

Conspectus
Precise control over reactivity and molecular structure is a fundamental goal of the chemical sciences. Billions of years of evolution by natural selection have resulted in chemical systems capable of information storage, self-replication, catalysis, capture and production of light, and even cognition. In all these cases, control over molecular structure is required to achieve a particular function: without structural control, function may be impaired, unpredictable, or impossible.
The search for molecules with a desired function is often achieved by synthesizing a combinatorial library, which contains many or all possible combinations of a set of chemical building blocks (BBs), and then screening this library to identify “successful” structures. The largest libraries made by conventional synthesis are currently of the order of 108 distinct molecules. To put this in context, there are 1013 ways of arranging the 21 proteinogenic amino acids in chains up to 10 units long. Given that we know that a number of these compounds have potent biological activity, it would be highly desirable to be able to search them all to identify leads for new drug molecules. Large libraries of oligonucleotides can be synthesized combinatorially and translated into peptides using systems based on biological replication such as mRNA display, with selected molecules identified by DNA sequencing; but these methods are limited to BBs that are compatible with cellular machinery. In order to search the vast tracts of chemical space beyond nucleic acids and natural peptides, an alternative approach is required.
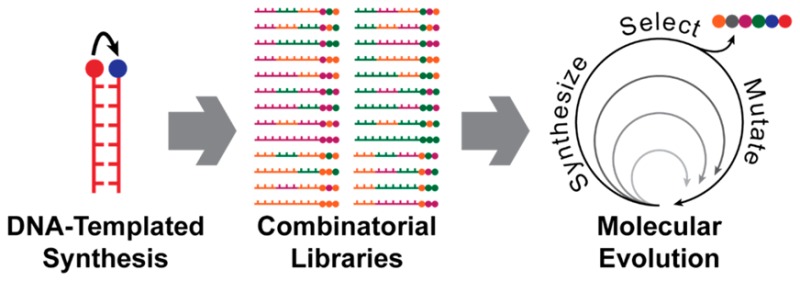
DNA-templated synthesis (DTS) could enable us to meet this challenge. DTS controls chemical product formation by using the specificity of DNA hybridization to bring selected reactants into close proximity, and is capable of the programmed synthesis of many distinct products in the same reaction vessel. By making use of dynamic, programmable DNA processes, it is possible to engineer a system that can translate instructions coded as a sequence of DNA bases into a chemical structure—a process analogous to the action of the ribosome in living organisms but with the potential to create a much more chemically diverse set of products. It is also possible to ensure that each product molecule is tagged with its identifying DNA sequence. Compound libraries synthesized in this way can be exposed to selection against suitable targets, enriching successful molecules. The encoding DNA can then be amplified using the polymerase chain reaction and decoded by DNA sequencing. More importantly, the DNA instruction sequences can be mutated and reused during multiple rounds of amplification, translation, and selection. In other words, DTS could be used as the foundation for a system of synthetic molecular evolution, which could allow us to efficiently search a vast chemical space. This has huge potential to revolutionize materials discovery—imagine being able to evolve molecules for light harvesting, or catalysts for CO2 fixation.
The field of DTS has developed to the point where a wide variety of reactions can be performed on a DNA template. Complex architectures and autonomous “DNA robots” have been implemented for the controlled assembly of BBs, and these mechanisms have in turn enabled the one-pot synthesis of large combinatorial libraries. Indeed, DTS libraries are being exploited by pharmaceutical companies and have already found their way into drug lead discovery programs. This Account explores the processes involved in DTS and highlights the challenges that remain in creating a general system for molecular discovery by evolution.
Introduction
Two centuries of research has furnished chemists with the ability to synthesize a huge variety of molecular architectures based on organic and inorganic components and to create materials with new functions ranging from therapeutics to solar cells. While the majority of new molecules with precisely defined structures are “small” (i.e., <1000 Da), solid-phase synthesis techniques have made it possible to produce monodisperse macromolecules such as DNA, peptides and their analogues,1,2 and advances in sequence-controlled polymerization continue.3 While much work remains to be done, we now have access to a very large chemical space. Searching this space for new molecules capable of meeting challenges in human health, energy, and security is of vital importance. However, even the largest combinatorial libraries are many orders of magnitude too small to search even the most synthetically accessible regions of chemical space effectively.4
A system capable of tackling the above challenge would need to (1) operate in parallel rather than in series, drastically reducing synthesis time; (2) use extremely small amounts of material, in order to bring costs down and render synthesis of very large libraries of compounds practical, while still allowing product selection and identification (typically below the detection limit of common analytical techniques such as mass spectrometry); (3) enable molecular evolution. Evolution is perhaps the most important innovation as it allows a very large chemical space to be sampled without the requirement to synthesize all possible molecules within that space. Sequential rounds of selection, mutation and resynthesis can allow for the identification of functional molecules that were not present in the initial compound library (Figure 1). While criterion (1) may be addressed by improvements in synthetic methods/technology, it is extremely difficult, if not impossible, to envisage how conventional combinatorial synthesis could address points (2) and (3).
Figure 1.

Molecular evolution allows large chemical spaces to be searched efficiently. A library of instructions is translated into the corresponding library of molecular products, which are then selected for target properties (Round 1). The instructions for the enriched products are then amplified, mutated and translated again to generate a library of new products (some or all of which may not have been present in the original product library) which can be selected against to identify products with improved properties (Round 2). Repeated cycles of translation, selection, amplification, and mutation can enable the system to identify on an optimized product (Round N) without the need for every possible library member to be synthesized.
One method that has been developed to allow functional evolution is messenger RNA (mRNA) display5,6 (Figure 2). Here, a combinatorial library of DNA sequences is converted by reverse transcription into the corresponding mRNA library. Each mRNA strand is then modified by ligation of a puromycin-modified DNA strand to its 3′ end and translated into the corresponding peptide by in vitro ribosomal peptide synthesis (RPS). When a ribosome reaches the RNA/DNA junction at the end of a mRNA template it stalls: at this point the terminal puromycin, a peptidyl acceptor antibiotic, can enter the active site causing the peptide product to be transferred to it. The resulting library of peptide products can comprise as many as 1013 unique members, each of which is attached to its encoding mRNA sequence. After subjecting the library to selection, the mRNA attached to successful products can be reverse transcribed to the corresponding DNA sequences and amplified using the polymerase chain reaction (PCR). Mutation can be achieved by cutting members of the DNA library using restriction enzymes and then randomly recombining the fragments, or by error-prone PCR. Multiple rounds of selection, mutation, and amplification allow many more peptide sequences, not present in the original library, to be explored. Eventually, a peptide that is highly optimized for a particular function can be identified.7 mRNA display, the related techniques of ribosome8 and phage9 display, and DNA aptamer libraries10 provide a solution to the selection and evolution problems identified above. However, techniques involving RPS are limited to peptides incorporating proteinogenic amino acids. Expanding the library of BBs to include non-natural amino acids is possible but difficult as it involves engineering the translation machinery of cells—a nontrivial undertaking.11 In order to truly revolutionize the way that we search chemical space, we need a system with the capacity of mRNA display for directed evolution but with fewer constraints on the chemical structures of the products. The purpose of this Account is to chart the development of just such a technology: DNA-Templated Synthesis (DTS).12
Figure 2.

Molecular evolution using mRNA display. aatRNA = aminoacyl tRNA.
The basic principle of DTS is illustrated in Figure 3. Reactive BBs are conjugated to short adapter strands of DNA. At suitably low concentrations (nM), reaction rates between BBs are negligible in the absence of DNA–DNA interactions.13 However, if two of the DNA adapters hybridize to form a duplex their attached BBs are brought into close proximity, greatly increasing their effective local concentration and hence the rate of reaction. This mechanism allows for the selective activation of reactions in the presence of many reactive species in the same mixture–a feat not ordinarily possible in conventional synthetic chemistry. The use of a nucleic acid template to control synthesis has a precedent in RPS (Figure 3c)14 in which peptide bond formation is directed by base-pairing between aminoacyl transfer RNAs (aatRNAs) and an mRNA template. Its ability to direct multiple reactions in parallel means that DTS is capable of addressing criterion (1), a key challenge in combinatorial synthesis.
Figure 3.

“Traditional” synthetic chemistry compared with DTS and RPS. a) The traditional approach requires separate synthesis of each distinct compound. DTS (b) and RPS (c) allow defined products to be synthesized in parallel within complex mixtures by using sequence-specific nucleic acid hybridization to control the proximity of reactive reactive building blocks, BBs.
As Gartner and Liu realized nearly 20 years ago, DTS also has the potential to address the more difficult questions of product identification and molecular evolution.13 The products of DTS are tagged with DNA: it is possible to design ribosome-inspired DTS systems that encode information about the order of addition of BBs in the base sequence of this DNA tag (Figure 4).15 Following selection against a target, DNA amplification and sequencing methods can be applied to “read off” the reaction sequence, from which the chemical structure of the successful product can be inferred. It is important to note that due to the expense associated with synthesizing BB-DNA adapters, it is usually practical to make only very small amounts of product by DTS (usually on the order of picomoles). However, since amplification by PCR requires, in principle, only a single DNA molecule, product detection is still possible even at such small reaction scales (criterion (2)). Finally, molecular evolution could be achieved by iterated cycles of DTS, selection, amplification and mutation13 (for example, by cutting or “restricting” the DNA into fragments then randomly recombining them). Translation is key to this process: the DNA tags attached to selected products must be capable of directing subsequent rounds of product synthesis.
Figure 4.

Principle of product encoding and molecular evolution enabled by DTS. The base sequence of a DNA tag directs the synthesis of a product and defines its chemical structure. Selection against a target followed by amplification, shuffling of the instructions encoded in the DNA tag (restriction and recombination), and then resynthesis by another round of DTS allows the production of new products with improved properties. Molecular evolution is therefore possible.
DTS thus has the potential for development into a tool to search efficiently and quickly a vast chemical space. In this Account, we outline the evolution of DTS toward this goal and the challenges associated with its development.
DNA-Templated Chemistry
A simple example of DTS is the use of a DNA template to facilitate ligation of two DNA strands through a native phosphodiester bond16 or a non-natural linkage,12 as pioneered by the groups of Orgel, Liu, and many others. Numerous examples of bond-forming and bond-breaking reactions directed by DNA templates have been reported (Figure 5),17 including Heck coupling, the copper-catalyzed azide–alkyne cycloaddition “click” reaction, transition metal-mediated catalysis, and synthesis of conductive polymers and macrocyclic drug-like molecules. Thanks to the work of Kool, Seitz, and others, there is a well-developed field of research into DNA/RNA probes based on fluorogenic reactions templated by a target nucleic acid.18
Figure 5.

Examples of reactions that have been performed on a DNA template. For acyl transfer X = S or N-hydroxysuccinimide.17
Three different architectures are commonly used to bring BBs into close proximity (Figure 6).19 In an “end of helix” design, the reactants coupled to each strand are brought together at the end of a double helix. In “cross nick”, reactions take place across a gap between DNA adapters held on a template strand. “Junction”-based designs template reactions in small volumes where multiple DNA strands intersect; an example is the YoctoReactor reported by Hansen and co-workers (see below).20
Figure 6.

DNA architectures employed in DTS.
For programmed, multistep synthesis, perhaps the most useful DTS reactions are transfer reactions in which bond formation is coordinated with cleavage from one of the DNA adapters (Figure 5, blue box). Transfer reactions can facilitate autonomous, multistep DTS as they avoid steric problems caused by the accumulation of DNA adapters. An exemplar from nature is RPS (Figure 3c). Here, amino acid BBs are linked to transfer RNAs (tRNAs) by activated ester bonds. As the ribosome scans from codon to codon along an mRNA, the growing peptide chain is continually passed to the incoming tRNA (selected by the next codon in the mRNA program) by means of an acyl transfer reaction that coordinates peptide bond formation with cleavage from the penultimate tRNA.
Relatively few DTS transfer reactions have been reported to date. The two predominant examples in the literature are acyl transfer and Wittig olefination. Acyl transfer is useful as it enables the creation of peptidomimetic molecules,21 and several research groups have used this reaction to create oligopeptides and for various other applications.17 The limited stability of the activated ester BBs in solution can cause problems, however.22,23 Wittig olefination results in the formation of a carbon–carbon double bond, so allows the exploration of a different region of chemical space. It has been used for DTS of macrocycles24−27 and linear oligomers (see ref (22) for a recent example). However, its broader application is limited by the stability of the phosphoryl BBs, which can be oxidized in water.22 Less commonly used transfer reactions include a modified Staudinger ligation,28 native chemical ligation,29 nucleophilic aromatic substitution30 and a tetrazine-transfer reaction.31 In combination, these reactions could be very useful for the introduction of specific functional groups during DTS. In our opinion, this avenue remains underexplored. However, with the current state of the art, multistep syntheses take around a day to complete, and the best yields per step are around 80%, resulting in rather low overall yields. Investigation of alternative transfer chemistries compatible with DTS conditions should be given high priority as the discovery of a highly efficient and versatile method for DNA-templated oligomer synthesis could make the development of autonomous systems analogous to the ribosome much more straightforward.
Product Encoding
The idea of encoding the identity of a small-molecule product using an attached DNA sequence was first proposed by Brenner and Lerner 25 years ago.15 DNA is an ideal identifying tag because it is straightforward to synthesize large libraries of unique oligonucleotides which can be sequenced to identify products. Its most useful feature, however, is its ability to be amplified by PCR, which has a limit of detection far below conventional analysis methods.
The original proposal was that solid-phase synthesis of a combinatorial library of target molecules (by repeatedly pooling then splitting support beads between different reactions) would proceed in parallel with the construction of DNA tags on each bead to encode the sequence of addition of BBs (Figure 7a). However, DNA serves only as a post hoc record of the reaction steps: it does not program synthesis, and cannot be used to direct the resynthesis of enriched products. As a result, this system is not suitable for the implementation of molecular evolution. An elegant alternative, termed “DNA routing”, was devised by Halpin and Harbury (Figure 7b):32 successive codons in a DNA “gene” are used to route a growing oligomer between reaction vessels, determining the sequence of BB coupling reactions and, therefore, the structure of the final product.
Figure 7.

Comparison of the reaction cycles of a) split and pool synthesis, b) DNA routing, and c) DTS.
Using DNA-encoded chemical libraries (DECLs) for molecular discovery is advantageous because compounds can be selected from a pooled library as opposed to serial screening, enabling a 106-fold increase in library size.32 Selection from DECLs has become a well-established method and has been used by pharmaceutical companies in drug discovery programs.4 In both split-and-pool and DNA-routed syntheses, each reaction occurs in a different reaction vessel without the direct involvement of the DNA tag. These methods are thus distinct from DTS, in which reactions occur in the same pot and are programed by DNA interactions. For this reason, we will not include them in our discussion below, but readers are directed to a recent paper illustrating the potential of DNA routing for molecular evolution.33
DTS has been employed in a number of ways to create products tagged with a unique identifying DNA sequence. These approaches fall into three categories, which we have termed “templated parallel”, “templated sequential” and “autonomous” (Figure 8). In each case, the use of DNA amplification and sequencing to confirm the identity of the DNA-tagged product oligomers has been demonstrated.20,22,34
Figure 8.

Methods for product encoding in DTS. (a) Templated parallel: BBs are arranged in sequence by hybridization of their adapters to the template, concatenated in a single step then cleaved from adapters. (b) Templated sequential: BBs attached to DNA adapters are sequentially transferred to a reactive site on the DNA template in response to an external stimulus; in the case illustrated, spent adapters are displaced by addition of the following adapter. (c) Autonomous systems. Upper: a simplified version of a DNA walker, reported by He and co-workers,35 which steps down a track (driven by ribozyme-catalyzed cleavage of the track anchorages) picking up BBs in a programmed order. Lower: the HCR system, developed in our laboratories,22 which coordinates programmed DNA polymerization with oligomer assembly. Complementary “toehold” domains, whose hybridization controls the reaction sequence, are identified by color.
In the templated parallel approach, BB-DNA adapters are arrayed in sequence by hybridization to a DNA template (Figure 8a). Template domains act as codons, each of which uniquely specifies a single BB. The BBs are then chemically linked to each other and released from the now-redundant adapters. Kleiner and co-workers elegantly demonstrated this idea by connecting BBs to peptide nucleic acid (PNA) adapters36 via cleavable linkers. Upon completion of the synthesis, the product was liberated while remaining tagged at one end with the templating DNA sequence.34 Zhu and co-workers have also applied the templated parallel approach to produce “nylon DNA” using amide condensation reactions.37
The templated sequential approach provides a more flexible but laborious route to oligomer synthesis. As in the templated parallel approach, the DNA template provides an ordered array of binding sites for BB-DNA adapters. However, the assembly of the BBs on the template, and hence the BB transfer reactions, occurs sequentially in this case–generally at the terminus of the template–and is controlled externally by strand-displacement reactions that bring successive reactants into close proximity with the growing oligomer (Figure 8b).38−41 Again, the product remains covalently attached to the DNA template, which can encode its chemical structure.
Finally, autonomous systems use a DNA “program” to control the sequential addition of BBs without the need for external intervention. One example of this approach couples motion of a DNA “walker” with chemical reactions between BBs (Figure 8c, upper scheme).35 The walker, a single strand of DNA, moves along a track consisting of an array of single-stranded anchorages. At each step the walker catalyzes cleavage of the anchorage to which it is bound, thereby initiating a strand-displacement reaction that transfers it to the next anchorage. In a sequence programmed by the track, BBs attached to the anchorages are transferred to the growing oligomer attached to the walker. In principle, the final product could be ligated to a template strand on which the track is built to enable the sequence of BBs to be read off.
We recently reported a second example of autonomous DTS, using a hybridization chain reaction (HCR) to bring BBs into proximity with the growing oligomer in sequence22 (Figure 8c, lower scheme). The DNA components are hairpins formed by partially self-complementary strands. A staggered duplex is formed by HCR between the hairpins, in which the sequence of hairpin addition is controlled by hybridization between complementary “toehold” domains. A set of “instruction” hairpins programs the sequence in which “chemistry” hairpins are incorporated and thus the sequence in which BBs coupled to these hairpins are added. The growing oligomer is carried forward on a strand of DNA that remains at the reactive end of the duplex. Ligation of the “instruction” hairpins creates a DNA-encoded record of the reaction sequence.
Extending the Length of Sequence-Controlled Oligomers
Extending the length of sequence-controlled oligomers that can be synthesized is important for two reasons. First, product diversity increases rapidly with oligomer length. For example, a library of 1 billion trimers requires 1000 distinct BBs, while a comparable library of decamers needs only eight. Second, sequence-controlled macromolecules are a “holy grail” of polymer science as perfect control may allow the discovery of synthetic polymers with well-defined folded conformations, analogous to native proteins, with greatly enhanced properties.
The mechanism and architecture used in DTS determines the maximum number of BBs that can be concatenated. For example, the YoctoReactor restricts the number of reactants that can be colocalized and thus cannot produce products longer than tetramers.20
By using the templated parallel approach, Niu and co-workers were able to synthesize long, precisely defined polymers by templating the concatenation of up to ten BBs that were themselves oligomers (of amino acids or ethylene glycol subunits).34 This method therefore makes it possible to explore the structure–function relationships of artificial polymers similar in length to proteins—this is extremely important for the development of artificial enzymes, for example. However, the system cannot encode variability within the oligomeric BBs, so only a fraction of possible sequences of the subunits could be explored.
Strategies for oligomer synthesis using the templated sequential method can be limited by the lengths of adapter and template that it is practical to synthesize, usually around 200 nucleotides. Alternative approaches in which the adapter length is kept constant usually result in the reactive site at the distal end of the oligomer moving further and further from the new BB as the synthesis proceeds, potentially limiting yield. As a result, hexamers are currently the longest oligomers that have been produced using these methods.40
To prevent the DNA mechanism from imposing limits on oligomer length we have developed a simpler strand-displacement mechanism (Figure 9a).42 Here, the growing oligomer is transferred to the incoming BB which remains attached to its adapter, as in the ribosome. The spent adapter is then removed by strand displacement, making way for hybridization of the next DNA-BB adapter. Adapters are distinguished by the unhybridized toehold domain used to drive their eventual displacement so they can all be designed to be the same length. Using this method we have so far been able to demonstrate the construction of decamers which, with those synthesized using the templated parallel method described above, are currently the longest oligomers constructed by DTS.43 However, control over reaction sequence requires the sequential addition of BBs. A more sophisticated system (Figure 9b) uses the serial addition of instruction strands to control reactions within a vessel containing a mixture of all BBs.44 Neither method lends itself to encoding the identity of the product in the final DNA tag, however.
Figure 9.

Methods for sequential DTS of oligomers based on DNA strand exchange. Oligomer sequence can be determined by controlling the sequence of addition of either (a) BBs or (b) separate instruction strands.
The autonomous systems described above have perhaps the greatest potential for the synthesis of long oligomers by DTS. The DNA walker35 and HCR22 systems have significant potential for optimization and extension, but they have yet to realize sequence-controlled synthesis of products longer than tetramers.
Combinatorial Synthesis by DTS
Gartner and Liu first demonstrated the potential of DTS for combinatorial synthesis by templating 1025 distinct thiol–maleimide additions in a single pot.13 The authors expanded this approach to produce a DNA-templated library of 65 macrocycles,24 and later larger libraries of 13 00026 and 160 000 similar molecules (Figure 10a).45 The library size in these and similar systems is ultimately limited by the number of orthogonal adapter sequences required, as a unique adapter is needed to encode not only the identity of each BB but also each possible position of that BB within the product (as with DNA routing33).
Figure 10.

Methods for combinatorial synthesis using DTS based on (a) linear templates;25 (b) a “universal” template;46 (c) the YoctoReactor;20 and (d) HCR.22
Li and co-workers exploited the non-natural base deoxyinosine, which forms base pairs almost indiscriminately with all four natural bases, in order to simplify the design of adapters.46 This enabled the use of a single “universal” template for the combinatorial synthesis of a model library of 114 688 distinct products, with the identity of the products encoded in DNA regions opposite deoxyinosine tracts (Figure 10b). However, the use of a universal template means that mutation is not possible.
The YoctoReactor has been used to generate libraries comprising more than 107 unique members.47 This method, as well as a related approach described by Cao and co-workers,41 simplifies adapter design by decoupling the DNA domain encoding BB identity from that directly involved in DNA templating (Figure 10c). However, in both cases a BB requires a different adapter for each position in the product. The preparation of all oligomers of length n therefore requires the synthesis of n different DNA-linked versions of the same BB, making this methodology time-consuming and limited in flexibility.
Our work on HCR goes some way toward solving issues related to adapter sequence design and BB interchangeability, since the identity of a BB is encoded in the base sequence of the chemistry hairpin loop and any chemistry hairpin can be added at any point in the sequence (Figure 10d). Using the HCR approach with a branching (nondeterministic) synthesis program we were able to demonstrate the combinatorial synthesis of a library of 12 different products22 and are working toward the synthesis of larger libraries.
Given that the largest combinatorial libraries synthesized by DTS are still at least an order of magnitude smaller than libraries generated by mRNA display,9 reaching libraries of this size remains a key target.
Selection of Functional Products
The principle of functional product selection from DNA-tagged libraries is illustrated in Figure 11. The most widely used method involves the incubation of a library with an immobilized target followed by stringent washing to remove any products that do not bind. The DNA tags of selected products are then amplified by PCR and sequenced: the chemical structures of the successful binders can be inferred from the DNA sequence. This approach has the advantage that it may not be necessary to remove unreacted DNA adapters/templates from the reaction mixture since these will be removed during the washing step, simplifying library synthesis.
Figure 11.
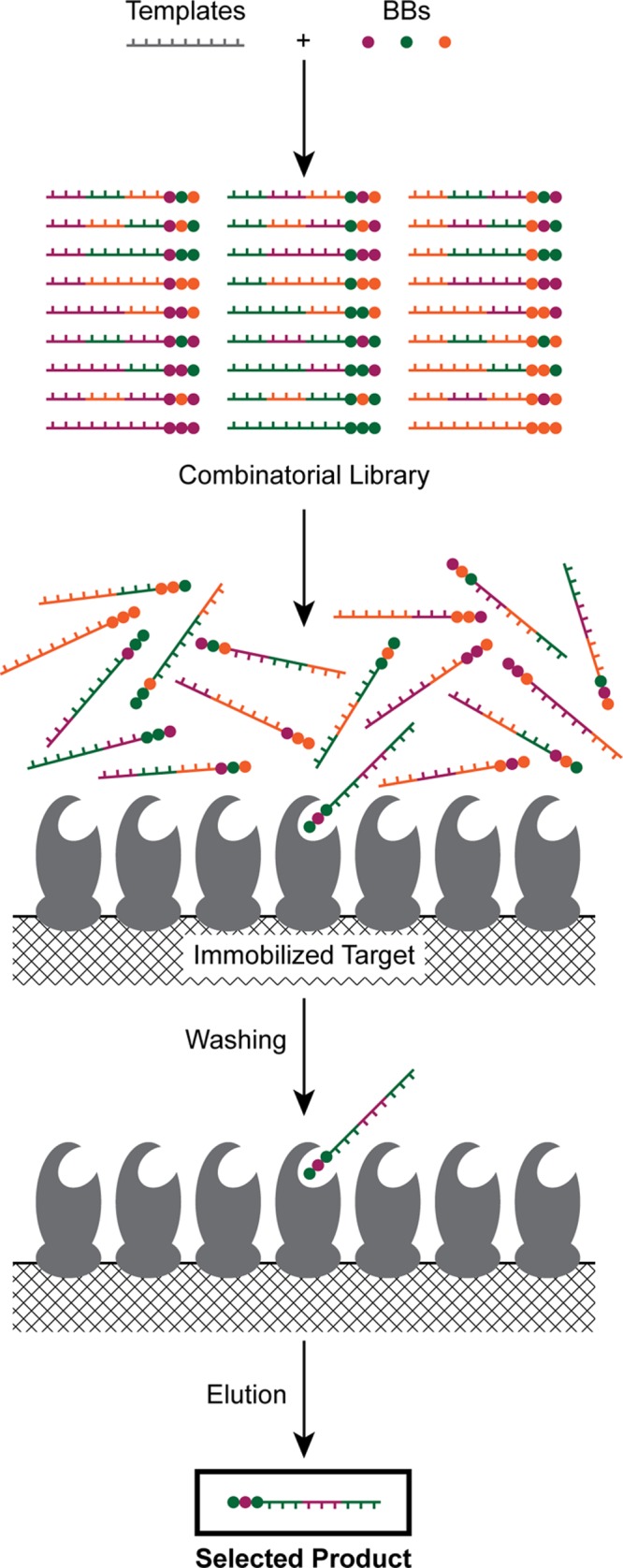
Principle of functional selection against DNA-tagged combinatorial libraries.
Many of the compound libraries produced by DTS have been exposed to selection experiments, resulting in the identification of inhibitors25,27,47 and antagonists45 of several important biological targets including kinases and apoptosis inhibitors. These examples demonstrate the great potential of DTS in drug discovery, but the libraries involved remain limited by their relatively small sizes. To our knowledge there are as yet no therapeutics discovered using DTS libraries that have made it to market, although both Vipergen and Ensemble Therapeutics are working toward this end.
Libraries produced by DTS are constrained by the requirement that product synthesis be water-compatible. In contrast, DECLs produced by solid phase methods encounter no such limitation since BB conjugation, DNA tag extension and hybridization reactions can be carried out in different solvents (Figure 7). Since comparable library sizes are achievable with both, it is perhaps not surprising that the adoption of DTS by the pharmaceutical industry has been slower, in spite of the promise shown by functional selection experiments.4
Conclusions and Future Challenges
Over the past 20 years, DTS has developed enormously. A diverse range of chemical reactions can now be directed by DNA templates. Different template architectures allow the synthesis of oligomeric and macrocyclic products. Mechanisms for autonomous DTS have been developed, and synthesis of large combinatorial libraries containing DNA-tagged molecules for selection against various biological targets is now possible. However, there remains much work to be done to identify water-stable yet reactive BBs, to develop autonomous DTS systems to the point where they can produce large combinatorial product libraries, and to diversify the range of targets against which selection experiments are performed. DTS has developed to meet most of the requirements of a system for molecular evolution—but not all. The capacity for mutation and resynthesis is still missing from all published DTS systems: we believe that this is a priority for those working in the field.
Some of the most exciting possibilities for DTS lie in non-natural materials discovery. The current approach to materials chemistry can largely be characterized by “make one, test one”—a material is made with a particular function in mind, it is tested, and then improvements are proposed based on the outcome. A DTS system capable of evolving molecules to meet challenges such as light harvesting or carbon sequestration would be truly revolutionary: this technology has the potential to usher in a new and exciting era of materials discovery.
Acknowledgments
We thank Annie Morton and Dr. Charlotte Zammit for providing feedback on the manuscript.
Biographies
Rachel O’Reilly was born in Holywood, Northern Ireland. She received her B.A. in Natural Sciences from the University of Cambridge in 1998, M.Sci. in Chemistry in 1999, and Ph.D. from Imperial College London in 2003. Following 2 years postdoctoral work in the United States, she returned to Cambridge as a Royal Society Fellow in 2005. In 2009, she moved to the University of Warwick as an EPSRC Fellow and was promoted to Professor in 2012. Her group undertakes research in the areas of catalysis, responsive polymers, nanostructure characterization, and DNA nanomaterials.
Andrew Turberfield was born in Wantage, U.K. He received his B.A. in Natural Sciences from the University of Cambridge in 1983 and D.Phil. from the University of Oxford in 1988. Since 1986, he has been employed at the University of Oxford, first as a Research Lecturer (Junior Research Fellow) at Christ Church then as a Lecturer and Professorial Research Fellow in the Department of Physics. He has worked on low-dimensional electron systems and photonic materials and now studies the science and applications of biomimetic self-assembly.
Tom Wilks was born in Brighton, U.K. He received his B.A./M.Sci. in Natural Sciences from the University of Cambridge in 2008 and Ph.D. from the University of Warwick in 2013. He was Midlands Director of The Brilliant Club, an educational charity placing researchers in schools to help children from disadvantaged backgrounds aspire to study at top universities. Since 2016, he has been a Research Fellow at the University of Warwick, focusing on applications for DNA- and nucleobase-containing materials.
Author Contributions
All authors wrote and have given approval to the final version of the manuscript.
RKOR thanks the ERC for funding. This research was supported by BBSRC sLoLa grant BB/J00054X/1 and EPSRC grants EP/F056605/1, EP/F008597/1, EP/F055803/1, and EP/F009062/2. A.J.T. was supported by a Royal Society–Wolfson Research Merit Award.
The authors declare no competing financial interest.
References
- Merrifield R. B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. 10.1021/ja00897a025. [DOI] [Google Scholar]
- Letsinger R.; Mahadevan V. Oligonucleotide Synthesis on a Polymer Support. J. Am. Chem. Soc. 1965, 87, 3526–3527. 10.1021/ja01093a058. [DOI] [PubMed] [Google Scholar]
- Lutz J.-F.; Lehn J.-M.; Meijer E. W.; Matyjaszewski K. From Precision Polymers to Complex Materials and Systems. Nat. Rev. Mater. 2016, 1, 16024. 10.1038/natrevmats.2016.24. [DOI] [Google Scholar]
- Goodnow R. A.; Dumelin C. E.; Keefe A. D. DNA-Encoded Chemistry: Enabling the Deeper Sampling of Chemical Space. Nat. Rev. Drug Discovery 2017, 16, 131–147. 10.1038/nrd.2016.213. [DOI] [PubMed] [Google Scholar]
- Roberts R. W.; Szostak J. W. RNA-Peptide Fusions for the in Vitro Selection of Peptides and Proteins. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 12297–12302. 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto N.; Miyamoto-Sato E.; Husimi Y.; Yanagawa H. In Vitro Virus: Bonding of mRNA Bearing Puromycin at the 3′-terminal End to the C-Terminal End of Its Encoded Protein on the Ribosome in Vitro. FEBS Lett. 1997, 414, 405–408. 10.1016/S0014-5793(97)01026-0. [DOI] [PubMed] [Google Scholar]
- Seelig B.; Szostak J. W. Selection and Evolution of Enzymes from a Partially Randomized Non-Catalytic Scaffold. Nature 2007, 448, 828–831. 10.1038/nature06032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanes J.; Plückthun A. In Vitro Selection and Evolution of Functional Proteins by Using Ribosome Display. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 4937–4942. 10.1073/pnas.94.10.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G. P.; Petrenko V. A. Phage Display. Chem. Rev. 1997, 97, 391–410. 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- Mairal T.; Cengiz Özalp V.; Lozano Sánchez P.; Mir M.; Katakis I.; O’Sullivan C. K. Aptamers: Molecular Tools for Analytical Applications. Anal. Bioanal. Chem. 2008, 390, 989–1007. 10.1007/s00216-007-1346-4. [DOI] [PubMed] [Google Scholar]
- Chin J. W. Expanding and Reprogramming the Genetic Code of Cells and Animals. Annu. Rev. Biochem. 2014, 83, 379–408. 10.1146/annurev-biochem-060713-035737. [DOI] [PubMed] [Google Scholar]
- Li X.; Liu D. R. DNA-Templated Organic Synthesis: Nature’s Strategy for Controlling Chemical Reactivity Applied to Synthetic Molecules. Angew. Chem., Int. Ed. 2004, 43, 4848–4870. 10.1002/anie.200400656. [DOI] [PubMed] [Google Scholar]
- Gartner Z. J.; Liu D. R. The Generality of DNA-Templated Synthesis as a Basis for Evolving Non-Natural Small Molecules. J. Am. Chem. Soc. 2001, 123, 6961–6963. 10.1021/ja015873n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen P.; Hansen J.; Ban N.; Moore P. B.; Steitz T. A. The Structural Basis of Ribosome Activity in Peptide Bond Synthesis. Science 2000, 289, 920–930. 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- Brenner S.; Lerner R. A. Encoded Combinatorial Chemistry. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 5381–5383. 10.1073/pnas.89.12.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgel L. E. Unnatural Selection in Chemical Systems. Acc. Chem. Res. 1995, 28, 109–118. 10.1021/ar00051a004. [DOI] [PubMed] [Google Scholar]
- Gorska K.; Winssinger N. Reactions Templated by Nucleic Acids: More Ways to Translate Oligonucleotide-Based Instructions into Emerging Function. Angew. Chem., Int. Ed. 2013, 52, 6820–6843. 10.1002/anie.201208460. [DOI] [PubMed] [Google Scholar]
- Silverman A. P.; Kool E. T. Detecting RNA and DNA with Templated Chemical Reactions. Chem. Rev. 2006, 106, 3775–3789. 10.1021/cr050057+. [DOI] [PubMed] [Google Scholar]
- Gartner Z. J.; Grubina R.; Calderone C. T.; Liu D. R. Two Enabling Architectures for DNA-Templated Organic Synthesis. Angew. Chem., Int. Ed. 2003, 42, 1370–1375. 10.1002/anie.200390351. [DOI] [PubMed] [Google Scholar]
- Hansen M. H.; Blakskjær P.; Petersen L. K.; Hansen T. H.; Højfeldt J. W.; Gothelf K. V.; Hansen N. J. V. A Yoctoliter-Scale DNA Reactor for Small-Molecule Evolution. J. Am. Chem. Soc. 2009, 131, 1322–1327. 10.1021/ja808558a. [DOI] [PubMed] [Google Scholar]
- Fosgerau K.; Hoffmann T. Peptide Therapeutics: Current Status and Future Directions. Drug Discovery Today 2015, 20, 122–128. 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Meng W.; Muscat R. A.; McKee M. L.; Milnes P. J.; El-Sagheer A. H.; Bath J.; Davis B. G.; Brown T.; O’Reilly R. K.; Turberfield A. J. An Autonomous Molecular Assembler for Programmable Chemical Synthesis. Nat. Chem. 2016, 8, 542–548. 10.1038/nchem.2495. [DOI] [PubMed] [Google Scholar]
- McKee M. L.; Evans A. C.; Gerrard S. R.; O’Reilly R. K.; Turberfield A. J.; Stulz E. Peptidomimetic Bond Formation by DNA-Templated Acyl Transfer. Org. Biomol. Chem. 2011, 9, 1661–1666. 10.1039/c0ob00753f. [DOI] [PubMed] [Google Scholar]
- Gartner Z. J.; Tse B. N.; Grubina R.; Doyon J. B.; Snyder T. M.; Liu D. R. DNA-Templated Organic Synthesis and Selection of a Library of Macrocycles. Science 2004, 305, 1601–1605. 10.1126/science.1102629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner R. E.; Dumelin C. E.; Tiu G. C.; Sakurai K.; Liu D. R. In Vitro Selection of a DNA-Templated Small-Molecule Library Reveals a Class of Macrocyclic Kinase Inhibitors. J. Am. Chem. Soc. 2010, 132, 11779–11791. 10.1021/ja104903x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse B. N.; Snyder T. M.; Shen Y.; Liu D. R. Translation of DNA into a Library of 13 000 Synthetic Small-Molecule Macrocycles Suitable for in Vitro Selection. J. Am. Chem. Soc. 2008, 130, 15611–15626. 10.1021/ja805649f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georghiou G.; Kleiner R. E.; Pulkoski-Gross M.; Liu D. R.; Seeliger M. A. Highly Specific, Bisubstrate-Competitive Src Inhibitors from DNA-Templated Macrocycles. Nat. Chem. Biol. 2012, 8, 366–374. 10.1038/nchembio.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J.; Li X.; Yue X.; Taylor J. S. Nucleic Acid-Triggered Fluorescent Probe Activation by the Staudinger Reaction. J. Am. Chem. Soc. 2004, 126, 16324–16325. 10.1021/ja0452626. [DOI] [PubMed] [Google Scholar]
- Grossmann T. N.; Seitz O. DNA-Catalyzed Transfer of a Reporter Group. J. Am. Chem. Soc. 2006, 128, 15596–15597. 10.1021/ja0670097. [DOI] [PubMed] [Google Scholar]
- Shibata A.; Uzawa T.; Nakashima Y.; Ito M.; Nakano Y.; Shuto S.; Ito Y.; Abe H. Very Rapid DNA-Templated Reaction for Efficient Signal Amplification and Its Steady-State Kinetic Analysis of the Turnover Cycle. J. Am. Chem. Soc. 2013, 135, 14172–14178. 10.1021/ja404743m. [DOI] [PubMed] [Google Scholar]
- Wu H.; Cisneros B. T.; Cole C. M.; Devaraj N. K. Bioorthogonal Tetrazine-Mediated Transfer Reactions Facilitate Reaction Turnover in Nucleic Acid-Templated Detection of MicroRNA. J. Am. Chem. Soc. 2014, 136, 17942–17945. 10.1021/ja510839r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin D. R.; Harbury P. B. DNA Display II. Genetic Manipulation of Combinatorial Chemistry Libraries for Small-Molecule Evolution. PLoS Biol. 2004, 2, e174. 10.1371/journal.pbio.0020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krusemark C. J.; Tilmans N. P.; Brown P. O.; Harbury P. B. Directed Chemical Evolution with an Outsized Genetic Code. PLoS One 2016, 11, e0154765. 10.1371/journal.pone.0154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J.; Hili R.; Liu D. R. Enzyme-Free Translation of DNA into Sequence-Defined Synthetic Polymers Structurally Unrelated to Nucleic Acids. Nat. Chem. 2013, 5, 282–292. 10.1038/nchem.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Liu D. R. Autonomous Multistep Organic Synthesis in a Single Isothermal Solution Mediated by a DNA Walker. Nat. Nanotechnol. 2010, 5, 778–782. 10.1038/nnano.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner R. E.; Brudno Y.; Birnbaum M. E.; Liu D. R. DNA-Templated Polymerization of Side-Chain-Functionalized Peptide Nucleic Acid Aldehydes. J. Am. Chem. Soc. 2008, 130, 4646–4659. 10.1021/ja0753997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.; Lukeman P. S.; Canary J. W.; Seeman N. C. Nylon/DNA: Single-Stranded DNA with a Covalently Stitched Nylon Lining. J. Am. Chem. Soc. 2003, 125, 10178–10179. 10.1021/ja035186r. [DOI] [PubMed] [Google Scholar]
- Gartner Z. J.; Kanan M. W.; Liu D. R. Multistep Small-Molecule Synthesis Programmed by DNA Templates. J. Am. Chem. Soc. 2002, 124, 10304–10306. 10.1021/ja027307d. [DOI] [PubMed] [Google Scholar]
- Snyder T. M.; Liu D. R. Ordered Multistep Synthesis in a Single Solution Directed by DNA Templates. Angew. Chem., Int. Ed. 2005, 44, 7379–7382. 10.1002/anie.200502879. [DOI] [PubMed] [Google Scholar]
- He Y.; Liu D. R. A Sequential Strand-Displacement Strategy Enables Efficient Six-Step DNA-Templated Synthesis. J. Am. Chem. Soc. 2011, 133, 9972–9975. 10.1021/ja201361t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C.; Zhao P.; Li Z.; Chen Z.; Huang Y.; Bai Y.; Li X. A DNA-Templated Synthesis of Encoded Small Molecules by DNA Self-Assembly. Chem. Commun. 2014, 50, 10997–10999. 10.1039/C4CC03380A. [DOI] [PubMed] [Google Scholar]
- McKee M. L.; Milnes P. J.; Bath J.; Stulz E.; Turberfield A. J.; O’Reilly R. K. Multistep DNA-Templated Reactions for the Synthesis of Functional Sequence Controlled Oligomers. Angew. Chem., Int. Ed. 2010, 49, 7948–7951. 10.1002/anie.201002721. [DOI] [PubMed] [Google Scholar]
- Milnes P. J.; McKee M. L.; Bath J.; Song L.; Stulz E.; Turberfield A. J.; O’Reilly R. K. Sequence-Specific Synthesis of Macromolecules Using DNA-Templated Chemistry. Chem. Commun. 2012, 48, 5614. 10.1039/c2cc31975f. [DOI] [PubMed] [Google Scholar]
- McKee M. L.; Milnes P. J.; Bath J.; Stulz E.; O’Reilly R. K.; Turberfield A. J. Programmable One-Pot Multistep Organic Synthesis Using DNA Junctions. J. Am. Chem. Soc. 2012, 134, 1446–1449. 10.1021/ja2101196. [DOI] [PubMed] [Google Scholar]
- Seigal B. A.; Connors W. H.; Fraley A.; Borzilleri R. M.; Carter P. H.; Emanuel S. L.; Fargnoli J.; Kim K.; Lei M.; Naglich J. G.; Pokross M. E.; Posy S. L.; Shen H.; Surti N.; Talbott R.; Zhang Y.; Terrett N. K. The Discovery of Macrocyclic XIAP Antagonists from a DNA-Programmed Chemistry Library, and Their Optimization To Give Lead Compounds with in Vivo Antitumor Activity. J. Med. Chem. 2015, 58, 2855–2861. 10.1021/jm501892g. [DOI] [PubMed] [Google Scholar]
- Li Y.; Zhao P.; Zhang M.; Zhao X.; Li X. Multistep DNA-Templated Synthesis Using a Universal Template. J. Am. Chem. Soc. 2013, 135, 17727–17730. 10.1021/ja409936r. [DOI] [PubMed] [Google Scholar]
- Petersen L. K.; Blakskjær P.; Chaikuad A.; Christensen A. B.; Dietvorst J.; Holmkvist J.; Knapp S.; Kořínek M.; Larsen L. K.; Pedersen A. E.; Röhm S.; Sløk F. A.; Hansen N. J. V. Novel p38α MAP Kinase Inhibitors Identified from yoctoReactor DNA-Encoded Small Molecule Library. MedChemComm 2016, 7, 1332–1339. 10.1039/C6MD00241B. [DOI] [Google Scholar]