c__Cytophagia;o__Cytophagales;f__Cytophagaceae;g__Hymenobacter | 0±0 | 0.12±0.12 | ↑ << | << |
| p__Firmicutes;c__Clostridia;o__Clostridiales;f__Veillonellaceae;g__Megamonas | 0±0 | 0.13±0.12 | ↑ << | << |
| p__Firmicutes;c__Bacilli;o__Lactobacillales;f__Leuconostocaceae;g__ | 0±0 | 0.13±0.06 | ↑ << | << |
| p__Firmicutes;c__Clostridia;o__Clostridiales;f__Eubacteriaceae;g__Anaerofustis | 0±0 | 0.13±0.1 | ↑ << | << |
| p__Bacteroidetes;c__Sphingobacteriia;o__Sphingobacteriales;f__Sphingobacteriaceae;g__ | 0±0 | 0.14±0.14 | ↑ << | << |
| p__Actinobacteria;c__Actinobacteria;o__Actinomycetales;f__Micrococcaceae;g__Kocuria | 0±0 | 0.14±0.08 | ↑ << | << |
| p__Proteobacteria;c__Alphaproteobacteria;o__Rhizobiales;f__Bartonellaceae;g__ | 0±0 | 0.14±0.1 | ↑ << | << |
| p__TM7;c__TM7-3;o__;f__;g__ | 0±0 | 0.16±0.11 | ↑ << | << |
| p__[Thermi];c__Deinococci;o__Deinococcales;f__Deinococcaceae;g__Deinococcus | 0±0 | 0.19±0.19 | ↑ << | << |
| p__Bacteroidetes;c__Flavobacteriia;o__Flavobacteriales;f__[Weeksellaceae];g__Cloacibacterium | 0±0 | 0.2±0.2 | ↑ << | << |
| p__Actinobacteria;c__Coriobacteriia;o__Coriobacteriales;f__Coriobacteriaceae;g__Collinsella | 0±0 | 0.35±0.17 | ↑ << | << |
| p__Firmicutes;c__Clostridia;o__Clostridiales;f__Veillonellaceae;g__Veillonella | 0±0 | 0.37±0.27 | ↑ << | << |
| p__Firmicutes;c__Bacilli;o__Lactobacillales;f__Streptococcaceae;g__Lactococcus | 0±0 | 0.67±0.62 | ↑ << | << |
| Summation | 29.9 | 52.2 | ||
The percent compositions of microbiomes at the genus level for which occupancy differed by more than one SD (one z) in Tg-APP/PS1 mice. Microbiomes with occupancies higher than 0.1% in either wild-type or Tg-APP/PS1 mice were included.
Data are presented as the mean percentage +/− SEM. The z-score is X − µ/σ, where µ=mean; X=individual score; σ=standard deviation (SD). The increase and decrease of the mean occupancy by more than one z in Tg-APP/PS1 are indicated by ↑ and ↓, respectively. « and » denote infinite increases and decreases, respectively.
Fig. 2. The genus members that were down-regulated or up-regulated in Tg-APP/PS1 mice. (A) Heat maps depicting the list of microbiota for which occupancies increased (red) or decreased (green) by more than one SD (one z) in Tg-APP/PS1 mice relative to the percent mean of wild-type controls. In total 130 genus members with occupancies higher than 0.1% in either wild-type or Tg-APP/PS1 mice were considered. Subsequently clustering of respective microbiomes in independent comparisons were overlaid. n/s, the microbiomes for which occupancy was not detected in wild-type or Tg-APP/PS1 mice. The z-score is expressed as X − µ/σ, where µ=mean; X=individual score; σ=standard deviation (SD). (B) The percent compositions of microbiomes at the genus level in wild-type and Tg-APP/PS1 mice. All genus members with occupancies higher than 0.1% in either wild-type (blue) or Tg-APP/PS1 mice (red) are presented (n=5, each).

DISCUSSION
Metagenome analysis of bacterial EVs in blood identified altered bodily microbiota compositions in Tg-APP/PS1 mice
We demonstrated in the present study that bodily microbiota compositions were altered in Tg-APP/PS1 mice compared to those of non-transgenic control. Of the alterations in phylum members, it is worth noting that the occupancy of p_Firmicutes increased from 34.7 to 57.5% in Tg-APP/PS1 mice (Fig. 1C). This increase was comprised change in the following genus members; g_Aerococcus (from 0.02 to 10.45%), g_Jeotgalicoccus (from 0.83 to 8.50%), an unclassified member of o_Clostridiales (from 4.66 to 7.03%), g_Blautia (from 0.12 to 1.37%), and an unclassified member of f_Ruminococcaceae (from 2.44 to 6.09%). In contrast to these genus members, g_Lactobacillus decreased (from 5.57 to 3.13%).
The total occupancy of p_Proteobacteria decreased from 30.5 to 20.7% in Tg-APP/PS1 mice. Although the occupancy of some genus members of p_Proteobacteria, including g_Amaricoccus, g_Sphingopyxis, g_Oligella, and g_Ralstonia, decreased (from 0.13~0.41% to 0%), no single genus member that occupied over than 1% in wild-type mice contributed to this decrease. On the other hand, g_Pseudomonas, a member of p_Proteobacteria, increased (from 2.78 to 4.13%) in Tg-APP/PS1 mice. It was notable that g_Corynebacterium, a genus member of p_Actinobacteria, decreased from 3.50 to 0.94%, and that an unclassified member of f_S24-7, a genus member of p_Bacteroidetes, decreased from 6.30 to 1.81%.
As summarized with the results of the heat-map analysis, certain genus members were newly detected in Tg-APP/PS1 mice, but not in wild-type mice, while other members were detected in wild-type mice, but lost in Tg-APP/PS1 mice. Although the relative occupancy of each of these genus members was not high, determining whether these genus members could can serve as microbiome markers for specific AD pathology will require further investigation.
In comparing the results of the present study with those published in the literature, it may be worth noting that the altered occupancy of p_Firmicutes in Tg-APP/PS1 mice (from 34.7 to 57.5%, z=3.45) in the present study was similar to a recently reported increase of this phylum in 5xFAD mice (from 34 to 49%, p=0.003) [9]. On the other hand, the decrease in occupancy of p_ Bacteroidetes was more prominent in 5xFAD mice (from 61 to 46%, p=0.003) [9] than in Tg-APP/PS1 mice (from 17.8 to 8.7%, z=−0.73). The occupancy of p_Proteobacteria decreased in Tg-APP/PS1 mice (from 30.5 to 20.7%, z=−1.56), whereas no information on this phylum member was available in 5xFAD mice (Table 3).
Table 3. Summary of microbiota composition characterized in the present study and comparison with that identified from fecal samples in recent studies.
| Present | Published | |||||
|---|---|---|---|---|---|---|
| APP/PS1a) (serum) | APP/PS1b) (fecal) | 3xTg (fecal) | ||||
| (Harach et al., 2017) | (Bonfili et al., 2017) | |||||
| <Microbial diversity> | WT vs. Tg | WT vs. Tg | WT vs. Tg | |||
| Alpha diversity | ↑ | ↑ | = | |||
The microbiomes for which the percent composition increased or decreased by more than one z score in Tg-APP/PS1 are marked by ↑ and ↓, respectively. « and » denote infinite increases and decreases, respectively. The increases and decreases of the indicated microbiota in the previous studies are also marked by ↑ and ↓, respectively.
The z-score is expressed as X − µ/σ, where µ=mean; X=individual score; σ=standard deviation (SD). a)APPswe/PSEN1dE9; b)APPswe/PSEN1-L166P.
At the genus level, Tg-APP/PS1 mice exhibited decreases in occupancy of g_Allobaculum, and g_Anaerostipes, and increases in occupancy of g_Odoribacter, g_Streptococcus and g_Dorea. Similar results were also found in 3xTg mouse fecal samples [7]. Tg-APP/PS1 mice displayed increases of g_Aerococcus (from 0.02 to 10.45%), g_Jeotgalicoccus (from 0.83 to 8.50%), g_Blautia (from 0.12 to 1.37%), and g_Pseudomonas (from 2.78 to 4.13%), while a decrease of g_Lactobacillus (from 5.57 to 3.13%). However, these genus members were not altered in the available studies of fecal samples (Table 3).
Although physiological significance of these changes are not known, it will be worthy of note the following genus members. A recent study reported that the occupancy of g_Odoribacter increased in the gut of aged mice [27], and in Tg-APP/PS1 (this study) and 3xTg mouse [7]. These results suggest that the increase of g_Odoribacter in Tg-APP/PS1 mice is likely related to AD pathology, rather than simple aging. Oral treatment with Lactobacillus helveticus and Bifidobacterium longum improved behavioral and cognitive impairments caused by chronic stress [28] and reversed stress-induced decreased BDNF expression in the hypothalamus [29]. Administration of Lactobacillus fermentum increased hippocampal mineralocorticoid receptor and NMDA receptor levels in rats with psychological changes induced by antibiotics [30]. Lactobacillus plantarum attenuated circulating TNF-α and IL-6 levels while reducing immobility time in the forced swim test and increasing sucrose preference in mice with early-life stress [31]. Lactobacillus rhamnosus attenuated increased IL-10+ regulatory T cells and decreased stress-induced anxiety-like behavior in mice with chronic social stress [32]. Lactobacillus reuteri restored VTA synaptic plasticity and social deficits in an autism mouse model induced by maternal high-fat diet treatment [33]. Regarding these results, it will be interesting to test whether the decrease of g_Lactobacillus in Tg-APP/PS1 is a risk factor for AD pathology.
Overall, these results suggest that the microbiota composition of AD patients and AD animal models is altered, although detailed microbiota profiles are not consistent. These results imply that the microbiota composition of AD or AD models in different physiological contexts is highly complex and dynamic. Considering the importance of understanding the interaction between the gut microbiota and the brain in AD, more systematic and comprehensive analyses of the microbiota compositions in AD are needed.
Bacterial EVs in blood represent a new avenue for metagenome analysis of microbiota in AD models and patients
The results of the present study raise some important issues regarding bodily microbiota in Tg-APP/PS1 mice, which are summarized as follows. First, it is worth noting that Tg-APP/PS1 mice and their wild-type controls were cultured in the same animal room environment with the same food from gestation and birth. Nonetheless, microbiota compositions of Tg-APP/PS1 mice were altered. This result emphasizes the importance of the relationship between microbiota and AD pathology. Second, detailed analysis of microbiome profiles indicated that the microbiota represented in blood EVs matched the gut microbiota reported in previous studies [34,35,36,37,38]. This is not surprising, because the main source of bodily microbiota is in the gut/gastrointestinal tract [39,40,41]. Microbiota that are metabolically active [42] or exist under pathogenic conditions [12,15] produce EVs. It will be worth investigating what proportions of bacteria-derived EVs in blood in Tg-APP/PS1 mice are metabolically active or exist under pathogenic conditions. Third, the specific microbiota identified to have altered occupancies in the present study were partly consistent with those assessed on the basis of fecal microbiomes, although the relative proportions of certain taxa differed from those in fecal samples (Table 3). As summarized in Table 3, the relative proportions of specific taxa identified from fecal samples have varied across different studies (eg., APPPS1 mice of [6] vs. 3xTg mice of [7]). Given the considerable variation among AD animal models, which are cultured in relatively defined environments, more comprehensive and systematic studies are necessary in AD patients to elaborate whether and how various genetic, ethnic, environmental and regional factors affect microbiota profiles. Fourth, to the best of our knowledge, this is the first study characterizing bodily microbiota on the basis of blood EVs. Unlike feces, blood is collected during normal medical examination. Therefore, the ability to use blood as a sample source will facilitate the rapid assessment of the microbiomes of AD patients in various physiological contexts.
In summary, we identified a number of microbiota from blood EVs whose composition was altered in an AD mouse model. Among the altered microbiota, a number of genus members were detected in Tg-APP/PS1 mice, but not in wild-type mice, while other genus members were identified in wild-type mice, but not in Tg-APP/PS1 mice. The results of the present study support that bacterial EVs in blood represent a new opportunity for metagenome analysis of microbiota in AD models and AD patients.
ACKNOWLEDGEMENTS
This research was supported by a grant (HI15C1834) from the Ministry of Health and Welfare, Republic of Korea.
References
- 1.Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13:701–712. doi: 10.1038/nrn3346. [DOI] [PubMed] [Google Scholar]
- 2.Rogers GB, Keating DJ, Young RL, Wong ML, Licinio J, Wesselingh S. From gut dysbiosis to altered brain function and mental illness: mechanisms and pathways. Mol Psychiatry. 2016;21:738–748. doi: 10.1038/mp.2016.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fung TC, Olson CA, Hsiao EY. Interactions between the microbiota, immune and nervous systems in health and disease. Nat Neurosci. 2017;20:145–155. doi: 10.1038/nn.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cattaneo A, Cattane N, Galluzzi S, Provasi S, Lopizzo N, Festari C, Ferrari C, Guerra UP, Paghera B, Muscio C, Bianchetti A, Volta GD, Turla M, Cotelli MS, Gennuso M, Prelle A, Zanetti O, Lussignoli G, Mirabile D, Bellandi D, Gentile S, Belotti G, Villani D, Harach T, Bolmont T, Padovani A, Boccardi M, Frisoni GB INDIA-FBP Group. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging. 2017;49:60–68. doi: 10.1016/j.neurobiolaging.2016.08.019. [DOI] [PubMed] [Google Scholar]
- 5.Emery DC, Shoemark DK, Batstone TE, Waterfall CM, Coghill JA, Cerajewska TL, Davies M, West NX, Allen SJ. 16S rRNA next generation sequencing analysis shows bacteria in Alzheimer’s post-mortem brain. Front Aging Neurosci. 2017;9:195. doi: 10.3389/fnagi.2017.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harach T, Marungruang N, Duthilleul N, Cheatham V, Mc Coy KD, Frisoni G, Neher JJ, Fåk F, Jucker M, Lasser T, Bolmont T. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci Rep. 2017;7:41802. doi: 10.1038/srep41802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonfili L, Cecarini V, Berardi S, Scarpona S, Suchodolski JS, Nasuti C, Fiorini D, Boarelli MC, Rossi G, Eleuteri AM. Microbiota modulation counteracts Alzheimer’s disease progression influencing neuronal proteolysis and gut hormones plasma levels. Sci Rep. 2017;7:2426. doi: 10.1038/s41598-017-02587-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen L, Liu L, Ji HF. Alzheimer's disease histological and behavioral manifestations in transgenic mice correlate with specific gut microbiome state. J Alzheimers Dis. 2017;56:385–390. doi: 10.3233/JAD-160884. [DOI] [PubMed] [Google Scholar]
- 9.Brandscheid C, Schuck F, Reinhardt S, Schäfer KH, Pietrzik CU, Grimm M, Hartmann T, Schwiertz A, Endres K. Altered gut microbiome composition and tryptic activity of the 5xFAD Alzheimer’s mouse model. J Alzheimers Dis. 2017;56:775–788. doi: 10.3233/JAD-160926. [DOI] [PubMed] [Google Scholar]
- 10.Kuehn MJ, Kesty NC. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 2005;19:2645–2655. doi: 10.1101/gad.1299905. [DOI] [PubMed] [Google Scholar]
- 11.Deatherage BL, Cookson BT. Membrane vesicle release in bacteria, eukaryotes, and archaea: a conserved yet underappreciated aspect of microbial life. Infect Immun. 2012;80:1948–1957. doi: 10.1128/IAI.06014-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kulp A, Kuehn MJ. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol. 2010;64:163–184. doi: 10.1146/annurev.micro.091208.073413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee EY, Choi DY, Kim DK, Kim JW, Park JO, Kim S, Kim SH, Desiderio DM, Kim YK, Kim KP, Gho YS. Gram-positive bacteria produce membrane vesicles: proteomics-based characterization of Staphylococcus aureus-derived membrane vesicles. Proteomics. 2009;9:5425–5436. doi: 10.1002/pmic.200900338. [DOI] [PubMed] [Google Scholar]
- 14.Brown L, Wolf JM, Prados-Rosales R, Casadevall A. Through the wall: extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat Rev Microbiol. 2015;13:620–630. doi: 10.1038/nrmicro3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ellis TN, Kuehn MJ. Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol Mol Biol Rev. 2010;74:81–94. doi: 10.1128/MMBR.00031-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maldonado R, Wei R, Kachlany SC, Kazi M, Balashova NV. Cytotoxic effects of Kingella kingae outer membrane vesicles on human cells. Microb Pathog. 2011;51:22–30. doi: 10.1016/j.micpath.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hong SW, Kim MR, Lee EY, Kim JH, Kim YS, Jeon SG, Yang JM, Lee BJ, Pyun BY, Gho YS, Kim YK. Extracellular vesicles derived from Staphylococcus aureus induce atopic dermatitis-like skin inflammation. Allergy. 2011;66:351–359. doi: 10.1111/j.1398-9995.2010.02483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang CS, Ban M, Choi EJ, Moon HG, Jeon JS, Kim DK, Park SK, Jeon SG, Roh TY, Myung SJ, Gho YS, Kim JG, Kim YK. Extracellular vesicles derived from gut microbiota, especially Akkermansia muciniphila, protect the progression of dextran sulfate sodium-induced colitis. PLoS One. 2013;8:e76520. doi: 10.1371/journal.pone.0076520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jang SC, Kim SR, Yoon YJ, Park KS, Kim JH, Lee J, Kim OY, Choi EJ, Kim DK, Choi DS, Kim YK, Park J, Di Vizio D, Gho YS. In vivo kinetic biodistribution of nano-sized outer membrane vesicles derived from bacteria. Small. 2015;11:456–461. doi: 10.1002/smll.201401803. [DOI] [PubMed] [Google Scholar]
- 20.Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- 21.Choi Y, Kwon Y, Kim DK, Jeon J, Jang SC, Wang T, Ban M, Kim MH, Jeon SG, Kim MS, Choi CS, Jee YK, Gho YS, Ryu SH, Kim YK. Gut microbe-derived extracellular vesicles induce insulin resistance, thereby impairing glucose metabolism in skeletal muscle. Sci Rep. 2015;5:15878. doi: 10.1038/srep15878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoo JY, Rho M, You YA, Kwon EJ, Kim MH, Kym S, Jee YK, Kim YK, Kim YJ. 16S rRNA gene-based metagenomic analysis reveals differences in bacteria-derived extracellular vesicles in the urine of pregnant and non-pregnant women. Exp Mol Med. 2016;48:e208. doi: 10.1038/emm.2015.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andreu Z, Rivas E, Sanguino-Pascual A, Lamana A, Marazuela M, González-Alvaro I, Sánchez-Madrid F, de la Fuente H, Yáñez-Mó M. Comparative analysis of EV isolation procedures for miRNAs detection in serum samples. J Extracell Vesicles. 2016;5:31655. doi: 10.3402/jev.v5.31655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seo JS, Lee KW, Kim TK, Baek IS, Im JY, Han PL. Behavioral stress causes mitochondrial dysfunction via ABAD up-regulation and aggravates plaque pathology in the brain of a mouse model of Alzheimer disease. Free Radic Biol Med. 2011;50:1526–1535. doi: 10.1016/j.freeradbiomed.2011.02.035. [DOI] [PubMed] [Google Scholar]
- 25.Kim TK, Han HE, Kim H, Lee JE, Choi D, Park WJ, Han PL. Expression of the plant viral protease NIa in the brain of a mouse model of Alzheimer's disease mitigates Aβ pathology and improves cognitive function. Exp Mol Med. 2012;44:740–748. doi: 10.3858/emm.2012.44.12.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JE, Han PL. An update of animal models of Alzheimer disease with a reevaluation of plaque depositions. Exp Neurobiol. 2013;22:84–95. doi: 10.5607/en.2013.22.2.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scott KA, Ida M, Peterson VL, Prenderville JA, Moloney GM, Izumo T, Murphy K, Murphy A, Ross RP, Stanton C, Dinan TG, Cryan JF. Revisiting Metchnikoff: age-related alterations in microbiota-gut-brain axis in the mouse. Brain Behav Immun. 2017;65:20–32. doi: 10.1016/j.bbi.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 28.Liang S, Wang T, Hu X, Luo J, Li W, Wu X, Duan Y, Jin F. Administration of Lactobacillus helveticus NS8 improves behavioral, cognitive, and biochemical aberrations caused by chronic restraint stress. Neuroscience. 2015;310:561–577. doi: 10.1016/j.neuroscience.2015.09.033. [DOI] [PubMed] [Google Scholar]
- 29.Ait-Belgnaoui A, Colom A, Braniste V, Ramalho L, Marrot A, Cartier C, Houdeau E, Theodorou V, Tompkins T. Probiotic gut effect prevents the chronic psychological stress-induced brain activity abnormality in mice. Neurogastroenterol Motil. 2014;26:510–520. doi: 10.1111/nmo.12295. [DOI] [PubMed] [Google Scholar]
- 30.Wang T, Hu X, Liang S, Li W, Wu X, Wang L, Jin F. Lactobacillus fermentum NS9 restores the antibiotic induced physiological and psychological abnormalities in rats. Benef Microbes. 2015;6:707–717. doi: 10.3920/BM2014.0177. [DOI] [PubMed] [Google Scholar]
- 31.Liu YW, Liu WH, Wu CC, Juan YC, Wu YC, Tsai HP, Wang S, Tsai YC. Psychotropic effects of Lactobacillus plantarum PS128 in early life-stressed and naïve adult mice. Brain Res. 2016;1631:1–12. doi: 10.1016/j.brainres.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 32.Bharwani A, Mian MF, Surette MG, Bienenstock J, Forsythe P. Oral treatment with Lactobacillus rhamnosus attenuates behavioural deficits and immune changes in chronic social stress. BMC Med. 2017;15:7. doi: 10.1186/s12916-016-0771-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buffington SA, Di Prisco GV, Auchtung TA, Ajami NJ, Petrosino JF, Costa-Mattioli M. Microbial reconstitution reverses maternal diet-induced social and synaptic deficits in offspring. Cell. 2016;165:1762–1775. doi: 10.1016/j.cell.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Claesson MJ, O'Sullivan O, Wang Q, Nikkilä J, Marchesi JR, Smidt H, de Vos WM, Ross RP, O'Toole PW. Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS One. 2009;4:e6669. doi: 10.1371/journal.pone.0006669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Doré J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, MetaHIT Consortium. Bork P, Ehrlich SD, Wang J. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stearns JC, Lynch MD, Senadheera DB, Tenenbaum HC, Goldberg MB, Cvitkovitch DG, Croitoru K, Moreno-Hagelsieb G, Neufeld JD. Bacterial biogeography of the human digestive tract. Sci Rep. 2011;1:170. doi: 10.1038/srep00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reid G, Younes JA, Van der Mei HC, Gloor GB, Knight R, Busscher HJ. Microbiota restoration: natural and supplemented recovery of human microbial communities. Nat Rev Microbiol. 2011;9:27–38. doi: 10.1038/nrmicro2473. [DOI] [PubMed] [Google Scholar]
- 40.Human Microbiome Project Consortium. A framework for human microbiome research. Nature. 2012;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bauman SJ, Kuehn MJ. Purification of outer membrane vesicles from Pseudomonas aeruginosa and their activation of an IL-8 response. Microbes Infect. 2006;8:2400–2408. doi: 10.1016/j.micinf.2006.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]