

Summary
The caspase activation and recruitment domain (CARD)-based inflammasome sensors NLRP1b and NLRC4 induce caspase-1-dependent pyroptosis independent of the inflammasome adaptor ASC. Here, we show that NLRP1b and NLRC4 trigger caspase-8-mediated apoptosis as an alternative cell death program in caspase-1−/− macrophages and intestinal epithelial organoids (IECs). The caspase-8 adaptor FADD was recruited to ASC specks, which served as cytosolic platforms for caspase-8 activation and NLRP1b/NLRC4-induced apoptosis. We further found that caspase-1 protease activity dominated over scaffolding functions in suppressing caspase-8 activation and induction of apoptosis of macrophages and IECs. Moreover, TLR-induced c-FLIP expression inhibited caspase-8-mediated apoptosis downstream of ASC speck assembly, but did not affect pyroptosis induction by NLRP1b and NLRC4. Moreover, unlike during pyroptosis, NLRP1b- and NLRC4-elicited apoptosis retained alarmins and the inflammasome-matured cytokines interleukin 1β (IL-1β) and IL-18 intracellularly. This work identifies critical mechanisms regulating apoptosis induction by the inflammasome sensors NLRP1b and NLRC4 and suggests converting pyroptosis into apoptosis as a paradigm for suppressing inflammation.
Keywords: cell death, inflammasome, caspase-1, ASC, caspase-8, apoptosis, pyroptosis, inflammation, NLRC4, NLRP1
Graphical Abstract
Highlights
-
•
NLRP1b and NLRC4 induce apoptosis in casp1−/− macrophages and epithelial organoids
-
•
NLRP1b/NLRC4-induced apoptosis requires ASC-dependent caspase-8 activation
-
•
Caspase-1 activity is dominant over scaffolding in suppressing apoptosis induction
-
•
TLR-induced c-FLIP levels inhibit NLRP1b/NLRC4-induced apoptosis in primed cells
Van Opdenbosch et al. find that CARD-based inflammasome sensors drive ASC- and caspase-8-dependent apoptosis as an alternative cell death program when caspase-1 activation is impaired. TLR-mediated upregulation of c-FLIP is identified as a second checkpoint that regulates ASC/caspase-8-mediated apoptosis. Moreover, apoptosis differs from pyroptosis in retaining inflammasome-dependent cytokines and alarmins intracellularly.
Introduction
Inflammasomes are cytosolic protein platforms with important roles in host-pathogen interactions, and mutations in several inflammasome components have been shown to predispose to chronic inflammatory diseases (Lamkanfi and Dixit, 2014, Van Gorp et al., 2017). Inflammasomes have long been recognized to contribute to inflammatory outcomes by promoting caspase-1-mediated maturation of the inflammatory cytokines interleukin (IL)-1β and IL-18. Activation of inflammatory caspase-1 and caspase-11 also triggers pyroptosis, which is a lytic cell death mode that has been characterized primarily in cells of the myeloid lineage (Jorgensen and Miao, 2015, Vande Walle and Lamkanfi, 2016). Pyroptosis is increasingly regarded as a mechanism that contributes importantly to the host’s ability to clear infections. It promotes passive release of the inflammasome-dependent cytokines IL-1β and IL-18 along with danger-associated molecular patterns (DAMPs), such as HMGB1, to attract and stimulate secondary immune cells at the infected tissue (He et al., 2015, Kayagaki et al., 2015, Liu et al., 2014). Pyroptosis is also thought to trap bacteria inside the pyroptotic cell corpse, which has been coined the “pyroptotic intracellular trap,” as a mechanism to counter pathogen spreading and to facilitate efferocytosis by infiltrating neutrophils (Jorgensen et al., 2016). Although its roles in chronic inflammatory diseases remain to be characterized, it is likely that pyroptosis contributes to detrimental inflammation and tissue destruction in this context. Thus, a better understanding of inflammasome-induced cell death regulation may reveal cell death checkpoints for altering the course of life-threatening infections and chronic inflammatory pathologies.
Notably, several studies have shown that pyroptosis induction by the Pyrin domain (PYD)-based NLRP3, AIM2, and Pyrin inflammasomes requires the inflammasome adaptor ASC (Mariathasan et al., 2004, Xu et al., 2014), and ASC has also been shown to induce caspase-8 activation and apoptosis in caspase-1-deficient macrophages that have been stimulated with NLRP3 and AIM2 agonists (Pierini et al., 2012, Sagulenko et al., 2013). Contrastingly, ASC is dispensable for induction of pyroptosis by the CARD-based inflammasome sensors NLRP1b and NLRC4, which may recruit caspase-1 directly through CARD-CARD interactions (Broz et al., 2010, Guey et al., 2014, Mariathasan et al., 2004, Van Opdenbosch et al., 2014). Accordingly, anthrax lethal toxin (LeTx)-elicited pyroptosis was unchanged in ASC-deficient macrophages (Van Opdenbosch et al., 2014). ASC was also not needed for NLRC4- and caspase-1-dependent pyroptosis of Salmonella enterica serovar Typhimurium (S. Typhimurium)-infected macrophages, although it did contribute to efficient IL-1β secretion and caspase-1 maturation under these conditions (Broz et al., 2010, Guey et al., 2014, Mariathasan et al., 2004, Van Opdenbosch et al., 2014). Other work established that caspase-8 co-localizes with caspase-1 in ASC specks of wild-type macrophages infected with Salmonella (S.) Typhimurium, although caspase-8 proved to be dispensable for S. Typhimurium-induced pyroptosis (Man et al., 2013). NLRC4 and ASC have also been shown recently to promote caspase-8 activation to induce gasdermin D-independent cell lysis of Legionella pneumophila-infected macrophages that were deficient for caspase-1 and caspase-11 (Mascarenhas et al., 2017). However, the role of caspase-8 in NLRP1b- and NLRC4-induced apoptosis and the regulatory mechanisms that control apoptosis induction by inflammasome sensors are largely unknown.
Here we showed that NLRP1b and NLRC4 induce apoptosis as an alternative cell death program in caspase-1−/− macrophages and intestinal epithelial organoids that requires ASC and caspase-8. Moreover, we delineated critical mechanisms that regulate caspase-8 activation and apoptosis induction by these CARD-based inflammasome sensors. Overall, this work suggests re-routing pyroptosis toward ASC- and caspase-8-dependent apoptosis as a paradigm for suppressing inflammation.
Results
Anthrax LeTx Induces Caspase-8-Dependent Apoptosis of Caspase-1-Deficient Macrophages
Anthrax LeTx induces ASC-independent pyroptosis in macrophages expressing a functional NLRP1b allele (Guey et al., 2014, Van Opdenbosch et al., 2014). To study the role of ASC specks in the absence of confounding induction of pyroptosis, we bred C57BL/6J (B6) mice that express a functional Nlrp1b transgene (B6Nlrp1b+) to mice lacking caspase-1 and caspase-11 (B6Nlrp1b+C1−/−C11−/−). As a complementary approach, C1−/−C11−/− mice were backcrossed to the BALB/c genetic background (BALB/c-C1−/−C11−/−), which encodes a functional NLRP1b allele and is naturally susceptible to LeTx-induced inflammasome activation (Boyden and Dietrich, 2006, Van Opdenbosch et al., 2014). Consistent with defective pyroptosis induction in B6Nlrp1b+C1−/−C11−/− macrophages, culture supernatants of LeTx-treated B6Nlrp1b+C1−/−C11−/− bone marrow-derived macrophages (BMDMs) lacked lactate dehydrogenase (LDH) activity, unlike caspase-1-sufficient B6Nlrp1b+ macrophages that had been exposed to LeTx (Figure 1A). Rather than being protected from cell death, however, microscopic analysis of LeTx-treated B6Nlrp1b+C1−/−C11−/− macrophages revealed these cells to display hallmark features of apoptosis, including cell shrinkage, nuclear condensation, and formation of membrane blebs and apoptotic bodies (Figure 1B). Fluorescence-activated cell sorting (FACS) analysis of annexin V (which binds to phosphatidylserine) and propidium iodide (PI) (a membrane-impermeant DNA-intercalating agent) staining confirmed cell death induction in LeTx-treated B6Nlrp1b+C1−/−C11−/− macrophages, with both B6Nlrp1b+ and B6Nlrp1b+C1−/−C11−/− macrophages displaying approximately 90% annexin V positivity 3 hr after LeTx treatment (Figures 1C and 1D). Microscopic analysis of BALB/c-C1−/−C11−/− macrophages confirmed the appearance of an apoptotic morphology after intoxication with LeTx akin to our results in B6Nlrp1b+C1−/−C11−/− macrophages (Figure 1E). In contrast, the morphology of LeTx-treated wild-type BALB/c macrophages (which express a functional NLRP1b and caspase-1) was characterized by features of pyroptosis, including cell rounding and swollen nuclei. Together, these results suggest that caspase-1 ablation converts LeTx-induced pyroptosis into apoptosis.
Figure 1.

LeTx Triggers Caspase-8 Activation and Apoptosis in Caspase-1-Deficient Macrophages
(A–D) B6Nlrp1b+ and B6Nlrp1b+C1−/−C11−/− BMDMs were treated with LeTx for 3 hr before supernatants were analyzed for LDH activity (A), bright-field images were acquired (B), and cells were stained for annexin V and PI followed by FACS analysis (C) and quanitification of annexin V-positive cells (D).
(E and H) Wild-type and caspase-1/11-deficient BALB/c BMDMs were stimulated with LeTx for 3 hr and then microscopically analyzed (E), and lysates were immunoblotted for caspase-1 and caspase-8 (H).
(F and G) BMDM lysates of the indicated genotypes were immunoblotted for RIPK3 and RIPK1 (F) and caspase- 1 and caspase-8 (G) following LeTx-treatment for the indicated durations.
(I–K) z-IETD-fmk-pretreated B6Nlrp1b+C1−/−C11−/− BMDMs were stimulated with LeTx for 3 hr before lysates were immunoblotted for caspase-8 (I). Cells were analyzed by FACS (J) and annexin V-positive cells (K) were quantified in parallel.
(L and M) BMDMs were incubated with TAT-CrmA for 10 min, followed by LeTx for 3 hr, before lysates were immunoblotted for caspase-1 and caspase-8 (L), and bright-field images were acquired (M).
All scale bars are set at 20 μm. All data are representative of results from three independent experiments. Data are shown as mean ± SD from a single representative experiment of at least three independent experiments, with each condition performed in triplicate.
Caspase-8 is the key initiator caspase that triggers apoptosis in the context of death receptor-induced apoptosis, and proteolysis of RIPK1 and RIPK3 is a biochemical hallmark of death receptor-induced caspase-8 activation (Feng et al., 2007, Martinon et al., 2000). We hypothesized that caspase-8 activation would induce similar RIPK1 and RIPK3 cleavage in LeTx-treated apoptotic macrophages. In agreement, we observed a significant accumulation of RIPK1 and RIPK3 cleavage fragments as early as 90 min after LeTx treatment in apoptotic but not pyroptotic macrophages (Figure 1F). The detection of significant levels of procaspase-8 maturation in apoptotic B6Nlrp1b+C1−/−C11−/− (Figure 1G) and BALB/c-C1−/−C11−/− macrophages (Figure 1H) further supported this notion. Notably, weak maturation of procaspase-8 was observed in pyroptotic macrophages, in agreement with published reports (Gurung et al., 2014, Man et al., 2013). The kinetics of caspase-8 maturation in apoptotic B6Nlrp1b+C1−/−C11−/− macrophages was akin to that of caspase-1 in pyroptotic B6Nlrp1b+ cells, with both being clearly detected 2 hr post-LeTx treatment (Figure 1G). Similar results were obtained when comparing caspase-1-sufficient BALB/c macrophages with caspase-1-deficient cells (Figure 1H). Because caspase-8-deficient mice are embryonically lethal (Varfolomeev et al., 1998), and caspase-8 is required for macrophage differentiation (Gurung et al., 2014, Kang et al., 2004), we initially took advantage of the peptidomimetic caspase-8 inhibitor benzyloxycarbonyl-Ile-Glu(OMe)-Thr-Asp(OMe)-fluoromethylketone (z-IETD-fmk) to further characterize the role of caspase-8 in LeTx-induced apoptosis. This inhibitor significantly inhibited (but did not abolish) caspase-8 maturation in LeTx-treated B6Nlrp1b+C1−/−C11−/− macrophages (Figure 1I), which corresponded with a 50% reduction in annexin V-positive cells (Figures 1J and 1K). Consistently, caspase-8-targeting small interfering RNAs (siRNAs) significantly reduced procaspase-8 expression levels in naive macrophages to about half of those of scrambled control siRNA-treated cells (Figure S1A). This corresponded with important reductions in caspase-3 activity, as measured by Asp-Glu-Val-Asp-ase (DEVD-ase) assay (Figure S1B), and caspase-8 and caspase-3 maturation levels (Figure S1C) in LeTx-treated B6Nlrp1b+C1−/−C11−/− macrophages. Moreover, the selective poxvirus-encoded caspase-1/8 inhibitor cytokine response modifier (CrmA) provided marked inhibition of caspase-8 maturation (Figure 1L) and apoptosis induction (Figure 1M) in B6Nlrp1b+C1−/−C11−/− macrophages. Overall, these results establish caspase-8 as a major driver of LeTx-induced apoptosis.
NLRP1b and ASC Specks Are Required for LeTx-Induced Caspase-8 Activation and Apoptosis
We next set out to investigate the mechanism of LeTx-induced caspase-8 maturation and apoptosis induction. The requirement for NLRP1b was addressed by comparing apoptotic responses in caspase-1/11-deficient macrophages that are sufficient (B6Nlrp1b+C1−/−C11−/−) or deficient (C1−/−C11−/−) for a functional Nlrp1b allele, respectively. Consistent with our earlier findings, B6Nlrp1b+C1−/−C11−/− macrophages efficiently activated caspase-8 and the apoptotic effector caspase-3 within 2 hr post-LeTx exposure (Figure 2A). Contrastingly, the absence of a functional Nlrp1b allele prevented maturation of caspase-3 and caspase-8 in LeTx-exposed C1−/−C11−/− macrophages (Figure 2A) and rendered these cells resistant to apoptosis induction (Figure 2B). As reported previously (Fink et al., 2008, Van Opdenbosch et al., 2014), the protease activity of LeTx was necessary for NLRP1b-mediated caspase-1 maturation and pyroptosis induction in B6Nlrp1b+ macrophages (Figures 2C and 2D). LeTx metalloprotease activity was also required for apoptosis induction because the catalytically inactive LFE687C mutant failed to activate caspase-8 (Figure 2C) and induce apoptosis (Figure 2D) in B6Nlrp1b+C1−/−C11−/− macrophages. The 26S proteasome inhibitor MG132 was shown previously to inhibit LeTx-induced caspase-1 activation in the J774A.1 cell line (Squires et al., 2007), and we extended this observation by demonstrating inhibition of caspase-1 and pyroptosis in primary B6Nlrp1b+ macrophages (Figure 2E; Figure S2A). Moreover, MG132 blocked caspase-8 activation and apoptosis in B6Nlrp1b+C1−/−C11−/− macrophages (Figure 2E; Figure S2A). As a control, MG132 did not interfere with the protease activities of LeTx, caspase-1, and caspase-8 (Figures S2B–S2D), suggesting that the proteasome is selectively required upstream of NLRP1b for LeTx-induced pyroptosis and apoptosis. In conclusion, these results suggest that the apoptotic and pyroptotic signaling pathways upstream of NLRP1b are conserved.
Figure 2.

NLRP1b and ASC Are Required for LeTx-Induced Caspase-8 Activation and Apoptosis
(A and B) BMDMs were treated with LeTx for the indicated durations before lysates were immunoblotted for caspase-8 and caspase-3 (A). Bright-field images were taken 3 hr after LeTx stimulation (scale bar, 20 μm) (B).
(C and D) BMDMs were stimulated with PA and LF or the catalytic LFE687C mutant before lysates were immunoblotted for caspase11 and caspase-8 (C), and bright-field images were acquired (scale bar, 20 μm) (D).
(E) BMDMs were treated with MG132 followed by LeTx before lysates were immunoblotted for caspase-1 and caspase-8.
(F–H) Crosslinked ASC oligomers were isolated from BMDMs following LeTx stimulation, and lysates were immunoblotted for ASC (F).
(G and H) ASC specks were quantified over multiple representative confocal micrographs depicting DAPI (blue), caspase-8 (green), and ASC (red) staining (scale bar, 10 μm) (G). Representative images are shown (H).
(I–K) BMDMs of the indicated genotypes were stimulated with LeTx before lysates were immunoblotted for caspase-1 and caspase-8 and ASC (I). Confocal images were acquired for DAPI (blue), caspase-8 (green), and ASC (red) (scale bar, 10 μm) (J), and bright-field images were taken (scale bar, 20 μm) (K).
All results are representative of at least three independent experiments, and data are shown as mean ± SD from a single representative experiment.
Based on these results, we then sought to address the role of ASC in LeTx-induced apoptosis. To this end, we first investigated whether ASC specks are formed in apoptotic B6Nlrp1b+C1−/−C11−/− macrophages. Both biochemical (Figure 2F) and microscopic (Figures 2G and 2H) analysis confirmed significantly more ASC specks being formed during LeTx-induced apoptosis compared with pyroptotic cells. Notably, an antibody specific for active caspase-8 weakly stained ASC specks in pyroptotic cells (Figure 2H), consistent with reported observations of caspase-8 recruitment to the NLRP3 (Gurung et al., 2014, Sagulenko et al., 2013), NLRC4 (Man et al., 2013), and AIM2 (Pierini et al., 2012) inflammasomes in cells undergoing pyroptosis. Relative to pyroptotic cells, however, active caspase-8 staining in apoptotic B6Nlrp1b+C1−/−C11−/− macrophages was markedly increased, and active caspase-8 was readily detected both within and outside of ASC specks during apoptosis (Figure 2H). Moreover, MG132 pretreatment blocked NLRP1b-mediated caspase-8 activation and ASC speck formation (Figure S2E), consistent with the 26S proteasome acting upstream of NLRP1b and ASC speck assembly. These results prompted us to delete ASC to dissect its contribution to caspase-8 activation during LeTx-induced apoptosis. Remarkably, ASC deletion in B6Nlrp1b+C1−/−C11−/− macrophages abolished LeTx-induced caspase-8 maturation, as evidenced by western blotting (Figure 2I) and immunostaining (Figure 2J) for active caspase-8. We previously showed that ASC was dispensable for LeTx-induced pyroptosis (Van Opdenbosch et al., 2014), but, contrastingly, ASC deletion rendered B6Nlrp1b+C1−/−C11−/− macrophages fully resistant to LeTx-induced apoptosis (Figure 2K). These results suggest that NLRP1b induces pyroptosis and apoptosis from different cytosolic platforms: ASC-independent NLRP1b inflammasomes engage caspase-1 directly for pyroptosis, whereas NLRP1b-induced caspase-8 activation and apoptosis originate from ASC specks.
NLRC4 Induces ASC- and Caspase-8-Mediated Apoptosis in Caspase-1−/− Macrophages and Intestinal Epithelial Cell Organoids
Akin to NLRP1b, NLRC4 contains a CARD that allows it to recruit caspase-1 directly and induce pyroptosis independently of ASC (Broz et al., 2010, Van Opdenbosch et al., 2014). The NAIP5/NLRC4 inflammasome can be engaged by Streptolysin O (SLO)-mediated delivery of recombinant S. Typhimurium flagellin into the cytosol of macrophages (Lamkanfi et al., 2007). As expected, cytosolic flagellin triggered pyroptosis in B6Nlrp1b+ macrophages (Figure 3A). However, B6Nlrp1b+C1−/−C11−/− macrophages responded with apoptosis, suggesting that agonists of the NAIP5/NLRC4 inflammasome activate caspase-8 when caspase-1 engagement fails. Paralleling our earlier results with NLRP1b, apoptosis induction by the NAIP5/NLRC4 inflammasome was abolished by further deletion of ASC in B6Nlrp1b+C1−/−C11−/− macrophages (Figure 3A). Consistent with flagellin-induced apoptosis being ASC- and caspase-8-dependent, we observed efficient caspase-8 maturation in B6Nlrp1b+C1−/−C11−/− BMDMs, which was abolished in congenic cells lacking ASC (Figure 3B). Notably, pyroptosis induction in flagellin-treated B6Nlrp1b+ macrophages was associated with weak caspase-8 maturation concomitant with efficient caspase-1 cleavage (Figure 3B), consistent with a previous report showing co-localization of caspases 1 and 8 in ASC specks of S. Typhimurium-infected B6 macrophages (Man et al., 2013). NAIP5/NLRC4 inflammasome-dependent pyroptosis can also be induced by stimulating macrophages with B. anthracis protective antigen (PA) in the presence of a chimeric fusion protein that consists of Legionella pneumophila flagellin (FlaA) and the N-terminal domain of B. anthracis lethal factor (LF) (von Moltke et al., 2012). As with SLO-mediated delivery of recombinant S. Typhimurium flagellin, stimulation with PA + LFn-FlaA (hereafter called FlaTox) triggered pyroptosis in B6Nlrp1b+ BMDMs, whereas ASC/caspase-8-mediated apoptosis was induced in B6Nlrp1b+C1−/−C11−/− BMDMs (data not shown). Akin to LeTx-induced apoptosis in macrophages of the latter genotype (Figure 2J), FlaTox-induced apoptosis was accompanied by abundant ASC speck assembly (Figure 3C). Notably, the bipartite death domain (DD)/death effector domain (DED) adaptor protein FADD also accumulated in ASC specks, suggesting that FADD bridges the interaction between ASC and caspase-8 (Figure 3C). To formally address the role of caspase-8 in NAIP5/NLRC4-mediated apoptosis, we infected macrophages with S. Typhimurium and quantified cell death in time using SYTOX Green incorporation and western blotting for caspase-1 and caspase-8. As expected, S. Typhimurium infection promptly induced caspase-1 maturation (Figure 3D) and pyroptosis (Figure 3E) of wild-type (B6) macrophages. Deficiency in caspase-1 and caspase-11 rerouted the cell death response toward apoptosis, as evidenced by efficient caspase-8 maturation and SYTOX Green incorporation that was delayed by 2 hr compared with pyroptotic cells (Figures 3D and 3E). Notably, deletion of the necroptosis regulator RIPK3 failed to rescue C1−/−C11−/− macrophages from S. Typhimurium-induced apoptosis, whereas further deletion of caspase-8 fully abolished cell death induction (Figures 3D and 3E). Together, these results establish that NAIP5/NLRC4 agonists induce ASC speck-dependent caspase-8 activation and apoptosis in macrophages lacking caspase-1.
Figure 3.

NAIP5/NLRC4-Induced Apoptosis Is Dependent on ASC and Caspase-8
(A and B) Recombinant flagellin was transfected in BMDMs using SLO before bright-field images were acquired (scale bar, 20 μm) (A) and lysates were immunoblotted for caspase-1 and caspase-8 and ASC (B).
(C) BMDMs were treated with FlaTox, and confocal micrographs for DAPI (blue), FADD (green), and ASC (red) were taken (scale bar, 10 μm).
(D and E) BMDMs of the indicated genotypes were infected with S. Typhimurium (50 MOI). Lysates were collected 2 hr post-infection and immunoblotted for caspases 1 and 8 and RIPK3 (D), and cell death was quantified by tracking SYTOX Green incorporation over time (E). Data are shown as mean ± SD from a single representative experiment, with each condition performed in duplicate.
(F and G) Primary intestinal epithelial cell (IEC) organoids of the indicated genotypes were stimulated with FlaTox and quantified for PI staining over a 16 hr time window (scale bar, 100 μm). Representative images are shown (F). Observed PI intensity was normalized to organoid surface (G).
All data are representative of results from three independent experiments.
Next to macrophages, the NAIP5/NLRC4 inflammasome mediates IL-1β and IL-18 secretion and expulsion of infected intestinal epithelial cells (IECs) in the gut (Rauch et al., 2017, Sellin et al., 2014). However, the role of the NAIP5/NLRC4 inflammasome in IEC death is not clear. Concordant with reported findings (Rauch et al., 2017) and paralleling our results in macrophages, we found that FlaTox induced a pyroptotic response in B6Nlrp1b+ IEC organoids, whereas caspase-1-deficient B6Nlrp1b+ IEC organoids responded with apoptosis (Figure 3F). Notably, FlaTox-induced apoptosis required ASC, as illustrated by micrographs (Figure 3F) and the absence of FlaTox-induced PI staining in IEC organoids lacking ASC and caspase-1 (Figure 3F; Movie S1). Cell death was FlaTox-induced because mock-treated IEC organoids of all analyzed genotypes had minimal PI uptake over the studies’ incubation periods (Figure S3; Movie S2). Together, these results demonstrate that NLRC4 elicits ASC-dependent caspase-8 activation and apoptosis in caspase-1-deficient macrophages and IEC organoids.
Caspase-1 Enzymatic and Scaffolding Functions Regulate ASC- and Caspase-8-Dependent Apoptosis
In addition to its enzymatic functions as a protease, caspase-1 might exert non-enzymatic scaffolding functions, as highlighted by the identification of autoinflammatory disease patients expressing caspase-1 variants with reduced or absent catalytic activity (Luksch et al., 2013, Luksch et al., 2015). We generated knockin mice that homozygously express an enzymatically inactive caspase-1C284A mutant from the endogenous casp1 locus to distinguish between caspase-1 enzymatic activity and caspase-1 scaffolding as mechanisms that suppress ASC- and caspase-8-dependent apoptosis (Figures S4A and S4B). Caspase-1C284A/C284A mice were born at Mendelian ratios and were phenotypically normal (data not shown). As expected, caspase-1C284A was expressed to normal levels in macrophages, but it failed to support FlaTox-induced caspase-1 automaturation (Figure 4A). Moreover, FlaTox-induced cleavage and secretion of IL-1β and IL-18 were abolished in caspase-1C284A/C284A macrophages, akin to caspase-1-deficient cells (Figures 4A–4C). Notably, FlaTox triggered marked caspase-3 and caspase-8 maturation in both caspase-1 knockout and caspase-1C284A/C284A macrophages (Figure 4D), in line with their apoptotic appearance (Figure 4E). However, when tracking DEVD-ase activity over time, we consistently noted reduced caspase-3/7 activity in caspase-1C284A/C284A macrophages relative to caspase-1-deficient cells (Figure 4F). FlaTox triggered IEC organoids of caspase-1C284A/C284A mice to die by apoptosis, which was characterized by slowed PI uptake relative to pyroptotic IEC organoids of wild-type control mice (Figures 4G and 4H; Movie S3). As expected, cell death was FlaTox-induced because mock-treated IEC organoids of all analyzed genotypes had minimal PI uptake over the studies’ incubation periods (Figure S4C; Movie S4). Moreover, FlaTox-induced pyroptosis was blunted in IEC organoids of NAIP5−/− and NLRC4−/− mice (Figures 4G and 4H; Movie S3). Together, these results reveal a dominant role for caspase-1 enzymatic activity, with an accessory contribution of caspase-1 scaffolding functions to suppression of ASC- and caspase-8-mediated apoptosis of macrophages and IEC organoids.
Figure 4.

Caspase-1 Protease and Scaffolding Functions Contribute to Suppression of ASC- and Caspase-8-Dependent Apoptosis
(A–E) BMDMs of the indicated genotypes were either primed or not primed with LPS for 3 hr and subsequently stimulated with FlaTox for another 2 hr. Lysates were immunoblotted for caspase-1, caspase-3, and caspase-8, IL-1β; and IL-18 (A and D). The levels of IL-1β (B) and IL-18 (C) were determined in culture supernatants, and bright-field micrographs were taken 2 hr post-stimulation (scale bar, 20 μm) (E). Data are shown as mean ± SD from a single representative experiment of three independent experiments, with each condition performed in triplicate.
(F) FlaTox-treated C1−/−and C1C284A/C284A BMDMs were incubated with CellEvent Caspase-3/7 Green and followed in time by IncuCyte technology. Data are shown as mean ± SD from a single representative experiment of three independent experiments, with each condition performed in duplicate.
(G and H) Primary intestinal epithelial organoids of the indicated genotypes were stimulated with FlaTox and followed during 16 hr for PI incorporation (scale bar, 100 μm) (G). Quantification was performed on PI intensity normalized to the surface of the organoid (H). All data are representative of results from at least three independent experiments.
ASC/Caspase-8-Induced Apoptosis Retains Inflammatory Cytokines and Alarmins Intracellularly
Because caspase-8 was shown to promote maturation and secretion of IL-1β under certain experimental conditions (Bossaller et al., 2012, Gringhuis et al., 2012, Maelfait et al., 2008, Moriwaki et al., 2015, Vince et al., 2012), we next examined the role of caspase-8 in maturation and secretion of IL-1β and IL-18 from LeTx-stimulated pyroptotic B6Nlrp1b+ and apoptotic B6Nlrp1b+C1−/−C11−/− macrophages. As expected (Van Opdenbosch et al., 2014), secretion of IL-1β from pyroptotic B6Nlrp1b+ cells required prior lipopolysaccharide (LPS) priming for transcriptional upregulation of proIL-1β, whereas proIL-18 is constitutively expressed, and IL-18 secretion by these cells occurred with or without prior LPS priming (Figures 5A and 5B). In marked contrast, culture supernatants of LeTx-treated B6Nlrp1b+C1−/−C11−/− macrophages were devoid of secreted IL-1β and IL-18 regardless of whether cells were primed with LPS (Figure 5A and 5B). Immunoblotting showed that, unlike in caspase-1-sufficient B6Nlrp1b+ BMDMs, intracellular IL-1β and IL-18 pools were not converted into mature cytokines in LeTx-treated B6Nlrp1b+C1−/−C11−/− macrophages (Figure 5C). Similar results were obtained in B6Nlrp1b+ BMDMs and B6Nlrp1b+C1−/−C11−/− macrophages that had been primed with the Toll-like receptor 1/2 (TLR1/2) agonist Pam3CSK4 or the TLR3 ligand poly(I:C) prior to LeTx challenge (Figures 5D and 5E). The alarmins HMGB1 and IL-1α were also selectively detected in conditioned medium of pyroptotic B6Nlrp1b+ macrophages (Figure 5F, G). They were absent from those of LeTx-treated B6Nlrp1b+C1−/−C11−/− macrophages (Figures 5F and 5G), although HMGB1 was released when cells underwent secondary necrosis (data not shown).
Figure 5.

Cytokines and Alarmins Are Not Released during NLRP1b- and NLRC4-Mediated Apoptosis
(A–C) BMDMs of the indicated genotypes were stimulated as depicted. Supernatants were analyzed for IL-1β (A) and IL-18 (B), and lysates were immunoblotted for IL-1β and IL-18 (C).
(D and E) BMDMs of the indicated genotypes were stimulated as depicted, and supernatants were analyzed for IL-1β (D) and IL-18 (E) by Luminex assay.
(F) B6Nlrp1b+ and B6Nlrp1b+C1−/−C11−/− BMDMs were stimulated with LeTx, and culture supernatants were immunoblotted for HMGB1 and albumin.
(G) LPS-primed or unprimed B6Nlrp1b+ and B6Nlrp1b+C1−/−C11−/− BMDMs were stimulated with LeTx. The supernatant was analyzed for IL-1α (G). Data are shown as mean ± SD from a single representative experiment of three experiments, with each condition performed in triplicate.
(H–M) BMDMs from B6, NLRC4−/−, ASC−/−, and C1−/−C11−/− mice were left unprimed (H, J, and L) or primed (I, K, and M) with LPS for 3 hr and subsequently infected with S. typhimurium (MOI 50). The supernatant was used to determine the levels of IL-1β (H and I), IL-18 (J and K), and IL-1α (L and M) by Luminex. Data are shown as mean ± SD from a single representative experiment of three experiments, with each condition performed in triplicate.
As expected (Broz et al., 2010, Van Opdenbosch et al., 2014), S. Typhimurium infection of wild-type (B6) BMDMs triggered significant secretion of IL-1β, which was partially dependent on ASC (Figure 5H). Prior LPS treatment significantly increased secreted IL-1β levels from wild-type and ASC−/− macrophages at both 1 and 3 hr post-infection (Figure 5I). In sharp contrast, NLRC4−/− and C1−/−C11−/− macrophages failed to secrete IL-1β regardless of whether they had been prestimulated with LPS (Figure 5H, I). IL-18 also was selectively secreted by S. Typhimurium-infected wild-type and ASC−/− macrophages that underwent pyroptosis but not by apoptotic C1−/−C11−/− macrophages (Figures 5J and 5K). In agreement with a pre-existing pool of proIL-18 being present in naive macrophages, IL-18 secretion from wild-type macrophages was already significant 1 hr post-infection, and it did not require prior LPS priming (Figures 5J and 5K). We similarly found that the alarmin IL-1α was released extracellularly by pyroptotic wild-type and ASC−/− macrophages but not by S. Typhimurium-infected NLRC4−/− and C1−/−C11−/− macrophages that do not undergo pyroptosis (Figures 5L and 5M). Together, these results show that pyroptotic cell death is associated with release of inflammatory cytokines and chemotactic alarmins, whereas ASC/caspase-8-mediated apoptosis largely retains these inflammatory mediators intracellularly.
c-FLIP Upregulation Suppresses Caspase-8 Activation and Apoptosis Downstream of ASC Speck Assembly
While examining possible reasons for the apparent lack of IL-1β and IL-18 maturation and alarmin secretion from LeTx-treated B6Nlrp1b+C1−/−C11−/− macrophages, we found that LPS priming inhibited LeTx-induced caspase-8 activation (Figure 6A) and apoptosis induction (Figure 6B), which we confirmed to occur in LeTx-treated BALB/c-C1−/−C11−/− macrophages as well (Figures S5A and S5B). LPS specifically inhibited LeTx-induced caspase-8 activation because it failed to inhibit LeTx-induced caspase-1 activation and pyroptosis in B6Nlrp1b+ (Figures 6A and S6B) and BALB/c (Figures S5A and S5B) macrophages. Interestingly, preventing de novo protein translation with cycloheximide restored LeTx-induced caspase-8 activation (Figure 6C) and apoptosis (Figure S5C) in LPS-primed B6Nlrp1b+C1−/−C11−/− macrophages. The TLR1/2 agonist Pam3CSK4 and the TLR3 ligand poly(I:C) also inhibited LeTx-induced caspase-8 maturation and apoptosis (Figure 6D; Figure S5D), suggesting that de novo protein synthesis by both the MyD88- and TRIF-dependent TLR signaling axes interfered with LeTx-induced caspase-8 activation. Notably, LPS priming prevented caspase-8 activation and apoptosis downstream of ASC speck formation (Figures 6E–6G). Together, these results implied that TLR signaling upregulated a factor that inhibited LeTx-induced apoptosis by blocking caspase-8 activation in ASC specks. c-FLIP is a nuclear factor κB (NF-kB)-regulated protein that is known to modulate caspase-8 activation (Micheau et al., 2002). In agreement, we only detected significant expression of known c-FLIP isoforms (named c-FLIPR and c-FLIPL) in B6Nlrp1b+C1−/−C11−/− macrophages when cells had been prestimulated with LPS (Figure 6H). Notably, LPS-primed macrophages expressed full-length c-FLIPL, which appeared to undergo cleavage into a 43-kDa product upon LeTx challenge (Figure 6H), reminiscent of its reported cleavage by caspase-8 upon death receptor engagement (Micheau et al., 2002). The c-FLIPL-caspase-8 heterodimer was shown to restrict caspase-8-mediated cleavage to locally available substrates, suppressing apoptosis induction while still allowing non-apoptotic caspase-8 signaling. We observed diminished LeTx-induced cleavage of the caspase-8 substrates caspase-3 and RIP1 in LPS-primed B6Nlrp1b+C1−/−C11−/− macrophages (Figure 6I), suggesting that these substrates may not be readily accessible to ASC speck-confined caspase-8. We employed siRNA-mediated knockdown of c-FLIP expression to further characterize the role of c-FLIP in the modulation of LeTx-induced caspase-8 activation and apoptosis induction. Unlike scrambled siRNA controls, c-FLIP-targeting siRNAs (individually and pooled) efficiently reduced c-FLIP expression in Pam3CSK4-stimulated B6Nlrp1b+C1−/−C11−/− macrophages (Figure 6J). Moreover, c-FLIP knockdown enabled “spontaneous” LPS-induced caspase-8 activation and apoptosis in B6Nlrp1b+C1−/−C11−/− macrophages, whereas cells pretreated with control siRNAs failed to induce these responses (Figures S5E and S5F and data not shown). Spontaneous LPS-induced caspase-8 activation was likely TRIF-mediated because Pam3CSK4 priming did not facilitate caspase-8 activation following c-FLIP downregulation (Figures 6K and 6L). However, c-FLIP downregulation did rescue LeTx-induced caspase-8 activity in Pam3CSK4-primed macrophages (Figures 6K and 6L), confirming the critical role of TLR-induced c-FLIP upregulation in modulating ASC- and caspase-8-dependent apoptosis.
Figure 6.

TLR-Induced Upregulation of c-FLIP Expression Inhibits LeTx-Induced Caspase-8 Activation Downstream of ASC Speck Formation
(A and B) BMDMs of the indicated genotypes were stimulated as depicted, lysates were immunoblotted for caspase-1 and caspase-8 (A), and bright-field micrographs were taken (scale bar, 20 μm) (B).
(C) Where indicated, B6Nlrp1b+C1−/−C11−/− BMDMs were pretreated with cycloheximide (CHX) prior to stimulation with LPS and LeTx for 3 hr, and lysates were immunoblotted for caspase-8.
(D) BMDMs of the indicated genotypes were primed with Pam3CSK4 or poly(I:C) before treatment with LeTx for 3 hr. Lysates were immunoblotted for caspase-1 and caspase-8.
(E–G) B6Nlrp1b+ and B6Nlrp1b+C1−/−C11−/− BMDMs were stimulated as indicated before confocal micrographs of DAPI (blue), caspase-8 (green), and ASC (red) staining were taken (scale bar, 10 μm) (E), and ASC specks were quantified over multiple representative confocal micrographs (F). Crosslinked ASC oligomers were isolated, and lysates were immunoblotted for ASC (G).
(H) B6Nlrp1b+C1−/−C11−/− BMDMs were stimulated as indicated, and lysates were immunoblotted for c-FLIP and β-actin.
(I) Lysates B6Nlrp1b+C1−/−C11−/− BMDMs were immunoblotted for the indicated proteins.
(J–L) Pam3CSK4-stimulated B6Nlrp1b+C1−/−C11−/− BMDMs were transfected with scrambled and c-FLIP siRNAs before lysates were probed for c-FLIP and β-actin expression (J). Pam3CSK4-stimulated BMDMs were treated as depicted before lysates were immunoblotted for caspase-8 (K), and analyzed for DEVD-amc conversion (L).
Data are shown as mean ± SD from a single representative experiment of at least three experiments.
c-FLIP Suppresses S. Typhimurium-Induced Caspase-8 Activation and Apoptosis Induction
S. Typhimurium is a pathogen that stimulates TLR4 while also engaging the NLRC4 inflammasome. We therefore addressed whether c-FLIP was upregulated in S. Typhimurium-infected macrophages and whether this also affected NLRC4-induced apoptosis. Consistent with our earlier findings, c-FLIP expression was below the detection limit in mock-treated B6Nlrp1b+C1−/−C11−/− macrophages, but expression of both the c-FLIPL and c-FLIPR isoforms was induced by S. Typhimurium infection (Figure 7A). As before, prior LPS priming further upregulated the expression levels of these c-FLIP isoforms (Figure 7A). LPS priming significantly reduced caspase-8 maturation in C1−/−C11−/− and B6Nlrp1b+C1−/−C11−/− cells, whereas it had no effect on S. Typhimurium-induced caspase-1 maturation in B6 (Figure 7B) and B6Nlrp1b+ macrophages (Figure S6A). Priming with the TLR1/2 agonist Pam3CSK4 also markedly attenuated caspase-8 activation upon introduction of S. Typhimurium flagellin in the cytosol of B6Nlrp1b+C1−/−C11−/− macrophages (Figure 7C). Unlike scrambled control siRNAs, c-FLIP-targeting siRNAs efficiently rescued cytosolic flagellin-induced caspase-8 activation in Pam3CSK4-primed macrophages (Figure 7C), demonstrating that c-FLIP upregulation in TLR-primed macrophages suppresses NLRC4-mediated caspase-8 activation.
Figure 7.

NLRC4-Induced Apoptosis Is Inhibited by LPS Priming
(A) Naive and LPS-primed B6Nlrp1b+C1−/−C11−/− BMDMs were infected with S. Typhimurium (50 MOI) for 2 hr before lysates were immunoblotted for c-FLIP and β-actin.
(B) BMDMs of the indicated genotypes were treated as depicted before lysates were immunoblotted for caspase-1 and caspase-8.
(C) B6Nlrp1b+C1−/−C11−/− BMDMs were transfected with scrambled control and c-FLIP-targeting siRNAs before stimulation with Pam3CSK4 (Pam) and SLO-mediated cytosolic delivery of recombinant flagellin. Lysates were immunoblotted for caspase-8.
(D–G) BMDMs of the indicated genotypes were left unprimed (●) or primed with LPS (▪) for 3 hr before infection with S. Typhimurium (50 MOI). Supernatants were analyzed for LDH activity (dash line), and annexin V/PI staining (full line) was performed by FACS. Data are shown as mean from a single representative experiment out of two independent experiments, with each condition performed in triplicate.
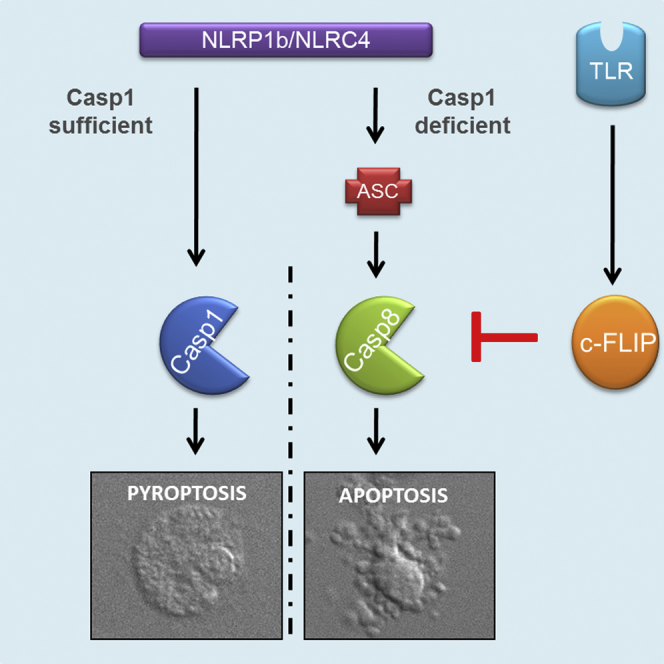
(H) Schematic representation of cell death signaling by the CARD-based inflammasome sensors NLRP1b and NLRC4. Engagement of NLRP1b or NLRC4 leads to caspase-1 maturation and induction of pyroptosis with release of alarmins and inflammatory cytokines, resulting in inflammation. In the absence of caspase-1, ASC specks serve as cytosolic platforms for efficient caspase-8 activation, which drives apoptosis characterized by the absence of alarmin and inflammatory cytokine release. TLR-mediated c-FLIP upregulation potently impedes caspase-8-dependent apoptosis but not pyroptosis.
To further address the role of TLR-induced c-FLIP upregulation in the regulation of NLRC4-mediated cell death responses, we compared cell death levels of naive and LPS-primed macrophages following S. Typhimurium infection. Apoptotic cells were defined as cells that became positive for annexin V in the absence of LDH release, whereas pyroptosis was associated with simultaneous annexin V staining and LDH release. Indicative of pyroptosis induction, S. Typhimurium-infected wild-type (B6) and ASC−/− macrophages concomitantly stained positive for annexin V and released LDH when analyzed 1 and 3 hr post-infection (Figures 7D and 7E; Figures S6B and S6C). As expected, NLRC4−/− macrophages were largely protected from S. Typhimurium-induced cell death at these time points (Figure 7F). In contrast, infected C1−/−C11−/− macrophages responded with delayed induction of apoptosis, as evidenced by annexin V positivity in the absence of LDH activity (Figure 7G; Figures S6B and S6C). Notably, LPS priming prior to S. Typhimurium infection inhibited apoptosis in infected C1−/−C11−/− macrophages, but not pyroptosis, in wild-type and ASC−/− macrophages (Figures 7D, 7E, and 7G; Figures S6B and S6C). Together, these results demonstrate that NLRC4-induced apoptosis is sensitive to c-FLIP-mediated inhibition whereas pyroptosis is not.
Discussion
Pyroptosis is a non-homeostatic, lytic regulated cell death mode that is increasingly recognized as a critical host defense mechanism for fighting microbial pathogens (Jorgensen and Miao, 2015). In addition to trapping intracellular pathogens and depriving them of their replicative niches, pyroptotic cell rupture is thought to release IL-1β, IL-18, and the nuclear alarmin HMGB1 to attract and stimulate neutrophils and other immune cells at the infectious locus (Kayagaki et al., 2015, Lamkanfi et al., 2010, Liu et al., 2014, Shi et al., 2015). A recent single-cell analysis of inflammasome activation supported this notion by demonstrating that IL-1β-secreting macrophages corresponded to pyroptotic cells (Liu et al., 2014). Consequently, many bacterial and viral pathogens express virulence factors that suppress inflammasome activation (Lamkanfi and Dixit, 2011). Under conditions of chronic inflammation, however, pyroptosis is likely to be detrimental to the host. Although this hypothesis awaits empirical validation, genetic deletion of caspase-1 has been shown to be significantly more effective than targeting downstream IL-1β and IL-18 signaling in controlling early perinatal lethality and inflammatory pathology of Nlrp3 knockin mice that model Muckle-Wells syndrome-associated disease (Brydges et al., 2013). Moreover, recent studies identified autoinflammatory disease patients with autosomal dominant NLRC4 and NLRP1 mutations (Bracaglia et al., 2015, Canna et al., 2014, Grandemange et al., 2017, Kawasaki et al., 2016, Kitamura et al., 2014, Romberg et al., 2014, Volker-Touw et al., 2016, Zhong et al., 2016). The clinical presentation of NLRC4- and NLRP1-associated autoinflammatory diseases may vary considerably, but IL-1β, IL-18, and pyroptosis are thought to contribute importantly to the etiology of these diseases. Unlike during pyroptosis, however, apoptotic cells are orderly packed in “apoptotic bodies” for efferocytosis by neighboring cells and professional phagocytes without spilling immunogenic factors in the extracellular space. Therefore, steering cell death responses away from pyroptosis and toward apoptotic cell death may represent an attractive approach to treat IL-1β/IL-18- and alarmin-induced inflammation and tissue damage in NLRP1- and NLRC4-associated autoinflammation and other chronic inflammatory diseases (Figure 7H).
Here we showed that the failure to activate caspase-1 following engagement of NLRP1b and NLRC4 results in caspase-8-driven apoptosis instead of pyroptosis. In support of our model, S. Typhimurium infection has been shown previously to trigger apoptosis in caspase-1/11-deficient macrophages, although the molecular mechanisms involved have not been fully explored (Puri et al., 2012). Our analysis of knockin mice that express a catalytically inactive caspase-1C284A mutant revealed a dominant role for caspase-1 protease activity and an accessory contribution of caspase-1 scaffolding for suppression of caspase-8 activation. Importantly, genetic deletion of ASC abolished NLRP1b- and NLRC4-induced apoptosis, thus establishing ASC specks as bona fide intracellular platforms for caspase-8-mediated apoptosis on par with death receptor-induced caspase-8-activating platforms. In agreement, we detected low levels of active caspase-8 in ASC specks of pyroptotic macrophages and showed that ASC specks of apoptotic macrophages contained markedly higher levels of active caspase-8. We further showed that TLRs stimulated de novo synthesis of c-FLIP, which inhibited ASC/caspase-8-mediated apoptosis but not inflammasome-induced pyroptosis. This implies that, in cells failing caspase-1 engagement, c-FLIP upregulation serves as a second checkpoint for modulating inflammasome-associated caspase-8 activation and apoptosis induction (Figure 7H). Moreover, unlike during pyroptosis, inflammasome-dependent cytokines and alarmins were retained intracellularly during NLRP1b- and NLRC4-elicited apoptosis until cells became secondarily necrotic. Overall, this work demonstrates that CARD-based inflammasome sensors induce c-FLIP-sensitive, non-inflammatory, ASC/caspase-8-dependent apoptosis when caspase-1 engagement fails and suggests that switching pyroptosis to ASC/caspase-8-mediated apoptosis might suppress inflammatory pathology in patients with gain-of-function mutations in NLRP1 and NLRC4 as well as in other patients suffering from chronic inflammation.
Experimental Procedures
Mice
B6Nlrp1b+ (Boyden and Dietrich, 2006), C1−/−C11−/− (Kuida et al., 1995), ASC−/− (Mariathasan et al., 2004), Nlrc4−/− (Mariathasan et al., 2004), C1−/− (Van Gorp et al., 2016), C8−/−RIPK3−/− (Newton et al., 2014) and Naip5−/− (Lightfield et al., 2008) mice on a C57BL/6 background have been described previously. C1−/−C11−/− mice have also been backcrossed to the BALB/c genetic background for 7 generations. C1C284A/C284A mice were produced by targeting exon 5 of Casp1 by CRISPR-Cas9 technology using the single guide RNA (sgRNA) 5′ CCGTGGAGGTAAGTGCTGA 3′ in C57BL/6 zygotes. The single-stranded DNA (ssDNA) sequence to introduce the C284A mutation was 5′ GATGAACACTTTGAAGTGCCCAAGCTTGAAAGACAAGCCCAAGGTGATCATTATTCAGGCCGCCCG TGGAGGTAAGTGCTGATTGTTTAAAATAACAGGGCATTCCCATTGAGACTTTATCATTTATAGATAGAGAGTACTCTT 3′. A silent HaeIII restriction site was created to genotype the mice. C57BL/6J and BALB/c mice were originally purchased from Charles River Laboratories and bred in-house. Mice of any sex and age were used for production of BMDMs and IECs. Animals were housed in individually ventilated cages under specific pathogen-free conditions, and all studies were conducted under protocols approved by the Ghent University Committee on the Use and Care of Animals.
Macrophage Differentiation and Stimulation
BMDMs were generated by culturing mouse bone marrow cells in L-cell-conditioned Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acids, and 1% penicillin-streptomycin for 6 days in a humidified atmosphere containing 5% CO2. BMDMs were seeded in 12-well plates and, the next day, either left untreated or stimulated with 500 ng/mL ultrapure LPS from Salmonella minnesota (InvivoGen), 1 μg/mL Pam3CSK4 (InvivoGen), or 1 μg/mL poly(I:C) (InvivoGen) for 3 hr prior to treatment with anthrax PA (500 ng/mL, Quadratech) and wild-type or proteolytically inactive LF (250 ng/mL, Quadratech). In some experiments, BMDMs were pretreated with the caspase-8 inhibitor z-IETD-fmk (50 μM, R&D Systems) or the proteasome inhibitor MG132 (10 μM, Calbiochem) for 30 min prior to LeTx incubation. Alternatively, BMDMs were pretreated with the translation inhibitor cycloheximide (CHX, 50 ng/mL, Sigma-Aldrich) for 15 min prior to LPS priming (500 ng/mL), followed by LeTx treatment. Otherwise, 10 min pretreatment with trans-activator of transcription of HIV-1 (TAT)-CrmA (2 μM, purified as described previously; Krautwald et al., 2010) was performed prior to LeTx stimulation. To stimulate the NLRC4 inflammasome, BMDMs were either primed or not primed with 500 ng/mL LPS for 3 hr, followed by S. Typhimurium infection (MOI 50) for 1 hr and 3 hr, respectively. Gentamycin (50 μg/mL, 10131-027, Life Technologies) was added 1 hr post-infection to kill extracellular bacteria. Alternatively, recombinant S. Typhimurium flagellin (fliC, 5 μg/mL) produced in E. coli (Van Maele et al., 2014) was delivered in the cytosol using SLO (25 μg/mL, S5265-25KU, Sigma) as described before (Lamkanfi et al., 2007). In other experiments, cells were treated with anthrax PA (500 ng/mL, Quadratech) and 1 μg/mL LFn-FlaA (Yang et al., 2013).
Primary Intestinal Epithelial Organoids
Primary intestinal epithelial organoids were grown as described before (Sato and Clevers, 2013). Briefly, the small intestine was flushed and cut into small pieces that were dissociated in PBS containing 2 mM EDTA for 30 min at 4°C. After extensive washing, the isolated crypts were pelleted and mixed with 50 μL of Matrigel (Corning) and put in a 24-well plate. After polymerization of the Matrigel, complete culture medium containing advanced DMEM/F12 (Gibco) supplemented with B27 supplement (0.02%, Invitrogen), N2 supplement (0.1%, Invitrogen), N-acetylcysteine (0.0025%, Sigma-Aldrich), mouse epidermal growth factor (mEGF; 0.001%, Invitrogen), and conditioned Rspondin and mNoggin medium. Organoids were seeded and imaged in an 8-well chamber (iBidi). Cell death was induced just before imaging and was monitored by an increase in PI positivity. Live-cell imaging was performed on an Axio Observer Z1 (Zeiss, Germany) equipped with a CSU-X1 spinning-disk head (Yokogawa) and AxioCam MRm (Zeiss), with a EC Plan-Neofluar 10× dry objective (numerical aperture [NA] 0.30). Images were acquired every 15 min for 16 hr. Data analysis and image reconstruction were performed with ImageJ (NIH).
Western Blotting
Cell lysates were incubated in cell lysis buffer (20 mM Tris HCl [pH 7.4], 200 mM NaCl, and 1% NP-40) and denatured in Laemmli buffer. Protein samples were boiled at 95°C for 10 min and separated by SDS-PAGE. Separated proteins were transferred to polyvinylidene fluoride (PVDF) membranes. Blocking, incubation with antibody, and washing of the membrane were done in PBS supplemented with 0.05% or 0.2% Tween 20 (v/v) and 3% or 1% (w/v) non-fat dry milk. Immunoblots were incubated overnight with primary antibodies against caspase-1 (AG-20B-0042-C100, Adipogen), caspase-8 (ALX-804-447-C100, 1G12, Enzo Life Sciences; 8592S, D5B2, Cell Signaling Technology), caspase-3 (9662, Cell Signaling Technology; 9664S, 5A1E, Cell Signaling Technology), ASC (AG-25B-0006, Adipogen), RIPK3 (ADI-905-242-100, Enzo Life Sciences), RIPK1 (610458, BD Biosciences), IL-1β (GTX74034, GeneTex), IL-18 (5180R-100, BioVision Technologies), c-FLIP (AG-20B-0005-C100, Adipogen), HMGB1 (ab18256, Abcam), albumin (sc-46291, Santa Cruz Biotechnology), and β-actin (sc-47778-HRP, Santa Cruz Biotechnology). Horseradish peroxidase-conjugated goat anti-mouse (115-035-146, Jackson ImmunoResearch Laboratories), anti-rabbit (111-035-144, Jackson ImmunoResearch Laboratories), or anti-rat (112-035-143, Jackson ImmunoResearch Laboratories) secondary antibody was used to detect proteins by enhanced chemiluminescence (Thermo Scientific).
Cell Death Kinetic Measurements
A plate-based fluorometric assay (FLUOstar, Omega BMG Labtech) was used to quantify cell permeabilization (5 μM SYTOX Green) and caspase-3 activation (10 μM Asp-Glu-Val-Asp-7-amino-4-methylcoumarin [DEVD-amc]). Briefly, stimulated cells were incubated in the presence of SYTOX Green and DEVD-amc in a CO2- and temperature-controlled environment that allowed measurement of fluorescent signals over a time span of 3 hr. The maximum slope of fluorescence over time (ΔF/minute) was used as a measure for cell death. Alternatively, the IncuCyte Zoom system (Essenbio) was used to acquire and analyze cell death rates over a period of time. Caspase-3/7 activation was detected using CellEvent Caspase3/7 Green substrate (Invitrogen) according to the manufacturer’s instructions.
siRNA Transfection
Viromer Green (Lipocalyx) was used according to the manufacturer’s instructions. The siRNAs directed against c-FLIP (SR400039) and caspase-8 (SR415523) were purchased from OriGene with a scrambled control present. After 24 hr of siRNA transfection, Pam3CSK4 (InvivoGen) or ultrapure LPS from Salmonella minnesota (InvivoGen) priming for 3 hr was followed by LeTx stimulation for another 3 hr. Alternatively, after 24 hr of siRNA transfection, purified fliC (5 μg/mL) was delivered in the cytosol using SLO for another 3 hr.
Fluorescence and Confocal Microscopy
B6Nlrp1b+, B6Nlrp1b+C1−/−C11−/−, and B6Nlrp1b+C1−/−C11−/−ASC−/− macrophages grown on coverslips were either left untreated (mock) or stimulated with LeTx for 105 min, fixed in 4% paraformaldehyde, and stained with antibodies against ASC (04-147, Millipore; pAL177 AG-25B-0006, Adipogen) and caspase-8 (8592S, D5B2, Cell Signaling Technology) or FADD (sc-6036, Santa Cruz Biotechnology). The secondary antibodies anti-mouse Alexa Fluor 594, anti-rabbit Alexa Fluor 488, or anti-goat Alexa Fluor 488 (Invitrogen) were used. Slides were mounted in ProLong Gold Antifade reagent with DAPI (Life Technologies). Confocal micrographs were taken on an Olympus microscope using a 60× objective lens. Alternatively, fixed cells were imaged on a Zeiss LSM780 confocal microscope (Zeiss, Germany) using a Plan-Apochromat 40× (NA 1.4) oil objective. The pinhole was set at 1 Airy unit (the theoretical point with optimal collection of the most “real” signal, with use of a pinhole to physically filter out most scattered light), and 4× frame averaging was applied to reduce noise.
Cytokine Analysis
Cytokine levels in cell culture medium were determined by magnetic bead-based multiplex assay using Luminex technology (Bio-Rad) according to the manufacturer’s instructions.
Cell Death Measurements
Cell death levels were determined by LDH assay (Promega) in culture medium. Alternatively, annexin V (BD Pharmingen) and PI staining on cells was performed according to the manufacturer’s instructions. Flow cytometry was used to measure stained cells, and data were analyzed with FlowJo software.
ASC Oligomer Analysis
ASC oligomers were purified as described before (Fernandes-Alnemri et al., 2010). Briefly, culture supernatants were discarded, and cells were lysed in 500 μL lysis buffer (20 mM potassium; 2-[4-(2-hydroxyethyl)piperazin-1-yl] ethanesulfonic acid; hydroxide [HEPES-KOH] [pH 7.5], 150 mM KCl, 1% NP40, 0.1 mM PMSF, and protease inhibitor cocktail) on ice. Lysates were prepared by syringing 10 times, followed by centrifugation for 10 min at 6,000 rpm at 4°C. Pellets were washed twice with PBS and resuspended in 500 μL PBS, followed by crosslinking with 2 mM fresh disuccinimidyl suberate (DSS) for 30 min at room temperature. Subsequently, cells were pelleted by centrifugation at 6,000 rpm for 10 min at 4°C, and pellets were resuspended in 60 μL 1× SDS buffer, followed by immunoblotting for ASC.
In Vitro Protease Activity Assays
In vitro caspase-1 activity was determined by incubating 1 IU recombinant mouse caspase-1 (1181-100, BioVision Technologies) with 50 μM fluorogenic caspase-1 substrate peptide Acetyl-Trp-Glu-His-Asp-7-Amino-4-methylcoumarin (Ac-WEHD-amc) (ALX-260-057-M005, Enzo Life Sciences) in 200 μL cell free system (CFS) buffer (10 mM HEPES [pH 7.4], 220 mM mannitol, 68 mM sucrose, 2 mM NaCl, 2.5 mM KH2PO4, 0.5 mM EGTA, 2 mM MgCl2, 0.5 mM sodium pyruvate, 0.5 mM L-glutamine, and 10 mM DTT). The release of fluorescent 7-amino-4-methylcoumarin in the presence of the indicated concentrations of the proteasome inhibitor MG132 (474791, Calbiochem), DMSO (Sigma-Aldrich), or the caspase-1 inhibitor Acetyl-Tyr-Val-Ala-Asp - chloromethylketone (Ac-YVAD-cmk) (ALX-260-028-M001, Enzo Life Sciences) was measured for 20 min at 1-min intervals by fluorometry (excitation at 360 nm and emission at 460 nm) on a microplate fluorescence reader (Bio-Tek Instruments), and the maximal rate of increase in fluorescence was calculated (ΔF/minute). The same was done for caspase-8 activity by incubating 100 ng recombinant mouse caspase-8 (ALX-201-163-C020, Enzo Life Sciences) with 50 μM fluorogenic caspase-8 substrate peptide Acetyl-Ile-Glu-Thr-Asp-7-Amino-4-methylcoumarin (Ac-IETD-amc) (ALX-260-042-M005, Enzo Life Sciences) in 50 μL CFS buffer in the presence of MG132, DMSO, or z-IETD-fmk (FMK0F07, R&D Systems). To determine anthrax LF activity, 50 nM recombinant LF (Quadratech) was incubated with 2 mM anthrax LF substrate III (176904, Calbiochem) in the presence of MG132, DMSO, or In-2-LF (176901, Calbiochem).
Acknowledgments
We thank Dr. William F. Dietrich (Harvard), Dr. Richard Flavell (Yale University), Dr. Russell Vance (University of California), and Dr. Vishva M. Dixit (Genentech) for the generous supply of mutant mice. N.V.O., F.V.H., and L.V.W. are supported by a postdoctoral fellowship of the Fund for Scientific Research-Flanders. T.-D.K. is supported by grants from the NIH (AR056296, CA163507, and AI101935) and the American Lebanese Syrian Associated Charities (ALSAC). This work was supported in part by grants from the European Research Council (281600 and 683144) and a Baillet Latour medical research grant (to M.L.).
Author Contributions
N.V.O. and M.L. designed the study. N.V.O., H.V.G., M.V., P.H.V.S., N.M.d.V., A.G., L.V.W., M.M., D.D., and F.V.H. performed the experiments. N.V.O., H.V.G., L.V.W., A.G., T-D.K., and M.L. analyzed the data. J.D., T.H., S.K., and T.-D.K. provided essential reagents. N.V.O. and M.L. wrote the manuscript with input from the other authors. M.L. oversaw the project.
Declaration of Interests
The authors declare no competing interests.
Published: December 19, 2017
Footnotes
Supplemental Information includes six figures and four movies and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.11.088.
Supplemental Information
Primary intestinal epithelial organoids from B6Nlrp1b+, B6Nlrp1b+C1−/−C11−/− and B6Nlrp1b+C1−/−C11−/−ASC−/− were stimulated with FlaTox and followed in time for 16 h in the presence of propidium iodide (PI). All data are representative of results from three independent experiments.
Primary intestinal epithelial organoids from B6Nlrp1b+, B6Nlrp1b+C1−/−C11−/− and B6Nlrp1b+C1−/−C11−/−ASC−/− were left unstimulated and followed in time for 16 h in the presence of PI. All data are representative of results from three independent experiments.
Primary intestinal epithelial organoids from B6, NAIP5−/−, NLRC4−/− and C1C284A/C284A were stimulated with FlaTox and followed in time for 16 h in the presence of PI. All data are representative of results from three independent experiments.
Primary intestinal epithelial organoids from B6, NAIP5−/−, NLRC4−/− and C1C284A/C284A were left unstimulated and followed in time for 16 h in the presence of PI. All data are representative of results from three independent experiments.
References
- Bossaller L., Chiang P.I., Schmidt-Lauber C., Ganesan S., Kaiser W.J., Rathinam V.A., Mocarski E.S., Subramanian D., Green D.R., Silverman N. Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 2012;189:5508–5512. doi: 10.4049/jimmunol.1202121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden E.D., Dietrich W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 2006;38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- Bracaglia C., Gatto A., Pardeo M., Lapeyre G., Ferlin W., Nelson R., de Min C., De Benedetti F. Anti interferon-gamma (IFNγ) monoclonal antibody treatment in a patient carrying an NLRC4 mutation and severe hemophagocytic lymphohistiocytosis. Pediatric Rheumatology Online J. 2015;13:O68. [Google Scholar]
- Broz P., von Moltke J., Jones J.W., Vance R.E., Monack D.M. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe. 2010;8:471–483. doi: 10.1016/j.chom.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brydges S.D., Broderick L., McGeough M.D., Pena C.A., Mueller J.L., Hoffman H.M. Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J. Clin. Invest. 2013;123:4695–4705. doi: 10.1172/JCI71543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canna S.W., de Jesus A.A., Gouni S., Brooks S.R., Marrero B., Liu Y., DiMattia M.A., Zaal K.J., Sanchez G.A., Kim H. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014;46:1140–1146. doi: 10.1038/ng.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S., Yang Y., Mei Y., Ma L., Zhu D.E., Hoti N., Castanares M., Wu M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal. 2007;19:2056–2067. doi: 10.1016/j.cellsig.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Fernandes-Alnemri T., Yu J.W., Juliana C., Solorzano L., Kang S., Wu J., Datta P., McCormick M., Huang L., McDermott E. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 2010;11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink S.L., Bergsbaken T., Cookson B.T. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc. Natl. Acad. Sci. USA. 2008;105:4312–4317. doi: 10.1073/pnas.0707370105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandemange S., Sanchez E., Louis-Plence P., Tran Mau-Them F., Bessis D., Coubes C., Frouin E., Seyger M., Girard M., Puechberty J. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis) Ann. Rheum. Dis. 2017;76:1191–1198. doi: 10.1136/annrheumdis-2016-210021. [DOI] [PubMed] [Google Scholar]
- Gringhuis S.I., Kaptein T.M., Wevers B.A., Theelen B., van der Vlist M., Boekhout T., Geijtenbeek T.B. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat. Immunol. 2012;13:246–254. doi: 10.1038/ni.2222. [DOI] [PubMed] [Google Scholar]
- Guey B., Bodnar M., Manié S.N., Tardivel A., Petrilli V. Caspase-1 autoproteolysis is differentially required for NLRP1b and NLRP3 inflammasome function. Proc. Natl. Acad. Sci. USA. 2014;111:17254–17259. doi: 10.1073/pnas.1415756111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung P., Anand P.K., Malireddi R.K., Vande Walle L., Van Opdenbosch N., Dillon C.P., Weinlich R., Green D.R., Lamkanfi M., Kanneganti T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014;192:1835–1846. doi: 10.4049/jimmunol.1302839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W.T., Wan H., Hu L., Chen P., Wang X., Huang Z., Yang Z.H., Zhong C.Q., Han J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I., Miao E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015;265:130–142. doi: 10.1111/imr.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I., Zhang Y., Krantz B.A., Miao E.A. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med. 2016;213:2113–2128. doi: 10.1084/jem.20151613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T.B., Ben-Moshe T., Varfolomeev E.E., Pewzner-Jung Y., Yogev N., Jurewicz A., Waisman A., Brenner O., Haffner R., Gustafsson E. Caspase-8 serves both apoptotic and nonapoptotic roles. J. Immunol. 2004;173:2976–2984. doi: 10.4049/jimmunol.173.5.2976. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y., Oda H., Ito J., Niwa A., Tanaka T., Hijikata A., Seki R., Nagahashi A., Osawa M., Asaka I. Pluripotent cell-based phenotypic dissection identifies a high-frequency somatic NLRC4 mutation as a cause of autoinflammation. Arthritis Rheumatol. 2016;69:447–459. doi: 10.1002/art.39960. [DOI] [PubMed] [Google Scholar]
- Kayagaki N., Stowe I.B., Lee B.L., O’Rourke K., Anderson K., Warming S., Cuellar T., Haley B., Roose-Girma M., Phung Q.T. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- Kitamura A., Sasaki Y., Abe T., Kano H., Yasutomo K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J. Exp. Med. 2014;211:2385–2396. doi: 10.1084/jem.20141091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krautwald S., Ziegler E., Rölver L., Linkermann A., Keyser K.A., Steen P., Wollert K.C., Korf-Klingebiel M., Kunzendorf U. Effective blockage of both the extrinsic and intrinsic pathways of apoptosis in mice by TAT-crmA. J. Biol. Chem. 2010;285:19997–20005. doi: 10.1074/jbc.M110.122127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K., Lippke J.A., Ku G., Harding M.W., Livingston D.J., Su M.S., Flavell R.A. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M., Dixit V.M. Modulation of inflammasome pathways by bacterial and viral pathogens. J. Immunol. 2011;187:597–602. doi: 10.4049/jimmunol.1100229. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M., Dixit V.M. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M., Amer A., Kanneganti T.D., Muñoz-Planillo R., Chen G., Vandenabeele P., Fortier A., Gros P., Núñez G. The Nod-like receptor family member Naip5/Birc1e restricts Legionella pneumophila growth independently of caspase-1 activation. J. Immunol. 2007;178:8022–8027. doi: 10.4049/jimmunol.178.12.8022. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M., Sarkar A., Vande Walle L., Vitari A.C., Amer A.O., Wewers M.D., Tracey K.J., Kanneganti T.D., Dixit V.M. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 2010;185:4385–4392. doi: 10.4049/jimmunol.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightfield K.L., Persson J., Brubaker S.W., Witte C.E., von Moltke J., Dunipace E.A., Henry T., Sun Y.H., Cado D., Dietrich W.F. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 2008;9:1171–1178. doi: 10.1038/ni.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Yamaguchi Y., Shirasaki Y., Shikada K., Yamagishi M., Hoshino K., Kaisho T., Takemoto K., Suzuki T., Kuranaga E. Single-cell imaging of caspase-1 dynamics reveals an all-or-none inflammasome signaling response. Cell Rep. 2014;8:974–982. doi: 10.1016/j.celrep.2014.07.012. [DOI] [PubMed] [Google Scholar]
- Luksch H., Romanowski M.J., Chara O., Tüngler V., Caffarena E.R., Heymann M.C., Lohse P., Aksentijevich I., Remmers E.F., Flecks S. Naturally occurring genetic variants of human caspase-1 differ considerably in structure and the ability to activate interleukin-1β. Hum. Mutat. 2013;34:122–131. doi: 10.1002/humu.22169. [DOI] [PubMed] [Google Scholar]
- Luksch H., Winkler S., Heymann M.C., Schulze F., Hofmann S.R., Roesler J., Rösen-Wolff A. Current knowledge on procaspase-1 variants with reduced or abrogated enzymatic activity in autoinflammatory disease. Curr. Rheumatol. Rep. 2015;17:45. doi: 10.1007/s11926-015-0520-5. [DOI] [PubMed] [Google Scholar]
- Maelfait J., Vercammen E., Janssens S., Schotte P., Haegman M., Magez S., Beyaert R. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J. Exp. Med. 2008;205:1967–1973. doi: 10.1084/jem.20071632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man S.M., Tourlomousis P., Hopkins L., Monie T.P., Fitzgerald K.A., Bryant C.E. Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1β production. J. Immunol. 2013;191:5239–5246. doi: 10.4049/jimmunol.1301581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S., Newton K., Monack D.M., Vucic D., French D.M., Lee W.P., Roose-Girma M., Erickson S., Dixit V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- Martinon F., Holler N., Richard C., Tschopp J. Activation of a pro-apoptotic amplification loop through inhibition of NF-kappaB-dependent survival signals by caspase-mediated inactivation of RIP. FEBS Lett. 2000;468:134–136. doi: 10.1016/s0014-5793(00)01212-6. [DOI] [PubMed] [Google Scholar]
- Mascarenhas D.P.A., Cerqueira D.M., Pereira M.S.F., Castanheira F.V.S., Fernandes T.D., Manin G.Z., Cunha L.D., Zamboni D.S. Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5/NLRC4/ASC inflammasome. PLoS Pathog. 2017;13:e1006502. doi: 10.1371/journal.ppat.1006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O., Thome M., Schneider P., Holler N., Tschopp J., Nicholson D.W., Briand C., Grütter M.G. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J. Biol. Chem. 2002;277:45162–45171. doi: 10.1074/jbc.M206882200. [DOI] [PubMed] [Google Scholar]
- Moriwaki K., Bertin J., Gough P.J., Chan F.K. A RIPK3-caspase 8 complex mediates atypical pro-IL-1β processing. J. Immunol. 2015;194:1938–1944. doi: 10.4049/jimmunol.1402167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K., Dugger D.L., Wickliffe K.E., Kapoor N., de Almagro M.C., Vucic D., Komuves L., Ferrando R.E., French D.M., Webster J. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- Pierini R., Juruj C., Perret M., Jones C.L., Mangeot P., Weiss D.S., Henry T. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 2012;19:1709–1721. doi: 10.1038/cdd.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri A.W., Broz P., Shen A., Monack D.M., Bogyo M. Caspase-1 activity is required to bypass macrophage apoptosis upon Salmonella infection. Nat. Chem. Biol. 2012;8:745–747. doi: 10.1038/nchembio.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch I., Deets K.A., Ji D.X., von Moltke J., Tenthorey J.L., Lee A.Y., Philip N.H., Ayres J.S., Brodsky I.E., Gronert K., Vance R.E. NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and -8. Immunity. 2017;46:649–659. doi: 10.1016/j.immuni.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romberg N., Al Moussawi K., Nelson-Williams C., Stiegler A.L., Loring E., Choi M., Overton J., Meffre E., Khokha M.K., Huttner A.J. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 2014;46:1135–1139. doi: 10.1038/ng.3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagulenko V., Thygesen S.J., Sester D.P., Idris A., Cridland J.A., Vajjhala P.R., Roberts T.L., Schroder K., Vince J.E., Hill J.M. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013;20:1149–1160. doi: 10.1038/cdd.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T., Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. 2013;340:1190–1194. doi: 10.1126/science.1234852. [DOI] [PubMed] [Google Scholar]
- Sellin M.E., Müller A.A., Felmy B., Dolowschiak T., Diard M., Tardivel A., Maslowski K.M., Hardt W.D. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe. 2014;16:237–248. doi: 10.1016/j.chom.2014.07.001. [DOI] [PubMed] [Google Scholar]
- Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H., Zhuang Y., Cai T., Wang F., Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- Squires R.C., Muehlbauer S.M., Brojatsch J. Proteasomes control caspase-1 activation in anthrax lethal toxin-mediated cell killing. J. Biol. Chem. 2007;282:34260–34267. doi: 10.1074/jbc.M705687200. [DOI] [PubMed] [Google Scholar]
- Van Gorp H., Saavedra P.H., de Vasconcelos N.M., Van Opdenbosch N., Vande Walle L., Matusiak M., Prencipe G., Insalaco A., Van Hauwermeiren F., Demon D. Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc. Natl. Acad. Sci. USA. 2016;113:14384–14389. doi: 10.1073/pnas.1613156113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gorp H., Van Opdenbosch N., Lamkanfi M. Inflammasome-Dependent Cytokines at the Crossroads of Health and Autoinflammatory Disease. Cold Spring Harb. Perspect. Biol. 2017:a028563. doi: 10.1101/cshperspect.a028563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Maele L., Fougeron D., Janot L., Didierlaurent A., Cayet D., Tabareau J., Rumbo M., Corvo-Chamaillard S., Boulenouar S., Jeffs S. Airway structural cells regulate TLR5-mediated mucosal adjuvant activity. Mucosal Immunol. 2014;7:489–500. doi: 10.1038/mi.2013.66. [DOI] [PubMed] [Google Scholar]
- Van Opdenbosch N., Gurung P., Vande Walle L., Fossoul A., Kanneganti T.D., Lamkanfi M. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat. Commun. 2014;5:3209. doi: 10.1038/ncomms4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Walle L., Lamkanfi M. Pyroptosis. Curr. Biol. 2016;26:R568–R572. doi: 10.1016/j.cub.2016.02.019. [DOI] [PubMed] [Google Scholar]
- Varfolomeev E.E., Schuchmann M., Luria V., Chiannilkulchai N., Beckmann J.S., Mett I.L., Rebrikov D., Brodianski V.M., Kemper O.C., Kollet O. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–276. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- Vince J.E., Wong W.W., Gentle I., Lawlor K.E., Allam R., O’Reilly L., Mason K., Gross O., Ma S., Guarda G. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36:215–227. doi: 10.1016/j.immuni.2012.01.012. [DOI] [PubMed] [Google Scholar]
- Volker-Touw C.M., de Koning H.D., Giltay J., de Kovel C., van Kempen T.S., Oberndorff K., Boes M., van Steensel M.A., van Well G.T., Blokx W.A. Erythematous nodes, urticarial rash and arthralgias in a large pedigree with NLRC4-related autoinflammatory disease, expansion of the phenotype. Br. J. Dermatol. 2016;176:244–248. doi: 10.1111/bjd.14757. [DOI] [PubMed] [Google Scholar]
- von Moltke J., Trinidad N.J., Moayeri M., Kintzer A.F., Wang S.B., van Rooijen N., Brown C.R., Krantz B.A., Leppla S.H., Gronert K., Vance R.E. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490:107–111. doi: 10.1038/nature11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Yang J., Gao W., Li L., Li P., Zhang L., Gong Y.N., Peng X., Xi J.J., Chen S. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513:237–241. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- Yang J., Zhao Y., Shi J., Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc. Natl. Acad. Sci. USA. 2013;110:14408–14413. doi: 10.1073/pnas.1306376110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong F.L., Mamai O., Sborgi L., Boussofara L., Hopkins R., Robinson K., Szeverenyi I., Takeichi T., Balaji R., Lau A. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell. 2016;167:187–202.e17. doi: 10.1016/j.cell.2016.09.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primary intestinal epithelial organoids from B6Nlrp1b+, B6Nlrp1b+C1−/−C11−/− and B6Nlrp1b+C1−/−C11−/−ASC−/− were stimulated with FlaTox and followed in time for 16 h in the presence of propidium iodide (PI). All data are representative of results from three independent experiments.
Primary intestinal epithelial organoids from B6Nlrp1b+, B6Nlrp1b+C1−/−C11−/− and B6Nlrp1b+C1−/−C11−/−ASC−/− were left unstimulated and followed in time for 16 h in the presence of PI. All data are representative of results from three independent experiments.
Primary intestinal epithelial organoids from B6, NAIP5−/−, NLRC4−/− and C1C284A/C284A were stimulated with FlaTox and followed in time for 16 h in the presence of PI. All data are representative of results from three independent experiments.
Primary intestinal epithelial organoids from B6, NAIP5−/−, NLRC4−/− and C1C284A/C284A were left unstimulated and followed in time for 16 h in the presence of PI. All data are representative of results from three independent experiments.