

Abstract
Contraction and relaxation of the heart result from cyclical changes of intracellular Ca2+ concentration ([Ca2+]i). The entry of Ca2+ into the cell via the L‐type Ca2+ current leads to the release of more from the sarcoplasmic reticulum (SR). Compared to other regulatory mechanisms such as phosphorylation, Ca2+ signalling is very rapid. However, since Ca2+ cannot be destroyed, Ca2+ signalling can only be controlled by pumping across membranes. In the steady state, on each beat, the amount of Ca2+ released from the SR must equal that taken back and influx and efflux across the sarcolemma must be equal. Any imbalance in these fluxes will result in a change of SR Ca2+ content and this provides a mechanism for regulation of SR Ca2+ content. These flux balance considerations also explain why simply potentiating Ca2+ release from the SR has no maintained effect on the amplitude of the Ca2+ transient. A low diastolic [Ca2+]i is essential for cardiac relaxation, but the factors that control diastolic [Ca2+]i are poorly understood. Recent work suggests that flux balance is also important here. In particular, decreasing SR function decreases the amplitude of the systolic Ca2+ transient and the resulting decrease of Ca2+ efflux results in an increase of diastolic [Ca2+]i to maintain total efflux.
Keywords: Calcium, Sarcoplasmic reticulum, flux
Introduction
It is well established that contraction of the heart is controlled by changes of cytoplasmic Ca2+ concentration ([Ca2+]i). [Ca2+]i must be high enough in systole to activate the contractile proteins in order to pump blood out of the heart. Equally importantly, it must fall during diastole to low enough levels that the muscle of the heart relaxes so that the chambers can refill with blood. The aim of this article is to discuss factors responsible for both the rise and fall of [Ca2+]i.
The importance of Ca2+ in contraction of the heart was originally demonstrated by Sydney Ringer (Ringer, 1883). I have recently described both the serendipitous nature of his discovery and the manner in which it was published (Eisner, 2014). In the subsequent 130 years it has become clear that changes of [Ca2+]i regulate the function of virtually all organs. It took, however, almost 100 years after Ringer's discovery to measure directly the transient elevation of [Ca2+]i (the Ca2+ transient) which underlies cardiac contraction (Allen & Blinks, 1978; Allen & Kurihara, 1980).
Calcium versus other signalling modalities
Calcium has its actions by binding to proteins and thereby changing their shape and properties (Fig. 1 A). In the case of striated muscle, Ca2+ binds to troponin and the resulting movement of tropomyosin allows actin and myosin to interact. There are, of course, many other ways to alter the properties of proteins including phosphorylation and nitrosylation. This immediately raises the question, what are the advantages and disadvantages of using Ca2+ to modify protein structure? One great advantage of Ca2+ is its speed of action. [Ca2+]i rises and Ca2+ ions bind to troponin very quickly. In cardiac muscle contraction can be 50% complete within 200 ms (Fig. 1 A). In contrast regulation by phosphorylation is much slower. Figure 1 B shows the effects of β‐adrenergic stimulation on the Ca2+ current. This takes about 20 s to be 50% complete (Frace et al. 1993). This slower time scale is due to the many steps involved; agonist binding to the receptor, generation of cyclic AMP, and, finally, the activation of protein kinase A which then phosphorylates the L‐type channel. Although signalling by phosphorylation is slower than that by Ca2+, it has the advantage that it can be regulated in a much more subtle way. Specific phosphodiesterases can break down cAMP and various regulators affect the activities of the kinases and phosphatases. Another significant advantage of control by phosphorylation is that it can be reversed simply by dephoshorylation. In contrast, Ca2+ ions cannot be destroyed. The only way to reverse Ca2+ binding is to pump the Ca2+ across a membrane, either out of the cell or into an intracellular store. Put simply, Ca2+ must be recycled and many of the results and conclusions of the remainder of this review follow from this.
Figure 1. Comparison of control by Ca2+ and phosphorylation.
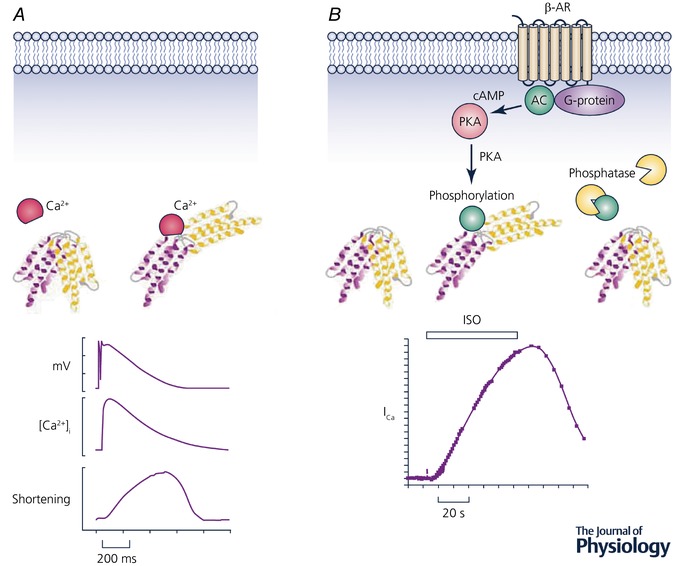
A, Ca2+. The upper part shows a schematic diagram of a protein changing shape when it binds Ca2+. The lower part shows time course of (from top to bottom): action potential, [Ca2+]i and cell shortening recorded from a sheep ventricular myocyte. (Record kindly provided by K. M. Dibb.) B, phosphorylation. The upper part shows the change of shape produced by phosphorylation and its reversal by dephosphorylaton with a phosphatase. The steps between binding to the receptor and phosphorylation are shown: agonist binds to the β‐adrenergic receptor (β‐AR) which, via a G‐protein, activates adenylate cyclase (AC) producing cyclic AMP (cAMP) which activates protein kinase A (PKA). (Illustrations by Jessica Caldwell.) The lower part shows the time course of the effects of isoprenaline (ISO) on the L‐type Ca2+ current in a frog ventricular myocyte. Trace reproduced with permission from Frace et al. (1993).
The above argument needs some qualification. Although it describes well the situation in striated muscle, where Ca2+ binding is followed rapidly by contraction, in other tissues the steps subsequent to Ca2+ binding may involve slower, enzymatic processes. This is, for example, the case in smooth muscle where Ca2+ binds to calmodulin, activating the enzyme myosin light chain kinase which, in turn, phosphorylates myosin. There is therefore a longer delay between the rise of [Ca2+]i and that of contraction (Shabir et al. 2004). It should also be noted that factors other than speed are also involved in the evolution of Ca2+ signalling (Case et al. 2007).
Flux balance
The events underlying Ca2+ cycling during the heartbeat are illustrated in Fig. 2 (see Bers (2001, 2008) and Eisner et al. (2017) for reviews). Ca2+ enters the cell through the L‐type Ca2+ channel resulting in a large increase of [Ca2+]i in the small dyadic space between the surface membrane/transverse tubule and the sarcoplasmic reticulum (SR). This Ca2+ binds to the SR Ca2+ release channel (ryanodine receptor; RyR) making it open and releasing a much greater amount of Ca2+ into the cytoplasm from the SR, a process known as Ca2+‐induced Ca2+ release (Fabiato, 1985). Relaxation requires that [Ca2+]i be lowered to resting levels. This occurs in two ways. (1) Ca2+ is pumped back into the SR by the sarco/endoplasmic reticulum Ca2+‐ATPase (SERCA). The activity of SERCA is regulated in part by [Ca2+]i and also by the inhibitory accessory protein phospholamban. Phosphorylation of phospholamban, as occurs during β‐adrenergic stimulation, relieves the inhibition and accelerates SERCA (Kirchberber et al. 1975). (2) Ca2+ is also removed from the cell, largely by the Na+/Ca2+ exchanger (NCX), which uses the entry of three Na+ to power the exit of one Ca2+ from the cell (Kimura et al. 1986). The resulting electrogenic current can be used to measure the activity of NCX in the intact cell. It is important to note that the various Ca2+ fluxes must be balanced on each cycle. In other words, in the steady state, the Ca2+ influx into the cell must exactly equal that pumped out. Likewise, the amount of Ca2+ released from the SR must equal that pumped back. This Ca2+ flux balance is an example of the need to recycle Ca2+ mentioned above.
Figure 2. Control points in cellular Ca2+ cycling.
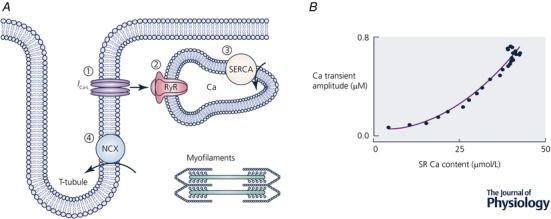
A, schematic diagram showing Ca2+ fluxes. (1) Ca2+ enters the cell via the L‐type Ca2+ current. (2) Ca2+ is released via the RyR from the SR. (The amplitude of the Ca2+ transient depends, therefore, on the sum of Ca2+ entry and release from the SR.) (3) Ca2+ is pumped back into the SR by SERCA and pumped out of the cell largely via NCX. B, the dependence of the amplitude of the Ca2+ transient on SR Ca content. (Redrawn from Trafford et al. (1997).)
Control of the amplitude of the Ca2+ transient
The size of the systolic Ca2+ transient is not fixed. It increases, for example, during exercise due to β‐adrenergic stimulation (Endoh & Blinks, 1988) and decreases in heart failure (Beuckelmann et al. 1992). Three of the steps indicated in Fig. 2 A are potential control points. (1) The size of the L‐type Ca2+ current; the larger the current, the greater the triggering of RyRs to open. (2) The ease with which the RyRs open. I will show later that this is probably not an important control point. (3) The amount of Ca2+ in the SR. Figure 2 B shows that, as SR Ca content increases, there is a steep increase of the amplitude of the Ca2+ transient (Bassani et al. 1995). On average, the amplitude of the Ca2+ transient is proportional to the cube of SR Ca content (Trafford et al. 2000). Several factors account for the steepness. An increase of SR luminal Ca2+ increases the driving force for Ca2+ to leave the SR. It will also increase the open probability of the RyR. This can occur either by Ca2+ binding directly to sites on the luminal part of the RyR or by the Ca2+ which flows out through the RyR binding to sites on the cytoplasmic surface (Guo et al. 2012). Whatever the origin of the steep relationship, it is of great significance. For example, a small reduction of SR Ca2+ content in cardiac hypertrophy will have a much larger fractional effect on the amplitude of the systolic Ca2+ transient (Díaz et al. 2004). Furthermore, the steep dependence means that regulation of the amplitude of the systolic Ca2+ transient requires exquisitely precise control of SR Ca content. This therefore raises the question of how SR Ca content is controlled.
Control of SR Ca content
Much work has shown that SR Ca content in the heart is controlled by a simple, negative feedback mechanism (Trafford et al. 1997). This depends on the fact that (i) the Ca2+ transient amplitude is a steep function of SR Ca content and (ii) fluxes of Ca2+ across the sarcolemma are sensitive to the amplitude of the Ca2+ transient. An example of this mechanism is shown in Fig. 3 A. A high (10 mm) concentration of caffeine had been applied to empty the SR. Following removal of caffeine, the cell was electrically stimulated. The Ca2+ transient was initially very small as a result of the low SR Ca content but then increased over the course of about 10 beats as the SR refilled. The underlying changes of sarcolemmal fluxes are illustrated in Fig. 3 B. When the Ca2+ transient is small (a), there is less Ca2+‐dependent inactivation of the L‐type current (Sipido et al. 1995) and therefore increased influx. There is also less efflux on NCX. The cell is therefore not in Ca2+ flux balance and gains Ca2+. The resulting increase of SR Ca content increases the amplitude of the Ca2+ transient making the L‐type current inactivate more quickly, decreasing Ca2+ influx and there is also more efflux on NCX such that a new steady state is reached.
Figure 3. Negative feedback control of SR Ca2+ content.
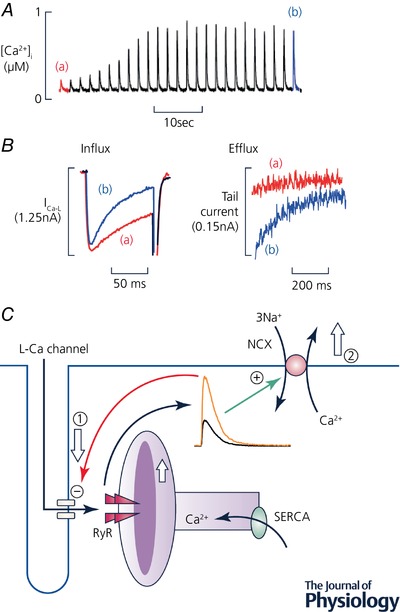
A, time course. The trace shows [Ca2+]i from a ferret ventricular myocyte stimulated with voltage‐clamp pulses. Before the record began, the cell had been exposed (in the absence of stimulation) to caffeine (10 mm) to empty the SR. Caffeine was then removed. The trace shows the effects of recommencing stimulation. B, expanded traces of L‐type current (left) and NCX (right). In both panels the traces correspond to the points in A; i.e. a is the first record after commencing stimulation and b the steady state. (Traces in A and B redrawn from Trafford et al. (1997).) C, schematic diagram of negative feedback loop. An increase of SR Ca2+ increases the amplitude of the Ca2+ transient. This decreases Ca2+ influx via the L‐type Ca2+ current (1) and increases efflux on NCX (2) leading to a decrease of cell and therefore SR Ca2+ content. (A and B redrawn from Trafford et al. (1997).)
These effects of the Ca2+ transient on sarcolemmal Ca2+ fluxes underpin a simple yet powerful negative feedback loop which controls SR Ca2+ content (Fig. 3 C). This can be illustrated by considering the consequence of an increase of SR Ca content. This will increase the amplitude of the Ca2+ transient which, in turn, will increase Ca2+ efflux and decrease influx, thereby decreasing cell and SR Ca content. Not only does this mechanism regulate SR Ca content but it is fundamental to ensuring that the fluxes of Ca2+ into and out of the cell and across the SR membrane are balanced on each beat. Any imbalance of Ca2+ flux across the surface membrane will result in a change of cell and SR Ca leading to an alteration of the amplitude of the Ca2+ transient and thence to changes of Ca2+ fluxes that will return the cell to Ca2+ flux balance. Ca2+ fluxes can, of course, be out of balance but only for short periods. One example has already been given: the effect of stimulating when the SR is empty. Under these conditions, the cell is initially not in Ca2+ flux balance; influx is greater than efflux until the SR Ca2+ content and Ca2+ efflux increase sufficiently that efflux equals influx and the cell is back in flux balance. Another, well‐known example is when the frequency of stimulation is changed or stimulation is recommenced after a pause. For example, in species other than rodents, when stimulation is stopped Ca2+ leaks out of the SR (Bridge, 1986). The SR refills during stimulation resulting in a gradual increase of the amplitude of the Ca2+ transient and contraction (Allen et al. 1976).
The effects of increasing RyR opening
A particularly striking demonstration of the consequences of the maintenance of Ca2+ flux balance is provided by the effects of increasing the open probability of the RyR. Figure 4 demonstrates that the application of 500 μm caffeine to increase RyR opening produces an immediate increase of the amplitude of the systolic Ca2+ transient. This increase is, however, short‐lived and the amplitude of the Ca2+ transient decays to basal levels within a few beats (Trafford et al. 2000). The explanation of this result is as follows. Under control conditions, Ca2+ efflux and influx are equal. The increase in the size of the systolic Ca2+ transient produced by caffeine increases Ca2+ efflux until it is greater than influx resulting in a decrease of SR Ca content. (See Shannon et al. (2005) for detailed mathematical modelling of this phenomenon.) The decrease of SR Ca2+ content in Fig. 4 was calculated from the change of NCX flux. More direct evidence for it comes from showing that the amount of SR Ca2+ that can be released by a high concentration (10 mm) of caffeine is also decreased (Trafford et al. 1998) and measuring the decrease of SR Ca2+ directly with a low affinity Ca2+ indicator (Greensmith et al. 2014). The decrease of SR Ca2+, in turn, decreases the amplitude of the Ca2+ transient until a new steady state is reached at which Ca2+ influx and efflux are again equal. Similar transient effects are seen for other manoeuvres which affect the opening of the RyR (Choi et al. 2000; for review see Eisner et al. 2009). This transient response of Ca2+ transient amplitude to an increase of RyR opening can be thought of as an emergent property of the system. It results because the various, independent Ca2+ transporters are effectively coupled by their dependence on cytoplasmic and SR [Ca2+].
Figure 4. The effects of potentiating RyR opening.
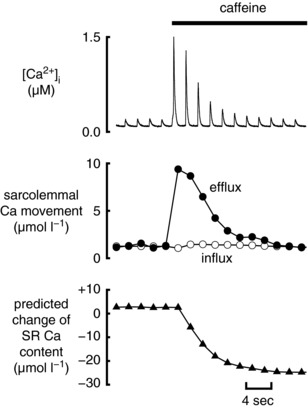
The effects of adding caffeine (500 μm) to increase RyR opening in a rat ventricular myocyte. Traces show (from top to bottom): [Ca2+]i; sarcolemmal Ca2+ fluxes (influx on L‐type Ca2+ current, efflux on NCX; predicted change of SR Ca content. (Redrawn from Trafford et al. (2000).)
The regulation of resting and diastolic [Ca2+]i
For the heart to work properly as a pump, diastolic [Ca2+]i must be sufficiently low that the ventricle is relaxed and can therefore refill with blood between beats. An increase of diastolic force, accompanied by impaired relaxation, is seen in many patients with heart failure. Furthermore, heart failure with preserved ejection fraction (HFpEF) is increasingly being regarded as an important clinical problem (Borlaug & Paulus, 2011). Such patients have symptoms of heart failure but normal ejection fractions and their major clinical problem is diastolic. While other factors may well also contribute to diastolic dysfunction, there is evidence that diastolic [Ca2+]i is increased in patients with heart failure (Gwathmey et al. 1987, 1991; Beuckelmann et al. 1992; Sipido et al. 1998; Runte et al. 2017). It is therefore important to understand how diastolic [Ca2+]i is regulated.
Control of resting [Ca2+]i
Many studies have investigated the regulation of [Ca2+]i in unstimulated (resting) cardiac preparations. Early work, performed before the introduction of Ca2+‐sensitive indicators, used resting force as a bio‐assay of [Ca2+]i. The role of the NCX, for example, was originally demonstrated by examining the effects of changing extracellular Na+ concentration (Niedergerke, 1963; Chapman & Tunstall, 1980). Subsequent work measuring intracellular Na+ concentration showed that the voltage‐dependent NCX played a major role in controlling resting force and therefore [Ca2+]i (Eisner et al. 1983). The advent of fluorescent Ca2+ indicators allowed more direct measurements and also identified a role for the plasma membrane Ca2+‐ATPase in the regulation of resting [Ca2+]i (Lamont & Eisner, 1996). What is less well established is the route by which Ca2+ enters a resting cell. One possibility might be the random opening of L‐type Ca2+ channels but the likely flux would be low at the normal resting potential where the open probability is low. Work on rat ventricular myocytes found evidence for three voltage‐sensitive mechanisms involved in the control of resting [Ca2+]i. (1) NCX; the greater the depolarization, the more the Ca2+ influx. (2) L‐type Ca2+ current; depolarization to about 0 mV increased influx but the magnitude decreased on further depolarization as the driving force for Ca2+ entry was reduced. (3) Finally, there was an increase of [Ca2+]i on hyperpolarization, attributed to increased driving force (Kupittayanant et al. 2006) on an un‐gated Ca2+ entry pathway. Gadolinium at a concentration of 250 μm was the only substance found to completely inhibit this Ca2+ entry leaving its identity unresolved. One of the classes of channels which are sensitive to gadolinium is the so called transient receptor potential (TRP). There has been considerable interest in the properties of such channels in the heart with their being implicated in the development of hypertrophy (Gao et al. 2012) and the cell damage in muscular dystrophy (Lorin et al. 2015). Another channel which may contribute to the background Ca2+ entry mechanism is that responsible for store‐operated Ca2+ entry since its inhibition has been reported to decrease resting [Ca2+]i in HL‐1 cells (Touchberry et al. 2011). The importance of store‐operated channels in normal adult cardiac myocytes is, however, still controversial (for review see Eisner et al. 2017).
Diastolic [Ca2+]i
At first sight one might think that the diastolic level of resting [Ca2+]i would be similar to the resting level in an unstimulated cell. In point of fact this is not the case and diastolic is higher (Fig. 5 and Dibb et al. 2007). This is because, in the steady state, in a resting cell, [Ca2+]i is controlled solely by the surface membrane with the sarcolemmal Ca2+ influx being exactly balanced by efflux (Eisner et al. 1984; Friel & Tsien, 1992; Rios, 2010). The question then is what does control diastolic [Ca2+]i? In recent work we have investigated the effects of interfering with SR function. This work was prompted by the fact that heart failure affects the SR by both making the RyR leaky and decreasing SERCA activity (for review see Bers, 2014).
Figure 5. Effects of caffeine and isoprenaline on diastolic and systolic [Ca2+]i .

A, time course (rat ventricular myocyte). The grey trace shows [Ca2+]i during rest and stimulation at 2 Hz. The blue is a low‐pass filtered version. Caffeine (Caf, 1 mm) and isoprenaline (ISO, 1 μm) were applied as shown. B, mean data showing normalized values for the amplitude of the Ca2+ transient, diastolic and average [Ca2+]i. In each group the unlabelled bar is control; C, caffeine; I, ISO; I+C, ISO + caffeine. (Reproduced from Sankaranarayanan et al. (2017).)
The experiment illustrated in Fig. 5 A shows the effects on diastolic [Ca2+]i of making the RyR leaky with caffeine (1 mm) (Sankaranarayanan et al. 2017). This is a higher concentration than was used in Fig. 4 and results in a release of Ca2+ from the SR even in the absence of stimulation indicating a marked decrease of SR Ca2+. The grey trace shows that, under basal conditions, the diastolic [Ca2+]i at 2 Hz stimulation is only slightly greater than resting [Ca2+]i. The subsequent application of caffeine decreased systolic [Ca2+]i and increased diastolic [Ca2+]i. After removing caffeine, application of the β‐adrenergic agonist isoprenaline (ISO) increased systolic [Ca2+]i. The subsequent addition of caffeine again decreased systolic [Ca2+]i and produced a more marked increase of diastolic [Ca2+]i than was seen in the absence of ISO. The average data (Fig. 5 B) confirm the effects of caffeine on diastolic and systolic [Ca2+]i. It is also noteworthy that, in the absence of caffeine, ISO produces a large increase of the amplitude of the Ca2+ transient with little or no effect on diastolic [Ca2+]i. In contrast, in the presence of caffeine, the effect of ISO on the amplitude is greatly diminished and that on diastolic is increased.
The maintained decrease of systolic [Ca2+]i contrasts with the transient potentiation with no steady state effect seen in Fig. 4. The explanation is that the higher concentration of caffeine results in a large release of Ca2+ from the SR thereby greatly decreasing SR Ca. If SR Ca2+ falls sufficiently then, even if RyR opening is potentiated enormously and all the Ca2+ is released from the SR, the Ca2+ transient will be smaller than in control (Negretti et al. 1993; Sankaranarayanan et al. 2016). A similar argument has been used to explain why RyR potentiation decreases the amplitude of the Ca2+ transient in heart failure (Belevych et al. 2007). An explanation of the associated increase of diastolic [Ca2+]i is that it is a direct result of the decrease of systolic [Ca2+]i and the need to preserve flux balance. Before caffeine is added, influx and efflux will be equal. In the presence of caffeine the smaller Ca2+ transient will decrease the systolic Ca2+ efflux to less than the influx leading to the cell gaining Ca2+. This is compensated by the increase of diastolic [Ca2+]i, which will increase efflux. Assuming that Ca2+ influx is unaffected by caffeine, then the total (systolic plus diastolic) efflux must be the same in the absence and presence of caffeine. If we assume that NCX activity is proportional to [Ca2+]i (Barcenas‐Ruiz et al. 1987), then one would expect the time‐averaged [Ca2+]i concentration to be constant. That this is indeed the case is demonstrated by the low‐pass filtered trace of Fig. 5 A and the mean data of Fig. 5 B, which show that average [Ca2+]i is unaffected by making the RyR leaky with caffeine (Sankaranarayanan et al. 2017). Figure 6 demonstrates the results of stimulating over a wider range of frequencies. The average level of [Ca2+]i is unaffected by addition of caffeine (Fig. 6 A – see Fig. 6 B for mean data). Increasing frequency increases average [Ca2+]i but in a manner that tends to saturation. The average level of [Ca2+]i can usefully be compared with the Ca2+ influx into the cell. As the frequency of stimulation is increased, the influx per pulse decreases due to increased inactivation (Fig. 6 C) (Sipido et al. 1998; Dibb et al. 2007). The influx per unit time, however, increases in a saturating manner with frequency. The linear relationships between influx per unit time and average [Ca2+]i are shown in Fig. 6 D. The lines are statistically identical in the absence and presence of caffeine. One noteworthy feature of Fig. 6 D is that, even when there is no Ca2+ influx, there is still a finite level of [Ca2+]i. The extrapolation of the fitted lines provides horizontal intercepts of the order of 4 μmol l−1 s−1 suggesting that, in addition to the L‐type Ca2+ current, there is a continuous influx of Ca2+ into the cell of this magnitude. This is a similar magnitude to the value obtained with a different method referred to above. It therefore appears that, at least under the conditions of these experiments, Ca2+ influx consists of two components: (i) background and (ii) L‐type Ca2+ current. There will doubtless be other routes including possible entry through reverse mode NCX but these are not resolved here. The tight correlation between Ca2+ entry and average [Ca2+]i demonstrates that it is the influx per unit time which sets the average level of [Ca2+]i; the properties of the SR simply determine the relative contributions of diastolic and systolic [Ca2+]i to the average. A good example of this is the effect of β‐adrenergic stimulation on [Ca2+]i. Under normal conditions (Fig. 5 B) this results in an increase of systolic [Ca2+]i with little or no effect on diastolic. In contrast, when the RyR is made leaky with ryanodine, the bulk of the increase is diastolic.
Figure 6. Interactions between RyR leak and stimulation frequency on diastolic, systolic and average [Ca2+]i .

A, time course (rat ventricular myocyte). Stimulation frequency was altered and 1 mm caffeine (Caf) applied as shown above. B, mean data showing (from top to bottom): amplitude, diastolic, average [Ca2+]i, Ca2+ influx per pulse, Ca2+ influx per second. C, top, specimen Ca2+ currents; bottom, integrated Ca2+ influx at the frequencies indicated. D, average [Ca2+]i as a function of Ca2+ influx per second. In all panels the red symbols indicate the presence of caffeine. (Reproduced from Sankaranarayanan et al. (2017).)
In addition to diastolic and systolic [Ca2+]i, Ca2+ waves also contribute to Ca2+ efflux and average [Ca2+]i. An increase of SR Ca2+ content increases the frequency of Ca2+ sparks resulting in the occurrence of waves of Ca2+‐induced Ca2+ release which propagate through the cell (Cheng et al. 1996). Some of the Ca2+ in these waves is pumped out of the cell by NCX and the resulting current has been shown to produce delayed afterdepolarizations and thereby initiate various arrhythmias (Ferrier et al. 1973; Mechmann & Pott, 1986). Despite their arrhythmogenic nature, these Ca2+ waves may serve a useful function in providing a route for Ca2+ efflux, in addition to elevated diastolic [Ca2+]i under conditions of Ca2+ overload (Díaz et al. 1997). More generally, at a given Ca2+ influx, the sum of the effluxes produced by the three components of efflux, systolic, diastolic and Ca2+ waves, must be constant. One example of the consequences of this is shown in Fig. 7. Under basal conditions (left hand panel), the systolic efflux (SE) balances Ca2+ influx. The application of a high concentration of isoprenaline (1 μm, middle panels), increases Ca2+ influx on the L‐type current (I) and results in Ca2+ waves. It is largely the Ca2+ efflux associated with the wave (DE) which balances the increased influx due to isoprenaline. Finally (right hand panel), tetracaine, which decreases RyR opening, abolishes the waves and the associated efflux. Ca2+ flux balance is now maintained by an increase of systolic efflux due to the increased Ca2+ transient (Venetucci et al. 2006).
Figure 7. Ca2+ efflux associated with Ca2+ waves.
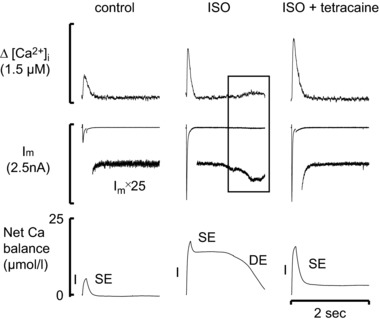
In each panel traces show (from top to bottom): [Ca2+]i; membrane current (both low gain and amplified); Ca2+ balance (the calculated change of total intracellular Ca2+ concentration). Panels show (from left to right): control; addition of isoprenaline (ISO,1 μm); addition of isoprenaline (1 μm) and tetracaine (50 μm). Experiment performed on a rat ventricular myocyte. I: influx; SE: Ca2+ efflux associated with the systolic Ca2+ transient; DE: Ca2+ efflux mediated by diastolic Ca2+ release. (Reproduced from Venetucci et al. (2006).)
The previous section has investigated the effects of disabling SR function by making the RyR leaky. We have also studied the effects of decreasing Ca2+ uptake into the SR by decreasing SERCA activity with thapsigargin. As has been shown previously (Janczewski & Lakatta, 1993; Negretti et al. 1993), the resulting decrease of SR Ca content decreases the amplitude of the Ca2+ transient. Figure 8 also shows that there is an increase of diastolic [Ca2+]i. This, in turn, will increase the diastolic efflux thereby compensating for the decrease of systolic efflux. Therefore, no matter whether the amplitude of the Ca2+ transient is decreased by making the RyR leaky or decreasing SERCA activity, the result is a compensatory increase of diastolic [Ca2+]i. Figure 8 B shows, again, that increasing stimulation frequency increases average [Ca2+]i. Thapsigargin has a much greater effect on diastolic [Ca2+]i at the higher stimulation rate. This is presumably because, at lower rates, there is time for [Ca2+]i to fall to close to the resting (unstimulated) level. Indeed, as shown in Fig. 8 A, in the absence of stimulation thapsigargin has no effect on resting [Ca2+]i.
Figure 8. The effects of SERCA inhibition on [Ca2+]i .

A, original record (rat ventricular myocyte). The cell was stimulated at the frequencies indicated and thapsigargin (1 μm) applied as shown. B, mean data showing the effects of stimulation frequency on (from top to bottom): amplitude, diastolic and average [Ca2+]i. (Reproduced from Sankaranarayanan et al. (2017).)
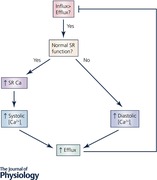
From these experiments, we therefore conclude that systolic [Ca2+]i is a major factor regulating diastolic [Ca2+]i. The overall regulation of diastolic [Ca2+]i is yet another example of the importance of considering Ca2+ flux balance. A given Ca2+ influx must be balanced by an equal efflux. Assuming that the properties of NCX are constant and the rate of NCX pumping is proportional to [Ca2+]i, then the level of Ca2+ influx sets the average level of [Ca2+]i. The relative diastolic and systolic levels that determine the average are determined by factors such as SR function. Making the RyR leaky or inhibiting SERCA will result in a situation with elevated diastolic and decreased systolic [Ca2+]i compared to the situation with a normal SR. All other things being equal, a decrease of systolic [Ca2+]i as occurs in heart failure would be expected to be associated with elevated diastolic [Ca2+]i. What actually happens will depend, also, on the Ca2+ influx. Although many studies in heart failure find that the L‐type current is unaffected, some work has reported a decrease (for review see Benitah et al. 2010). The latter case should be associated with a decrease of average [Ca2+]i thereby mitigating the expected rise of diastolic [Ca2+]i. It should, however, also be noted that the situation is potentially more complex. Firstly, the decrease of the Ca2+ transient may slow Ca2+‐dependent inactivation of the L‐type current and thereby maintain or even increase Ca2+ influx. Secondly, the prolongation of the action potential which occurs in heart failure will increase Ca2+ entry through the L‐type current.
Conclusions
I have referred extensively to the fact that the ventricular myocyte is in Ca2+ flux balance: on each beat, in the steady state, the amount of Ca2+ that enters the cell equals that which leaves. As I have reviewed, this has important consequences for cellular Ca2+ cycling and cardiac function. It underlies the regulation of SR Ca content. It explains why an increase in RyR open probability has no effect, in the steady state, on the amplitude of the Ca2+ transient. Finally, it explains the inverse relationship between the levels of diastolic and systolic [Ca2+]i as well as the importance of average [Ca2+]i.
Additional information
Competing interests
None.
Funding
Work from the author's laboratory was supported by a grant from the British Heart Foundation (grant number: CH/2000004/12801).
Acknowledgements
I am very grateful to the many colleagues who have participated in this research and made it all fun. I would particularly like to thank Andrew Trafford for his collaboration over many years.
Biography
David Eisner is The British Heart Foundation Professor of Cardiac Physiology at The University of Manchester. He studied Natural Sciences in Cambridge and then obtained his DPhil in Oxford in the laboratory of Denis Noble. He has held academic positions at University College London and Liverpool University before moving to Manchester. His research focuses on the regulation of intracellular calcium both in normal conditions and in the alterations that lead to cardiac arrhythmias and heart failure.

Edited by: Ole Petersen & Livia Hool
This review article is based on the 2017 Annual Review Prize Lecture of The Physiological Society, presented at the 38th World Congress of the International Union of Physiological Sciences held in Rio de Janeiro, Brazil, 1–5 August 2017.
This is an Editor's Choice article from the 1 January 2018 issue.
References
- Allen DG & Blinks JR (1978). Calcium transients in aequorin‐injected frog cardiac muscle. Nature 273, 509–513. [DOI] [PubMed] [Google Scholar]
- Allen DG, Jewell BR & Wood EH (1976). Studies of the contractility of mammalian myocardium at low rates of stimulation. J Physiol 254, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen DG & Kurihara S (1980). Calcium transients in mammalian ventricular muscle. Eur Heart J 1(Suppl A), 5–15. [DOI] [PubMed] [Google Scholar]
- Barcenas‐Ruiz L, Beuckelmann DJ & Wier WG (1987). Sodium‐calcium exchange in heart: membrane currents and changes in [Ca2+]i . Science 238, 1720–1722. [DOI] [PubMed] [Google Scholar]
- Bassani JW, Yuan W & Bers DM (1995). Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am J Physiol Cell Physiol 268, C1313–C1319. [DOI] [PubMed] [Google Scholar]
- Belevych A, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA & Györke S (2007). Enhanced ryanodine receptor‐mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys J 93, 4083–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitah JP, Alvarez JL & Gomez AM (2010). L‐type Ca2+ current in ventricular cardiomyocytes. J Mol Cell Cardiol 48, 26–36. [DOI] [PubMed] [Google Scholar]
- Bers DM (2001). Excitation‐Contraction Coupling and Cardiac Contractile Force. Kluwer Academic, Dordrecht. [Google Scholar]
- Bers DM (2008). Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70, 23–49. [DOI] [PubMed] [Google Scholar]
- Bers DM (2014). Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol 76, 107–127. [DOI] [PubMed] [Google Scholar]
- Beuckelmann DJ, Näbauer M & Erdmann E (1992). Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation 85, 1046–1055. [DOI] [PubMed] [Google Scholar]
- Borlaug BA & Paulus WJ (2011). Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J 32, 670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridge JH (1986). Relationships between the sarcoplasmic reticulum and sarcolemmal calcium transport revealed by rapidly cooling rabbit ventricular muscle. J Gen Physiol 88, 437–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case RM, Eisner D, Gurney A, Jones O, Muallem S & Verkhratsky A (2007). Evolution of calcium homeostasis: from birth of the first cell to an omnipresent signalling system. Cell Calcium 42, 345–350. [DOI] [PubMed] [Google Scholar]
- Chapman RA & Tunstall J (1980). The interaction of sodium and calcium ions at the cell membrane and the control of contractile strength in frog atrial muscle. J Physiol 305, 109–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Lederer MR, Lederer WJ & Cannell MB (1996). Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am J Physiol Cell Physiol 270, C148–C159. [DOI] [PubMed] [Google Scholar]
- Choi HS, Trafford AW, Orchard CH & Eisner DA (2000). The effect of acidosis on systolic Ca2+ and sarcoplasmic reticulum calcium content in isolated rat ventricular myocytes. J Physiol 529, 661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz ME, Graham HK & Trafford AW (2004). Enhanced sarcolemmal Ca2+ efflux reduces sarcoplasmic reticulum Ca2+ content and systolic Ca2+ in cardiac hypertrophy. Cardiovasc Res 62, 538–547. [DOI] [PubMed] [Google Scholar]
- Díaz ME, Trafford AW, O'Neill SC & Eisner DA (1997). Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J Physiol 501, 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibb KM, Eisner DA & Trafford AW (2007). Regulation of systolic [Ca2+]i and cellular Ca2+ flux balance in rat ventricular myocytes by SR Ca2+, L‐type Ca2+ current and diastolic [Ca2+]i . J Physiol 585, 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner D (2014). Calcium in the heart: from physiology to disease. Exp Physiol 99, 1273–1282. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Caldwell JL, Kistamás K & Trafford AW (2017). Calcium and excitation‐contraction coupling in the heart. Circ Res 121, 181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner DA, Kashimura T, O'Neill SC, Venetucci LA & Trafford AW (2009). What role does modulation of the ryanodine receptor play in cardiac inotropy and arrhythmogenesis? J Mol Cell Cardiol 46, 474–481. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Lederer WJ & Vaughan‐Jones RD (1983). The control of tonic tension by membrane potential and intracellular sodium activity in the sheep cardiac Purkinje fibre. J Physiol 335, 723–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner DA, Orchard CH & Allen DG (1984). Control of intracellular ionized calcium concentration by sarcolemmal and intracellular mechanisms. J Mol Cell Cardiol 16, 137–146. [DOI] [PubMed] [Google Scholar]
- Endoh M & Blinks JR (1988). Actions of sympathomimetic amines on the Ca2+ transients and contractions of rabbit myocardium: reciprocal changes in myofibrillar responsiveness to Ca2+ mediated through alpha‐ and beta‐adrenoceptors. Circ Res 62, 247–265. [DOI] [PubMed] [Google Scholar]
- Fabiato A (1985). Time and calcium dependence of activation and inactivation of calcium‐induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol 85, 247–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrier GR, Saunders JH & Mendez C (1973). A cellular mechanism for the generation of ventricular arrhythmias by acetylstrophanthidin. Circ Res 32, 600–609. [DOI] [PubMed] [Google Scholar]
- Frace AM, Mery PF, Fischmeister R & Hartzell HC (1993). Rate‐limiting steps in the beta‐adrenergic stimulation of cardiac calcium current. J Gen Physiol 101, 337–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friel DD & Tsien RW (1992). A caffeine‐ and ryanodine‐sensitive Ca2+ store in bullfrog sympathetic neurones modulates effects of Ca2+ entry on [Ca2+]i . J Physiol 450, 217–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Wang F, Wang W, Makarewich CA, Zhang H, Kubo H, Berretta RM, Barr LA, Molkentin JD & Houser SR (2012). Ca2+ influx through L‐type Ca2+ channels and transient receptor potential channels activates pathological hypertrophy signaling. J Mol Cell Cardiol 53, 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greensmith DJ, Galli GLJ, Trafford AW & Eisner DA (2014). Direct measurements of SR free Ca reveal the mechanism underlying the transient effects of RyR potentiation under physiological conditions. Cardiovasc Res 103, 554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Gillespie D & Fill M (2012). Ryanodine receptor current amplitude controls Ca2+ sparks in cardiac muscle. Circ Res 111, 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W & Morgan JP (1987). Abnormal intracellular calcium handling in myocardium from patients with end‐stage heart failure. Circ Res 61, 70–76. [DOI] [PubMed] [Google Scholar]
- Gwathmey JK, Warren SE, Briggs GM, Copelas L, Feldman MD, Phillips PJ, Callahan M Jr, Schoen FJ, Grossman W & Morgan JP (1991). Diastolic dysfunction in hypertrophic cardiomyopathy. Effect on active force generation during systole. J Clin Invest 87, 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janczewski AM & Lakatta EG (1993). Thapsigargin inhibits Ca2+ uptake, and Ca2+ depletes sarcoplasmic reticulum in intact cardiac myocytes. Am J Physiol Heart Circ Physiol 265, H517–H522. [DOI] [PubMed] [Google Scholar]
- Kimura J, Noma A & Irisawa H (1986). Na‐Ca exchange current in mammalian heart cells. Nature 319, 596–597. [DOI] [PubMed] [Google Scholar]
- Kirchberber MA, Tada M & Katz AM (1975). Phospholamban: a regulatory protein of the cardiac sarcoplasmic reticulum. Recent Adv Stud Cardiac Struct Metab 5, 103–115. [PubMed] [Google Scholar]
- Kupittayanant P, Trafford AW, Díaz ME & Eisner DA (2006). A mechanism distinct from the L‐type Ca current or Na‐Ca exchange contributes to Ca entry in rat ventricular myocytes. Cell Calcium 39, 417–423. [DOI] [PubMed] [Google Scholar]
- Lamont C & Eisner DA (1996). The sarcolemmal mechanisms involved in the control of diastolic intracellular calcium in isolated rat cardiac trabeculae. Pflugers Arch 432, 961–969. [DOI] [PubMed] [Google Scholar]
- Lorin C, Vogeli I & Niggli E (2015). Dystrophic cardiomyopathy: role of TRPV2 channels in stretch‐induced cell damage. Cardiovasc Res 106, 153–162. [DOI] [PubMed] [Google Scholar]
- Mechmann S & Pott L (1986). Identification of Na‐Ca exchange current in single cardiac myocytes. Nature 319, 597–599. [DOI] [PubMed] [Google Scholar]
- Negretti N, O'Neill SC & Eisner DA (1993). The effects of inhibitors of sarcoplasmic reticulum function on the systolic Ca2+ transient in rat ventricular myocytes. J Physiol 468, 35–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedergerke R (1963). Movements of Ca in frog heart ventricles at rest and during contractures. J Physiol 167, 515–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringer S (1883). A further contribution regarding the influence of the different constituents of the blood on the contraction of the heart. J Physiol 4, 29–42.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios E ( 2010). The cell boundary theorem: a simple law of the control of cytosolic calcium concentration. J Physiol Sci 60, 81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runte KE, Bell SP, Selby DE, Häußler TN, Ashikaga T, LeWinter MM, Palmer BM & Meyer M (2017). Relaxation and the role of calcium in isolated contracting myocardium from patients with hypertensive heart disease and heart failure with preserved ejection fraction. Circ Heart Fail 10, e004311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan R, Kistamas K, Greensmith DJ, Venetucci LA & Eisner DA (2017). Systolic [Ca2+]i regulates diastolic levels in rat ventricular myocytes. J Physiol 595, 5545–5555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan R, Li Y, Greensmith DJ, Eisner DA & Venetucci L (2016). Biphasic decay of the Ca transient results from increased sarcoplasmic reticulum Ca leak. J Physiol 594, 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabir S, Borisova L, Wray S & Burdyga T (2004). Rho‐kinase inhibition and electromechanical coupling in rat and guinea‐pig ureter smooth muscle: Ca2+‐dependent and ‐independent mechanisms. J Physiol 560, 839–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon TR, Wang F & Bers DM (2005). Regulation of cardiac sarcoplasmic reticulum Ca release by luminal [Ca] and altered gating assessed with a mathematical model. Biophys J 89, 4096–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipido KR, Callewaert G & Carmeliet E (1995). Inhibition and rapid recovery of Ca2+ current during Ca2+ release from sarcoplasmic reticulum in guinea pig ventricular myocytes. Circ Res 76, 102–109. [DOI] [PubMed] [Google Scholar]
- Sipido KR, Stankovicova T, Flameng W, Vanhaecke J & Verdonck F (1998). Frequency dependence of Ca2+ release from the sarcoplasmic reticulum in human ventricular myocytes from end‐stage heart failure. Cardiovasc Res 37, 478–488. [DOI] [PubMed] [Google Scholar]
- Touchberry CD, Elmore CJ, Nguyen TM, Andresen JJ, Zhao X, Orange M, Weisleder N, Brotto M, Claycomb WC & Wacker MJ (2011). Store‐operated calcium entry is present in HL‐1 cardiomyocytes and contributes to resting calcium. Biochem Biophys Res Commun 416, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trafford AW, Díaz ME & Eisner DA (1998). Stimulation of Ca‐induced Ca release only transiently increases the systolic Ca transient: measurements of Ca fluxes and sarcoplasmic reticulum Ca. Cardiovasc Res 37, 710–717. [DOI] [PubMed] [Google Scholar]
- Trafford AW, Díaz ME, Negretti N & Eisner DA (1997). Enhanced Ca2+ current and decreased Ca2+ efflux restore sarcoplasmic reticulum Ca2+ content after depletion. Circ Res 81, 477–484. [DOI] [PubMed] [Google Scholar]
- Trafford AW, Díaz ME, Sibbring GC & Eisner DA (2000). Modulation of CICR has no maintained effect on systolic Ca2+: simultaneous measurements of sarcoplasmic reticulum and sarcolemmal Ca2+ fluxes in rat ventricular myocytes. J Physiol 522, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venetucci LA, Trafford AW, Díaz ME, O'Neill SC & Eisner DA (2006). Reducing ryanodine receptor open probability as a means to abolish spontaneous Ca2+ release and increase Ca2+ transient amplitude in adult ventricular myocytes. Circ Res 98, 1299–1305. [DOI] [PubMed] [Google Scholar]