

Abstract
Alzheimer's disease (AD) is marked by the presence of extracellular amyloid beta (Aβ) plaques, intracellular neurofibrillary tangles (NFTs) and gliosis, activated glial cells, in the brain. It is thought that Aβ plaques trigger NFT formation, neuronal cell death, neuroinflammation and gliosis and, ultimately, cognitive impairment. There are increased numbers of reactive astrocytes in AD, which surround amyloid plaques and secrete proinflammatory factors and can phagocytize and break down Aβ. It was thought that neuronal cells were the major source of Aβ. However, mounting evidence suggests that astrocytes may play an additional role in AD by secreting significant quantities of Aβ and contributing to overall amyloid burden in the brain. Astrocytes are the most numerous cell type in the brain, and therefore even minor quantities of amyloid secretion from individual astrocytes could prove to be substantial when taken across the whole brain. Reactive astrocytes have increased levels of the three necessary components for Aβ production: amyloid precursor protein, β-secretase (BACE1) and γ-secretase. The identification of environmental factors, such as neuroinflammation, that promote astrocytic Aβ production, could redefine how we think about developing therapeutics for AD.
Keywords: Alzheimer's disease, astrogliosis, neuroinflammation, amyloid beta
1. Alzheimer's disease
Alzheimer's disease (AD), the most common form of dementia, is characterized by diminished cognitive function, specifically dysfunction of memory and judgement. With a rapidly ageing population, AD has become a major public health concern.
Pathologically, AD is marked by the presence of extracellular amyloid plaques, intracellular neurofibrillary tangles (NFTs) and gliosis [1] in the brain. The extracellular amyloid plaques are mainly composed of aggregated β-amyloid peptide (Aβ), whereas the NFTs are intracellular and are composed of hyperphosphorylated tau, a microtubule-binding protein [2]. Gliosis is a non-specific phenomenon that occurs in response to any injury to the CNS and involves the activation, and often proliferation, of glial cells. In AD, gliosis is marked by increases in activated microglia and reactive astrocytes near the sites of amyloid plaques [3]. Reactive astrocytes surrounding amyloid beta plaques contribute to the local inflammatory response and modulate calcium signalling [4,5].
2. Amyloid beta
The widely accepted amyloid cascade hypothesis states that AD is driven by Aβ accumulation [6]. It is thought that Aβ aggregates trigger a cascade of reactions, involving NFT formation, neuronal cell death, neuroinflammation and gliosis and, ultimately, cognitive impairment. It is important to note that Aβ exists in many forms: monomers, dimers, oligomers, fibrils and plaques [7].
The amyloid cascade hypothesis has significant genetic support. Autosomal dominant AD (ADAD) mutations have been identified in amyloid precursor protein (APP) [8] and presenilin (PS) [9,10], two necessary genes for Aβ production. Specifically, 30 APP mutations, 9 APP duplications, 211 PS1 mutations and 33 PS2 mutations have been identified that cause early onset ADAD [11]. Furthermore, Down's syndrome patients with trisomy of chromosome 21 and individuals with small internal chromosome 21 duplications have an additional copy of APP and greatly increased risk of developing AD [12]. While these mutations provide insight into disease aetiology, they account for a very small percentage of AD cases. In addition, an APP mutation that reduces Aβ production protects against AD and age-related cognitive decline [13], providing another line of support for the Aβ hypothesis. It is worth mentioning that how these PS1 and PS2 mutations contribute to the pathogenesis of AD, such as if they are loss-of-function or gain-of-function, is the subject of considerable debate [14–19].
Most commonly, AD presents as a sporadic multi-factorial condition. ADAD studies strongly indicate that Aβ plays a critical role in AD pathogenesis. APP is a highly conserved integral membrane protein thought to play a role in synapse formation and neural plasticity, but its primary function has yet to be described. It can be processed in two separate pathways. In the amyloidogenic pathway, APP is first cleaved by β-secretase (BACE) followed by γ-secretase cleavage and release of Aβ peptides and APP intracellular domain (AICD) (figure 1) [20–23]. In the non-amyloidogenic pathway, α-secretase and then γ-secretase cleave APP sequentially; this does not result in the generation of Aβ species and acts as a negative feedback on γ-secretase activity [24,25].
Figure 1.
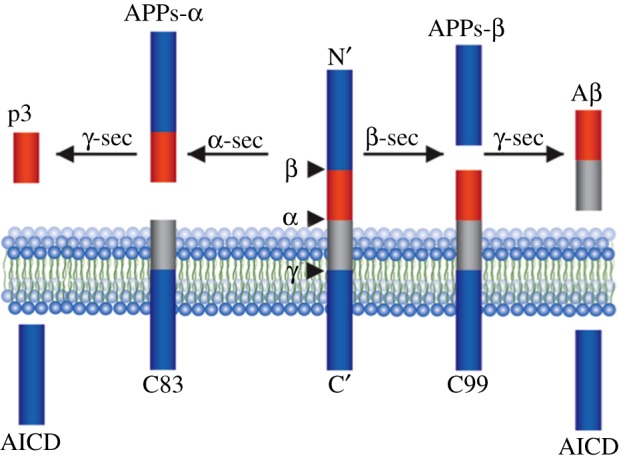
Aβ production. In the amyloidogenic pathway (right), APP is cleaved by BACE followed by γ-secretase which releases Aβ peptides and APP intracellular domain (AICD), which generate the N and C termini of Aβ, respectively. In the non-amyloidogenic pathway (left), APP is cleaved sequentially by α-secretase and γ-secretase, which does not result in the generation of Aβ species.
BACE1, an aspartyl protease, is tethered to the membrane by a long tail; it is found in the endoplasmic reticulum and Golgi and functions to prune proteins. Many of its substrates are involved in neural function, including neuregulin and voltage-gated sodium channels [20–23,26].
PS is the catalytic subunit of γ-secretase [27,28]. Three other necessary γ-secretase subunits have also been identified: nicastrin (Nct), anterior pharynx-defective-1 (Aph1) and presenilin enhancer-2 (Pen2) [29,30]. These four components constitute the mature γ-secretase complex [31,32], and their stepwise assembly, followed by endoproteolysis of PS into amino-terminal (PS-NTF) and carboxy-terminal fragments (PS-CTF), is necessary for active complex formation [33]. Therefore, γ-secretase activity is regulated by the abundance of the four essential subunits and their assembly. Additionally, only a small fraction of γ-secretase in the cell is actually catalytically active [34–37]. This suggests that additional events are necessary to activate the inactive complex [38].
3. Astrogliosis and neuroinflammation
Aβ accumulation triggers a neuroinflammatory state that plays a significant role in the progression of AD [39,40]. Levels of Aβ in the brain are regulated by an innate immune response. Aβ, thought to be primarily produced by neurons, can activate an inflammatory response that ultimately drives microglia and astrocytes to uptake and clear it from the brain [41–43]. Genetic studies identifying single-nucleotide polymorphisms in inflammatory genes that are associated with the risk of AD underline the involvement of inflammation in AD [44–48]. Furthermore, AD patients have higher levels of proinflammatory cytokines and activated inflammasomes [49].
Astrocytes are key regulators of the brain's inflammatory response and, as mentioned previously, reactive astrogliosis is a universally acknowledged feature of AD. Marked by cellular hypertrophy and an increase in glial fibrillary acidic protein (GFAP) and S100B expression, astrogliosis is observed in post-mortem tissues from AD patients and mouse models. Moreover, the degree of astrogliosis is correlated with cognitive decline [50–52].
Astroglia are found throughout the CNS and are thought to be the most prevalent cell type in the brain [53]. Astrocytes function in territorial domains in which they are connected to the vasculature through processes terminating in the endfoot. Furthermore, their processes also envelope neuronal synapses [54]. This intricate system of connections enables astrocytes to exert control over many necessary brain functions including regulation of the blood–brain barrier, delivering nutrients to nervous tissue and maintaining ion and metabolite balance. Astrocytes can propagate calcium currents, release gliotransmitters and signal with neurons [55]. Specifically, astrocytes release neurotransmitters such as glutamate, GABA and ATP, neuromodulators d-serine and kynurenic acid, and growth factors and inflammatory mediators [56–58]. A chief role of astrocytes in the brain is to protect in all manners against CNS injury and to repair nervous tissue after injury. This is primarily achieved through astrogliosis, an evolutionarily conserved event that contributes to the neuroprotection and isolation of damaged tissue through the formation of a glial scar and removal of pathogens from the CNS [59].
Astrogliosis occurs when astrocytes respond to injuries to the CNS by undergoing a spectrum of molecular and morphological changes. Inflammatory mediators released by microglia, neurons, oligodendrocytes, endothelial cells, leucocytes and other astrocytes in response to injury initiate the changes associated with an astrocyte becoming reactive. Molecular changes entail a wide spectrum of genes resulting in differing expression of structural proteins, transcriptional regulators, extracellular matrix components, inflammatory regulators, vascular regulators and synaptic modulators (figure 2).
Figure 2.

Cellular stress can trigger astrogliosis—increased numbers of reactive astrocytes, which are characterized by hypertrophy of processes. Astrocytes undergo many molecular changes when activated and can secrete a plethora of proinflammatory cytokines.
Interestingly, these molecular changes are highly context specific. While there is a core group of genes that are consistently upregulated across different reactive models, approximately 50% of the altered gene expression varies depending on the initiating injury [60,61]. The primary morphological change during reactive astrogliosis is hypertrophy of processes, which is linked to increased expression of intermediate filament, most notably GFAP [62,63]. The functional consequences of this increased GFAP expression are not yet fully understood; however, it appears to be critical in limiting Aβ plaque build-up.
The impact of reactive astrogliosis in disease is complex: reactive astrocytes can be both harmful and beneficial to surrounding cells and may worsen or resolve the initial CNS injury. Notably, reactive astrocytes are necessary for scar formation, which helps to contain the spread of inflammatory cells, and also for repairing insults to the blood–brain barrier. Reactive astrocytes surround Aβ plaques in a manner similar to glial scarring and express receptors such as RAGE, low-density lipoprotein receptor-like protein, membrane-associated proteoglycans and scavenger receptor-like receptors that are known to bind Aβ [3].
Conversely, reactive astrocytes may be neurotoxic when producing reactive oxygen species or some inflammatory cytokines [64]. Further research into the mechanisms regulating the balance between when reactive gliosis is neuroprotective and when it is neurotoxic is critical to understanding the functional consequence of reactive astrocytes in AD. However, a caveat to much of this research is the reliance on rodent models, as human astrocytes are much larger and have a multitude more processes than their rodent counterparts [65]. To summarize, reactive astrogliosis is a complicated and diverse phenomenon yet is ubiquitous across various CNS pathologies.
4. Astrocytes in Alzheimer's disease
Astrocytes undergo complex and conflicting region-specific changes during the course of AD. The number of astrocytes is thought to remain constant throughout the brain during the progression of disease; however, some populations, those in proximity to amyloid beta plaques, become reactive, while conversely, large numbers of astrocytes atrophy [66]. In the 3XTg AD model, the initial astroglial phenotype is general dystrophy [66].
Astrodegeneration, defined as reduced astrocyte volume and surface area and a decrease in protoplasmic processes, has been observed in mouse models of AD. Beginning early in pathology, prior to amyloid beta presence, 3XTg AD mice exhibit astrodegeneration in the medial prefrontal cortex (mPFC), entorhinal cortex (EC) and hippocampus [3,67,68]. Similarly, astroglial atrophy is observed in the hippocampus prior to amyloid beta presence in the PDAPP mouse model [69]. At the later stages of disease, the presence of Aβ triggers a secondary astroglial response by activating astrocytes, resulting in reactive astrogliosis in areas surrounding the plaques [3,70].
Both resting and reactive astrocytes are key regulators of the brain's inflammatory response and are capable of releasing, and responding to, a spectrum of immune mediators [71]. Specifically, astrocytes secrete many cytokines capable of inducing inflammation, notably IFNγ, IL-1β, TNFα, IL-6 and TGFβ [72–75]. Many of these proinflammatory cytokines are upregulated in human AD brain samples and in transgenic mouse models of AD [76–79].
IFNγ is a potent regulatory cytokine that activates microglia, promotes inflammation and is upregulated in the AD brain [80]. It is primarily produced by T cells and natural killer cells but can also be secreted by microglia and astrocytes [81,82]. TNFα, a cytokine involved in inducing acute-phase inflammation, is elevated in AD serum, CSF and cortex [83]. Tg2576 mice deficient in CD40 (a TNF receptor gene) have reduced BACE activity, Aβ load and gliosis compared with normal Tg2576 mice, highlighting the importance of TNF in AD progression [79]. IL-6 can be both proinflammatory and anti-inflammatory and has been reported to be elevated in the plasma, cerebrospinal fluid, and the brain of AD [84–89]. IL-1β, one of the first cytokines secreted in response to injury, is an important mediator of inflammatory response as well as cell proliferation, differentiation and apoptosis. It is found at high levels near the sites of amyloid plaques [87,90,91].
A genetic polymorphism in transforming growth factor β1 (TGFB1), an immunosuppressive cytokine, is associated with the risk of developing AD [92]. Additionally, post-mortem AD brains contain increased levels of TGFβ, specifically in plaques, suggesting it may play a role in pathology [93,94]. In agreement, aged mice overexpressing TGFβ in astrocytes displayed Aβ deposition, and astrocytes containing TGFB1 are found in close proximity to Aβ deposits in mice overexpressing APP with the Swedish mutation. This suggests that the mechanism by which TGFβ contributes to pathology is astrocyte specific [78,95–97].
5. Astrocytes contribute to Aβ load
It was long thought that neurons were the only cell type that expressed high levels of BACE1 and, therefore, were the only cell capable of producing Aβ [98]. However, subsequent studies have demonstrated that astrocytes express BACE1 at sufficient levels to generate Aβ, and that expression can be increased by cellular stress [99–104]. Additionally, stressors can upregulate APP expression and, therefore, Aβ secretion. The effect of cellular stress on the activity of γ-secretase, the third necessary factor for amyloid production, in astrocytes has yet to be fully elucidated. As astrocytes substantially outnumber neurons in the brain, the identification of environmental factors (i.e. inflammation), which promote astrocytic Aβ production, could redefine how we think about developing therapeutics for AD.
As mentioned previously, FAD mutations in APP and PS have been extensively studied to gain insight into the mechanisms underlying AD. However, the majority of these studies have focused on neurons. In order to determine the effect of FAD PS1 mutations on Aβ production from cell types other than neurons, Veeraraghavalu et al. [105] selectively inactivated PS1ΔE9 in postnatal forebrain excitatory neurons, which are thought to be the primary source of Aβ, by crossing PS1ΔE9flox mice with CaMKIICre mice. They determined that at 10–12 months of age, total Aβ burden in these mice was indistinguishable from mice expressing PS1ΔE9 in all cell types. This suggests that the FAD mutation drives high Aβ load by increasing Aβ secretion in other types of neurons or glial cells. To identify these other cellular sources of Aβ, they dissociated primary astrocytes and microglia from the brains of newborn PS1ΔE9flox or APPswe mice or 8-week-old APPswe/PS1ΔE9flox mice. Both astrocytes and microglia secreted detectable Aβ. Treatment with a γ-secretase inhibitor prevented Aβ secretion and resulted in the accumulation of the βCTF fragment [105]. This suggests that not only are astrocytes capable of producing Aβ, but that they do so in levels substantial enough to contribute significantly to total amyloid load. If this is the case, then astrocytes must express appreciable levels of APP, BACE and γ-secretase. In support, Grolla et al. [106] detected APP, BACE1 and γ-secretase subunits PS1, PS2, PEN2 and NCT expression in primary rat hippocampal astrocyte cultures.
6. Amyloid precursor protein expression in astrocytes
APP is expressed in all tissues; however, the relative amount of APP in different cell types varies [107–109]. Astrocytic expression of APP has been demonstrated. APP695, APP751 and APP770 mRNA have been identified in non-neuronal cells in the human brain [110] and rat astrocytes [111]. This is supported in primary microglial and astrocyte culture of newborn rat pups, which express mRNA for all three APP isoforms [112]. When normalized to beta-actin mRNA levels, primary rat astrocytes expressed 94% of the amount of APP as neurons [113]. Additionally, inflammatory mediators have been shown to regulate APP levels [114–116].
Multiple proinflammatory cytokines have been shown to upregulate APP in the mouse brain and in human neuroblastoma cells and non-neuronal cells such as human astrocyte cultures [116]. This implies that in the neuroinflammatory context of AD, reactive astrocytes express higher levels of APP than when at rest and, therefore, could produce more Aβ. Lipopolysaccharides (LPS) treatment induces chronic neuroinflammation [117,118] and can contribute to learning and memory deficits [119–121]. It has been established that stress from injections of LPS, a known activator of CNS glia [122], can induce a twofold increase in APP expression in the whole brains of APPswe mice. Dramatically, LPS treatment resulted in an 18-fold increase in βCTF, suggesting massively increased BACE activity, and ultimately a threefold upregulation of both Aβ40 and Aβ42. While this study looked at the whole brain, and not specific cell types, it is worth noting that LPS treatment also increased the levels of GFAP-positive astrocytes in the cortex and hippocampus [123].
APP expression can be upregulated by the transcription factor AP-1, which is found in the promoter region of most acute-phase proteins that are induced by IL-1β and IL-6, suggesting APP may be regulated by these specific cytokines [114,115]. Supporting this, IL-1β has been shown to upregulate APP in human astrocytes and the U373MG human astrocytoma cell line [124,125].
Zhao et al. [103] demonstrated that in primary mouse astrocytes, stimulation with several proinflammatory cytokine combinations (LPS + IFNγ, TNFα + IFNγ and TNFα + IL-1β + IFNγ) markedly increases expression of APP. The same combinations of cytokines also induce increases in BACE1 protein by up to eightfold. The downstream consequence is a 20–40% increase in Aβ40 secretion [103]. A combination of IFNγ and TNFα has also been shown to induce Aβ secretion from primary human astrocytes and the U373 cell line [104]. To conclude, systemic inflammation from LPS treatment and specific AD-associated inflammatory mediators can upregulate APP expression in astrocytes.
As previously mentioned, TGFβ is associated with AD development [92]. Lesné et al. [126] confirmed this effect in a mouse model that overexpresses TGFβ under the GFAP promoter, driving astrocytic expression. They found increased APP and soluble Aβ40 and Aβ42 in the whole brain. To investigate whether the increase in Aβ was produced by the astrocytes, or whether the TGFβ overexpressing astrocytes released a secondary mediator that, in turn, induced neuronal Aβ production, they cultured primary neurons and astrocytes with TGFβ. They found an increase in APP and Aβ40 and Aβ42 in the astrocyte culture, but not in the neuronal culture, indicating that the increased Aβ in the mice was produced by astrocytes [126]. Furthermore, astrocytoma cell lines and normal human astrocytes have increased expression of APP when exposed to TGFβ [127–129]. Taken together, this suggests that TGFβ increases Aβ in the AD brain by inducing APP upregulation in astrocytes and subsequently inducing astrocytic Aβ secretion.
7. BACE1 expression in astrocytes
It has been thought that astrocytes do not contribute to Aβ load due to a lack of BACE1 activity, which is highly expressed in the brain but primarily in neurons [22]. There is little evidence to suggest that nonreactive astrocytes express significant levels of BACE1 [99,130,131]. Zhao et al. [132] generated two mouse models overexpressing the APP751. In one line, APP was under the NSE promoter driving neuronal expression, while in the other line the GFAP promoter was used to drive astrocytic expression. They determined that primary neurons from the NSE-APP mice produced large amounts of βCTF, suggesting high BACE1 activity. However, primary astrocytes from the GFAP–APP mice had very little BACE1 activity and no detectable Aβ production [20–23,132].
Studies propose that resting astrocytes express BACE1 mRNA but not protein, indicting a translational block may inhibit Aβ production in astrocytes [131]. This suggests that stress may be able to upregulate BACE1 activity by overturning this translational block. Hartlage-Rubsamen et al. [130] demonstrated that activation of glial cells induces BACE1 expression in six rat models of acute stress known to induce reactive gliosis: LPS + IFNγ treatment, intraaccumbal α,β-methylene adenosine 5′-triphosphate (α,β-meATP) treatment, middle cerebral artery occlusion, experimental autoimmune encephalomyelitis or Borna disease virus infection. Interestingly, they found marked increases in BACE1 expression in GFAP-positive astrocytes in the chronic models of stress, but not in the acute inflammation resulting from LPS + IFNγ or α,β-meATP [130]. This suggests that in the context of AD, neuroinflammatory stress my upregulate BACE expression in astrocytes. In fact, BACE1 expression has been demonstrated in reactive astrocytes and in astrocytes in human AD patients [99–102].
BACE expression is observed in reactive astrocytes around amyloid plaques. The number of these BACE1-positive reactive astrocytes was increased in AD patients compared to old age controls, particularly in the entorhinal cortex [100]. In Tg2576 mice, which overexpress APP695 with the Swedish mutation, the level of BACE1 protein is correlated to the level of Aβ in astrocytes. Additionally, reactive astrocytes surrounding Aβ plaques always stain positively for BACE1 protein, whereas resting astrocytes do not [99,101].
Similar to APP expression, inflammation has also been shown to upregulate BACE1 [133]. Specifically, proinflammatory cytokines upregulate BACE1 activity [134]. Many proinflammatory cytokines signal through JAK/STAT pathways to ultimately influence transcriptional changes. STAT1 directly binds to the BACE1 promoter, suggesting a possible mechanism by which inflammation induces BACE1 expression. Specifically, IFNγ induces BACE1 expression in the U373 cell line and in primary mouse astrocytes [135]. In support of this, it has been shown that IFNγ and TNFα regulated BACE1 expression and Aβ production in APPswe transgenic mice. Furthermore, APP overexpressing mice, with the IFNγ receptor knocked out, have reduced Aβ deposition compared with APP transgenic mice. This is paired with reduced numbers of astrocytes and microglia in the cortex and hippocampus. Primary astrocytes overexpressing APP with the Swedish mutation (via adenovirus) secrete higher levels of TNFα than wild-type (WT) astrocytes, and this effect is abolished in IFNGR KO astrocytes, indicating that IFNγ signalling is critical for TNFα secretion. Furthermore, TNFα induces BACE1 expression and Aβ production in astrocytes in a dose-dependent manner. This effect was enhanced by the addition of IFNγ [136]. Taken together, we can conclude that IFNγ and TNFα upregulate BACE1 expression in astrocytes and ultimately increase Aβ secretion. Subsequently, Cho et al. [137] demonstrated that this upregulation was mediated by the activation of JAK2 and ERK1/2 signalling.
Other inflammatory mediators have also been shown to upregulate BACE1 expression. NF-κB is a protein complex of DNA transcription factors that plays a role in cytokine production and cell survival. In the aged and AD brain, there are increased levels of NF-κB and NF-κB transcription factor-mediated responses to stress are enhanced [138–140]. Aβ stimulates NF-κB activation in primary rat astrocytes in a dose- and time-dependent manner [141]. The rat and human BACE1 promoters have an NF-κB binding site [142]. Deletion studies suggest that the NF-κB binding site suppresses BACE1 expression and Aβ secretion in neurons when occupied by NF-κB [143,144]. It has been demonstrated that NF-κB suppresses BACE1 expression in nonreactive astrocytes; however, it has the opposite effect in TNFα-activated primary rat astrocytes. We can conclude, that in the context of inflammation, when astrocytes are reactive, NF-κB can induce BACE1 expression [145].
Other specific stress-induced pathways that upregulate BACE1 have been identified. The transcription factor Ying Yang 1 (YY1) functions in glucose metabolism, DNA repair and notch signalling and can bind to the BACE1 promoter to induce BACE1 activity in primary rat astrocytes and neurons. When permanent cerebral ischaemia was induced in rats by coagulation of the middle cerebral artery [146] in order to stress astrocytes, primary cultured astrocytes from these rats robustly expressed YY1. This suggests that YY1 can upregulate BACE1 activity in astrocytes under stressful conditions [147].
Furthermore, astrocytes also express BACE2, a close homologue of BACE1. Whether BACE2 activity results in APP cleavage, and ultimately Aβ production, is still up for debate. Ablation of BACE1 and BACE2 in a mouse model had reduced Aβ production compared to just a BACE1 knockout, suggesting BACE2 does, in fact, contribute to Aβ load [148]. BACE2 activity is detectable in nonreactive primary rat astrocytes and levels of activity actually decrease when the astrocyte is activated [103,104,131].
Aβ itself could be considered a proinflammatory mediator due to its ability to induce inflammation [149]. Additionally, it is well established that Aβ can stimulate proinflammatory cytokine release from astrocytes [150,151] Therefore, we can conclude that in AD, Aβ itself can upregulate BACE1 expression in astrocytes by stimulating an inflammatory response. Additionally, Aβ may cause neurotoxicity by disrupting intracellular calcium homeostasis in neurons and in glial cells. Disrupted calcium homeostasis is observed in the brains of AD patients; however, the mechanism of this Aβ-induced deregulation is unclear [152]. Nuclear factor of activated T cells (NFAT) is a transcription factor that regulates BACE1 expression by directly binding to its promoter region in response to signalling by the calcium- and calmodulin-dependent phosphatase calcineurin. BACE1 expression is enhanced in primary neuronal cells and SH-SY5Y neuroblastoma cells after stimulation by a calcium ionophore. This upregulation can be blocked by pretreatment with either an inhibitor of calcineurin or a calcium chelator. Aβ treatment stimulates activation and nuclear translocation of NFAT1 resulting in increased BACE1 expression. Additionally, NFAT1 activation is observed in APPswe mouse brains. Taken together, Aβ induces increases in intracellular calcium that can stimulate BACE1 expression, inducing further Aβ generation [153].
Jin et al. [101] demonstrated Aβ1–42 or Aβ25–35 treatment enhances BACE1 promoter activity and BACE1 protein levels in U373 cells, and this can be blocked by pretreatment with a calcineurin inhibitor. This increase in BACE1 levels resulted in increased Aβ secretion that could also be prevented by pretreatment with a calcineurin inhibitor. Furthermore, this Aβ-induced BACE1 upregulation can be blocked by preventing calcium influx through treatment with 2APB, an inhibitor of IP3-dependent calcium release, and U73122, an inhibitor of PLC. Aβ can form pores in cell membranes that may be permeable to calcium influx, which can be blocked by Zn2+ [154]. Jin et al. [101] used pretreatment with ZnCl2 to prevent calcium influx through Aβ-induced pores and found that Aβ no longer enhanced BACE1 expression.
Dal Prà et al. [155] found little Aβ secretion from resting normal adult human astrocytes. However, when these cells are activated by exposure to Aβ25–35, an Aβ42 proxy which contains its active site [155], there was a translocation of HIFα to the nucleus. This upregulated BACE1 and increased γ-secretase activity, ultimately leading to significant Aβ42 secretion [156]. These observations indicate that neuronally secreted Aβ could induce Aβ production in reactive astrocytes through HIF1α.
8. γ-Secretase activity in astrocytes
As previously mentioned, γ-secretase activity is not correlated with the quantity of catalytic subunit; this makes it difficult to quantify, so it is not surprising that little is known about γ-secretase activity in astrocytes in AD. PS1 mRNA is highly expressed in astrocytes [10], and PS1 protein expression has been confirmed in glial cells in primate brain; however, staining is weak compared to neuronal cells [157]. Similar to APP and BACE1 expression, PS1 is elevated in reactive astrocytes in the AD brain [158,159]. Specifically, TGFβ may upregulate PS1 mRNA in the human U87 MG astrocytoma cell line [160]. However, as γ-secretase protein levels do not correlate to γ-secretase activity levels, these studies do not fully elucidate the role of γ-secretase-mediated Aβ production in astrocytes.
9. Other stressors
In addition to neuroinflammation, there may be other physiologically relevant cellular stressors that trigger APP and BACE and promote Aβ generation in astrocytes. Under stress, the activation of the hypothalamic–pituitary–adrenal axis results in glucocorticoid (GC) secretion from the adrenal cortex. Elevated GC is associated with cognitive impairment and has been implicated in AD pathology [161,162]. There are GC response elements in the APP and BACE1 promoter, indicating that GC signalling can upregulate their expression [163,164]. Wang et al. [165] demonstrated that GCs promote Aβ40 and Aβ42 secretion from primary mouse astrocytes. They attributed this to an increase in both APP and BACE1 mRNA and protein. Next, they demonstrated a similar increase in APP, BACE and Aβ production in 9-month-old mice treated with dexamethasone. Furthermore, this treatment induced reactive astrocytes that stained positively for both APP and BACE, indicating that this change occurs primarily in reactive cells [165].
Other stressors, such as tissue damage, have been shown to induce APP expression in astrocytes. Hippocampal lesions stimulate APP expression in nearby astrocytes [166]. Brain injury has been shown to enhance astrocytic APP expression [167,168]. Traumatic brain injury has long been linked to the risk of developing AD [169,170] and is associated with accelerated Aβ deposition in AD [171]. Significant evidence suggests that acute brain injury can induce PS1 expression in mice [172], rats [173] and in human brains following cerebral infarcts [174,175]. Importantly, these studies indicate that brain injury induces PS1 expression in astrocytes. Nadler et al. [176] used three models to induce brain injury closed head injury (CHI), a well-established model for head trauma which is accompanied by neuroinflammation [177], brain stabbing or intracerebroventricular injection of LPS. In each incidence, the brain trauma resulted in more reactive astrocytes and increased expression of presenilin-1 and nicastrin.
Inorganic arsenic (iAs), a toxic metalloid, can contaminate drinking water and is associated with cognitive impairment [178,179]. Cells process iAs to a highly toxic monomethylarsonous acid, MMAIII, which has been suggested to be associated with neurodegenerative disorders, although epidemiological studies have failed to demonstrate association. Primary rat astrocytes exposed to MMAIII have increased mRNA levels of a plethora of AD-related cytokines including IL-1β, IL-6 and TNFα. Additionally, MMAIII induces a roughly threefold increase in APP and BACE1 mRNA expression [180].
These studies fit with the wider narrative that chronic stress can induce Aβ production [181,182]. Taken together, this suggests that a feed-forward mechanism is at play. Amyloid beta, perhaps initially from neurons, stimulates proinflammatory cytokine release from microglia and astrocytes, which in turn leads to upregulation of APP and BACE expression and possibly γ-secretase activity to drive astrocytic Aβ secretion (figure 3). This inflammation-induced Aβ then stimulates further neuroinflammation and ultimately additional amyloid production.
Figure 3.

A feed-forward mechanism of Aβ secretion by reactive astrocytes. Cellular stressors and proinflammatory cytokines upregulate APP, BACE and γ-secretase in astrocytes resulting in astrocytic Aβ production. In turn, this Aβ initiates further stress and inflammation driving subsequent Aβ production.
Furthermore, Aβ produced by astrocytes may be more pathogenic than that of neurons. A large portion of the Aβ species comprising amyloid beta plaques are N-truncated [183–185]. Aβ peptides beginning at Glu3 and at Phe4 are abundant in plaques. Studies suggest that these N-truncated species arise because other enzymes either compete with or modify BACE1 cleavage of APP [186–188]. The proportion of N-truncated peptides making up Aβ plaques seems to increase with disease progression and Braak stage [189]. N-truncation may affect the pathogenicity of the peptide; for example, Aβ42 with a truncated N-terminus is highly prone to aggregation [190]. Oberstein et al. [191] observed that astrocytes produced sevenfold less Aβ40 than neurons; however, they found that 60% of the Aβ secreted by astrocytes was N-truncated, compared to 20% from neurons.
10. Astrocytes and ageing
As the primary risk factor for AD is ageing, it is important to understand the effect of ageing on astrocytes. However, currently, little is known about the effect of ageing on metabolic, biochemical and morphological changes in astrocytes. Some research has demonstrated that ageing is associated with increased astroglial proliferation and reactivity, measured as an increase in GFAP expression, particularly in the CA1 region of the hippocampus [192–196]. In this model of ‘inflammageing’, reactive astrocytes contribute to chronic neuroinflammation throughout the brain [197]. This suggests that there is the potential for ageing to induce reactive astrogliosis, which could result in astrocyte Aβ production. Therefore, ageing-induced neuroinflammation may be an initiating factor of late onset AD.
Studies in other regions of the brain have found evidence of age-associated astroglial atrophy [198–200]. Using GFAP, glutamine synthetase, and s100β as markers for astroglia, Rodriguez et al. [66] confirmed ageing is associated with both reactivity and atrophy in different brain regions. Owing to the heterogeneity of astrocytes throughout the brain, it is possible for ageing to be associated with both astroglial dystrophy and reactivity. To conclude, ageing-dependent changes in astroglia are context and region specific, and more research into the downstream consequences of astroglial ageing needs to be done as a framework to fully understand the role of astrocytes in AD.
11. Aβ as an anti-microbial protein
Aβ is generally described as having no normal physiological role. However, it has recently been suggested that Aβ may act as anti-microbial protein (AMP) in the brain as first line of defence against invading pathogens. AMPs, also known as host defence peptides, are broad spectrum antibiotics that are active against a host of pathogens, including bacteria, fungi and viruses [201]. If this is the case, then astrocytes, as mediators of innate immunity, may secrete Aβ in response to stress as an innate defence mechanism.
BACE1 and BACE2 double knockout mice have higher neonatal mortality rates than WT mice and this is not due to maternal care issues or deficient active immunity. This increased mortality disappears when the mice are housed in a pathogen-free facility, suggesting they may have increased susceptibility to pathogens [148]. If this is the case, their compromised innate immune system may be due to the lack of Aβ in these mice.
Soscia et al. [202] compared the ability of Aβ to inhibit the growth of pathogens to that of LL-37, an established human AMP in the cathelicidin family [202,203]. Aβ was active against 8 of 12 pathogens, in rates similar to that of LL-37. Furthermore, homogenate from the temporal lobe of AD patients had 24% greater activity against Candida albicans than that of control subjects. This activity returned to that of control patients when they immunodepleted Aβ from the homogenate [202].
Aβ has also been shown to be protective against viral infections such as Herpes simplex virus 1 (HSV-1) and H3N2 and H1N1 influenza A virus (IAV). HSV-1 is a known AD risk factor and viral particles colocalize with amyloid plaques [204,205]. Pretreatment of fibroblast, epithelial and neuronal cell lines with either Aβ40 or Aβ42 reduced HSV-1 replication [206]. In vitro, Aβ42 reduces epithelial cell uptake of IAV and causes aggregation of viral particles. Aβ42 also reduced viral protein synthesis monocytes and decreased IL-6 secretion [207].
After intracerebral injection of Salmonella Typhimurium, 4-week-old 5XFAD transgenic mice, which overexpress both human APP and PS1 with five FAD mutations, and therefore express high levels of Aβ, had increased survival compared with WT mice. In the same assay, APP knockout mice that do not produce Aβ fared worse than non-transgenic mice. Consistent with this, the human brain neuroglioma cell line H4, stably overexpressing either Aβ40 or Aβ42, has increased survival when challenged with C. albicans compared to WT H4 cells. The mechanism behind this seems to be reduced adhesion to Aβ overproducing cells and increased microbial agglutination [208]. Taken together, this suggests that Aβ may play a role as an AMP in the innate immune system and this may explain why astrocytes secrete Aβ in response to cellular stressors.
12. Interplay with neurons and microglia
If inflammation-induced astrocytic Aβ production plays a significant role in AD pathology, then the relationship between microglia, astrocytes and neurons needs to be redefined. Microglia play a central role in the immune system of the CNS and produce and respond to a variety of inflammatory mediators that are implicated in AD [209]. Genetic studies support the significance of microglia in pathology: it is well established that mutations in triggering receptor expressed on myeloid cells 2 (TREM2) and CD33 increase the risk for AD [45–48,210].
Similar to astrocytes, microglia undergo substantial changes in response to stimulus and may be resting or activated depending on the cellular environment [211]. Microglia can be activated by Aβ from either neurons or astrocytes and in response secrete proinflammatory cytokines [212,213]. It has been suggested that there is a delicate balance between harmful and beneficial microglial cytokine production in AD. Inflammation is necessary to promote efficient microglial clearance of Aβ, but excessive inflammation may accelerate disease by causing neuronal and glial cell death [214,215]. We argue that microglial cytokine production also contributes to disease progression by inducing astrocytic Aβ production.
Taken together, this suggests a complicated relationship between neurons, microglia and astrocytes in the context of AD. Neurons contribute to total amyloid load, which consequently activates microglia and astrocytes. Microglia respond to Aβ by producing proinflammatory cytokines, which in turn activate astrocytes, inducing APP and BACE1 expression and further Aβ production. At the same time, astrocytes are also capable of clearing and degrading amyloid and secreting inflammatory mediators [216–218]. Downstream, inflammation may be a triggering event for the neuronal death seen in AD and contribute to cognitive decline (figure 4).
Figure 4.

In AD, neurons secrete Aβ, which activates resting microglia and astrocytes. In turn, activated microglia and reactive astrocytes secrete proinflammatory mediators, which may induce neuronal death. Additionally, neuroinflammation also induces APP and BACE1 expression in astrocytes resulting in further Aβ production.
13. Conclusion
Several of the causal and risk factor genes for AD—amyloid precursor protein (APP), presenilin-1, presenilin-2, ApoE, clusterin (CLU), phosphatidylinositol-binding clathrin assembly protein (PICALM), triggering receptor expressed on myeloid cells 2 (TREM2)—are expressed not only by neurons but also, if not predominantly, by astrocytes [102], corroborating the idea that astrocytes are important players in AD pathogenesis. Neurons are often considered the lone source of Aβ in AD, yet there is plenty of evidence that astrocytes also contribute to Aβ load [111]. In particular, astrocytes activated by a multitude of cellular stressors upregulate the necessary machinery for Aβ production. This may be part of an innate immune response where Aβ functions as an AMP. Furthermore, astrocytes can be stimulated by Aβ from nearby neurons to make and secrete Aβ. In this cycle, Aβ-exposed astrocytes act as vectors to spread Aβ production in a self-sustaining way [219] that may drive AD pathology.
Acknowledgement
We thank Lauren Jonas for proofreading the manuscript.
Data accessibility
This article has no additional data.
Authors' contributions
G.R.F. and Y.-M.L. wrote and edited the manuscript.
Competing interests
We declare we have no competing interests.
Funding
This work is supported by NIH grant R01AG026660 (Y.-M.L.), R01NS076117 (Y.-M.L.), R01NS096275 (Y.-M.L.), the JPB Foundation (Y.-M.L.), the MetLife Foundation (Y.-M.L.), Cure Alzheimer's Fund (Y.-M.L.), The Edward and Della L. Thome Memorial Foundation (Y.-M.L.) and Coins for the Alzheimer's Research Trust (Y.-M.L.). The authors also acknowledge the MSK Cancer Center Support Grant/Core Grant (grant no. P30 CA008748), Mr William H. Goodwin and Mrs Alice Goodwin and the Commonwealth Foundation for Cancer Research, the Experimental Therapeutics Center of MSKCC, and the William Randolph Hearst Fund in Experimental Therapeutics.
References
- 1.Beach TG, Walker R, McGeer EG. 1989. Patterns of gliosis in Alzheimer's disease and aging cerebrum. Glia 2, 420–436. (doi:10.1002/glia.440020605) [DOI] [PubMed] [Google Scholar]
- 2.Morris M, Maeda S, Vossel K, Mucke L. 2011. The many faces of tau. Neuron 70, 410–426. (doi:10.1016/j.neuron.2011.04.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olabarria M, Noristani HN, Verkhratsky A, Rodríguez JJ. 2010. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer's disease. Glia 58, 831–838. (doi:10.1002/glia.20967) [DOI] [PubMed] [Google Scholar]
- 4.Nagele RG, D'Andrea MR, Lee H, Venkataraman V, Wang H-Y. 2003. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 971, 197–209. (doi:10.1016/S0006-8993(03)02361-8) [DOI] [PubMed] [Google Scholar]
- 5.Rodríguez JJ, Olabarria M, Chvatal A, Verkhratsky A. 2009. Astroglia in dementia and Alzheimer's disease. Cell Death Differ. 16, 378–385. (doi:10.1038/cdd.2008.172) [DOI] [PubMed] [Google Scholar]
- 6.Selkoe DJ, Hardy J. 2016. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 8, 595–608. (doi:10.15252/emmm.201606210) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardy J, Selkoe DJ. 2002. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. (doi:10.1126/science.1072994) [DOI] [PubMed] [Google Scholar]
- 8.Goate A, et al. 1991. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 349, 704–706. (doi:10.1038/349704a0) [DOI] [PubMed] [Google Scholar]
- 9.Levy-Lahad E, et al. 1995. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 269, 973–977. (doi:10.1126/science.7638622) [DOI] [PubMed] [Google Scholar]
- 10.Sherrington R, et al. 1995. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 375, 754–760. (doi:10.1038/375754a0) [DOI] [PubMed] [Google Scholar]
- 11.Cruts M, Theuns J, Van Broeckhoven C. 2012. Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 33, 1340–1344. (doi:10.1002/humu.22117) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiseman FK, Al-Janabi T, Hardy J, Karmiloff-Smith A, Nizetic D, Tybulewicz VLJ, Fisher EMC, Strydom A. 2015. A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 16, 564–574. (doi:10.1038/nrn3983) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jonsson T, et al. 2012. A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature 488, 96–99. (doi:10.1038/nature11283) [DOI] [PubMed] [Google Scholar]
- 14.Shen J, Kelleher RJ III. 2007. The presenilin hypothesis of Alzheimer's disease: evidence for a loss-of-function pathogenic mechanism. Proc. Natl Acad. Sci. USA 104, 403–409. (doi:10.1073/pnas.0608332104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolfe MS. 2007. When loss is gain: reduced presenilin proteolytic function leads to increased Aβ42/Aβ40. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 8, 136–140. (doi:10.1038/sj.embor.7400896) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Strooper B. 2007. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 8, 141–146. (doi:10.1038/sj.embor.7400897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xia D, Kelleher RJ, Shen J, Shen J. 2016. Loss of Aβ43 production caused by presenilin-1 mutations in the knockin mouse brain. Neuron 90, 417–422. (doi:10.1016/j.neuron.2016.03.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chávez-Gutiérrez L, et al. 2012. The mechanism of γ-secretase dysfunction in familial Alzheimer disease. EMBO J. 31, 2261–2274. (doi:10.1038/emboj.2012.79) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun L, Zhou R, Yang G, Shi Y. 2017. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc. Natl Acad. Sci. USA 114, E476–E485. (doi:10.1073/pnas.1618657114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hussain I, et al. 1999. Identification of a novel aspartic protease (Asp 2) as β-secretase. Mol. Cell. Neurosci. 14, 419–427. (doi:10.1006/mcne.1999.0811) [DOI] [PubMed] [Google Scholar]
- 21.Vassar R, et al. 1999. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. (doi:10.1126/science.286.5440.735) [DOI] [PubMed] [Google Scholar]
- 22.Sinha S, et al. 1999. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 402, 537–540. (doi:10.1038/990114) [DOI] [PubMed] [Google Scholar]
- 23.Yan R, et al. 1999. Membrane-anchored aspartyl protease with Alzheimer's disease β-secretase activity. Nature 402, 533–537. (doi:10.1038/990107) [DOI] [PubMed] [Google Scholar]
- 24.Tian Y, Crump CJ, Li Y-M. 2010. Dual role of alpha-secretase cleavage in the regulation of gamma-secretase activity for amyloid production. J. Biol. Chem. 285, 32 549–32 556. (doi:10.1074/jbc.M110.128439) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tian Y, Bassit B, Chau D, Li Y-M. 2010. An APP inhibitory domain containing the Flemish mutation residue modulates γ-secretase activity for Aβ production. Nat. Struct. Mol. Biol. 17, 151–158. (doi:10.1038/nsmb.1743) [DOI] [PubMed] [Google Scholar]
- 26.Willem M, et al. 2006. Control of peripheral nerve myelination by the ß-secretase BACE1. Science 314, 664–666. (doi:10.1126/science.1132341) [DOI] [PubMed] [Google Scholar]
- 27.Li Y-M, et al. 2000. Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405, 689–694. (doi:10.1038/35015085) [DOI] [PubMed] [Google Scholar]
- 28.Ahn K, Shelton CC, Tian Y, Zhang X, Gilchrist ML, Sisodia SS, Li Y-M. 2010. Activation and intrinsic γ-secretase activity of presenilin 1. Proc. Natl Acad. Sci. USA 107, 21 435–21 440. (doi:10.1073/pnas.1013246107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.St George-Hyslop P, et al. 2000. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 407, 48–54. (doi:10.1038/35024009) [DOI] [PubMed] [Google Scholar]
- 30.Goutte C, Tsunozaki M, Hale VA, Priess JR. 2002. APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc. Natl Acad. Sci. USA 99, 775–779. (doi:10.1073/pnas.022523499) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Strooper B. 2003. Aph-1, Pen-2, and nicastrin with presenilin generate an active γ-secretase complex. Neuron 38, 9–12. (doi:10.1016/S0896-6273(03)00205-8) [DOI] [PubMed] [Google Scholar]
- 32.Selkoe DJ, Wolfe MS. 2007. Presenilin: running with scissors in the membrane. Cell 131, 215–221. (doi:10.1016/j.cell.2007.10.012) [DOI] [PubMed] [Google Scholar]
- 33.Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T. 2003. The role of presenilin cofactors in the γ-secretase complex. Nature 422, 438–441. (doi:10.1038/nature01506) [DOI] [PubMed] [Google Scholar]
- 34.Beher D, et al. 2003. In Vitro characterization of the presenilin-dependent γ-secretase complex using a novel affinity ligand. Biochemistry 42, 8133–8142. (doi:10.1021/bi034045z) [DOI] [PubMed] [Google Scholar]
- 35.Lai M-T, et al. 2003. Presenilin-1 and Presenilin-2 exhibit distinct yet overlapping γ-secretase activities. J. Biol. Chem. 278, 22 475–22 481. (doi:10.1074/jbc.M300974200) [DOI] [PubMed] [Google Scholar]
- 36.Gu Y, et al. 2004. The presenilin proteins are components of multiple membrane-bound complexes that have different biological activities. J. Biol. Chem. 279, 31 329–31 336. (doi:10.1074/jbc.M401548200) [DOI] [PubMed] [Google Scholar]
- 37.Villa JC, et al. 2014. Nontranscriptional role of Hif-1α in activation of γ-secretase and notch signaling in breast cancer. Cell Rep. 8, 1077–1092. (doi:10.1016/j.celrep.2014.07.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gertsik N, Chiu D, Li Y-M. 2014. Complex regulation of γ-secretase: from obligatory to modulatory subunits. Front. Aging Neurosci. 6, 342 (doi:10.3389/fnagi.2014.00342) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akiyama H, et al. 2017. Inflammation and Alzheimer's disease. Neurobiol. Aging 21, 383–421. (doi:10.1016/S0197-4580(00)00124-X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wyss-Coray T, Mucke L. 2002. Inflammation in neurodegenerative disease—a double-edged sword. Neuron 35, 419–432. (doi:10.1016/S0896-6273(02)00794-8) [DOI] [PubMed] [Google Scholar]
- 41.Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. 2006. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J. Neuroinflamm. 3, 27 (doi:10.1186/1742-2094-3-27) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jekabsone A, Mander PK, Tickler A, Sharpe M, Brown GC. 2006. Fibrillar beta-amyloid peptide Abeta1–40 activates microglial proliferation via stimulating TNF-alpha release and H2O2 derived from NADPH oxidase: a cell culture study. J. Neuroinflammation 3, 24 (doi:10.1186/1742-2094-3-24) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morgan D. 2006. Modulation of microglial activation state following passive immunization in amyloid depositing transgenic mice. Neurochem. Int. 49, 190–194. (doi:10.1016/j.neuint.2006.03.017) [DOI] [PubMed] [Google Scholar]
- 44.Malik M, et al. 2015. Genetics ignite focus on microglial inflammation in Alzheimer's disease. Mol. Neurodegener. 10, 52 (doi:10.1186/s13024-015-0048-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jin SC, et al. 2015. TREM2 is associated with increased risk for Alzheimer's disease in African Americans. Mol. Neurodegener. 10, 19 (doi:10.1186/s13024-015-0016-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heslegrave A, et al. 2016. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer's disease. Mol. Neurodegener. 11, 3 (doi:10.1186/s13024-016-0071-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wunderlich P, Glebov K, Kemmerling N, Tien NT, Neumann H, Walter J, Bu G. 2013. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and γ-secretase-dependent intramembranous cleavage. J. Biol. Chem. 288, 33 027–33 036. (doi:10.1074/jbc.M113.517540) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Painter MM, Atagi Y, Liu C-C, Rademakers R, Xu H, Fryer JD, Bu G. 2015. TREM2 in CNS homeostasis and neurodegenerative disease. Mol. Neurodegener. 10, 43 (doi:10.1186/s13024-015-0040-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saresella M, et al. 2016. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer's disease. Mol. Neurodegener. 11, 23 (doi:10.1186/s13024-016-0088-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beach TG, Mcgeer EG. 1988. Lamina-specific arrangement of astrocytic gliosis and senile plaques in Alzheimer's disease visual cortex. Brain Res. 463, 357–361. (doi:10.1016/0006-8993(88)90410-6) [DOI] [PubMed] [Google Scholar]
- 51.Kashon ML, et al. 2004. Associations of cortical astrogliosis with cognitive performance and dementia status. J. Alzheimer’s Dis. 6, 595–604. (doi:10.3233/JAD-2004-6604) [DOI] [PubMed] [Google Scholar]
- 52.Mrak RE, Sheng JG, Griffin WS. 1996. Correlation of astrocytic S100 beta expression with dystrophic neurites in amyloid plaques of Alzheimer's disease. J. Neuropathol. Exp. Neurol. 55, 273–279. (doi:10.1097/00005072-199603000-00002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sofroniew M V, Vinters H V. 2010. Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. (doi:10.1007/s00401-009-0619-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nedergaard M, Ransom B, Goldman SA. 2003. New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci. 26, 523–530. (doi:10.1016/j.tins.2003.08.008) [DOI] [PubMed] [Google Scholar]
- 55.Figley CR, Stroman PW. 2011. The role(s) of astrocytes and astrocyte activity in neurometabolism, neurovascular coupling, and the production of functional neuroimaging signals. Eur. J. Neurosci. 33, 577–588. (doi:10.1111/j.1460-9568.2010.07584.x) [DOI] [PubMed] [Google Scholar]
- 56.Choi SS, Lee HJ, Lim I, Satoh J, Kim SU. 2014. Human astrocytes: secretome profiles of cytokines and chemokines. PLoS ONE 9, e92325 (doi:10.1371/journal.pone.0092325) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martineau M, Parpura V, Mothet J-P. 2014. Cell-type specific mechanisms of D-serine uptake and release in the brain. Front. Synaptic Neurosci. 6, 12 (doi:10.3389/fnsyn.2014.00012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malarkey EB, Parpura V. 2008. Mechanisms of glutamate release from astrocytes. Neurochem. Int. 52, 142–154. (doi:10.1016/j.neuint.2007.06.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Verkhratsky A, Olabarria M, Noristani HN, Yeh C-Y, Rodriguez JJ. 2010. Astrocytes in Alzheimer's Disease. Neurotherapeutics 7, 399–412. (doi:10.1016/j.nurt.2010.05.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. 2012. Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. (doi:10.1523/JNEUROSCI.6221-11.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Daginakatte GC, Gadzinski A, Emnett RJ, Stark JL, Gonzales ER, Yan P, Lee J-M, Cross AH, Gutmann DH. 2008. Expression profiling identifies a molecular signature of reactive astrocytes stimulated by cyclic AMP or proinflammatory cytokines. Exp. Neurol. 210, 261–267. (doi:10.1016/j.expneurol.2007.10.016) [DOI] [PubMed] [Google Scholar]
- 62.Wilhelmsson U, Bushong EA, Price DL, Smarr BL, Phung V, Terada M, Ellisman MH, Pekny M. 2006. Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc. Natl Acad. Sci. USA 103, 17 513–8. (doi:10.1073/pnas.0602841103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kamphuis W, et al. 2012. GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS ONE 7, e42823 (doi:10.1371/journal.pone.0042823) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sofroniew MV. 2009. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647. (doi:10.1016/j.tins.2009.08.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oberheim NA, et al. 2009. Uniquely hominid features of adult human astrocytes. J. Neurosci. 29, 3276–3287. (doi:10.1523/JNEUROSCI.4707-08.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rodríguez JJ, Yeh C-Y, Terzieva S, Olabarria M, Kulijewicz-Nawrot M, Verkhratsky A. 2014. Complex and region-specific changes in astroglial markers in the aging brain. Neurobiol. Aging 35, 15–23. (doi:10.1016/j.neurobiolaging.2013.07.002) [DOI] [PubMed] [Google Scholar]
- 67.Yeh C-Y, Vadhwana B, Verkhratsky A, Rodríguez JJ. 2011. Early astrocytic atrophy in the entorhinal cortex of a triple transgenic animal model of Alzheimer's disease. ASN Neuro 3, 271–279. (doi:10.1042/AN20110025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kulijewicz-Nawrot M, Verkhratsky A, Chvátal A, Syková E, Rodríguez JJ. 2012. Astrocytic cytoskeletal atrophy in the medial prefrontal cortex of a triple transgenic mouse model of Alzheimer's disease. J. Anat. 221, 252–262. (doi:10.1111/j.1469-7580.2012.01536.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beauquis J, Vinuesa A, Pomilio C, Pavía P, Galván V, Saravia F. 2014. Neuronal and glial alterations, increased anxiety, and cognitive impairment before hippocampal amyloid deposition in PDAPP mice, model of Alzheimer's disease. Hippocampus 24, 257–269. (doi:10.1002/hipo.22219) [DOI] [PubMed] [Google Scholar]
- 70.Hou L, Liu Y, Wang X, Ma H, He J, Zhang Y, Yu C, Guan W, Ma Y. 2011. The effects of amyloid-β42 oligomer on the proliferation and activation of astrocytes in vitro. Vitr. Cell. Dev. Biol. Anim. 47, 573–580. (doi:10.1007/s11626-011-9439-y) [DOI] [PubMed] [Google Scholar]
- 71.Farina C, Aloisi F, Meinl E. 2007. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 28, 138–145. (doi:10.1016/j.it.2007.01.005) [DOI] [PubMed] [Google Scholar]
- 72.Constam DB, Philipp J, Malipiero U V, ten Dijke P, Schachner M, Fontana A. 1992. Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J. Immunol. 148, 1404–1410. [PubMed] [Google Scholar]
- 73.McGeer PL, McGeer EG. 1995. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Brain Res. Rev. 21, 195–218. (doi:10.1016/0165-0173(95)00011-9) [DOI] [PubMed] [Google Scholar]
- 74.Hu J, Akama KT, Krafft GA, Chromy BA, Van Eldik LJ. 1998. Amyloid-β peptide activates cultured astrocytes: morphological alterations, cytokine induction and nitric oxide release. Brain Res. 785, 195–206. (doi:10.1016/S0006-8993(97)01318-8) [DOI] [PubMed] [Google Scholar]
- 75.Johnstone M, Gearing AJ, Miller KM. 1999. A central role for astrocytes in the inflammatory response to beta-amyloid; chemokines, cytokines and reactive oxygen species are produced. J. Neuroimmunol. 93, 182–193. (doi:10.1016/S0165-5728(98)00226-4) [DOI] [PubMed] [Google Scholar]
- 76.Abbas N, et al. 2002. Up-regulation of the inflammatory cytokines IFN-gamma and IL-12 and down-regulation of IL-4 in cerebral cortex regions of APP(SWE) transgenic mice. J. Neuroimmunol. 126, 50–57. (doi:10.1016/S0165-5728(02)00050-4) [DOI] [PubMed] [Google Scholar]
- 77.Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR. 2017. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol. Aging 20, 581–589. (doi:10.1016/S0197-4580(99)00065-2) [DOI] [PubMed] [Google Scholar]
- 78.Apelt J, Schliebs R. 2001. Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res. 894, 21–30. (doi:10.1016/S0006-8993(00)03176-0) [DOI] [PubMed] [Google Scholar]
- 79.Tan J, Town T, Crawford F, Mori T, DelleDonne A, Crescentini R, Obregon D, Flavell RA, Mullan MJ. 2002. Role of CD40 ligand in amyloidosis in transgenic Alzheimer's mice. Nat. Neurosci. 5, 1288–1293. (doi:10.1038/nn968) [DOI] [PubMed] [Google Scholar]
- 80.Huberman M, Shalit F, Roth-Deri I, Gutman B, Brodie C, Kott E, Sredni B. 1994. Correlation of cytokine secretion by mononuclear cells of Alzheimer patients and their disease stage. J. Neuroimmunol. 52, 147–152. (doi:10.1016/0165-5728(94)90108-2) [DOI] [PubMed] [Google Scholar]
- 81.Fultz MJ, Barber SA, Dieffenbach CW, Vogel SN. 1993. Induction of IFN-gamma in macrophages by lipopolysaccharide. Int. Immunol. 5, 1383–1392. (doi:10.1093/intimm/5.11.1383) [DOI] [PubMed] [Google Scholar]
- 82.De Simone R, Levi G, Aloisi F. 1998. Interferon γ gene expression in rat central nervous system glial cells. Cytokine 10, 418–422. (doi:10.1006/cyto.1997.0314) [DOI] [PubMed] [Google Scholar]
- 83.Tarkowski E, Blennow K, Wallin A, Tarkowski A. 1999. Intracerebral production of tumor necrosis factor-alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J. Clin. Immunol. 19, 223–230. (doi:10.1023/A:1020568013953) [DOI] [PubMed] [Google Scholar]
- 84.Shibata N, Ohnuma T, Takahashi T, Baba H, Ishizuka T, Ohtsuka M, Ueki A, Nagao M, Arai H. 2002. Effect of IL-6 polymorphism on risk of Alzheimer disease: genotype–phenotype association study in Japanese cases. Am. J. Med. Genet. 114, 436–439. (doi:10.1002/ajmg.10417) [DOI] [PubMed] [Google Scholar]
- 85.Ershler WB, Keller ET. 2000. Age-associated increased interleukin-6 gene expression, late-life diseases, and frailty. Annu. Rev. Med. 51, 245–270. (doi:10.1146/annurev.med.51.1.245) [DOI] [PubMed] [Google Scholar]
- 86.Licastro F, Grimaldi LME, Bonafè M, Martina C, Olivieri F, Cavallone L, Giovanietti S, Masliah E, Franceschi C. 2003. Interleukin-6 gene alleles affect the risk of Alzheimer's disease and levels of the cytokine in blood and brain. Neurobiol. Aging 24, 921–926. (doi:10.1016/S0197-4580(03)00013-7) [DOI] [PubMed] [Google Scholar]
- 87.Licastro F, Pedrini S, Caputo L, Annoni G, Davis LJ, Ferri C, Casadei V, Grimaldi LME. 2000. Increased plasma levels of interleukin-1, interleukin-6 and α-1-antichymotrypsin in patients with Alzheimer's disease: peripheral inflammation or signals from the brain? J. Neuroimmunol. 103, 97–102. (doi:10.1016/S0165-5728(99)00226-X) [DOI] [PubMed] [Google Scholar]
- 88.Galimberti D, et al. 2008. Intrathecal levels of IL-6, IL-11 and LIF in Alzheimer's disease and frontotemporal lobar degeneration. J. Neurol. 255, 539–544. (doi:10.1007/s00415-008-0737-6) [DOI] [PubMed] [Google Scholar]
- 89.Baranowska-Bik A, Bik W, Wolinska-Witort E, Martynska L, Chmielowska M, Barcikowska M, Baranowska B. 2008. Plasma beta amyloid and cytokine profile in women with Alzheimer's disease. Neuro Endocrinol. Lett. 29, 75–79. [PubMed] [Google Scholar]
- 90.Das S, Potter H. 1995. Expression of the Alzheimer amyloid-promoting factor antichymotrypsin is induced in human astrocytes by IL-1. Neuron 14, 447–456. (doi:10.1016/0896-6273(95)90300-3) [DOI] [PubMed] [Google Scholar]
- 91.Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, Araoz C. 1989. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl Acad. Sci. USA 86, 7611–7615. (doi:10.1073/pnas.86.19.7611) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Luedecking EK, DeKosky ST, Mehdi H, Ganguli M, Kamboh MI. 2000. Analysis of genetic polymorphisms in the transforming growth factor-beta1 gene and the risk of Alzheimer's disease. Hum. Genet. 106, 565–569. (doi:10.1007/s004390000313) [DOI] [PubMed] [Google Scholar]
- 93.Chao CC, Hu S, Frey WH, Ala TA, Tourtellotte WW, Peterson PK, Peterson PK. 1994. Transforming growth factor beta in Alzheimer's disease. Clin. Diagn. Lab. Immunol. 1, 109–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van der Wal EA, Gómez-Pinilla F, Cotman CW. 1993. Transforming growth factor-beta 1 is in plaques in Alzheimer and Down pathologies. Neuroreport 4, 69–72. (doi:10.1097/00001756-199301000-00018) [DOI] [PubMed] [Google Scholar]
- 95.Wyss-Coray T, Borrow P, Brooker MJ, Mucke L. 1997. Astroglial overproduction of TGF-beta 1 enhances inflammatory central nervous system disease in transgenic mice. J. Neuroimmunol. 77, 45–50. (doi:10.1016/S0165-5728(97)00049-0) [DOI] [PubMed] [Google Scholar]
- 96.Peress NS, Perillo E. 1995. Differential expression of TGF-beta 1, 2 and 3 isotypes in Alzheimer's disease: a comparative immunohistochemical study with cerebral infarction, aged human and mouse control brains. J. Neuropathol. Exp. Neurol. 54, 802–811. (doi:10.1097/00005072-199511000-00007) [DOI] [PubMed] [Google Scholar]
- 97.Wyss-Coray T, Lin C, Yan F, Yu G-Q, Rohde M, McConlogue L, Masliah E, Mucke L. 2001. TGF-β1 promotes microglial amyloid-β clearance and reduces plaque burden in transgenic mice. Nat. Med. 7, 612–618. (doi:10.1038/87945) [DOI] [PubMed] [Google Scholar]
- 98.Laird FM, et al. 2005. BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J. Neurosci. 25, 11 693–11 709. (doi:10.1523/JNEUROSCI.2766-05.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rossner S, Lange-Dohna C, Zeitschel U, Perez-Polo JR. 2005. Alzheimer's disease beta-secretase BACE1 is not a neuron-specific enzyme. J. Neurochem. 92, 226–234. (doi:10.1111/j.1471-4159.2004.02857.x) [DOI] [PubMed] [Google Scholar]
- 100.Leuba G, Wernli G, Vernay A, Kraftsik R, Mohajeri MH, Saini KD. 2005. Neuronal and nonneuronal quantitative BACE immunocytochemical expression in the entorhinohippocampal and frontal regions in Alzheimer's disease. Dement. Geriatr. Cogn. Disord. 19, 171–183. (doi:10.1159/000083496) [DOI] [PubMed] [Google Scholar]
- 101.Jin SM, Cho HJ, Kim YW, Hwang JY, Mook-Jung I. 2012. Aβ-induced Ca2+ influx regulates astrocytic BACE1 expression via calcineurin/NFAT4 signals. Biochem. Biophys. Res. Commun. 425, 649–655. (doi:10.1016/j.bbrc.2012.07.123) [DOI] [PubMed] [Google Scholar]
- 102.Orre M, Kamphuis W, Osborn LM, Jansen AHP, Kooijman L, Bossers K, Hol EM. 2014. Isolation of glia from Alzheimer's mice reveals inflammation and dysfunction. Neurobiol. Aging 35, 2746–2760. (doi:10.1016/j.neurobiolaging.2014.06.004) [DOI] [PubMed] [Google Scholar]
- 103.Zhao J, O'Connor T, Vassar R. 2011. The contribution of activated astrocytes to Aβ production: implications for Alzheimer's disease pathogenesis. J. Neuroinflamm. 8, 150 (doi:10.1186/1742-2094-8-150) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Blasko I, Veerhuis R, Stampfer-Kountchev M, Saurwein-Teissl M, Eikelenboom P, Grubeck-Loebenstein B. 2000. Costimulatory effects of interferon-γ and interleukin-1β or tumor necrosis factor α on the synthesis of Aβ1–40 and Aβ1–42 by human astrocytes. Neurobiol. Dis. 7, 682–689. (doi:10.1006/nbdi.2000.0321) [DOI] [PubMed] [Google Scholar]
- 105.Veeraraghavalu K, Zhang C, Zhang X, Tanzi RE, Sisodia SS. 2014. Age-dependent, non-cell-autonomous deposition of amyloid from synthesis of β-amyloid by cells other than excitatory neurons. J. Neurosci. 34, 3668–3673. (doi:10.1523/JNEUROSCI.5079-13.2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Grolla AA, Fakhfouri G, Balzaretti G, Marcello E, Gardoni F, Canonico PL, DiLuca M, Genazzani AA, Lim D. 2013. Aβ leads to Ca2+ signaling alterations and transcriptional changes in glial cells. Neurobiol. Aging 34, 511–522. (doi:10.1016/j.neurobiolaging.2012.05.005) [DOI] [PubMed] [Google Scholar]
- 107.Busciglio J, Gabuzda DH, Matsudaira P, Yankner BA. 1993. Generation of beta-amyloid in the secretory pathway in neuronal and nonneuronal cells. Proc. Natl Acad. Sci. USA 90, 2092–2096. (doi:10.1073/pnas.90.5.2092) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Seubert P, et al. 1993. Secretion of β-amyloid precursor protein cleaved at the amino terminus of the β-amyloid peptide. Nature 361, 260–263. (doi:10.1038/361260a0) [DOI] [PubMed] [Google Scholar]
- 109.Shoji M, et al. 1992. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 258, 126–129. (doi:10.1126/science.1439760) [DOI] [PubMed] [Google Scholar]
- 110.Golde TE, Estus S, Usiak M, Younkin LH, Younkin SG. 1990. Expression of beta amyloid protein precursor mRNAs: recognition of a novel alternatively spliced form and quantitation in Alzheimer's disease using PCR. Neuron 4, 253–267. (doi:10.1016/0896-6273(90)90100-T) [DOI] [PubMed] [Google Scholar]
- 111.LeBlanc AC, Papadopoulos M, Bélair C, Chu W, Crosato M, Powell J, Goodyer CG. 1997. Processing of amyloid precursor protein in human primary neuron and astrocyte cultures. J. Neurochem. 68, 1183–1190. (doi:10.1046/j.1471-4159.1997.68031183.x) [DOI] [PubMed] [Google Scholar]
- 112.Haass C, Hung AY, Selkoe DJ. 1991. Processing of beta-amyloid precursor protein in microglia and astrocytes favors an internal localization over constitutive secretion. J. Neurosci. 11, 3783–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Forloni G, Demicheli F, Giorgi S, Bendotti C, Angeretti N. 1992. Expression of amyloid precursor protein mRNAs in endothelial, neuronal and glial cells: modulation by interleukin-1. Brain Res. Mol. Brain Res. 16, 128–134. (doi:10.1016/0169-328X(92)90202-M) [DOI] [PubMed] [Google Scholar]
- 114.Goldgaber D, Harris HW, Hla T, Maciag T, Donnelly RJ, Jacobsen JS, Vitek MP, Gajdusek DC. 1989. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc. Natl Acad. Sci. USA 86, 7606–7610. (doi:10.1073/pnas.86.19.7606) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Heinrich PC, Castell J V, Andus T. 1990. Interleukin-6 and the acute phase response. Biochem. J. 265, 621–636. (doi:10.1042/bj2650621) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Brugg B, Dubreuil YL, Huber G, Wollman EE, Delhaye-Bouchaud N, Mariani J. 1995. Inflammatory processes induce beta-amyloid precursor protein changes in mouse brain. Proc. Natl Acad. Sci. USA 92, 3032–3035. (doi:10.1073/pnas.92.7.3032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hauss-Wegrzyniak B, Dobrzanski P, Stoehr JD, Wenk GL. 1998. Chronic neuroinflammation in rats reproduces components of the neurobiology of Alzheimer's disease. Brain Res. 780, 294–303. (doi:10.1016/S0006-8993(97)01215-8) [DOI] [PubMed] [Google Scholar]
- 118.Sugaya K, Chou S, Xu SJ, McKinney M. 1998. Indicators of glial activation and brain oxidative stress after intraventricular infusion of endotoxin. Brain Res. Mol. Brain Res. 58, 1–9. (doi:10.1016/S0169-328X(97)00365-3) [DOI] [PubMed] [Google Scholar]
- 119.Tanaka S, Ide M, Shibutani T, Ohtaki H, Numazawa S, Shioda S, Yoshida T. 2006. Lipopolysaccharide-induced microglial activation induces learning and memory deficits without neuronal cell death in rats. J. Neurosci. Res. 83, 557–566. (doi:10.1002/jnr.20752) [DOI] [PubMed] [Google Scholar]
- 120.Lee J, Lee Y, Yuk D, Choi D, Ban S, Oh K, Hong J. 2008. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflamm. 5, 37 (doi:10.1186/1742-2094-5-37) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Deng X-H, Ai W-M, Lei D-L, Luo X-G, Yan X-X, Li Z. 2012. Lipopolysaccharide induces paired immunoglobulin-like receptor B (PirB) expression, synaptic alteration, and learning–memory deficit in rats. Neuroscience 209, 161–170. (doi:10.1016/j.neuroscience.2012.02.022) [DOI] [PubMed] [Google Scholar]
- 122.Kalehua AN, Taub DD, Baskar P V, Hengemihle J, Muñoz J, Trambadia M, Speer DL, De Simoni MG, Ingram DK. 2017. Aged mice exhibit greater mortality concomitant to increased brain and plasma TNF-alpha levels following intracerebroventricular injection of lipopolysaccharide. Gerontology 46, 115–128. (doi:10.1159/000022146) [DOI] [PubMed] [Google Scholar]
- 123.Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE. 2003. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol. Dis. 14, 133–145. (doi:10.1016/S0969-9961(03)00069-X) [DOI] [PubMed] [Google Scholar]
- 124.Rogers JT, Leiter LM, McPhee J, Cahill CM, Zhan SS, Potter H, Nilsson LN. 1999. Translation of the Alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 5′-untranslated region sequences. J. Biol. Chem. 274, 6421–6431. (doi:10.1074/jbc.274.10.6421) [DOI] [PubMed] [Google Scholar]
- 125.Machein U, Lieb K, Hüll M, Fiebich BL. 1995. IL-1β and TNFα, but not IL-6, induce α1-antichymotrypsin expression in the human astrocytoma cell line U373 MG. Neuroreport. See http://journals.lww.com/neuroreport/abstract/1995/11270/il_1_beta__and_tnf_alpha_,_but_not_il_6,_induce.4.aspx (accessed on 12 April 2017). [DOI] [PubMed]
- 126.Lesné S, et al. 2003. Transforming growth factor-beta 1 potentiates amyloid-beta generation in astrocytes and in transgenic mice. J. Biol. Chem. 278, 18 408–18 418. (doi:10.1074/jbc.M300819200) [DOI] [PubMed] [Google Scholar]
- 127.Burton T, Liang B, Dibrov A, Amara F. 2002. Transcriptional activation and increase in expression of Alzheimer's beta-amyloid precursor protein gene is mediated by TGF-beta in normal human astrocytes. Biochem. Biophys. Res. Commun. 295, 702–712. (doi:10.1016/S0006-291X(02)00724-6) [DOI] [PubMed] [Google Scholar]
- 128.Gray CW, Patel AJ. 1993. Regulation of beta-amyloid precursor protein isoform mRNAs by transforming growth factor-beta 1 and interleukin-1 beta in astrocytes. Brain Res. Mol. Brain Res. 19, 251–256. (doi:10.1016/0169-328X(93)90037-P) [DOI] [PubMed] [Google Scholar]
- 129.Amara FM, Junaid A, Clough RR, Liang B. 1999. TGF-beta(1), regulation of Alzheimer amyloid precursor protein mRNA expression in a normal human astrocyte cell line: mRNA stabilization. Brain Res. Mol. Brain Res. 71, 42–49. (doi:10.1016/S0169-328X(99)00158-8) [DOI] [PubMed] [Google Scholar]
- 130.Hartlage-Rübsamen M, et al. 2003. Astrocytic expression of the Alzheimer's disease β-secretase (BACE1) is stimulus-dependent. Glia 41, 169–179. (doi:10.1002/glia.10178) [DOI] [PubMed] [Google Scholar]
- 131.Bettegazzi B, Mihailovich M, Di Cesare A, Consonni A, Macco R, Pelizzoni I, Codazzi F, Grohovaz F, Zacchetti D. 2011. β-Secretase activity in rat astrocytes: translational block of BACE1 and modulation of BACE2 expression. Eur. J. Neurosci. 33, 236–243. (doi:10.1111/j.1460-9568.2010.07482.x) [DOI] [PubMed] [Google Scholar]
- 132.Zhao J, et al. 1996. Beta-secretase processing of the beta-amyloid precursor protein in transgenic mice is efficient in neurons but inefficient in astrocytes. J. Biol. Chem. 271, 31 407–31 411. [DOI] [PubMed] [Google Scholar]
- 133.Roßner S, Sastre M, Bourne K, Lichtenthaler SF. 2006. Transcriptional and translational regulation of BACE1 expression—implications for Alzheimer's disease. Prog. Neurobiol. 79, 95–111. (doi:10.1016/j.pneurobio.2006.06.001) [DOI] [PubMed] [Google Scholar]
- 134.Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, Heneka MT. 2003. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J. Neurosci. 23, 9796–9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hong HS, Hwang EM, Sim HJ, Cho H-J, Boo JH, Oh SS, Kim SU, Mook-Jung I. 2003. Interferon gamma stimulates beta-secretase expression and sAPPbeta production in astrocytes. Biochem. Biophys. Res. Commun. 307, 922–927. (doi:10.1016/S0006-291X(03)01270-1) [DOI] [PubMed] [Google Scholar]
- 136.Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T. 2007. Interferon-γ and tumor necrosis factor-α regulate amyloid-β plaque deposition and β-secretase expression in Swedish mutant APP transgenic mice. Am. J. Pathol. 170, 680–692. (doi:10.2353/ajpath.2007.060378) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cho HJ, Kim S-K, Jin SM, Hwang E-M, Kim YS, Huh K, Mook-Jung I. 2007. IFN-γ-induced BACE1 expression is mediated by activation of JAK2 and ERK1/2 signaling pathways and direct binding of STAT1 to BACE1 promoter in astrocytes. Glia 55, 253–262. (doi:10.1002/glia.20451) [DOI] [PubMed] [Google Scholar]
- 138.Chen C-H, Zhou W, Liu S, Deng Y, Cai F, Tone M, Tone Y, Tong Y, Song W. 2012. Increased NF-κB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer's disease. Int. J. Neuropsychopharmacol. 15, 77–90. (doi:10.1017/S1461145711000149) [DOI] [PubMed] [Google Scholar]
- 139.Giardina C, Hubbard AK. 2002. Growing old with nuclear factor-κB. Cell Stress Chaperones Cell Stress Soc. Int. 7, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bales KR, Du Y, Holtzman D, Cordell B, Paul SM, Mariani J. 1998. Neuroinflammation and Alzheimer's disease: critical roles for cytokine/Abeta-induced glial activation, NF-kappaB, and apolipoprotein E. Neurobiol. Aging 21, 427–432. (doi:10.1016/S0197-4580(00)00143-3) [DOI] [PubMed] [Google Scholar]
- 141.Buggia-Prevot V, Sevalle J, Rossner S, Checler F. 2008. NFκB-dependent control of BACE1 promoter transactivation by Aβ42. J. Biol. Chem. 283, 10 037–10 047. (doi:10.1074/jbc.M706579200) [DOI] [PubMed] [Google Scholar]
- 142.Sambamurti K, Kinsey R, Maloney B, Ge Y-W, Lahiri DK. 2004. Gene structure and organization of the human β-secretase (BACE) promoter. FASEB J. 18, 1034–1036. (doi:10.1096/fj.03-1378fje) [DOI] [PubMed] [Google Scholar]
- 143.Chami L, Buggia-Prévot V, Duplan E, Del Prete D, Delprete D, Chami M, Peyron J-F, Checler F. 2012. Nuclear factor-κB regulates βAPP and β- and γ-secretases differently at physiological and supraphysiological Aβ concentrations. J. Biol. Chem. 287, 24 573–24 584. (doi:10.1074/jbc.M111.333054) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lange-Dohna C, Zeitschel U, Gaunitz F, Perez-Polo JR, Bigl V, Roßner S. 2003. Cloning and expression of the rat BACE1 promoter. J. Neurosci. Res. 73, 73–80. (doi:10.1002/jnr.10639) [DOI] [PubMed] [Google Scholar]
- 145.Bourne KZ, Ferrari DC, Lange-Dohna C, Roßner S, Wood TG, Perez-Polo JR. 2007. Differential regulation of BACE1 promoter activity by nuclear factor-κB in neurons and glia upon exposure to β-amyloid peptides. J. Neurosci. Res. 85, 1194–1204. (doi:10.1002/jnr.21252) [DOI] [PubMed] [Google Scholar]
- 146.Brint S, Jacewicz M, Kiessling M, Tanabe J, Pulsinelli W. 1988. Focal brain ischemia in the rat: methods for reproducible neocortical infarction using tandem occlusion of the distal middle cerebral and ipsilateral common carotid arteries. J. Cereb. Blood Flow Metab. 8, 474–485. (doi:10.1038/jcbfm.1988.88) [DOI] [PubMed] [Google Scholar]
- 147.Nowak K, Lange-Dohna C, Zeitschel U, Günther A, Lüscher B, Robitzki A, Perez-Polo R, Roßner S. 2006. The transcription factor Yin Yang 1 is an activator of BACE1 expression. J. Neurochem. 96, 1696–1707. (doi:10.1111/j.1471-4159.2006.03692.x) [DOI] [PubMed] [Google Scholar]
- 148.Dominguez D, et al. 2005. Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J. Biol. Chem. 280, 30 797–30 806. (doi:10.1074/jbc.M505249200) [DOI] [PubMed] [Google Scholar]
- 149.Maezawa I, Zimin PI, Wulff H, Jin L-W. 2011. Amyloid-β protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J. Biol. Chem. 286, 3693–3706. (doi:10.1074/jbc.M110.135244) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.White JA, Manelli AM, Holmberg KH, Van Eldik LJ, LaDu MJ. 2005. Differential effects of oligomeric and fibrillar amyloid-β1–42 on astrocyte-mediated inflammation. Neurobiol. Dis. 18, 459–465. (doi:10.1016/j.nbd.2004.12.013) [DOI] [PubMed] [Google Scholar]
- 151.Craft JM, Watterson DM, Frautschy SA, Eldik LJV. 2004. Aminopyridazines inhibit β-amyloid-induced glial activation and neuronal damage in vivo. Neurobiol. Aging 25, 1283–1292. (doi:10.1016/j.neurobiolaging.2004.01.006) [DOI] [PubMed] [Google Scholar]
- 152.Palotás A, Kálmán J, Palotás M, Juhász A, Janka Z, Penke B. 2002. Fibroblasts and lymphocytes from Alzheimer patients are resistant to beta-amyloid-induced increase in the intracellular calcium concentration. Prog. Neuropsychopharmacol. Biol. Psychiatry 26, 971–974. (doi:10.1016/S0278-5846(02)00214-2) [DOI] [PubMed] [Google Scholar]
- 153.Cho HJ, Jin SM, Youn HD, Huh K, Mook-Jung I. 2008. Disrupted intracellular calcium regulates BACE1 gene expression via nuclear factor of activated T cells 1 (NFAT 1) signaling. Aging Cell 7, 137–147. (doi:10.1111/j.1474-9726.2007.00360.x) [DOI] [PubMed] [Google Scholar]
- 154.Abramov AY, Canevari L, Duchen MR. 2003. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J. Neurosci. 23, 5088–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Arif M, Chikuma T, Ahmed MM, Nakazato M, Smith MA, Kato T. 2009. Effects of memantine on soluble Αβ25–35-induced changes in peptidergic and glial cells in Alzheimer's disease model rat brain regions. Neuroscience 164, 1199–1209. (doi:10.1016/j.neuroscience.2009.08.063) [DOI] [PubMed] [Google Scholar]
- 156.Dal Prà I, Whitfileld JF, Pacchiana R, Bonafini C, Talacchi A, Chakravarthy B, Armato U, Chiarini A. 2011. The amyloid-β42 proxy, amyloid-β25-35, induces normal human cerebral astrocytes to produce amyloid-β42. J. Alzheimers. Dis. 24, 335–347. (doi:10.3233/JAD-2011-101626) [DOI] [PubMed] [Google Scholar]
- 157.Lah JJ, Heilman CJ, Nash NR, Rees HD, Yi H, Counts SE, Levey AI. 1997. Light and electron microscopic localization of presenilin-1 in primate brain. J. Neurosci. 17, 1971–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Diehlmann A, Ida N, Weggen S, Grünberg J, Haass C, Masters CL, Bayer TA, Beyreuther K. 1999. Analysis of presenilin 1 and presenilin 2 expression and processing by newly developed monoclonal antibodies. J. Neurosci. Res. 56, 405–419. [DOI] [PubMed] [Google Scholar]
- 159.Huynh DP, Vinters HV, Ho DH, Ho VV, Pulst SM. 1997. Neuronal expression and intracellular localization of presenilins in normal and Alzheimer disease brains. J. Neuropathol. Exp. Neurol. 56, 1009–1017. (doi:10.1097/00005072-199709000-00006) [DOI] [PubMed] [Google Scholar]
- 160.Ren RF, Lah JJ, Diehlmann A, Kim ES, Hawver DB, Levey AI, Beyreuther K, Flanders KC. 1999. Differential effects of transforming growth factor-βs and glial cell line-derived neurotrophic factor on gene expression of presenilin-1 in human post-mitotic neurons and astrocytes. Neuroscience 93, 1041–1049. (doi:10.1016/S0306-4522(99)00215-8) [DOI] [PubMed] [Google Scholar]
- 161.Green KN, Billings LM, Roozendaal B, Mcgaugh JL, Laferla FM. 2006. Neurobiology of disease glucocorticoids increase amyloid-β and tau pathology in a mouse model of Alzheimer's disease. J. Neurosci. 26, 9047–9056. (doi:10.1523/JNEUROSCI.2797-06.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Catania C, Sotiropoulos I, Silva R, Onofri C, Breen KC, Sousa N, Almeida OFX. 2009. The amyloidogenic potential and behavioral correlates of stress. Mol. Psychiatry 14, 95–105. (doi:10.1038/sj.mp.4002101) [DOI] [PubMed] [Google Scholar]
- 163.Honer C, et al. 2003. Glucocorticoid receptor antagonism by cyproterone acetate and RU486. Mol. Pharmacol. 63, 1012–1020. (doi:10.1124/mol.63.5.1012) [DOI] [PubMed] [Google Scholar]
- 164.Lahiri DK. 2004. Functional characterization of amyloid β precursor protein regulatory elements: rationale for the identification of genetic polymorphism. Ann. NY Acad. Sci. 1030, 282–288. (doi:10.1196/annals.1329.035) [DOI] [PubMed] [Google Scholar]
- 165.Wang Y, Li M, Tang J, Song M, Xu X, Xiong J, Li J, Bai Y. 2011. Glucocorticoids facilitate astrocytic amyloid-β peptide deposition by increasing the expression of APP and BACE1 and decreasing the expression of amyloid-β-degrading proteases. Endocrinology 152, 2704–2715. (doi:10.1210/en.2011-0145) [DOI] [PubMed] [Google Scholar]
- 166.Siman R, Card JP, Davis LG. 1990. Proteolytic processing of beta-amyloid precursor by calpain I. J. Neurosci. 10, 2400–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Katsurabayashi S, et al. 2016. Overexpression of Swedish mutant APP in aged astrocytes attenuates excitatory synaptic transmission. Physiol. Rep. 4, 12665 (doi:10.14814/phy2.12665) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Siman R, Patrick Card J, Nelson RB, Davis LG. 1989. Expression of β-amyloid precursor protein in reactive astrocytes following neuronal damage. Neuron 3, 275–285. (doi:10.1016/0896-6273(89)90252-3) [DOI] [PubMed] [Google Scholar]
- 169.Roberts GW, Gentleman SM, Lynch A, Graham DI. 1991. βA4 amyloid protein deposition in brain after head trauma. Lancet 338, 1422–1423. (doi:10.1016/0140-6736(91)92724-G) [DOI] [PubMed] [Google Scholar]
- 170.Guo Z, et al. 2000. Head injury and the risk of AD in the MIRAGE study. Neurology 54, 1316–1323. (doi:10.1212/WNL.54.6.1316) [DOI] [PubMed] [Google Scholar]
- 171.Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. 1994. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry 57, 419–425. (doi:10.1136/JNNP.57.4.419) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cribbs DH, Chen LS, Cotman CW, LaFerla FM. 1996. Injury induces presenilin-1 gene expression in mouse brain. Neuroreport 7, 1773–1776. (doi:10.1097/00001756-199607290-00016) [DOI] [PubMed] [Google Scholar]
- 173.Ramirez MJ, Heslop KE, Francis PT, Rattray M. 2001. Expression of amyloid precursor protein, tau and presenilin RNAs in rat hippocampus following deafferentation lesions. Brain Res. 907, 222–232. (doi:10.1016/S0006-8993(01)02580-X) [DOI] [PubMed] [Google Scholar]
- 174.Miake H, Tsuchiya K, Nakamura A, Ikeda K, Levesque L, Fraser PE, St.-George Hyslop PH, Mizusawa H, Uchihara T. 1999. Glial expression of presenilin epitopes in human brain with cerebral infarction and in astrocytoma. Acta Neuropathol. 98, 337–340. (doi:10.1007/s004010051090) [DOI] [PubMed] [Google Scholar]
- 175.Yamada T, Takashima A. 1997. Presenilin 1 immunostaining using well-characterized antibodies in human tissues. Exp. Neurol. 148, 10–12. (doi:10.1006/exnr.1997.6661) [DOI] [PubMed] [Google Scholar]
- 176.Nadler Y, Alexandrovich A, Grigoriadis N, Hartmann T, Rao KSJ, Shohami E, Stein R. 2008. Increased expression of the γ-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia 56, 552–567. (doi:10.1002/glia.20638) [DOI] [PubMed] [Google Scholar]
- 177.Chen Y, Constantini S, Trembovler V, Weinstock M, Shohami E. 1996. An experimental model of closed head injury in mice: pathophysiology, histopathology, and cognitive deficits. J. Neurotrauma 13, 557–568. (doi:10.1089/neu.1996.13.557) [DOI] [PubMed] [Google Scholar]
- 178.Baum L, et al. 2010. Serum zinc is decreased in Alzheimer's disease and serum arsenic correlates positively with cognitive ability. Biometals 23, 173–179. (doi:10.1007/s10534-009-9277-5) [DOI] [PubMed] [Google Scholar]
- 179.Brinkel J, Khan MH, Kraemer A. 2009. A systematic review of arsenic exposure and its social and mental health effects with special reference to Bangladesh. Int. J. Environ. Res. Public 6, 1609–1619. (doi:10.3390/ijerph6051609) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Escudero-Lourdes C, Uresti-Rivera EE, Oliva-González C, Torres-Ramos MA, Aguirre-Bañuelos P, Gandolfi AJ. 2016. Cortical astrocytes acutely exposed to the monomethylarsonous acid (MMAIII) show increased pro-inflammatory cytokines gene expression that is consistent with APP and BACE-1: over-expression. Neurochem. Res. 41, 2559–2572. (doi:10.1007/s11064-016-1968-z) [DOI] [PubMed] [Google Scholar]
- 181.Dong H, Yuede CM, Yoo H-S, Martin M V, Deal C, Mace AG, Csernansky JG. 2008. Corticosterone and related receptor expression are associated with increased beta-amyloid plaques in isolated Tg2576 mice. Neuroscience 155, 154–163. (doi:10.1016/j.neuroscience.2008.05.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jeong YH, Park CH, Yoo J, Shin KY, Ahn S-M, Kim H-S, Lee SH, Emson PC, Suh Y-H. 2006. Chronic stress accelerates learning and memory impairments and increases amyloid deposition in APPV717I-CT100 transgenic mice, an Alzheimer's disease model. FASEB J. 20, 729–731. (doi:10.1096/fj.05-4265fje) [DOI] [PubMed] [Google Scholar]
- 183.Schieb H, Kratzin H, Jahn O, Mobius W, Rabe S, Staufenbiel M, Wiltfang J, Klafki HW. 2011. β-Amyloid peptide variants in brains and cerebrospinal fluid from amyloid precursor protein (APP) transgenic mice: comparison with human Alzheimer amyloid. J. Biol. Chem. 286, 33 747–33 758. (doi:10.1074/jbc.M111.246561) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Moore BD, et al. 2012. Overlapping profiles of Aβ peptides in the Alzheimer's disease and pathological aging brains. Alzheimers. Res. Ther. 4, 18 (doi:10.1186/alzrt121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bayer TA, Wirths O. 2014. Focusing the amyloid cascade hypothesis on N-truncated Aβ peptides as drug targets against Alzheimer's disease. Acta Neuropathol. 127, 787–801. (doi:10.1007/s00401-014-1287-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Liao MQ, Tzeng YJ, Chang LYX, Huang HB, Lin TH, Chyan CL, Chen YC. 2007. The correlation between neurotoxicity, aggregative ability and secondary structure studied by sequence truncated Aβ peptides. FEBS Lett. 581, 1161–1165. (doi:10.1016/j.febslet.2007.02.026) [DOI] [PubMed] [Google Scholar]
- 187.Sevalle J, Amoyel A, Robert P, Fournié-Zaluski M-C, Roques B, Checler F. 2009. Aminopeptidase A contributes to the N-terminal truncation of amyloid β-peptide. J. Neurochem. 109, 248–256. (doi:10.1111/j.1471-4159.2009.05950.x) [DOI] [PubMed] [Google Scholar]
- 188.Bien J, Jefferson T, Causevic M, Jumpertz T, Munter L, Multhaup G, Weggen S, Becker-Pauly C, Pietrzik CU. 2012. The metalloprotease meprin generates amino terminal-truncated amyloid peptide species. J. Biol. Chem. 287, 33 304–33 313. (doi:10.1074/jbc.M112.395608) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Güntert A, Döbeli H, Bohrmann B. 2006. High sensitivity analysis of amyloid-beta peptide composition in amyloid deposits from human and PS2APP mouse brain. Neuroscience 143, 461–475. (doi:10.1016/j.neuroscience.2006.08.027) [DOI] [PubMed] [Google Scholar]
- 190.Bouter Y, et al. 2013. N-truncated amyloid β (Aβ) 4–42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol. 126, 189–205. (doi:10.1007/s00401-013-1129-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Oberstein TJ, et al. 2015. Astrocytes and microglia but not neurons preferentially generate N-terminally truncated Aβ peptides. Neurobiol. Dis. 73, 24–35. (doi:10.1016/j.nbd.2014.08.031) [DOI] [PubMed] [Google Scholar]
- 192.Wu Y, Zhang A-Q, Yew DT. 2005. Age related changes of various markers of astrocytes in senescence-accelerated mice hippocampus. Neurochem. Int. 46, 565–574. (doi:10.1016/j.neuint.2005.01.002) [DOI] [PubMed] [Google Scholar]
- 193.Hayakawa N, Kato H, Araki T. 2007. Age-related changes of astorocytes, oligodendrocytes and microglia in the mouse hippocampal CA1 sector. Mech. Ageing Dev. 128, 311–316. (doi:10.1016/j.mad.2007.01.005) [DOI] [PubMed] [Google Scholar]
- 194.Goss JR, Finch CE, Morgan DG. 1991. Age-related changes in glial fibrillary acidic protein mRNA in the mouse brain. Neurobiol. Aging 12, 165–170. (doi:10.1016/0197-4580(91)90056-P) [DOI] [PubMed] [Google Scholar]
- 195.Kohama SG, Goss JR, Finch CE, McNeill TH. 1995. Increases of glial fibrillary acidic protein in the aging female mouse brain. Neurobiol. Aging 16, 59–67. (doi:10.1016/0197-4580(95)80008-F) [DOI] [PubMed] [Google Scholar]
- 196.Diniz DG, et al. 2010. Environmental impoverishment and aging alter object recognition, spatial learning, and dentate gyrus astrocytes. Eur. J. Neurosci. 32, 509–519. (doi:10.1111/j.1460-9568.2010.07296.x) [DOI] [PubMed] [Google Scholar]
- 197.Franceschi C. 2007. Inflammaging as a major characteristic of old people: can it be prevented or cured? Nutr. Rev. 65, S173–S176. (doi:10.1301/nr.2007.dec.S173-S176) [DOI] [PubMed] [Google Scholar]
- 198.Lasn H, Winblad B, Bogdanovic N. 2006. Neuroglia in the inferior olivary nucleus during normal aging and Alzheimer's disease. J. Cell. Mol. Med. 10, 145–156. (doi:10.1111/j.1582-4934.2006.tb00296.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Mansour H, Chamberlain CG, Weible MW II, Hughes S, Chu Y, Chan-Ling T. 2008. Aging-related changes in astrocytes in the rat retina: imbalance between cell proliferation and cell death reduces astrocyte availability. Aging Cell 7, 526–540. (doi:10.1111/j.1474-9726.2008.00402.x) [DOI] [PubMed] [Google Scholar]
- 200.Cerbai F, Lana D, Nosi D, Petkova-Kirova P, Zecchi S, Brothers HM, Wenk GL, Giovannini MG. 2012. The neuron–astrocyte–microglia triad in normal brain ageing and in a model of neuroinflammation in the rat hippocampus. PLoS ONE 7, e45250 (doi:10.1371/journal.pone.0045250) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zaiou M. 2007. Multifunctional antimicrobial peptides: therapeutic targets in several human diseases. J. Mol. Med. 85, 317–329. (doi:10.1007/s00109-006-0143-4) [DOI] [PubMed] [Google Scholar]
- 202.Soscia SJ, et al. 2010. The Alzheimer's disease-associated amyloid β-protein is an antimicrobial peptide. PLoS ONE 5, e9505 (doi:10.1371/journal.pone.0009505) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zanetti M. 2004. Cathelicidins, multifunctional peptides of the innate immunity. J. Leukoc. Biol. 75, 39–48. (doi:10.1189/jlb.0403147) [DOI] [PubMed] [Google Scholar]
- 204.Wozniak M, Mee A, Itzhaki R. 2009. Herpes simplex virus type 1 DNA is located within Alzheimer's disease amyloid plaques. J. Pathol. 217, 131–138. (doi:10.1002/path.2449) [DOI] [PubMed] [Google Scholar]
- 205.Miklossy J. 2011. Emerging roles of pathogens in Alzheimer disease. Expert Rev. Mol. Med. 13, e30 (doi:10.1017/S1462399411002006) [DOI] [PubMed] [Google Scholar]
- 206.Bourgade K, Garneau H, Giroux G, Le Page AY, Bocti C, Dupuis G, Frost EH, Fülöp T. 2015. β-Amyloid peptides display protective activity against the human Alzheimer's disease-associated herpes simplex virus-1. Biogerontology 16, 85–98. (doi:10.1007/s10522-014-9538-8) [DOI] [PubMed] [Google Scholar]
- 207.White MR, Kandel R, Tripathi S, Condon D, Qi L, Taubenberger J, Hartshorn KL. 2014. Alzheimer's associated β-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS ONE 9, e101364 (doi:10.1371/journal.pone.0101364) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kumar DKV, et al. 2016. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci. Transl. Med. 8, 340ra72 (doi:10.1126/scitranslmed.aaf1059) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Smith JA, Das A, Ray SK, Banik NL. 2012. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 87, 10–20. (doi:10.1016/j.brainresbull.2011.10.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ma L, Allen M, Sakae N, Ertekin-Taner N, Graff-Radford NR, Dickson DW, Younkin SG, Sevlever D. 2016. Expression and processing analyses of wild type and p.R47H TREM2 variant in Alzheimer's disease brains. Mol. Neurodegener. 11, 72 (doi:10.1186/s13024-016-0137-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Mrak RE. 2012. Microglia in Alzheimer brain: a neuropathological perspective. Int. J. Alzheimers. Dis. 2012, 1–6. (doi:10.1155/2012/165021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Dewapriya P, Li Y-X, Himaya SWA, Pangestuti R, Kim S-K. 2013. Neoechinulin A suppresses amyloid-β oligomer-induced microglia activation and thereby protects PC-12 cells from inflammation-mediated toxicity. Neurotoxicology 35, 30–40. (doi:10.1016/j.neuro.2012.12.004) [DOI] [PubMed] [Google Scholar]
- 213.Liu Y, et al. 2005. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer's amyloid peptide. Brain 128, 1778–1789. (doi:10.1093/brain/awh531) [DOI] [PubMed] [Google Scholar]
- 214.Block ML, Zecca L, Hong J-S. 2007. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. (doi:10.1038/nrn2038) [DOI] [PubMed] [Google Scholar]
- 215.Qin L, Liu Y, Cooper C, Liu B, Wilson B, Hong J-S. 2002. Microglia enhance beta-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J. Neurochem. 83, 973–983. (doi:10.1046/j.1471-4159.2002.01210.x) [DOI] [PubMed] [Google Scholar]
- 216.Thal DR. 2012. The role of astrocytes in amyloid β-protein toxicity and clearance. Exp. Neurol. 236, 1–5. (doi:10.1016/j.expneurol.2012.04.021) [DOI] [PubMed] [Google Scholar]
- 217.Koistinaho M, et al. 2004. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat. Med. 10, 719–726. (doi:10.1038/nm1058) [DOI] [PubMed] [Google Scholar]
- 218.Bélanger M, Magistretti PJ. 2009. The role of astroglia in neuroprotection. Dialogues Clin. Neurosci. 11, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Armato U, Chiarini A, Chakravarthy B, Chioffi F, Pacchiana R, Colarusso E, Whitfield JF, Dal Prà I. 2013. Calcium-sensing receptor antagonist (calcilytic) NPS 2143 specifically blocks the increased secretion of endogenous Aβ42 prompted by exogenous fibrillary or soluble Aβ25–35 in human cortical astrocytes and neurons—therapeutic relevance to Alzheimer's disease. Biochim. Biophys. Acta. Mol. Basis Dis. 1832, 1634–1652. (doi:10.1016/j.bbadis.2013.04.020) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.