

Abstract
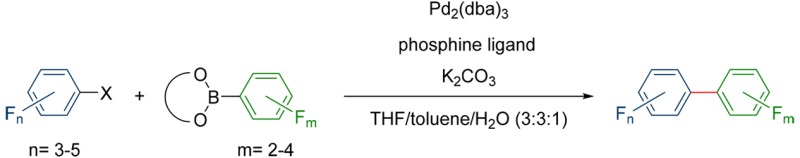
Polyfluorinated biphenyls are interesting and promising substrates for many different applications. Unfortunately, all current methods for the syntheses of these compounds only work for a hand full of molecules or only in very special cases. Thus, many of these compounds are still inaccessible to date. Here we report a general strategy for the synthesis of a wide range of highly fluorinated biphenyls. In our studies we investigated crucial parameters, such as different phosphine ligands and the influence of various nucleophiles and electrophiles with different degrees of fluorination. These results extend the scope of the already very versatile Suzuki–Miyaura reaction toward the synthesis of very electron-poor products, making these more readily accessible. The presented methodology is scalable and versatile without the need for elaborate phosphine ligands or Pd-precatalysts.
Introduction
Fluorinated biphenyls and their derivatives are interesting building blocks for many different applications because of their electron-poor nature, intrinsic rigidity, and chemical stability. One example is the development of liquid crystalline materials, which are important for liquid crystal displays (LCDs).1−3 Perfluorinated biphenyls have also been considered for the development of organic light emitting diodes (OLEDs)4 and organic semiconductors,5 making use of their electronic properties. Particularly, but not exclusively, the rigidity of these compounds is interesting for applications in crystal engineering,6,7 metal–organic frameworks (MOFs),8 polymers of intrinsic microporosity (PIMs),9,10 organic polymers of intrinsic microporosity (OMIMs),11 and supramolecular chemistry.12 In our group different polyfluorinated polyphenyl compounds have served as scaffold for halogen bond donors to build up structures for molecular recognition13 and halogen bond mediated catalysis.14 In these studies, the fluorine substituents play an important role since they ensure strong halogen bonds due to the electron withdrawing nature of the fluorine atoms.
The synthesis of highly fluorinated polyphenyl compounds is challenging since there are only few methods to synthesize these structures. The standard way is to synthesize them by nucleophilic substitution.15,16 However, this method has the major drawback that it is only applicable to symmetric compounds, or systems involving strong electron withdrawing substituents, such as CF3 or nitro groups that serve as directing group.17 Additionally, acidic protons can cause problems due to the requirement of basic metalorganic reagents, such as lithium or zinc organyls. Also the formation of arynes following M-F elimination has to be taken into consideration in highly fluorinated aromatic systems. Both issues can result in the formation of complex product mixtures and need to be addressed in the design of the syntheses.
Besides nucleophilic substitution, different metal mediated reactions have been used for the synthesis of fluorinated biaryl compounds. One approach by T. Suzuki and co-workers was to use Ullmann couplings during their synthesis of perfluorinated oligo(p-phenylene)s.18 The most prominent synthetic methods for building up biaryl structures are without doubt the Negishi,19 Suzuki–Miyaura,20,21 and Stille coupling.22 Concerning the latter, examples of couplings containing fluorinated aryl compounds are very rare and most examples refer to substrates which are only fluorinated on the electrophilic side.23−28 To our knowledge only one example is known in which two aryl sp2 carbons are coupled to each other.29 Nevertheless the Stille coupling with fluorinated stannane derivatives is also known.30 In the case of Negishi couplings also only few examples are known and the fluorinated products only contain one or two fluorine atoms.31−33 In contrast, more examples can be found for the Suzuki–Miyaura coupling. The first goes back to M. Hird and co-workers in 1999, in which they synthesized fluorinated terphenyls as part of liquid crystals. These contained up to four fluorine atoms, distributed to both, the electrophilic and nucleophilic side of the cross-coupling reaction.34 And although this is the first known example in that respect, it is until date one of only a few where both coupling partners are fluorinated. Much more research has been conducted on compounds where only one side of the biaryl is fluorinated. For example, Langer and co-workers have shown that selective monoarylation of fluorinated dibromobenzenes is possible.35−37 Also, M. Schlosser and C. Heiss have shown that polyfluorinated brominated compounds are suitable electrophiles in the Suzuki–Miyaura coupling.38 For the coupling of mono-, di-, and trifluorinated boronic acids to nonfluorinated electrophiles, different palladium catalysts have been found to be active.34,39−46 Much less examples are found with higher fluorinated compounds. For instance, in the case of tetrafluorophenylboronic acids, there are only two known examples. In one case 2,3,4,5-tetrafluoroboronic acid is coupled to 2-bromopyridine43 and in the other one the corresponding neopentylester is coupled three times to 1,3,5-trifluoro-2,4,6-triiodobenzene.47 Pentafluoroboronic acid is a very special case due to its inactivity under most Suzuki-Miyaura coupling conditions. It can only be coupled in the presence of CsF and Ag2O.48 The main problem of these fluorinated phenylboronic acids is the rapid deboronation under basic conditions which can be circumvented by elaborate precatalysts such as second generation palladacycles developed by Buchwald and co-workers.49 As an alternative to polyfluorophenylboronic acids, lithium polyfluorophenyltrimethoxyborates50 and potassium polyfluorophenyltrifluoroborates51−53 were proven to be effective coupling partners in cross coupling reactions and therefore were also able to introduce C6F5-groups into polyaryl compounds. Since highly fluorinated compounds are easier targets for hydrodefluorination compared to less fluorinated compounds, this has been exploited in C–F activation.54,55 The high acidity of the corresponding hydrogens in these compounds can be used for C–H activation56−59 and in combination with arenediazonium salts this was used for copper-catalyzed cross-coupling.60 During the preparation of this article, Carrow and co-workers published a study that used arenediazonium salts and fluorinated boronic acids via a cationic transmetalation pathway in a Suzuki–Miyaura coupling.61 Very recently Pd-nanoparticals62 and photocatalysis63,64 have been applied to the synthesis of polyfluorinated biphenyls. Especially the latter is very useful for the introduction of perfluoro phenyl moieties. However, these methods seem to be restricted to electron-rich counterparts or in the case of the Pd-nanoparticles to only a low degree of fluorination.
To summarize the state of the art, to date no general method for the synthesis of highly fluorinated biaryls has been published (Scheme 1). While former investigations have found ways to introduce one fluorinated phenyl ring into a biaryl or similar system, there has not been much progress in the synthesis of fluorinated biaryls with fluorine atoms on both phenyl rings. This is particularly true for biaryl systems containing more than four fluorine atoms. In our previous studies, it was already shown for these isolated cases that it is possible to synthesize polyfluorinated terphenyls14 and quaterphenyl13 with very high fluorine content and we were interested if these results can also be applied to the synthesis of biphenyl systems. In that respect, our motivation was not only to find a general way to synthesize polyfluorinated biphenyls and making these compounds better accessible, or depending on the structure, accessible at all, but also to find a methodology which is reliable, reproducible, scalable, easy to handle, and needs no elaborate and expensive reagents or precatalysts. Likewise, our emphasis was also to avoid any side products, which in general results in easy workups. Consequently, the Suzuki–Miyaura coupling was selected for our studies, because of the mild reaction conditions, the easy preparation and general stability of the boronic acid derivatives, and the low toxicity of the involved boron compounds and their byproducts.65 In addition the Suzuki–Miyaura coupling exhibits a wide functional group tolerance which is important for the transferability of the method to different systems.
Scheme 1. Synthesis of Polyfluorinated Biphenyls via the Suzuki–Miyaura Cross Coupling.

In this article the synthesis of various polyfluorinated biphenyls is presented which are in most cases unknown in literature. Reaction conditions were optimized and a set of catalyst-ligand systems was developed which performed the desired cross coupling reactions and avoided side reactions. Our goal was to introduce a synthetic toolkit to various electron poor bi- and polyphenyl compounds which has applicability beyond the presented results.
Results and Discussion
In the first part of this article we will have a closer look on the palladium catalyzed reaction between iodinated compound 1 and boronic acid 2 (Scheme 2) and explain how different reaction parameters influence this reaction. In the second part these results are extended toward the coupling of various polyfluorinated iodine and boronic acid compounds. Further we investigate the application of different classes of organoboron compounds to circumvent different side reaction and improve the methodology further. Finally, we have a look at the electrophile and discuss the boundaries of the presented methodology.
Scheme 2. Initial Benchmark Reaction.

Benchmark Reaction Reaction between 2,3,4,5-Tetrafluoro-1-iodobenzene and 3,4,5-Trifluorophenyl-boronic Acid
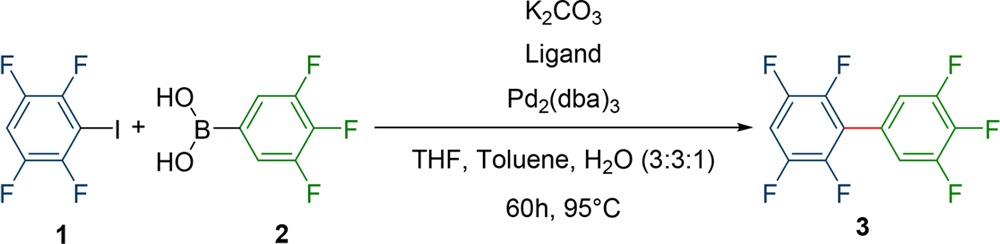
Since the aim was to find a general way to synthesize polyfluorinated aromatic polyphenyls, we were interested in the single coupling between two reaction partners to build up a biaryl system. Compared to previous research in which polyfluorinated ter- and quaterphenyl compounds were synthesized, we encountered the problem that the formed side products were not separable from the product by using standard purification methods. Even more elaborate techniques, such as HPLC, did not give satisfying results and are far from practicable on a larger scale. Therefore, conditions had to be found in which all side reactions can be avoided and both coupling partners can be used in a 1:1 ratio. This should make this methodology more appealing for more complex structures, since the synthesis of the coupling partners can be a multistep synthesis, too. Also, using one coupling partner in excess has the potential of promoting the formation of side products.
Our initial experiment was to couple tetrafluoroiodobenzene 1 with trifluoroboronic acid 2 which should result in the biphenyl 3. This reaction was chosen since the product 3 was easily distinguishable from other side products, foremost homocoupling, by GC/MS due to their mass difference and by 19F-NMR because of the symmetry in the side products. As palladium precatalyst Pd2(dba)3 with XPhos (Figure 1) as ligand and Na2CO3 as base was selected (Scheme 2). This was done in a THF/toluene/H2O mixture (3:3:1) at 95 °C for 16 h.66 These conditions were analogous to the conditions for the synthesis of our previously published terphenyls. However, the yields were much lower (36% for 3 compared to 90% for the terphenyl), although only a single coupling had to be performed compared to two couplings in the terphenyl case. The bigger issue, however, was the occurring homocoupling of the boronic acid to biphenyl 4. This compound was not separable from the product by any common purification technique, due to its similar physical properties and primarily polarity. Especially the apolar character makes normal phase column chromatography impossible.
Figure 1.

Ligands used in the screening of the benchmark reaction.
In a first step toward the optimization, the influence of the base with three different phosphine ligands, e.g., XPhos, SPhos, and XantPhos (Figure 1) were investigated. As bases Na2CO3, K2CO3, and Cs2CO3 were chosen. While the use of SPhos (up to 60%) and XPhos (up to 44%) resulted in product formation (Table 1), with XantPhos no conversion occurred.
Table 1. Initial Screening of the Reaction Conditions for the Benchmark Reaction.
| entrya | ligand | base | time | Pd/ligand ratiob | homocouplingd | yield |
|---|---|---|---|---|---|---|
| 1 | SPhos | Na2CO3 | 14 h | 1:2.5 | 5% | 36% |
| 2 | SPhos | K2CO3 | 14 h | 1:2.5 | 2% | 60% |
| 3 | SPhos | Cs2CO3 | 14 h | 1:2.5 | 3% | 39% |
| 4 | XPhos | Na2CO3 | 14 h | 1:2.5 | 2% | 44% |
| 5 | XPhos | K2CO3 | 14 h | 1:2.5 | 35% | |
| 6 | XPhos | Cs2CO3 | 14 h | 1:2.5 | 1% | 44% |
| 7c | XPhos | K2CO3 | 60 h | 1:2.5 | 85% | |
| 8c | SPhos | K2CO3 | 60 h | 1:2.5 | 2% | 90% |
| 9 | XPhos | K2CO3 | 60 h | 1:1.5 | 99% | |
| 10 | SPhos | K2CO3 | 60 h | 1:1.5 | 89% |
Reactions (except entry 10 and 11) were performed on a 0.18 mmol scale with 1 equiv of 1 and 2 and 2.2 equiv of the base at 95 °C.
As Pd source 5% Pd2(dba)3 was used resulting in 10% catalyst load.
These reactions were performed on a 1.8 mmol scale with 1 equiv of 1 and 2 and 2.2 equiv of the base and 3% Pd2(dba)3.
Homocoupling was determined by GC or 19F-NMR.
Although only different carbonate bases were tested (Table 1), the impact of the cation was meaningful. In the cases of XPhos, as well as SPhos, the lowest amount of the homocoupling side product 4 was found with K2CO3 as base (entries 2 and 5). And even if the differences are small, they still are significant and important to obtain pure product. In the case of XPhos with K2CO3 no side products were found. In contrast to K2CO3, the reaction with Na2CO3 exhibited the highest side product formation, although still very low with only 2%. Cs2CO3 was found to be in between Na2CO3 and K2CO3. Concerning product formation, K2CO3 was better in the case of SPhos (60%) compared to XPhos (35%). Na2CO3 and Cs2CO3 were in terms of yield very similar in both the XPhos (44 vs 44%) and the SPhos (36 vs 39%) case. Therefore, K2CO3 was identified as the best in our selection, because it had a positive influence on reducing the homocoupling. This is in accordance with Amatore and Jutand who have shown that the palladium-catalyzed Suzuki–Miyaura reaction can be modulated by the countercation and that K+ as countercation has the least rate decelerating effect in the transmetalation reaction for carbonate bases.67 Stronger bases were not investigated, since fluorinated phenylboronic acids are more likely to undergo hydrodeboronation under more basic conditions even at room temperature.68 As alternatives fluoride ions could be of interest, which was shown by Amatore and Jutand, but were not tested.69 Buchwald and co-workers have shown that different 3-fold fluorinated boronic acids deboronate in THF with K3PO4 as base within 10 min, stressing the point of an efficient catalytic system for these kind of reactions.
What also should be taken into consideration is the influence of the solvent system, which also plays a significant role, especially since a biphasic system was chosen. Toluene for example might lower the solubility of iodine salts in the organic phase which could be important for the acceleration of the reaction. The role of THF potentially could be that it stabilizes the electron-poor boronic acid and therefore slows down the deboronation. Also important for reducing the homocoupling was a long prestirring of the Pd-precatalyst with the ligand before adding the reactants and the exclusion of air to avoid oxidative homocoupling.70−72
In further experiments the catalyst load was reduced to 3 mol % Pd2(dba)3 instead of 5% (Table 1, entries 4 and 5). In the case of SPhos 1 was completely converted after 14 h, while with XPhos 1 was still present (determined by GC/MS). This suggest that the reaction with XPhos is slower and therefore the reaction time was extended to 60 h. This led to improved yields with both SPhos (90%) and XPhos (85%). The longer reaction time is not too surprising since XPhos is much more sterically hindered.73 Based on these results the mole fraction of Pd2(dba)3 was increased to 5 mol % which equals 10 mol % catalyst load. This high catalyst load is justified by the mentioned instability of the fluorinated phenylboronic acids under basic conditions. Another improvement was made by shifting the ligand/precatalyst ratio. In the first experiments a 1:2.5 ratio was used as suggested by standard Suzuki–Miyaura procedures.74 However, a 1:1.5 metal to ligand ratio resulted in higher yields and less formation of the homocoupling product (Table 1, entries 5 and 6). This has already been reported for various palladium catalyzed cross-coupling reactions75,76 and has led to the development of different generations of palladacyclic precatalysts.77−79
Benchmark Reaction Ligand Screening
In the beginning, the ligand screening focused on different Buchwald ligands (Figure 1) since XPhos and SPhos showed promising results and because Buchwald ligands, with their bulky groups and therefore large cone angles,80−82 are beneficial for the reductive elimination step.83−85 However, the screening was also extended to other monodentate and bidentate ligands. Every tested ligand induced product formation, even though very different types of phosphines (Figure 1) were used (Table 2, entry 1–17). The yield however varied widely from very low (14%) to 99%. This clearly demonstrates the key role of the respective ligand. More importantly, the choice of ligand is also crucial for the prevention of side product formation.
Table 2. Ligand Screening for Benchmark Reaction.
| scale | homocouplingb | |||||
|---|---|---|---|---|---|---|
| entrya | ligand | –PR22 | (mmol) | 1 (I) | 2 (B) | yield |
| 1 | SPhos | Cy | 0.18 | 89% | ||
| 2 | XPhos | Cy | 0.18 | 99% | ||
| 3 | DavePhos | Cy | 0.18 | 70% | ||
| 4 | MePhos | Cy | 0.18 | 82% | ||
| 5 | RuPhos | Cy | 0.18 | 80% | ||
| 6 | CyJohnPhos | Cy | 0.18 | 98% | ||
| 7 | JohnPhos | t-Bu | 0.18 | 5% | 8% | 38% |
| 8 | PhDavePhos | Ph | 0.18 | 3% | 87% | |
| 9 | tBuDavePhos | t-Bu | 0.18 | 3% | 23% | 31% |
| 10 | Me4tBuXphos | t-Bu | 0.18 | 20% | 11% | 14% |
| 11 | tBuXPhos | t-Bu | 0.18 | 24% | 39% | |
| 12 | sSPhos | Cy | 0.18 | 6% | 88% | |
| 13 | tBuMePhos | t-Bu | 0.18 | 4% | 17% | 48% |
| 14 | rac-BINAP | Ph | 0.18 | 6% | 51% | |
| 15 | P(pCF3Ph)3 | pCF3Ph | 0.18 | 1% | 3% | 95% |
| 16 | dppbz | Ph | 0.18 | 33% | 29% | |
| 17 | dppe | Ph | 0.18 | 11% | 74% | |
| 18 | PCy2Ph | Cy | 0.18 | 83% | ||
| 19 | PCy3 | Cy | 0.18 | 11% | 78% | |
| 20 | CyJohnPhos | Cy | 32 | 95% | ||
| 21 | CyJohnPhos | Cy | 32 | 99% | ||
Reactions were performed with 1 equiv. 1 and 2 and 2.2 equiv of Na2CO3 with 5 mol % Pd2(dba)3 and a 15 mol % of the corresponding ligand at 95 °C for 60 h on a 0.18 mmol scale.
Homocoupling was determined by GC or 19F-NMR (given as percent of product mixture).
As a general trend, it was found that ligands containing PCy2 are less likely to form side products. Six different Buchwald-ligands, containing this motif, formed no or only traces of side products (Table 2, entry 1–6). The only exception was sSPhos which is difficult to compare to the other ligands due to the sulfonic acid group resulting in a better water solubility. In the biphasic solvent system, this effectively reduces the catalyst concentration in the organic phase, which could favor side product formation. However, the yield of product (88%) is still comparable to the original SPhos (89%). The tested ligands which contained either PPh2 or P(t-Bu)2 groups generally showed side product formation, though varying over a large range from 3% in the case of PhDavePhos to 33% in the case of dppbz. To some extent the yield correlates negatively to the side product formation. Minor exceptions are rac-BINAP and JohnPhos which exhibit very low yields (51 and 38%) although comparably little side product formation (6 and 13%) was observed. This also applies to DavePhos which exhibits a yield of 70% even though no side product is formed with that ligand. The low conversion is probably due to a low turnover of the catalyst. Hence, deboronation of the boronic acid can take place and reduces the yield. This could of course be solved by adding more boronic acid or adding it slowly over time. However, this would be less practicable and can potentially even enable side product formation. Still DavePhos is a valuable ligand for the coupling of this type of compounds since the avoidance of side product formation is of utter importance due to the separation issue mentioned before. On the other hand, there are also ligands which perform very well in terms of yield, e.g., P(pCF3Ph)3, PhDavePhos and sSPhos (95, 87, and 88%), but are still less suitable because of side product formation (4, 3, and 6%). Looking at the formed side products, predominantly the boronic acid is homocoupled. In only few instances homocoupling of the iodine compound occurred and only with Me4tBuXphos it surpassed the homocoupling of the boronic acid.
An important question which should be answered is whether the side product formation originates from the steric or electronic properties of the ligands used. In two comparison experiments PCy2Ph or PCy3 were used as ligands (Table 2, entries 18 and 19). Both ligands gave good yields (83 and 78%), but only with PCy2Ph no side products were formed. This indicates that the steric influence of the biphenyl residue in the Buchwald-ligands is not the decisive part in terms of side product formation. However, it still could be an important factor in the acceleration of the reaction, since some of the Buchwald-ligands generated higher yields. Since PCy3 also created side products, it can be concluded that the combination of two cyclohexanes and one phenyl ring is very favorable for the selective product formation. Whether this originates from the steric influence of the cyclohexene rings or their electronic properties cannot be answered. Considering that the very sterically demanding XPhos and the simpler CyJohnPhos result in a very similar outcome of nearly quantitative yield, it seems that the electronic properties play a more substantial role. In our final experiments, we were interested in the scalability and reproducibility of our benchmark reaction under optimized conditions. Therefore, CyJohnPhos was tested on a 32 mmol scale, which is roughly a scale up of 18000%. Although XPhos performs slightly better during the screening, CyJohnPhos was chosen for the scale up simply out of economic reasons, since CyJohnPhos is cheaper per mol compared to XPhos. In two identical experiments 95 and 99% yield were obtained, respectively. This shows that this reaction is scalable and reproducible with high yields, and still no side products were formed.
Screening of Boronic Acid Derivatives as Nucleophiles
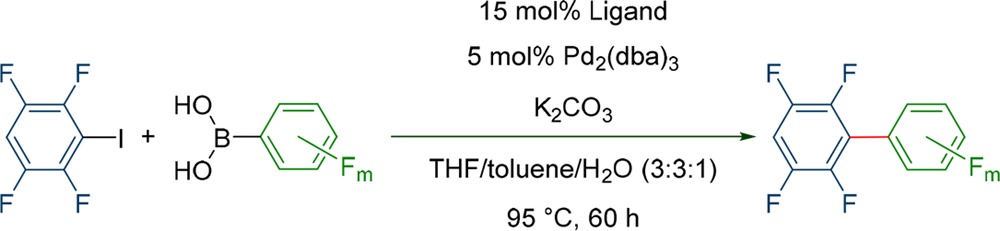
In the next step, the investigation was extended toward other boronic acid derivatives. To this end, different fluorinated boronic acids were chosen which comprise from two to five fluorine atoms with different substitution patterns. As electrophile 1 was used for all experiments to get a better understanding on the influence of the boronic acid, while still obtaining very electron poor biphenyls.
At first, only CyJohnPhos was tested for the coupling of different boronic acids as it was found to be a very efficient ligand in the benchmark reaction (Table 3). However, although CyJohnPhos lead to virtually quantitative product formation with boron acid 2 (98%), it did not perform well in all reactions. While the coupling of 1 and 5 still yielded 88% of the corresponding biphenyl, the coupling with boronic acids 6–8 only yielded 31 to 40% (Table 3). The coupling with 9 did not even form any product at all. Especially interesting was the coupling with the boronic acid 6 and 7, because the formation of the corresponding homocoupling compounds was observed.
Table 3. Coupling of Different Fluorinated Boronic Acids to 2,3,5,6-Tetrafluoroiodo-benzene.
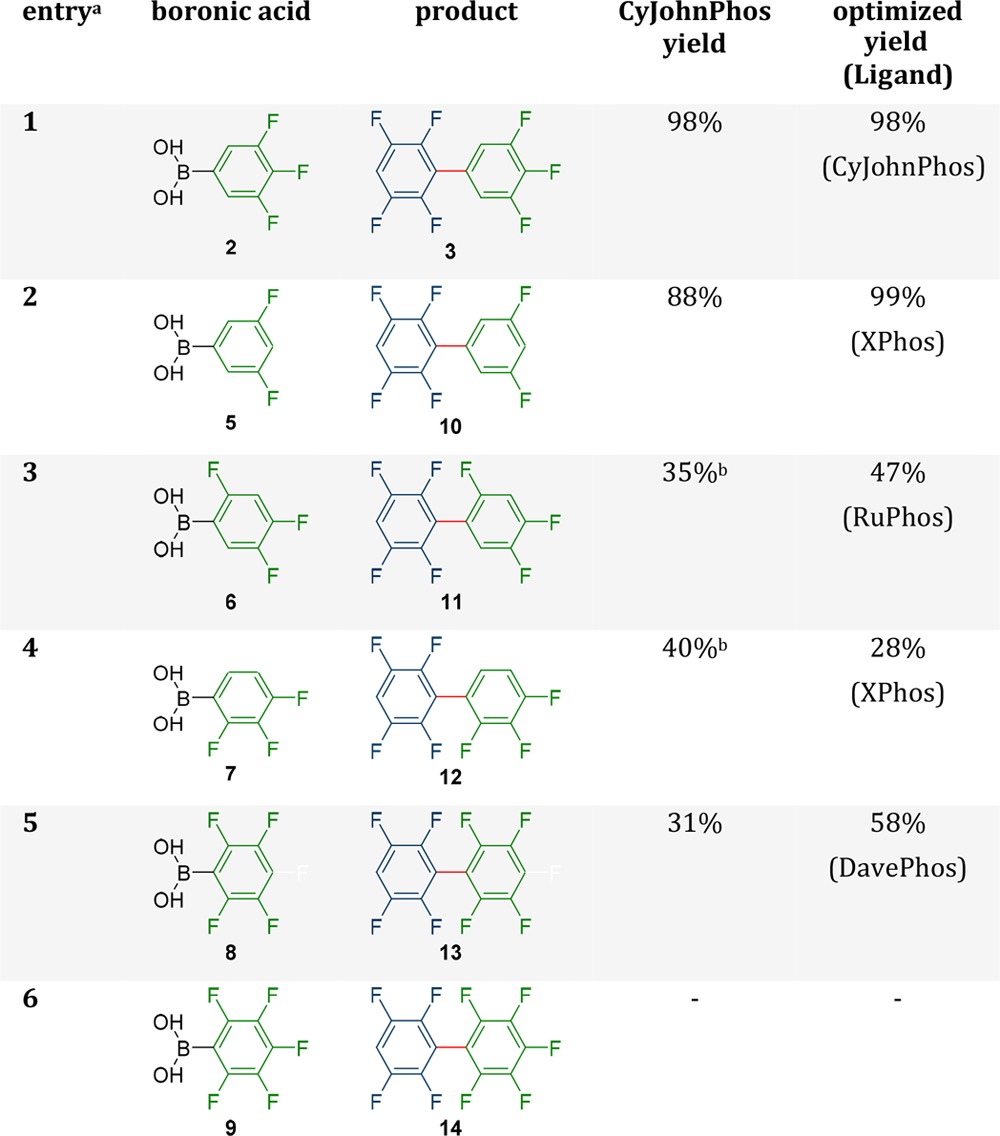
Reactions were performed with 1 equiv. 1 and the corresponding boronic acid and 2.2 equiv of Na2CO3 with 5 mol % Pd2(dba)3 and a 15 mol % of the corresponding ligand at 95 °C for 60 h on a 0.18 mmol scale.
Contains significant amounts of a homocoupling side product (>2%).
For the explanation of these results two considerations must be taken into account: On the one hand, the hydrodeboronation rate and on the other hand, the transmetalation rate of the boronic acids. Both are influenced by the electron withdrawing fluorine atoms, which means higher hydrodeboronation and higher transmetalation rate with increasing fluorine substitution. This is even more challenging if the fluorine atoms are present in ortho position to the boronic acid. The latter results in a faster transmetalation reaction, which was investigated by Buchwald and co-workers.49 They found that compared to phenylboronic acid, even one fluorine atom in ortho position can potentially increase the relative transmetalation rate 42-fold. This effect is less distinct in the meta (2-fold) and para (5-fold) position, but this can add. Opposing to that the competing hydrodeboronation is also accelerated by electron withdrawing fluorine atoms. Frohn and co-workers reported for 2 that it does not hydrodeboronate in the presence of 9% pyridine in water, even at 100 °C.68 Yet, if a fluorine atom in the ortho position is added (2,3,4,5-tetrafluorophenylboronic acid), this results in a deboronation within 50 min and in the case of 8 it only takes 60 min at 32 °C. Even very low fluorinated derivatives, such as 2,4-difluorophenylboronic acid, decompose over time (53% in 19 h). This is of course most be predominant in the case of 9, which decomposes within 2–5 min at 25 °C. This explains why it was not possible to couple 9.94
Since CyJohnPhos did not perform well in all reactions, the screening was extended toward other Buchwald ligands which contain the PCy2 group due to their low side product formation in the benchmark reaction. The chosen ligands were MePhos, RuPhos, XPhos, SPhos, and DavePhos. In most cases the screening improved the yields significantly and more important, ligands were found which exhibit no side product formation (Table 3). All obtained screening data can be found in the Supporting Information (Table S1).
With 6 the coupling exhibited much lower yields, but still, the screening of the ligands improved the yield significantly from 35% with CyJohnPhos to 48% with SPhos. More importantly, the screening of the ligands solved the homocoupling issues. In the case of RuPhos (Table 3, entry 3) and XPhos no homocoupling was found and similar yields were obtained with 47 and 45%. The homocoupling products which were found in the other cases were due to the homocoupling of compound 1 resulting in product 13. This suggests that the hydrodeboronation is very fast resulting in the homocoupling of the electrophile instead of the nucleophile.
The importance of the ligand choice can also be seen in the reaction of 7 and 8 with 1. With 7 as electrophile the yield could be increased from 40 to 86%. However, the obtained product 12 was not pure and 8% of the homocoupled biphenyl (electrophile) was found. This still could be viable, if the yield of the product is more important than the purity, but if purer product is needed, CyJohnPhos can be used to obtain 40% product without any side product formation. For the synthesis of 13 with boronic acid 8 the yields varied from no yield at all with MePhos to 58% with DavePhos (Table 3, entry 5). In the case of SPhos and RuPhos unknown side product were formed which could not be identified. In the case of DavePhos pure product was obtained and the yield with 58% was still very good considering the coupling of a boronic acid with two fluorine atoms in ortho position. Since it is not possible to differentiate between homocoupling and cross coupling, a comparison experiment was done without 1 as electrophile. In this control experiment only 19% of the homocoupled product was obtained which suggests that mainly cross coupling was responsible for the product formation. Interestingly, MePhos did not provide any product even though with CyJohnPhos 31% was obtained, which is noteworthy considering the structural resemblance of both ligands. However, these results were reproducible. In the case of boronic acid 9 the ligand screening did not lead to product 14.
Influence of Boronic Acid Protecting Groups on the Coupling of Electron Poor Biphenyls
Since it can be assumed that one main issue is the already discussed instability of the boronic acid derivatives, the screening was extended toward different organoboron electrophiles, namely neopentyl glycol boronic esters86 and MIDA(N-methyliminodiacetic acid)-boronates.87 Alternative boron nucleophiles are known to be more stable under most Suzuki–Miyaura conditions than the corresponding boronic acids. However, it is not known how the actual reaction mechanism proceeds and whether they react either directly as nucleophile or via the boronate pathway.88 Since our solvent system contains large quantities of water, our working hypothesis was that the protected boronic acid is hydrolyzed over time, generating the free boronic acid which afterward reacts as commonly known.89
Initially both the neopentyl glycol boronic ester 15 and the MIDA-boronate 16 of boronic acid 7 were tested since this boronic acid was especially susceptible toward homocoupling90 (Table 4, entries 1 and 2). In all cases tested with both protecting groups, no homocoupling was found and the yields were significantly better compared to the coupling of the free boronic acid. The best yields were obtained with 15 as nucleophile and DavePhos as ligand (84%). Although even more stable, the corresponding MIDA-derivative did not improve the reaction further (80% yield with RuPhos or CyJohnPhos). Complete screening data can be found in the Supporting Information (Table S2).
Table 4. Optimized Results after Ligand Screening for the Coupling of Different Fluorinated Protected Boronic Acids to 2,3,5,6-Tetrafluoroiodo-benzene.
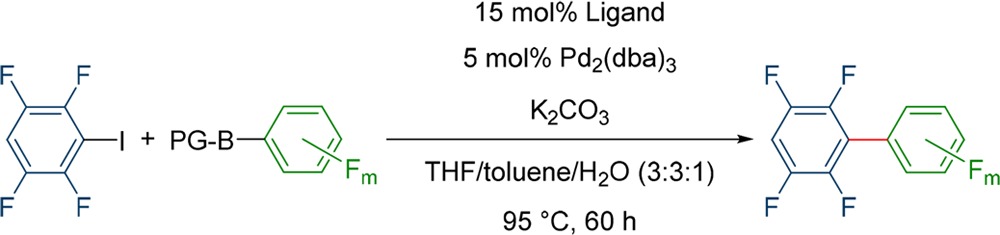
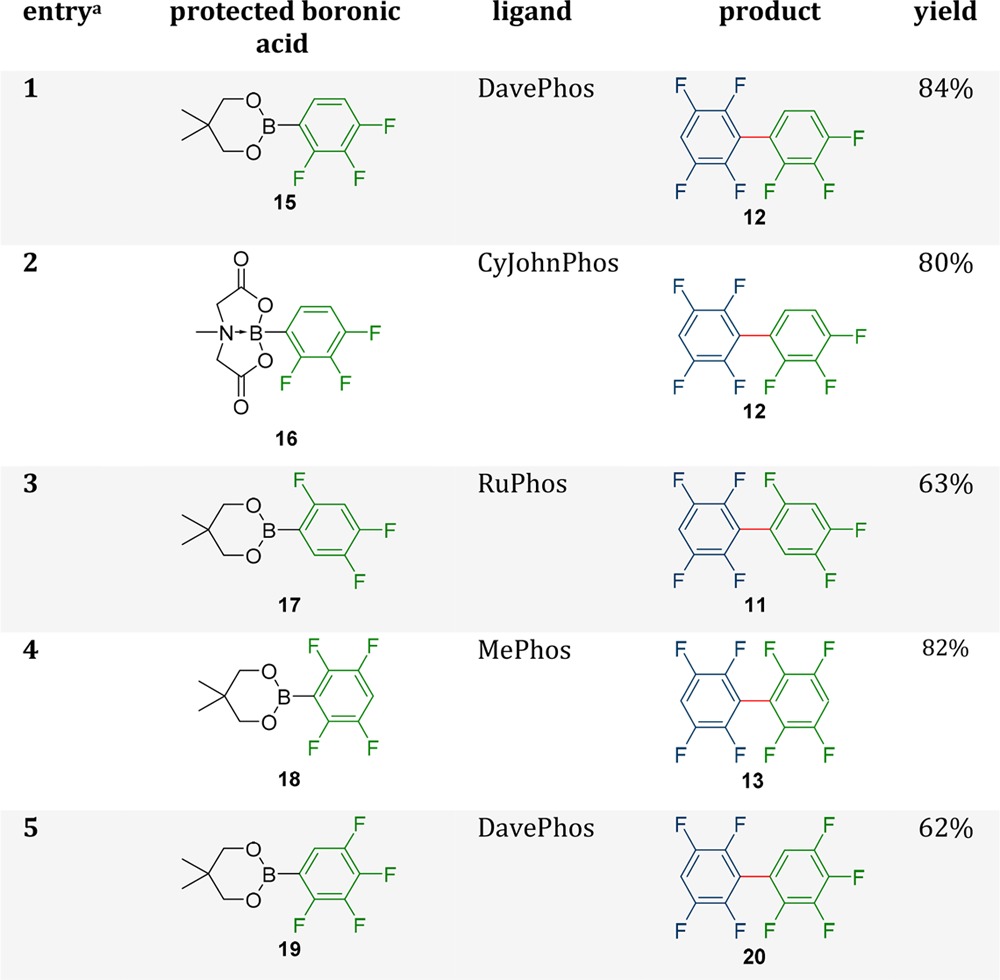
Reactions were performed with 1 equiv. 1 and the corresponding boronic acid and 2.2 equiv of Na2CO3 with 5 mol % Pd2(dba)3 and a 15 mol % of the corresponding ligand at 95 °C for 60 h on a 0.18 mmol scale.
These results encouraged us to extend this strategy to other boronic acids (Table 4). Since the neopentyl glycol boronic esters and the MIDA-boronate were very similar in their outcome, but the neopentyl glycol boronic esters are easier to prepare, we chose them for our further investigations. However, the MIDA-boronates could still be valuable for similar couplings. In the case of boronic acid 6, problems occurred with the homocoupling of 1 with four out of the six ligands. The use of the boronic acid ester did not solve this problem for these ligands, but still, the actual amount of the homocoupling decreased significantly. For example, in the case of CyJohnPhos the homocoupling was reduced from 30 to 7% while the yield increased from 35 to 78%. The yield also increased for MePhos, DavePhos, and RuPhos, while with XPhos and SPhos around 20% lower yields were observed. A similar observation was already made in the case of product 12, where the performances of XPhos and SPhos (51–61%) were weaker compared to the other ligands (68–84%) (see Supporting Information, Table S2, entries 1–12). For the synthesis of 11 with RuPhos no side product formation and the yield went up from 27 to 63%. This advocates that increasing the stability of the boronic acid is a useful strategy.
The synthesis of biaryl 13 was also improved significantly by the use of the neopentyl glycol boronic ester (Table 4, entry 4). While the best yield with boronic acid 8 was 58% with DavePhos, the yield was increased to 82% with 18 as nucleophile and MePhos as ligand. CyJohnPhos, DavePhos, and RuPhos showed much lower yields with 10–39%. In the cases of XPhos and SPhos in the presence of the ester no yield was observed. In general, it was observed that XPhos and SPhos, if used in combination with a boronic ester, are the worst performing ligands within our selection. In most cases they perform significantly worse with the esters as with the free boronic acids. This result is very interesting since it means that the protected boronic acid could also be directly involved in the transmetalation and not necessarily only the boronic acid. Therefore, very bulky ligands like XPhos and SPhos could slow down the transmetalation in combination with more sterically demanding protecting groups. All screening results can be found in the Supporting Information (Table S2 and S3).
Since the use of the neopentyl boronic ester has been proven to be a valuable strategy for the cross coupling of electron-poor biphenyls, it was interesting to see if this strategy can be applied to synthesize an even more challenging biphenyl. As coupling partners, 19 and 1 were chosen. From previous experiments in our studies with ter- and quaterphenyl compounds we already knew that the corresponding boronic acid is too instable for the coupling and even the ester had to be used in large excess for reasonable yields.13 However, DavePhos produced good yields (62%) even in a 1:1 stoichiometry. All other ligands performed much weaker with only 10–24%, again emphasizing how important the ligand screening is.
Extending the Screening toward Different Electrophiles
In our last experiments, we wanted to have a look how our obtained results can be applied to other electrophiles. Since our method failed to couple boronic acid 9, it was examined if pentafluoroiodobenzene can be used as electrophile to introduce a C6F5 group and whether there is an upper limit in terms of the fluorination grade. The ligands were chosen according to our previous experiments based on the boronic acid or ester that was used, with the hypothesis that the interplay between the boronic acid and the ligand is the decisive factor. This approach should be a viable starting point for all future couplings. (Table 5, entries 1–10). Additionally, it was of interest to find out if it is possible to substitute the iodine in the electrophiles with bromine (Table 5, entries 11–18) or chlorine (Table 5, entry 3), since the bromine compound is often the precursor for the iodine compound and brominated compounds are more often commercially available. In the end we also extended our scope to different highly fluorinated pyridine compounds to include heterocycles.
Table 5. Influence of the Electrophile in Different Suzuki–Miyaura Couplings for the Synthesis of Highly Fluorinated Biphenyls.
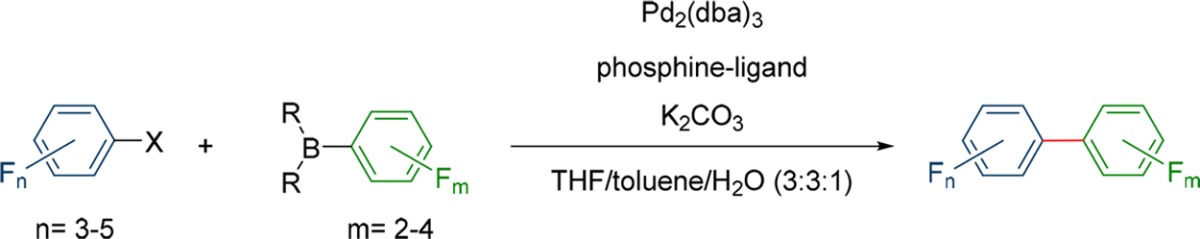
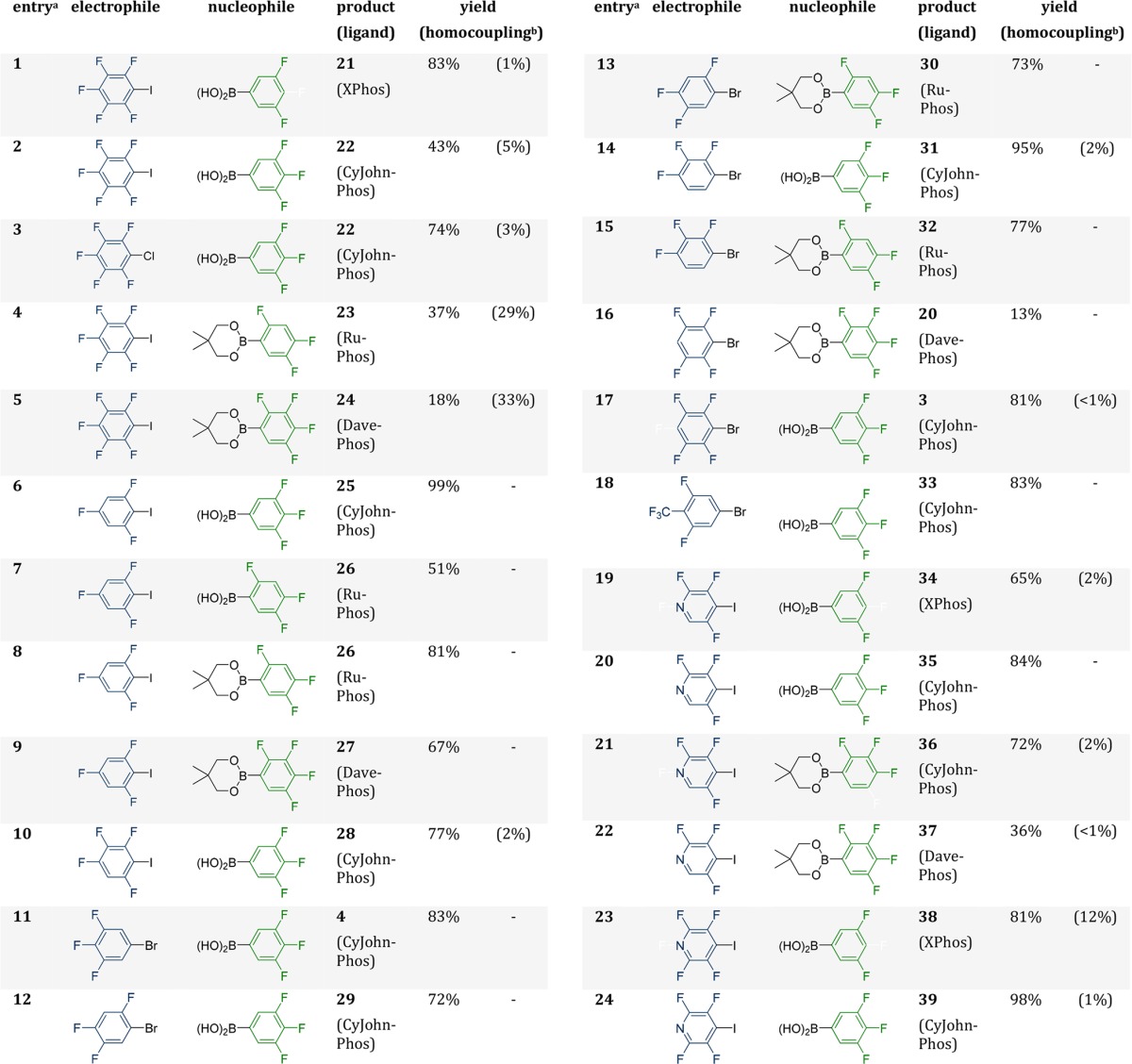
Reactions were performed with 1 equiv of the electrophile and the nucleophile and 2.2 equiv of Na2CO3 with 5 mol % Pd2(dba)3 and a 15 mol % of the corresponding ligand at 95 °C for 60 h.
Homocoupling determined by GC and 19F-NMR (percentage in the product mixture).
In most experiments moderate to excellent yields were obtained with only minor side product formation (<5%). In general, the method performs well in the synthesis of biphenyls with six or seven fluorine atoms, the side product formation is very minor (<2%) and the yields are very good (>51%). In these cases, it also seems that bromine derivatives can be used without major disadvantages in terms of yield and side product formation (Table 5, entries 11–18). However, iodine derivatives are still viable as can be seen in entry 16 of Table 5 where the coupling with the corresponding bromine compound is not nearly as good as the corresponding iodine derivative (62% vs 13%) which corresponds with the instability of the used boronic acid. It was also possible to introduce the C6F5 moiety into the biphenyl compounds (Table 5, entry 1, 5, and 6), but the yields decrease with the instability of the boronic acid derivative and the amount of homocoupling increases. With more stable boronic acids (2) even chlorinated substrates can be used for the coupling. With nine fluorine atoms in total (Table 5, entry 6) the obtained yield was only 18% with 33% of the mixture being the homocoupling product. Still, this is the first time a coupling of a biphenyl with nine fluorine atoms is reported under Suzuki–Miyaura conditions. In addition we extended our scope toward polyfluorinated pyridines and obtained between 36 and 98% yield, depending on the substitution pattern (Table 5, entries 19–24). Only in one case homocoupling was observed (12%).
Conclusion
The main goal of this research was to find a general and reliable way to synthesize various polyfluorinated biphenyls. By vigorous testing of different phosphines and reaction conditions we found a set of different ligands which were not only capable of synthesizing our target molecules, but also of producing them without any major side products, resulting in pure product after a simple work up. For every fluorinated boronic acid or ester a ligand was found which can couple it to various electrophiles. Especially the ligands containing an Aryl-PCy2 group were very suitable for these couplings and we showed how different boronic acid protecting groups can improve yields and reduce side product formation. We also found that a viable starting point for every coupling is to choose the ligand that is best for the respective boronic acid or ester. This should facilitate the synthesis of various unknown and more functionalized polyfluorinated biphenyls.
In summary, a wide range of different polyfluorinated biphenyls, many of them unknown in literature, have been synthesized by using Suzuki–Miyaura cross-coupling. By extending the accessibility of various polyfluorinated biphenyls they can now be used as building blocks for various applications in material science and supramolecular chemistry.
Experimental Section
General Methods
All reactions except the boresterfications were performed under an atmosphere of argon by using standard Schlenk techniques. 1H, 11B, 19F, and 13C–NMR spectra were recorded on either Bruker Avance III 300, Avance III 400 DPX 250, or DRX 400 spectrometers at 22 °C. All shift values are in ppm and all coupling constants (J) are printed in Hertz (Hz) with their multiplicity: s (singlet), bs (broad singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublet). GC/MS spectra were measured on a Hewlett-Packard 5972 GC/MS System equipped with a Phenomenex Zebron ZB-5HT Inferno (25 m) column. Mass spectra of GC/MS unsuitable molecules were measured on a Jeol AccuTOF GCv with electron ionization (EI) as ionization technique. Infrared spectra were obtained on a Shimadzu FTIR-8400s spectrometer equipped with a Specac Quest ATR through attenuated total reflection (ATR). Elemental analysis were performed on a vario MICRO cube from Elementar Analysensysteme GmbH.
Materials
For all cross-coupling reactions solvents were deoxygenated with the freeze–pump–thaw technique. Anhydrous tetrahydrofuran (THF) and diethyl ether (DEE) were obtained as technical grade, distilled prior use, dried, and purified with an MBraun MB-SPS-800 solvent purification system and stored over molecular sieves (3 Å). The THF and toluene for the cross-coupling reactions were purchased as p. a. grade from Fisher Chemical and used without further purification. Double distilled H2O (ddH2O) was used for all cross-coupling reactions. All other solvents were obtained as technical grade and used after simple distillation. Deuterated solvents for NMR were purchased from Euriso-top or Deutero and used as received. Boronic acids 5 [(3,5-difluorophenyl)boronic acid]91 and 8 [(2,3,5,6-tetrafluorophenyl)boronic acid]92 were synthesized according to literature procedures. The boronic acid ester (20) was synthesized as previously described by our group.13 All other chemicals were obtained from either Sigma-Aldrich, Carbolution, Fluorochem, Alfa-Aeser, ACROS, ABCR, or TCI Europe and used without prior purification.
General Procedure for Suzuki–Miyaura coupling
Into an argon-purged 10 mL Schlenk-tube deoxygenated THF (1 mL) and toluene (1 mL) was filled and 9 μmol Pd2(dba)3 (8,3 mg, 5 mol %) and 30 μmol of the corresponding phosphine ligand (15 mol %) were dissolved. The reaction mixture was stirred at 50 °C for 2 h to prepare the active catasslyst. This is indicated through a color change from deep red to yellow, orange or green depending on the used phosphine ligand. Afterward 0.33 mL deoxygenated ddH2O and 0.36 mmol K2CO3 (50.1 mg, 200 mol %) was added, resulting in a biphasic reaction mixture. Then the corresponding polyfluorinated phenylboronic acid or ester (0.18 mmol) and the polyfluoro- iodobenzene or bromobenzene (0.18 mmol) were added. The flask was sealed with a glass stopper and heated to 95 °C for 60 h. After cooling to room temperature, the aqueous phase was removed with a Pasteur pipet and the organic solvents were removed in vacuo. The residue is resuspended with 5 mL pentane and silica is added to the mixture. Afterward the solvents are again removed in vacuo. This should be done with care due to the volatile nature of the corresponding products. The residue is filtered through a plug of silica with pentane as eluent. Pentane was removed in vacuo and the corresponding polyfluorinated biphenyl was obtained as white solid. For all tested ligands please refer to the Supporting Information. In the following only the best ligands in each case are listed.
2,3,3′,4′,5,5′,6-Heptafluoro-1,1′-biphenyl (3)
Following the general procedure with 1,2,4,5-tetrafluoro-3-iodobenzene (1), (3,4,5-trifluorophenyl)boronic acid (2), and XPhos as phosphine ligand. The desired product was obtained as white solid (50,2 mg, 99%). Alternative procedure: Following the general procedure with 3-bromo-1,2,4,5-tetrafluorobenzene and CyJohnPhos as phosphine ligand. The reaction was done on a 0.44 mmol scale. The desired product was obtained as white solid (99 mg, 81%). 1H NMR: (250 MHz, chloroform-d) δ 7.15–6.93 (m). 13C{1H}NMR: (101 MHz, chloroform-d) δ 151.4 (ddd, J = 251.0, 10.1, 4.2 Hz), 146.4 (dddd, J = 249.2, 14.7, 10.3, 4.1 Hz), 143.7 (ddt, J = 248.7, 14.3, 4.0 Hz), 140.4 (dt, J = 255.5, 15.1 Hz), 123.8–122.7 (m), 118.6 (t, J = 15.8 Hz), 115.4–114.3 (m), 106.2 (t, J = 22.6 Hz). 19F{1H}NMR: (235 MHz, chloroform-d) δ −133.34 (d, J = 20.5 Hz), −137.41 – −138.40 (m), −142.83 – −144.21 (m), −158.26 (t, J = 20.5 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc’d for C12H3F7 [M]+: 280.0; found 279.9. EA: calc’d C, 51.45; H, 1.08; N, 0.00 found C, 51.51; H; 0.998; N, 0.00. Rf: 0.9 (pentane).
2,3,3′,5,5′,6-Hexafluoro-1,1′-biphenyl (10)
Following the general procedure with 1,2,4,5-tetrafluoro-3-iodobenzene (1), (3,5-difluorophenyl)boronic acid (5) and XPhos as phosphine ligand. The desired product was obtained as white solid (47 mg, 99%). 1H NMR: (250 MHz, chloroform-d) δ 7.13 (tt, J = 9.7, 7.4 Hz, 1H), 7.02 (ddd, J = 6.5, 2.3, 1.2 Hz, 2H), 6.93 (tt, J = 8.9, 2.3 Hz, 1H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 163.0 (dd, J = 249.4, 12.9 Hz), 146.4 (dddd, J = 248.9, 14.7, 10.3, 4.1 Hz), 143.8 (ddt, J = 248.8, 14.3, 4.1 Hz), 130.3 (tt, J = 10.4, 2.4 Hz), 119.4 (tt, J = 16.1, 2.4 Hz), 113.8–113.3 (m), 106.1 (t, J = 22.6 Hz), 105.0 (t, J = 25.1 Hz).. 19F{1H}NMR: (235 MHz, chloroform-d) δ −108.98 (s, 2F), −138.28 (dd, J = 21.8, 12.8 Hz, 2F), −143.32 (dd, J = 21.9, 12.9 Hz, 2F). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C12H4F6 [M]+: 262.0; found 262.0. EA: Anal. Calc'd for C12H4F6: C, 54.98; H, 1.54; N, 0.00; found C, 55.29; H; 1.507; N, 0.00. Rf: 0.9 (pentane).
2,2′,3,4′,5,5′,6-Heptafluoro-1,1′-biphenyl (11)
Following the general procedure with 1,2,4,5-tetrafluoro-3-iodobenzene (1), (2,4,5-trifluorophenyl)boronic acid (6) and RuPhos as phosphine ligand. The desired product was obtained as white solid (22.3 mg, 47%). Alternative procedure: Following the general procedure with 1,2,4,5-tetrafluoro-3-iodobenzene (1), 5,5-dimethyl-2-(2,4,5-trifluorophenyl)-1,3,2-dioxaborinane (17) and RuPhos as phosphine ligand. The desired product was obtained as white solid (32.0 mg, 63%). 1H NMR: (250 MHz, chloroform-d) δ 7.62–6.90 (m). 13C{1H}NMR: (101 MHz, chloroform-d) δ 155.3 (ddd, J = 250.3, 9.6, 2.7 Hz), 151.1 (ddd, J = 255.0, 14.2, 12.0 Hz), 146.8 (ddd, J = 246.7, 12.8, 3.8 Hz), 146.0 (dddd, J = 249.0, 14.6, 10.4, 4.2 Hz), 143.8 (ddt, J = 249.7, 14.3, 4.1 Hz), 119.66 (ddd, J = 20.4, 3.9, 1.9 Hz), 113.9 (t, J = 17.7 Hz), 111.4–111.1 (m), 106.6 (t, J = 21.5 Hz), 106.2 (d, J = 21.2 Hz). 19F{1H}NMR: (235 MHz, chloroform-d) δ −113.47 (dtd, J = 14.5, 11.0, 5.5 Hz), −130.09 (dd, J = 21.5, 5.5 Hz), −137.77 – −138.54 (m), −140.60 – −140.98 (m), −141.61 (dd, J = 21.5, 14.5 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C12H3F7 [M]+: 280.0; found 279.9. EA: Anal. Calc'd for C12H3F7: C, 51.45; H, 1.08; N, 0.00; found C, 52.65; H; 0.96; N, 0.00. Rf: 0.9 (pentane).
2,2′,3,3′,4,5′,6′-Heptafluoro-1,1′-biphenyl (12)
Following the general procedure with 1,2,4,5-tetrafluoro-3-iodobenzene (1), (2,3,4-trifluorophenyl)boronic acid (7) and XPhos as phosphine ligand. The desired product was obtained as white solid (14.2 mg, 28%). Alternative procedure: Following the general procedure with 1,2,4,5-tetrafluoro-3-iodobenzene (1), 5,5-dimethyl-2-(2,3,4-trifluorophenyl)-1,3,2-dioxaborinane (15) and DavePhos as phosphine ligand. The desired product was obtained as white solid (42.4 mg, 84%). Alternative Procedure: Following the general procedure with 1,2,4,5-tetrafluoro-3-iodobenzene (1) and the MIDA-boronate of (2,3,4-trifluorophenyl)boronic acid (16) and CyJohnPhos or RuPhos as phosphine ligand. The desired product was obtained as white solid (40.4 mg, 80%). 1H NMR: (250 MHz, chloroform-d) δ 7.25–7.05 (m, 3H). 13C{1H}NMR: (63 MHz, chloroform-d) δ 152.3 (ddd, J = 252.6, 9.9, 2.3 Hz), 149.5 (ddd, J = 254.4, 10.9, 3.4 Hz), 148.6–143.9 (m), 144.0 (ddt, J = 249.2, 14.2, 4.1 Hz), 140.7 (dt, J = 253.0, 15.3 Hz), 125.9–125.5 (m), 114.6–113.6 (m), 113.1 (dt, J = 4.8, 2.5 Hz), 112.73 (dd, J = 17.9, 3.9 Hz), 107.25–106.27 (m). 19F{1H}NMR: (235 MHz, chloroform-d) δ −131.43 (dd, J = 20.6, 9.2 Hz), −132.14 (ddd, J = 19.9, 10.4 Hz), −138.30 – −138.52 (m), −140.81 – −141.09 (m), −158.90 (t, J = 20.4 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C12H3F7 [M]+: 278.0; found 279.9. EA: Anal. Calc'd for C12H3F7: C, 51.45; H, 1.08; N, 0.00; found C, 51.44; H; 1.08; N, 0.00. Rf: 0.9 (pentane).
2,2′,3,3′,5,5′,6,6′-Octafluoro-1,1′-biphenyl (13)
Following the general procedure with 1,2,4,5-tetrafluoro-3-iodobenzene (1), (2,3,5,6-tetrafluorophenyl)boronic acid (8) and DavePhos as phosphine ligand. The desired product was obtained as white solid (31.3 mg, 58%). Alternative procedure: Following the general procedure with 1,2,4,5-tetrafluoro-3-iodobenzene (1), 5,5-dimethyl-2-(2,3,5,6-tetrafluorophenyl)-1,3,2-dioxaborinane (18) and MePhos as phosphine ligand. The desired product was obtained as white solid (44.4 mg, 82%). 1H NMR: (250 MHz, chloroform-d) δ 7.36–7.15 (m, 2H). 13C{1H}NMR: (63 MHz, chloroform-d) δ 148.6–143.7 (m), 146.4–141.9 (m), 108.1 (t, J = 22.1 Hz), one C missing. 19F{1H}NMR: NMR (377 MHz, chloroform-d) δ −137.66 – −137.85 (m,4F), −138.20 – −138.42 (m, 4F). FTIR (ATR): 3068 (w), 1603 (m), 1510 (m), 1479 (s), 1437 (m)0, 1437 (m), 1227 (m), 1179 (m), 962 (m), 910 (m), 853 (m), 704 (m), 685 (m), 665 (m). MS (EI+): calc'd for C12H2F8 [M]+: 298.0; found 297.9. EA: Anal. Calc'd for C12H2F8: C, 48.34; H, 0.68; N, 0.00; found C, 48.35; H; 0.67; N, 0.00. Rf: 0.9 (pentane).
2,2′,3,3′,4,5,5′,6′-Octafluoro-1,1′-biphenyl (20)
Following the general procedure with 1,2,4,5-tetrafluoro-3-iodobenzene (1), 5,5-dimethyl-2-(2,3,4,5-tetrafluorophenyl)-1,3,2-dioxaborinane (19) and DavePhos as phosphine ligand. The desired product was obtained as white solid (33.5 mg, 62%). Alternative procedure: Following the general procedure with 1,2,4,5-tetrafluoro-3-bromobenzene, 5,5-Dimethyl-2-(2,3,4,5-tetrafluorophenyl)-1,3,2-dioxborinane (19) and DavePhos as phosphine ligand. The desired product was obtained as white solid (7.0 mg, 13%). 1H NMR: (250 MHz, acetone-d6) δ 7.62 (tt, J = 10.3, 7.6 Hz, 1H), 7.44 (tdd, J = 10.6, 7.1, 2.5 Hz, 1H). 13C{1H}NMR: (63 MHz, chloroform-d) δ 147.4 (dddd, J = 248.5, 10.4, 3.8, 2.5 Hz), 148.2–143.5 (m), 148.8–144.1 (m), 144.1 (ddt, J = 250.1, 14.5, 4.1 Hz), 144.2–139.2 (m), 113.3 (ddt, J = 20.5, 3.7, 2.0 Hz), 113.2–112.1 (m), 111.7 (dddd, J = 13.0, 7.2, 4.5, 2.4 Hz), 107.3 (t, J = 22.6 Hz). 19F{1H}NMR: (235 MHz, chloroform-d) δ −137.25 (dddd, J = 20.1, 11.1, 5.4 Hz, 1F), −138.38 – −138.67 (m, 2F), −139.01 (ddd, J = 20.7, 12.0, 3.1 Hz, 1F), −140.81 – −141.58 (m, 2F), −153.17 (ddd, J = 20.8, 19.4, 5.4 Hz, 1F), −154.73 (td, J = 19.7, 3.1 Hz, 1F). MS (EI+): calc'd for C12H2F8 [M]+: 298.1; found 298.1. EA: Anal. Calc'd for C12H2F8: C, 48.34; H, 0.68; N, 0.00 found C, 48.36; H; 0.89; N, 0.00. Rf: 0.9 (pentane).
2,3,3′,4,5,5′,6-Heptafluoro-1,1′-biphenyl (21)
Following the general procedure with pentafluoroiodobenzene, (3,5-difluorophenyl)boronic acid (5) and XPhos as phosphine ligand. The reaction was done on a 0.36 mmol scale. The reaction was done on a 0.36 mmol scale. The desired product was obtained as white solid (87.4 mg, 95%). The product contained <1% of the corresponding homocoupling product. 1H NMR: (400 MHz, chloroform-d) δ 6.98 (ddt, J = 7.7, 2.4, 1.2 Hz, 2H), 6.93 (tt, J = 8.8, 2.3 Hz, 1H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 163.1 (dd, J = 249.8, 12.8 Hz), 145.7–142.8 (m), 141.2 (dtt, J = 255.7, 13.4, 5.1 Hz), 139.6–136.6 (m), 129.2 (tdd, J = 10.5, 3.9, 1.8 Hz), 114.2–113.9 (m), 113.8–113.4 (m), 105.2 (t, J = 25.1 Hz). 19F{1H}NMR: (235 MHz, chloroform-d) δ −108.59 (s, 2F), −142.15 – −143.59 (m, 2F), −153.35 (t, J = 20.9 Hz, 1F), −160.30 – −162.60 (m, 2F). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C12H3F7 [M]+: 280.0; found 279.9. EA: Anal. Calc'd for C12H3F7: C, 51.45; H, 1.08; N, 0.00 found C, 51.54; H; 1.09; N, 0.00. Rf: 0.9 (pentane).
2,3,3′,4,4′,5,5′,6-Octafluoro-1,1′-biphenyl (22)
Following the general procedure with pentafluoroiodobenzene, (3,4,5-trifluorophenyl)boronic acid (2) and CyJohnPhos as phosphine ligand. The reaction was done on a 0.36 mmol scale. The desired product was obtained as white solid (51.8 mg, 43%). The product also contained 5% of the corresponding homocoupling product. Alternative Procedure: Following the general procedure with chloropentafluorobenzene, (3,4,5-trifluorophenyl)boronic acid (2) and CyJohnPhos as phosphine ligand. The reaction was done on a 0.49 mmol scale. The product also contained 3% of the corresponding homocoupling product. The desired product was obtained as white solid (109 mg, 74%). 1H NMR: (250 MHz, chloroform-d) δ 7.14–7.05 (m, 2H). 19F{1H}NMR: (235 MHz, chloroform-d) δ −132.95 (d, J = 20.5 Hz, 2F), −142.54 – −142.85 (m, 2F), −152.76 – −153.12 (m, 1F), −157.90 (t, J = 20.6 Hz, 1F), −160.75 – −161.06 (m, 2F). 13C{1H}NMR: (101 MHz, chloroform-d) δ 151.5 (ddd, J = 251.5, 10.1, 4.2 Hz), 144.2 (dddt, J = 249.7, 10.9, 7.3, 4.0 Hz), 141.3 (dtt, J = 256.3, 13.4, 5.1 Hz), 140.6 (dt, J = 255.9, 15.1 Hz), 139.7–136.6 (m), 122.2 (dddd, J = 8.5, 7.1, 5.2, 1.7 Hz), 115.3–114.8 (m), 113.2 (tdd, J = 16.5, 3.6, 1.7 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C12H2F8 [M]+: 298.0; found 297.9. EA: Anal. Calc’d for C12H2F8: C, 48.34; H, 0.68; N, 0.00; found C, 48.34; H; 0.46; N, 0.00. Rf: 0.9 (pentane).
2,2′,3,4,4′,5,5′,6-Octafluoro-1,1′-biphenyl (23)
Following the general procedure with pentafluoroiodobenzene, (2,4,5-trifluorophenyl)boronic acid and RuPhos as phosphine ligand. The reaction was done on a 0.34 mmol scale. The desired product was obtained as white solid (54,6 mg, 37%) in a mixture with 2,2′,4,4′,5,5′-hexafluoro-1,1′-biphenyl (30; 29%). 1H NMR: (400 MHz, chloroform-d) δ 7.17–7.09 (m, 1H), 7.04 (ddd, J = 10.0, 8.8, 6.5 Hz, 1H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 155.4 (ddd, J = 250.3, 9.5, 2.4 Hz), 151.2 (ddd, J = 255.4, 14.3, 12.3 Hz), 148.3–145.5 (m), 145.7–142.95 (m), 143.03–140.01 (m), 139.5–136.2 (m), 119.7 (d, J = 21.1 Hz), 110.2 (d, J = 18.6 Hz), 108.3 (td, J = 18.3, 4.1 Hz), 106.5 (dd, J = 28.0, 21.2 Hz). 19F{1H}NMR: (235 MHz, chloroform-d) δ −113.29 – −113.94 (m), −129.75 (dd, J = 21.4, 5.5 Hz), −139.88 – −140.40 (m), −141.36 (dd, J = 21.5, 14.5 Hz), −152.58 (tt, J = 20.8, 2.2 Hz), −160.97 – −161.76 (m). MS (EI+): calc’d for C12H2F8 [M]+: 298.0; found 297.9. Rf: 0.9 (pentane).
2,2′,3,3′,4,4′,5,5′,6-Nonafluoro-1,1′-biphenyl (24)
Following the general procedure with pentafluoroiodobenzene, 5,5-dimethyl-2-(2,3,4,5-tetrafluorophenyl)-1,3,2-dioxaborinane (19) and DavePhos as phosphine ligand. The reaction was done on a 0.36 mmol scale. The desired product was obtained as white solid (27.4 mg, 33%). The product contained 33% of the corresponding homocoupling product. 19F{1H}NMR: (377 MHz, chloroform-d) δ −136.54 (dddd, J = 22.6, 20.9, 10.7, 5.6 Hz), −137.95 (ddd, J = 21.2, 12.0, 3.2 Hz), −139.76 (dddd, J = 23.2, 9.5, 6.7, 3.1 Hz), −151.35 (tt, J = 20.7, 2.4 Hz), −151.81 (td, J = 20.5, 5.5 Hz), −153.45 (td, J = 20.1, 3.4 Hz), −160.56 – −160.76 (m). MS (EI+): calc’d for C12H1F9 [M]+: 316.0; found 315.9. Rf: 0.9 (pentane).
2,3′,4,4′,5′,6-Hexafluoro-1,1′-biphenyl (25)
Following the general procedure with 1,3,5-trifluoro-2-iodobenzene, (3,4,5-trifluorophenyl)boronic acid (2) and CyJohnPhos as phosphine ligand. The reaction was done on a 0.39 mmol scale. The desired product was obtained as white solid (101 mg, 99%). 1H NMR: (400 MHz, chloroform-d) δ 7.01–6.95 (m, 2H), 6.75–6.62 (m, 2H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 164.5–161.4 (m), 160.6 (ddd, J = 250.9, 14.7, 8.9 Hz), 151.6 (ddd, J = 249.7, 10.0, 4.2 Hz), 140.3 (dt, J = 253.8, 15.1 Hz), 124.6 (td, J = 9.0, 5.1 Hz), 115.5–114.9 (m), 112.8 (td, J = 19.5, 3.6 Hz), 102.0–100.8 (m). 19F{1H}NMR: (235 MHz, chloroform-d) δ −106.69 (t, J = 6.5 Hz), −111.16 (d, J = 6.6 Hz), −134.42 (d, J = 20.5 Hz), −160.14 (t, J = 20.5 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc’d for C12H4F6 [M]+: 262.0; found 261.9. EA: Anal. Calc'd for C12H4F6: C, 54.98; H, 1.54; N, 0.00; found C, 55.01; H; 1.29; N, 0.00 Rf: 0.9 (pentane).
2,2′,4,4′,5,6′-Hexafluoro-1,1′-biphenyl (26)
Following the general procedure with 1,3,5-trifluoro-2-iodobenzene, (3,4,5-trifluorophenyl)boronic acid and RuPhos as phosphine ligand. The reaction was done on a 0.39 mmol scale. The desired product was obtained as white solid (51 mg, 51%). Alternative procedure: Following the general procedure with 5,5-dimethyl-2-(2,4,5-trifluorophenyl)-1,3,2-dioxaborinane and RuPhos as phosphine ligand. The desired product was obtained as white solid (82 mg, 81%). 1H NMR: (400 MHz, chloroform-d) δ 7.19 (ddd, J = 10.3, 8.7, 6.4 Hz, 1H), 7.06 (ddd, J = 10.2, 8.9, 6.6 Hz, 1H), 6.84–6.74 (m, 2H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 163.0 (dt, J = 251.9, 15.6 Hz), 160.4 (ddd, J = 251.4, 15.0, 9.4 Hz), 155.4 (ddd, J = 248.9, 9.5, 2.2 Hz), 150.6 (ddd, J = 253.7, 13.9, 12.0 Hz), 146.7 (ddd, J = 245.7, 12.5, 3.7 Hz), 119.9 (dd, J = 19.9, 4.0 Hz), 112.3 (dt, J = 18.6, 5.6 Hz), 107.4 (td, J = 20.0, 4.7 Hz), 106.0 (dd, J = 28.2, 21.0 Hz), 101.1–100.1 (m). 19F{1H}NMR: (235 MHz, chloroform-d) δ −106.33 (t, J = 6.9 Hz), −109.05 (dd, J = 10.8, 6.9 Hz), −114.09 (dtd, J = 14.6, 10.8, 5.0 Hz), −131.81 (dd, J = 21.5, 5.0 Hz), −142.46 (dd, J = 21.5, 14.6 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc’d for C12H4F6 [M]+: 262.0; found 261.9. EA: Anal. Calc'd for C12H4F6: C, 54.98; H, 1,54; N, 0.00; found C, 54.97; H; 1.22; N, 0.00 Rf: 0.9 (pentane).
2,2′,3,4,4′,5,6′-Heptafluoro-1,1′-biphenyl (27)
Following the general procedure with 1,3,5-trifluoro-2-iodobenzene, 5,5-dimethyl-2-(2,3,4,5-tetrafluorophenyl)-1,3,2-dioxaborinane (19) and DavePhos as phosphine ligand. The reaction was done on a 0.39 mmol scale. The desired product was obtained as white solid (73 mg, 67%). 1H NMR: (400 MHz, chloroform-d) δ 7.04–6.95 (m, 1H), 6.86–6.76 (m, 2H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 163.5 (dt, J = 252.2, 15.1 Hz), 160.5 (ddd, J = 252.2, 14.8, 8.9 Hz), 148.5–145.7 (m), 147.2–144.2 (m), 142.9–139.3 (m), 113.4 (d, J = 20.1 Hz), 112.6 (ddd, J = 14.6, 8.3, 4.4 Hz), 106.6 (t, J = 20.1 Hz), 101.8–100.3 (m). 19F{1H}NMR: (235 MHz, chloroform-d) δ −105.3 (t, J = 7.1 Hz), −108.9 (dd, J = 10.6, 7.0 Hz), −137.1 – −137.5 (m), −139.3 (ddd, J = 20.9, 12.0, 2.9 Hz), −154.1 (td, J = 20.3, 4.8 Hz), −154.9 (td, J = 20.2, 2.9 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C12H3F7 [M]+: 280.0; found 279.9. EA: Anal. Calc'd for C12H3F7: C, 51.45; H, 1.08; N, 0.00; found C, 51.54; H; 0.81; 0.00, Rf: 0.9 (pentane).
2,3,3′,4,4′,5′,6-Heptafluoro-1,1′-biphenyl (28)
Following the general procedure with 1,2,3,5-Tetrafluoro-4-iodobenzene, (3,4,5-trifluorophenyl)boronic acid (2) and CyJohnPhos as phosphine ligand. The reaction was done on a 0.36 mmol scale. The desired product was obtained as white solid (74.7 mg, 77%). The product contained <2% of the corresponding homocoupling Product. 1H NMR: (400 MHz, chloroform-d) δ 7.09 (ddd, J = 7.5, 6.4, 1.3 Hz, 2H), 6.91 (tdd, J = 9.8, 6.1, 2.5 Hz, 1H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 156.1–153.1 (m), 151.7 (ddd, J = 250.8, 10.1, 4.3 Hz), 152.5–149.4 (m), 150.8–147.5 (m), 140.6 (dt, J = 254.6, 15.1 Hz), 139.6–136.5 (m), 123.8–123.4 (m), 115.6–114.9 (m), 114.0–113.4 (m), 101.9 (ddd, J = 28.6, 21.4, 3.9 Hz). 19F{1H}NMR: (377 MHz, chloroform-d) δ −117.81 (dd, J = 11.1, 2.2 Hz, 1F), −130.78 (ddd, J = 21.7, 6.4, 2.4 Hz, 1F), −133.65 (d, J = 20.5 Hz, 2F), −135.04 (dd, J = 21.3, 6.3 Hz, 1F), −158.96 (t, J = 20.6 Hz, 1F), −163.61 (ddd, J = 21.2, 10.9 Hz, 1F). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C12H3F7 [M]+: 278.0; found 279.9. EA: Anal. Calc'd for C12H3F7: C, 51.45; H, 1.08; N, 0.00; found C, 51.56; H; 1.25; N, 0.00. Rf: 0.9 (pentane).
3,3′,4,4′,5,5′-Hexafluoro-1,1′-biphenyl (4)
Following the general procedure with 3,4,5-trifluoro-1-bromobenzene, (3,4,5-trifluorophenyl)boronic acid (2) and CyJohnPhos as phosphine ligand. The reaction was done on a 0.36 mmol scale. The reaction was done on a 0.36 mmol scale. The desired product was obtained as white solid (75.8 mg, 83%). 1H NMR: (400 MHz, chloroform-d) δ 7.22–6.96 (m, 4H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 151.6 (ddd, J = 251.2, 10.1, 4.3 Hz), 139.9 (dt, J = 254.1, 15.3 Hz), 134.6–134.0 (m), 111.4–110.8 (m). 19F{1H}NMR: (377 MHz, chloroform-d) δ −132.75 (d, J = 20.7 Hz), −160.30 (t, J = 20.4 Hz FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C12H4F6 [M]+: 262.0; found 261.9. EA: Anal. Calc'd for C12H4F6: C, 54.98; H, 1.54; N, 0.00; found C, 55.29; H; 1.51; N, 0.00. Rf: 0.9 (pentane).
2,3′,4,4′,5,5′-Hexafluoro-1,1′-biphenyl (29)
Following the general procedure with 1-bromo-2,4,5-trifluorobenzene, (3,4,5-trifluorophenyl)boronic acid (2) and CyJohnPhos as phosphine ligand. The reaction was done on a 0.47 mmol scale. The desired product was obtained as white solid (89 mg, 72%). 1H NMR: (400 MHz, chloroform-d) δ 7.19 (ddd, J = 10.3, 8.7, 6.4 Hz, 1H), 7.06 (ddd, J = 10.2, 8.9, 6.6 Hz, 1H), 6.84–6.74 (m, 2H).13C{1H}NMR: (101 MHz, chloroform-d) δ 154.6 (ddd, J = 248.6, 9.2, 2.6 Hz), 151.2 (ddd, J = 250.8, 10.1, 4.3 Hz), 150.0 (ddd, J = 253.8, 14.1, 12.5 Hz), 147.1 (ddd, J = 246.4, 12.8, 3.8 Hz), 139.7 (dt, J = 253.8, 15.0 Hz), 129.7 (dd, J = 7.8 Hz), 122.4 (d, J = 14.9 Hz), 117.8 (dd, J = 20.1, 4.2 Hz), 113.4–112.9 (m), 106.53 (dd, J = 28.8, 21.0 Hz). 19F{1H}NMR: (235 MHz, chloroform-d) δ −118.89 (dd, J = 15.3, 4.5 Hz), −132.60 (dd, J = 21.6, 4.4 Hz), −133.68 (dd, J = 20.5, 1.5 Hz), −141.60 (dd, J = 21.5, 15.1), −160.36 (t, J = 20.5). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C12H4F6 [M]+: 262.0; found 261.9. EA: Anal. Calc'd for C12H4F6: C, 54.98; H, 1.54; N, 0.00; found C, 54.90; H; 1.15; N, 0.00 Rf: 0.9 (pentane).
2,2′,4,4′,5,5′-Hexafluoro-1,1′-biphenyl (30)
Following the general procedure with 1-bromo-2,4,5-trifluorobenzene, 5,5-dimethyl-2-(2,4,5-trifluorophenyl)-1,3,2-dioxaborinane (17) and RuPhos as phosphine ligand. The reaction was done on a 0.47 mmol scale. The desired product was obtained as white solid (90 mg, 73%). 1H NMR: (400 MHz, chloroform-d) δ 7.17–7.07 (m, 2H), 7.02–6.91 (m, 2H).13C{1H}NMR: (101 MHz, chloroform-d) δ 155.1 (dd, J = 249.7, 9.6 Hz), 152.3–148.7 (m), 146.9 (ddd, J = 245.8, 12.7, 2.7 Hz), 119.4–119.0 (m), 118.1–117.7 (m), 106.9–105.7 (m). 19F{1H}NMR: (235 MHz, chloroform-d) δ −116.00 – −116.22 (m), −132.05 – −132.39 (m), −141.87 – −142.35 (m). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C12H4F6 [M]+: 262.0; found 261.9. EA: Anal. Calc'd for C12H4F6: C, 54.98; H, 1.54; N, 0.00; found C, 54.98; H; 1.13; N, 0.00 Rf: 0.9 (pentane).
2,3,3′,4,4′,5′-Hexafluoro-1,1′-biphenyl (31)
Following the general procedure with 2,3,4-trifluoro-1-bromobenzene, (3,4,5-trifluorophenyl)boronic acid (2) and CyJohnPhos as phosphine ligand. The reaction was done on a 0.36 mmol scale. The reaction was done on a 0.36 mmol scale. The desired product was obtained as white solid (86.5 mg, 95%). The product contained <2% of the corresponding homocoupling product. 1H NMR: (250 MHz, chloroform-d) δ 7.21–7.00 (m, 1H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 152.7–149.7 (m), 150.1–147.3 (m), 140.4 (dt, J = 252.6, 15.6 Hz), 139.8 (dt, J = 253.9, 15.2 Hz), 124.1–123.7 (m), 123.5 (dt, J = 7.6, 3.7 Hz), 113.2 (ddd, J = 16.3, 6.5, 3.3 Hz), 112.7 (dd, J = 17.6, 4.1 Hz). 19F{1H}NMR: (235 MHz, chloroform-d) δ −133.2 (dd, J = 20.4, 8.0 Hz), −133.6 (d, J = 20.6 Hz), −138.2 (dd, J = 20.2, 8.0 Hz), −158.8 (t, J = 20.3 Hz), −160.3 (t, J = 20.6 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C12H2F8 [M]+: 262.0; found 261.9. EA: Anal. Calc'd for C12H2F8: C, 54.98; H, 1.54; N, 0.00; found C, 54.59; H; 1.38; N, 0.00. Rf: 0.9 (pentane).
2,2′,3,4,4′,5′-Hexafluoro-1,1′-biphenyl (32)
Following the general procedure with 1-bromo-2,3,4-trifluorobenzene, 5,5-dimethyl-2-(2,4,5-trifluorophenyl)-1,3,2-dioxaborinane (17) and RuPhos as phosphine ligand. The reaction was done on a 0.47 mmol scale. The desired product was obtained as white solid (96 mg, 77%). 1H NMR: (400 MHz, chloroform-d) δ 7.25–7.14 (m, 1H), 7.13–7.01 (m, 3H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 155.1 (ddd, J = 248.7, 9.4, 2.5 Hz), 151.6 (ddd, J = 242.8, 10.1, 3.2 Hz), 150.5 (ddd, J = 253.7, 14.4, 12.6 Hz), 150.5–147.7 (m), 147.0 (ddd, J = 246.1, 12.8, 3.9 Hz), 140.5 (dt, J = 252.1, 15.2 Hz), 125.1–124.8 (m), 119.6–119.4 (m), 119.4–119.0 (m), 118.3–117.9 (m), 112.5 (dd, J = 17.5, 4.0 Hz), 106.3 (dd, J = 28.4, 21.2 Hz). 19F{1H}NMR: (235 MHz, chloroform-d) δ −115.95 (ddd, J = 16.3, 14.9, 4.8 Hz), −132.28 (dd, J = 21.5, 4.8 Hz), −133.21 (dd, J = 20.5, 8.3 Hz), −134.70 (ddd, J = 20.4, 16.3, 8.3 Hz), −142.17 (dd, J = 21.5, 14.9 Hz), −159.40 (t, J = 20.4 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C12H4F6 [M]+: 262.0; found 261.9. EA: Anal. Calc'd for C12H4F6: C, 54.98; H, 1.54; N, 0.00; found C, 55.08; H; 1.05; N, 0.00 Rf: 0.9 (pentane).
3,3′,4,5,5′-Pentafluoro-4′-(trifluoromethyl)-1,1′-biphenyl (33)
Following the general procedure with 5-bromo-1,3-difluoro-2-(trifluoromethyl)benzene, (3,4,5-trifluorophenyl)boronic acid (2) and CyJohnPhos as phosphine ligand. The reaction was done on a 0.38 mmol scale. The desired product was obtained as colorless oil (99 mg, 83%). 1H NMR: (400 MHz, chloroform-d) δ 7.20 (dd, J = 8.1, 6.4 Hz, 1H), 7.16 (d, J = 10.1 Hz, 1H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 160.5 (dd, J = 259.7, 5.8 Hz), 151.9 (ddd, J = 252.0, 10.1, 4.2 Hz), 144.5 (t, J = 10.9 Hz), 140.7 (dt, J = 255.6, 15.3 Hz), 133.6, 121.7 (q, J = 274.1 Hz), 111.8–111.4 (m), 111.4–111.0 (m), 108.4–107.0 (m). 19F{1H}NMR: (235 MHz, chloroform-d) δ −56.54 (t, J = 21.8 Hz), −109.22 (q, J = 21.9 Hz), −132.25 (d, J = 20.7 Hz), −158.58 (t, J = 20.2 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc’d for C13H4F8 [M]+: (312.0); found (311.9). EA: Anal. Calc'd for C13H4F8: C, 50.02; H, 1.29; N, 0.00; found C, 49.80; H; 1.03; N, 0.02 Rf: 0,1 (pentane).
4-(3,5-Difluorophenyl)-2,3,5-trifluoropyridine (34)
Following the general procedure with 2,3,5-trifluoro-4-iodopyridine, 3,5-difluorophenyl boronic acid and XPhos as phosphine ligand. The reaction was done on a 0.39 mmol scale. The desired product was obtained as white solid (62 mg, 65%). The product also contained 2% of the corresponding homocoupling product. 1H NMR: (400 MHz, chloroform-d) δ 8.02–7.99 (m, 1H), 7.12–7.04 (m, 2H), 6.98 (tt, J = 8.8, 2.3 Hz, 1H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 161.9 (dd, J = 250.2, 12.8 Hz), 152.5 (dd, J = 257.5, 4.3 Hz), 147.8 (ddd, J = 237.2, 15.2, 2.2 Hz), 145.8 (d, J = 162.1 Hz), 141.0 (ddd, J = 267.0, 31.7, 2.8 Hz), 128.3–127.8 (m), 127.8–127.6 (m), 112.7–111.7 (m), 104.7 (t, J = 25.0 Hz). 19F{1H}NMR: (377 MHz, chloroform-d) δ −88.00 (dd, J = 29.2, 25.3 Hz), −108.10, −132.10 (dd, J = 29.0, 1.6 Hz), −139.92 (dd, J = 25.8, 1.9 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C11H4F5N [M]+: (245.0); found (244.9). EA: Anal. Calc'd for C11H4F5N: C, 53.89; H, 1.64; N, 5.71; found C, 54.32; H; 3.84; N, 3.73 Rf: 0,1 (pentane).
2,3,5-Trifluoro-4-(3,4,5-trifluorophenyl)pyridine (35)
Following the general procedure with 2,3,5-trifluoro-4-iodopyridine, 3,4,5-trifluorophenyl boronic acid and CyJohnPhos as phosphine ligand. The reaction was done on a 0.39 mmol scale. The desired product was obtained as white solid. (85 mg, 84%). 1H NMR: (400 MHz, chloroform-d) δ 7.94 (t, J = 1.8 Hz, 1H), 7.18–7.10 (m, 2H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 154.9–152.1 (m), 151.5 (ddd, J = 251.9, 10.2, 4.1 Hz), 150.3–147.6 (m), 142.1 (ddd, J = 266.8, 31.7, 2.2 Hz), 141.0 (dt, J = 257.1, 15.0 Hz), 129.2 (ddd, J = 28.8, 14.3, 6.6 Hz), 126.8 (t, J = 13.9 Hz), 122.2–121.6 (m), 115.4–114.4 (m). 19F{1H}NMR: (377 MHz, chloroform-d) δ −87.68 (dd, J = 29.3, 25.5 Hz), −132.35 (dd, J = 29.0, 1.4 Hz), −132.47 (d, J = 20.5 Hz), −140.16 (d, J = 25.1 Hz), −156.38 (t, J = 20.4 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C11H3F6N [M]+: (263.0); found (262.9). EA: Anal. Calc'd for C11H3F6N: C, 50.21; H, 1.15; N, 5.32; found C, 49.73; H; 2.28; N, 4.50 Rf: 0,7 (pentane).
2,3,5-Trifluoro-4-(2,3,4-trifluorophenyl)pyridine (36)
Following the general procedure with 2,3,5-trifluoro-4-iodopyridine, 2,3,4-trifluorophenyl boronic acid and CyJohnPhos as phosphine ligand. The reaction was done on a 0.39 mmol scale. The desired product was obtained as colorless oil (72 mg, 72%%). The product also contained 2% of the corresponding homocoupling product.. 1H NMR: (400 MHz, chloroform-d) δ 8.01 (dd, J = 2.2, 1.3 Hz, 1H), 7.21–7.09 (m, 2H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 154.0 (dd, J = 258.4, 4.2 Hz), 152.7 (ddd, J = 254.9, 9.9, 3.3 Hz), 149.4 (ddd, J = 256.1, 11.2, 3.7 Hz), 148.6 (ddd, J = 237.3, 14.9, 2.3 Hz), 142.5 (ddd, J = 267.8, 31.8, 2.7 Hz), 140.7 (dt, J = 253.9, 15.2 Hz), 128.9 (ddd, J = 28.0, 14.3, 6.5 Hz), 125.6–125.1 (m), 123.1–122.4 (m), 113.09 (dd, J = 18.1, 3.8 Hz), 111.8 (dt, J = 12.9, 3.4 Hz). 19F{1H}NMR: (377 MHz, chloroform-d) δ −88.08 (dd, J = 29.1, 25.3 Hz), −129.78 (dd, J = 20.5, 10.0 Hz), −129.95 (dd, J = 29.2, 12.2 Hz), −130.95 – −131.42 (m), −136.97 (dd, J = 25.2, 12.2 Hz), −158.20 (t, J = 20.4 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C11H3F6N [M]+: (263.0); found (262.9). EA: Anal. Calc'd for C11H3F6N: C, 50.21; H, 1.15; N, 5.32; found C, 48.07; H; 1.94; N, 5.09 Rf: 0,1 (pentane).
2,3,5-Trifluoro-4-(2,3,4,5-tetrafluorophenyl)pyridine (37)
Following the general procedure with 2,3,5-trifluoro-4-iodopyridine, 5,5-dimethyl-2-(2,3,4,5-tetrafluorophenyl)-1,3,2-dioxaborinane and DavePhos as phosphine ligand. The reaction was done on a 0.39 mmol scale. The desired product was obtained as yellow oil7 (39 mg, 36%) The product also contained <1% of the corresponding homocoupling product.. 1H NMR: (400 MHz, chloroform-d) δ 7.97 (dd, J = 2.2, 1.3 Hz, 1H), 7.06–6.94 (m, 1H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 152.6 (dd, J = 259.3, 4.1 Hz), 147.4 (ddd, J = 237.8, 14.8, 2.3 Hz), 146.2 (dddd, J = 249.5, 10.3, 3.6, 2.3 Hz), 144.4 (dddd, J = 253.1, 11.6, 3.7, 1.4 Hz), 142.5–139.4 (m), 142.9–139.6 (m), 140.4 (dddd, J = 255.9, 16.6, 12.4, 4.0 Hz), 127.9 (ddd, J = 27.8, 14.3, 6.6 Hz), 120.6 (t, J = 16.5 Hz), 111.6 (ddd, J = 20.8, 4.1, 2.1 Hz), 109.2 (t, J = 7.6 Hz). 19F{1H}NMR(377 MHz, chloroform-d) δ −87.45 (dd, J = 29.3, 25.3 Hz), −129.71 (dd, J = 29.3, 12.0 Hz), −135.52 – −135.88 (m), −136.48 (dd, J = 25.3, 12.7 Hz), −137.41 (ddd, J = 21.1, 12.2, 3.6 Hz), −150.66 (td, J = 20.4, 6.1 Hz), −153.04 (td, J = 20.0, 3.4 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C11H2F7N [M]+: (281.0); found (280.9). EA: Anal. Calc'd for C11H2F7N: C, 47.00; H, 0.72; N, 4.98; found C, 45.56; H; 2.33; N, 4.34 Rf: 0,1 (pentane).
4-(3,5-Difluorophenyl)-2,3,5,6-tetrafluoropyridine (38)
Following the general procedure with 2,3,5,6-tetrafluoro-4-iodopyridine, (3,5-difluorophenyl) boronic acid and XPhos as phosphine ligand. The reaction was done on a 0.36 mmol scale. The desired product was obtained as white solid (77 mg, 72%). The product mixture contained 12% of the corresponding homocoupling product (3,3′,5,5′-tetrafluoro-1,1′-biphenyl). 1H NMR: (400 MHz, chloroform-d) δ 7.11–7.08 (m, 1H), 7.01 (tt, J = 8.7, 2.3 Hz, 1H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 163.0 (dd, J = 250.9, 12.7 Hz), 144.0 (dddd, J = 246.2, 16.6, 13.0, 3.0 Hz), 140.6–137.4 (m), 131.2–130.5 (m), 128.3 (t, J = 10.6 Hz), 113.4–113.0 (m), 106.3 (t, J = 25.0 Hz). 19F{1H}NMR: (377 MHz, chloroform-d) δ −89.34 – −89.57 (m), −107.57, −144.18 – −144.40 (m). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C11H3F6N [M]+: (263.0); found (262.9). EA: Anal. Calc'd for C11H3F6N: C, 50.21; H, 1.15; N, 5.32; found C, 51.47; H; 1.338; N, 4.54 Rf: 0,4 (pentane).
2,3,5,6-Tetrafluoro-4-(3,4,5-trifluorophenyl)pyridine (39)
Following the general procedure with 2,3,5,6-tetrafluoro-4-iodopyridine, (3,4,5-trifluorophenyl)boronic acid and CyJohnPhos as phosphine ligand. The reaction was done on a 0.36 mmol scale. The desired product was obtained as colorless oil (98 mg, 98%). The product also contained 2% of the corresponding homocoupling product. 1H NMR: (400 MHz, chloroform-d) δ 7.18–7.11 (m, 2H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 151.7 (ddd, J = 252.8, 10.1, 4.1 Hz), 144.2 (dddd, J = 246.4, 16.8, 12.8, 3.0 Hz), 141.3 (dt, J = 258.1, 14.9 Hz), 140.8–137.6 (m), 130.3 (t, J = 14.1 Hz), 121.7–121.3 (m), 115.2–114.7 (m).19F{1H}NMR: (377 MHz, chloroform-d) δ −89.04 – −89.27 (m), −131.96 (d, J = 20.4 Hz), −144.43 – −144.68 (m), −155.48 (t, J = 20.5 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C11H2F7N [M]+: (281.0); found (280.9). EA: Anal. Calc'd for C11H2F7N: C, 47.00; H, 0.72; N, 4.98; found C, 46.84; H; 0.91; N, 4.88 Rf: 0,3 (pentane).
Large Scale Procedure (36 mmol) for Suzuki–Miyaura coupling of 2,3,3′,4′,5,5′,6-Heptafluoro-1,1′-biphenyl (3)
An argon-purged 500 mL Schlenk-flask was charged with THF (100 mL) and toluene (100 mL) and deoxygenated via the freeze–pump–thaw technique. Afterward 1.91 g CyJohnPhos (5.4 mmol, 15 mol %) and 1.66 g Pd2(dba)3 (1.8 mmol, 5 mol %) were added. The solution was stirred at 50 °C for 2 h to prepare the active catalyst. During that time, the color of the mixture changed from dark red to dark orange. Subsequently 33 mL ddH2O, 10.02 g K2CO3 (72,5 mmol, 200 mol %), 10.00 g 1,2,4,5-tetrafluoro-3-iodobenzene (36 mol), and 6.37 g 3,4,5-trifluorophenylboronic acid (36 mmol) were added. The flask was equipped with a reflux condenser, septum, and an argon filled balloon and the biphasic mixture was refluxed at 95 °C for 60 h. After cooling to room temperature, the aqueous phase was separated and the organic phase was removed in vacuo. The residue is resuspended with pentane and silica is added to the mixture. The solvents are removed in vacuo. This should be done with care due to the volatile nature of the corresponding products. The residue is filtered through a plug of silica with pentane as eluent. The pentane was removed in vacuo and the product was obtained as white solid (10.06 g, 35.7 mmol, 99% yield).
1,2,4,5-Tetrafluoro-3-iodobenzene (1)
A flame-dried, two-necked 500 mL Schlenk-flask, equipped with an inner thermometer was filled with 200 mL dry DEE and 11.2 mL 1,2,4,5-tetrafluorobenzene (15.0 g, 100 mmol) under an argon atmosphere. The solution was cooled to −78 °C and 40 mL n-BuLi (2,5 M in hexanes) was added dropwise with a syringe pump at a rate of 40 mL/h. It is important that the temperature of the reaction mixture does not exceed −40 °C due to the otherwise occurring LiF formation. After the addition is completed, the mixture is stirred for additional 2 h. In a second flame-dried Schlenk-flask under an argon atmosphere 25.3 g I2 is dissolved in 100 mL dry THF. The resulting solution is transferred to the reaction mixture via cannula under a positive pressure of argon, stirred for an additional 10 min and warmed to room temperature overnight. The reaction mixture was quenched with H2O and the organic phase was washed with saturated Na2S2O3. The organic phase was dried with MgSO4 and the organic solvents were evaporated for the most part. After fractional distillation, the product was obtained as very light pink liquid (20.0 g 72,5%). 1H NMR: (250 MHz, chloroform-d) δ 7.16 (tt, J = 9.5, 7.2 Hz, 1H). 13C{1H}NMR: (63 MHz, chloroform-d) δ 147.1 (ddt, J = 245.1, 14.9, 4.1 Hz), 147.9–143.0 (m), 106.7 (t, J = 22.9 Hz), 73.4 (t, J = 27.3 Hz). 19F{1H}NMR: (235 MHz, chloroform-d) δ −119.73 – −120.76 (m, 2F), −136.20 – −137.61 (m, 2F). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C6HF4I [M]+: 275.9; found 275.9. b.p.: 56 °C (12 mbar).
1,2,3,5-Tetrafluoro-4-iodobenzene (40)
This compound was synthesized analogous to 1,2,4,5-tetrafluoro-3-iodobenzene (1) described above with 1,2,3,5-tetrafluorobenzene as starting material. After fractional distillation, the product was obtained as lightly pink liquid (4.5 g, 49% yield). The product contains traces of the diiodinated side product (<2%). 1H NMR: (250 MHz, chloroform-d) δ 7.12–6.67 (m, 1H). 13C{1H}NMR: (63 MHz, chloroform-d) δ 157.1 (dddd, J = 244.9, 12.4, 6.4, 3.8 Hz), 154.5–148.5 (m), 136.9 (dddd, J = 252.7, 17.5, 15.6, 5.5 Hz), 100.9 (ddd, J = 29.9, 22.1, 3.8 Hz), 66.0 (ddd, J = 31.7, 26.6, 5.0 Hz). 19F{1H}NMR: (235 MHz, chloroform-d) δ −96.19 (d, J = 9.7 Hz, 1F), −111.72 (ddd, J = 22.3, 5.2, 2.6 Hz, 1F), −131.07 (ddd, J = 20.4, 4.8, 2.4 Hz, 1F), −161.84 (ddd, J = 21.8, 21.3, 9.7 Hz, 1F). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C6HF4I [M]+: 275.9; found 275.9. b.p.: 59 °C (15 mbar)
2,3,5-Trifluoro-4-iodopyridine (41)
A flame-dried, two-necked 250 mL Schlenk-flask was filled with 50 mL dry THF and cooled to −78 °C. 7.5 mL n-BiLi (2.5 M in hexanes, 18.8 mmol) and 2.65 mL (18.8 mmol) diiosopropylamine were added to the solvent. The mixture was stirred for 15 min. Subsequently 1.8 mL 2,3,5-trifluoropyridine (18.8 mmol) was added dropwise within 30 min with a syringe pump. After the addition was completed, the mixture was stirred for additional 2.5 h. In a second flame-dried Schlenk-flask under an argon atmosphere 4.77 g I2 (18.8 mmol) was dissolved in 50 mL dry THF. The resulting solution is transferred to the reaction mixture via cannula under a positive pressure of argon, stirred for an additional 10 min and warmed to room temperature overnight. The reaction mixture was quenched with H2O and the organic phase was washed with saturated Na2S2O3. The organic phase was dried with anhydrous Na2SO4 and the organic solvents were evaporated for the most part. After fractional distillation, the product was obtained as very white crystalline solid (2.62 g, 53.85%). 1H NMR: (250 MHz, chloroform-d) δ 7.83–7.79 (m, 1H). 13C{1H}NMR: (101 MHz, chloroform-d) δ 157.7 (ddd, J = 254.9, 4.4, 1.7 Hz), 147.4 (ddd, J = 239.5, 16.4, 2.3 Hz), 146.6 (ddd, J = 263.4, 32.1, 2.3 Hz), 127.7 (ddd, J = 29.5, 14.4, 6.2 Hz), 85.8 (ddd, J = 28.8, 23.3, 3.0 Hz). 19F{1H}NMR: (235 MHz, chloroform-d) δ −87.89 (t, J = 26.4 Hz), −111.85 (d, J = 27.2 Hz), −116.94 (d, J = 24.7 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C5H1F3IN [M]+: (258.9); found (258.9). EA: Anal. Calc'd for C5H1F3IN: C, 23.19; H, 0.39; N, 5.41; found C, 23.0; H; 0.45; N, 5.41.
2,3,5,6-Tetrafluoro-4-iodopyridine (42)
This compound was synthesized analogous to 2,3,5-trifluoro-4-iodopyridine (41) described above with 2,3,5,6-pyridine as starting material. The reaction was performed on a 6.6 mmol scale. The desired product was obtained as off-white solid (760 mg, 41%). 13C{1H}NMR: (63 MHz, chloroform-d) δ 146.0–141.0 (m), 145.1–140.2 (m), 88.6 (tt, J = 25.8, 2.3 Hz). 19F{1H}NMR: (235 MHz, chloroform-d) δ −89.38 (dq, J = 28.0, 12.9 Hz), −122.33 – −123.18 (m). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C5F4IN [M]+: (276.9); found (276.9). EA: Anal. Calc'd for C5F4IN: C, 21.68; H, 0.00; N, 5.06; found C, 20.54; H; 0.13; N, 4.83.
(2,4,5-Trifluorophenyl)boronic Acid (6)
A flame-dried, 500 mL Schlenk-flask, equipped charged with 200 mL dry DEE and 5.54 mL 2,4,5-trifluorobromobenzene (10 g, 47.4 mmol) under an argon atmosphere. The solution was cooled to −78 °C and 18 mL n-BuLi (2,5 M in hexanes) was added dropwise within. It is important that the temperature of the reaction mixture does not exceed −40 °C due to the otherwise occurring LiF formation. After the addition is completed, the mixture is stirred for an additional hour. B(OMe)3 is added and the mixture is stirred for an additional 10 min and warmed to room temperature overnight. The reaction mixture was quenched with 2 M aq. HCl and the mixture is stirred for 2 h. The aqueous phase is separated and extracted twice with DEE. The combined organic phases are washed with aq. sat. NaHCO3. The organic phase is dried with anhydrous Na2SO4 and the organic solvents are removed in vacuo. The crude product was washed with pentane and dried. The product was obtained as white solid (5.80 g, 70% yield). 1H NMR: (250 MHz, acetone-d6) δ 7.59 (ddd, J = 11.0, 9.7, 5.5 Hz, 1H), 7.38 (d, J = 1.6 Hz, 2H), 7.16 (ddd, J = 10.9, 8.8, 6.1 Hz, 1H). 13C{1H}NMR: (63 MHz, Acetone-d6) δ 162.6 (dd, J = 244.7, 9.5 Hz), 151.8 (dt, J = 251.6, 14.2 Hz), 147.0 (dd, J = 243.4, 10.5 Hz), 123.4 (dd, J = 17.4, 10.8 Hz), 121.14–114.58 (m), 105.6 (dd, J = 32.1, 20.3 Hz). 19F{1H}NMR: (235 MHz, Acetone-d6) δ 69.66 (dd, J = 17.0, 6.3 Hz), 44.75 (dd, J = 21.0, 6.4 Hz), 31.29 (dd, J = 18.9 Hz). FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C6H4BF3O2 [M]+: 276.0; found 276.0. EA: Anal. Calc'd for C6H4BF3O2: C, 40.97; H, 2.29; N, 0.00 found C, 41.08; H; 2.26; N, 0.00.
(2,3,4-Trifluorophenyl)boronic Acid (7)
This compound was synthesized analogous to (2,4,5-trifluorophenyl)boronic acid (6) described above with 2,3,4-trifluorobromobenzene as starting material. The product was obtained as white solid (3.65 g, 87% yield). 1H NMR: (250 MHz, acetone-d6) δ 7.52 (ddd, J = 8.8, 5.5, 2.4 Hz, 1H), 7.44 (d, J = 1.1 Hz, 2H), 7.16 (dddd, J = 10.4, 8.6, 6.6, 1.9 Hz, 1H). 13C{1H}NMR: (63 MHz, acetone-d6) δ 156.5 (dd, J = 161.6, 6.1 Hz), 152.6 (dd, J = 163.1, 6.9 Hz), 140.2 (ddd, J = 248.5, 17.9, 14.7 Hz), 13010 (q, J = 8.2 Hz), 124.1–116.3 (m), 113.1 (d, J = 16.6 Hz). 19F{1H}NMR: (235 MHz, acetone-d6) δ 48.08 (dd, J = 21.0, 9.2 Hz, 1F), 42.72 (dd, J = 19.4, 9.0 Hz, 1F), 12.12 (t, J = 20.1 Hz). 11B NMR (80 MHz, acetone-d6) δ 27.50. FTIR (ATR): see Supporting Information. MS (GC/MS; EI+): calc'd for C6H4BF3O2 [M]+: 276.0; found 276.0. EA: Anal. Calc'd for C6H4BF3O2: C, 40.97; H, 2.29; N, 0.00; found C, 40.98; H; 1.93; N, 0.00.
General Procedure for the Boresterfication
These compounds were prepared according to a modified procedure.13 In a 100 mL round-bottom flask 5.7 mmol of the corresponding boronic acid and 2.2 g 2,2-dimethyl-1,3-propanediol (6.3 mmol, 110 mol %) were dissolved in 50 mL dry THF. 3.4 g MgSO4 (28.4 mmol, 500 mol %) was added and the suspension was stirred for 16 h at room temperature. The MgSO4 was filtered off and washed with THF. The solvents of the filtrate were evaporated in vacuo. The crude product was purified filtered through a plug of silica (hexanes/DEE 1:1).
5,5-Dimethyl-2-(2,3,4-trifluorophenyl)-1,3,2-dioxaborinane (15)
With (2,3,4-trifluorophenyl)boronic acid (7) as starting material. The product was obtained as a white solid (1.03 g, 72% yield). 1H NMR: (250 MHz, chloroform-d) δ 7.45 (dddd, J = 8.7, 6.3, 2.5 Hz, 1H), 7.29 (s, 0H), 6.95 (dddd, J = 9.5, 8.5, 6.4, 1.8 Hz, 0H), 3.82 (s, 1H), 1.07 (s, 1H). 13C{1H}NMR: (63 MHz, Acetone-d6) δ 156.5 (dd, J = 161.6, 6.1 Hz), 152.6 (dd, J = 163.1, 6.9 Hz), 140.2 (ddd, J = 248.5, 17.9, 14.7 Hz), 131.0 (q, J = 8.2 Hz), 124.1–116.3 (m), 113.1 (d, J = 16.6 Hz), C–B missing. 19F{1H}NMR: (235 MHz, chloroform-d) δ −127.11 (dd, J = 20.9, 10.5 Hz), −131.52 (dd, J = 19.9, 10.4 Hz), −162.44 (t, J = 20.4 Hz). FTIR (ATR): see Supporting Information. 11B NMR: (80 MHz, Acetone-d6) δ 27.50. MS (EI+): calc'd for C11H12BF3O2 [M]+: 244.1; found 244.1. EA: Anal. Calc'd for C11H12BF3O2: C, 54.14; H, 4.87; N, 0.00; found C, 54.14; H; 4.85; N, 0.00. Rf: 0.5 (cyclohexane/EtOAc 5:1)
5,5-Dimethyl-2-(2,4,5-trifluorophenyl)-1,3,2-dioxaborinane (17)
With (2,4,5-trifluorophenyl)boronic acid (6) as starting material. The product was obtained as a white solid (363 mg, 52% yield). 1H NMR: (250 MHz, chloroform-d) δ 7.50 (ddd, J = 10.7, 9.7, 5.5 Hz, 1H), 6.85 (ddd, J = 10.5, 8.7, 6.1 Hz, 1H), 3.78 (s, 4H), 1.03 (s, 6H). 13C{1H}NMR: (75 MHz, chloroform-d) δ 162.5 (ddd, J = 249.6, 9.4, 2.1 Hz), 152.1 (ddd, J = 254.0, 14.5, 13.2 Hz), 146.9 (ddd, J = 244.1, 11.9, 3.6 Hz), 123.4 (ddd, J = 17.6, 10.3, 1.9 Hz), 105.6 (dd, J = 31.1, 19.9 Hz), 72.6, 32.0, 22.0 C–B missing. 19F{1H}NMR: (235 MHz, chloroform-d) δ −106.19 (ddd, J = 16.8, 6.9, 1.6 Hz, 1F), −130.14 (dd, J = 21.3, 6.9 Hz, 1F), −144.71 (dd, J = 21.3, 16.8 Hz, 1F). 11B NMR: (80 MHz, chloroform-d) δ 26.00. FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C11H12BF3O2 [M]+: 244.1; found 244.1. EA: Anal. Calc'd for C11H12BF3O2: C, 54.14; H, 4.87; N, 0.00; found C, 54.22; H; 4.87; N, 0.00. Rf: 0.4 (cyclohexane/EtOAc 5:1)
5,5-Dimethyl-2-(2,3,5,6-tetrafluorophenyl)-1,3,2-dioxaborinane (18)
With (2,3,5,6-tetrafluorophenyl)boronic acid (8) as starting material. The product was obtained as a white solid (3.68 g, 54% yield). 1H NMR: (250 MHz, chloroform-d) δ 7.04 (tt, J = 9.6, 7.4 Hz, 1H), 3.82 (s, 4H), 1.07 (s, 6H). 13C{1H}NMR: (75 MHz, chloroform-d) δ 147.9 (dddd, J = 245.8, 13.0, 11.1, 3.6 Hz), 145.6 (dddd, J = 248.3, 16.5, 8.8, 3.8 Hz), 114.9–110.7 (m), 107.2 (tt, J = 22.6, 1.6 Hz), 72.7, 31.8, 21.4. 19F{1H}NMR: (235 MHz, chloroform-d) δ −133.33 (dd, J = 22.5, 14.7 Hz), −139.91 (dd, J = 22.4, 14.7 Hz). 11B NMR: (80 MHz, chloroform-d) δ 25.88. FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C11H11BF4O2 [M]+: 262.1; found 262.0. EA: Anal. Calc'd for C11H11BF4O2: C, 50.43; H, 4.23; N, 0.00; found C, 49.95; H; 4.03; N, 0.00. Rf: 0.2 (cyclohexane/EtOAc 5:1)
6-Methyl-2-(2,3,4-trifluorophenyl)-1,3,6,2-dioxazaborocane-4,8-dione (16)
This molecule was synthesized after a modified procedure of Burke and co-workers.93 In 100 mL round-bottom flask 1g 2,3,4-trilfuoroboronic acid (7) and 920 mg Methyliminodiacetic acid were dissolved in a mixture of 50 mL toluene and 5 mL DMSO. The mixture was refluxed for 20 h in an Dean–Stark apparatus. The solvents were removed in vacuo and the residue was purified by column chromatography (hexane/EtOAc 1:2). The product was obtained as white solid (524 mg, 32% yield). 1H NMR: (250 MHz, DMSO-d6) δ 7.39–7.20 (m, 2H), 4.43 (dd, J = 17.3, 0.9 Hz, 2H), 4.12 (d, J = 17.3 Hz, 2H), 2.65 (s, 3H). 13C{1H}NMR: (63 MHz, DMSO-d6) δ 169.3, 153.8 (ddd, J = 244.6, 8.7, 3.1 Hz), 151.6 (ddd, J = 248.5, 9.8, 3.6 Hz), 139.3 (ddd, J = 249.0, 18.1, 14.7 Hz), 129.5–129.0 (m), 113.2 (dd, J = 16.2, 3.3 Hz), 62.8 (d, J = 1.5 Hz), 48.0s, C–B missing. 19F{1H}NMR: (235 MHz, DMSO-d6) δ −128.90 (dd, J = 23.0, 8.2 Hz), −134.33 (dd, J = 21.0, 8.2 Hz), −163.02 (dd, J = 23.0, 20.9 Hz). 11B NMR: (80 MHz, DMSO-d6) δ 10.74. FTIR (ATR): see Supporting Information. MS (EI+): calc'd for C11H9BF3NO4 [M]+: 287.1; found 287.0. EA: Anal. Calc'd for C11H9BF3NO4: C, 46.03; H, 3.16; N, 4.88; found C, 46.14; H; 3.23; N, 5.03.
Acknowledgments
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (Grant no. 638337). This work is supported by the Cluster of Excellence RESOLV (EXC 1069) funded by the Deutsche Forschungsgemeinschaft.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b02267.
Additional screening data and spectroscopic data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kirsch P.Halofluorocarbons, Hydrofluorocarbons, and Related Compounds. In Modern Fluoroorganic Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA, 2013; pp 245–298. [Google Scholar]
- Kirsch P.; Hahn A. Eur. J. Org. Chem. 2005, 2005, 3095–3100. 10.1002/ejoc.200500125. [DOI] [Google Scholar]
- Kirsch P., 8-Fluorinated Nematic Liquid Crystals: Design, Synthesis, and Properties A2-Tressaud, Alain. In Photonic and Electronic Properties of Fluoride Materials; Poeppelmeier K., Ed.; Elsevier: Boston, 2016; pp 159–176. [Google Scholar]
- Jang M.-S.; Song S.-Y.; Shim H.-K. Polymer 2000, 41, 5675–5679. 10.1016/S0032-3861(99)00788-0. [DOI] [Google Scholar]
- Facchetti A.; Yoon M. H.; Stern C. L.; Katz H. E.; Marks T. J. Angew. Chem., Int. Ed. 2003, 42, 3900–3903. 10.1002/anie.200351253. [DOI] [PubMed] [Google Scholar]
- Lieffrig J.; Niassy A. G.; Jeannin O.; Fourmigue M. CrystEngComm 2015, 17, 50–57. 10.1039/C4CE01935K. [DOI] [Google Scholar]
- Espallargas G. M.; Recuenco A.; Romero F. M.; Brammer L.; Libri S. CrystEngComm 2012, 14, 6381–6383. 10.1039/c2ce26131f. [DOI] [Google Scholar]
- Chen T. H.; Popov I.; Zenasni O.; Daugulis O.; Miljanic O. S. Chem. Commun. 2013, 49, 6846–6848. 10.1039/c3cc41564c. [DOI] [PubMed] [Google Scholar]
- Budd P. M.; Makhseed S. M.; Ghanem B. S.; Msayib K. J.; Tattershall C. E.; McKeown N. B. Mater. Today 2004, 7, 40–46. 10.1016/S1369-7021(04)00188-9. [DOI] [PubMed] [Google Scholar]
- Budd P. M.; Ghanem B. S.; Makhseed S.; McKeown N. B.; Msayib K. J.; Tattershall C. E. Chem. Commun. 2004, 230–231. 10.1039/b311764b. [DOI] [PubMed] [Google Scholar]
- Taylor R. G. D.; Carta M.; Bezzu C. G.; Walker J.; Msayib K. J.; Kariuki B. M.; McKeown N. B. Org. Lett. 2014, 16, 1848–1851. 10.1021/ol500591q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi H.; Albrecht M.; Valkonen A.; Rissanen K. New J. Chem. 2015, 39, 746–749. 10.1039/C4NJ01654H. [DOI] [Google Scholar]
- Jungbauer S. H.; Bulfield D.; Kniep F.; Lehmann C. W.; Herdtweck E.; Huber S. M. J. Am. Chem. Soc. 2014, 136, 16740–16743. 10.1021/ja509705f. [DOI] [PubMed] [Google Scholar]
- Kniep F.; Jungbauer S. H.; Zhang Q.; Walter S. M.; Schindler S.; Schnapperelle I.; Herdtweck E.; Huber S. M. Angew. Chem., Int. Ed. 2013, 52, 7028–7032. 10.1002/anie.201301351. [DOI] [PubMed] [Google Scholar]
- Chen L. S.; Chen G. J.; Tamborski C. J. Organomet. Chem. 1981, 215, 281–291. 10.1016/S0022-328X(00)92605-6. [DOI] [Google Scholar]
- Respess W. L.; Tamborski C. J. Organomet. Chem. 1970, 22, 251–263. 10.1016/S0022-328X(00)86040-4. [DOI] [Google Scholar]
- Vinogradov A. S.; Platonov V. E. Russ. J. Org. Chem. 2015, 51, 1388–1394. 10.1134/S107042801510005X. [DOI] [Google Scholar]
- Heidenhain S. B.; Sakamoto Y.; Suzuki T.; Miura A.; Fujikawa H.; Mori T.; Tokito S.; Taga Y. J. Am. Chem. Soc. 2000, 122, 10240–10241. 10.1021/ja002309o. [DOI] [Google Scholar]
- Negishi E.; King A. O.; Okukado N. J. Org. Chem. 1977, 42, 1821–1823. 10.1021/jo00430a041. [DOI] [Google Scholar]
- Miyaura N.; Suzuki A. J. Chem. Soc., Chem. Commun. 1979, 866–867. 10.1039/c39790000866. [DOI] [Google Scholar]
- Miyaura N.; Yamada K.; Suzuki A. Tetrahedron Lett. 1979, 20, 3437–3440. 10.1016/S0040-4039(01)95429-2. [DOI] [Google Scholar]
- Milstein D.; Stille J. K. J. Am. Chem. Soc. 1978, 100, 3636–3638. 10.1021/ja00479a077. [DOI] [Google Scholar]
- Casado A. L.; Espinet P. J. Am. Chem. Soc. 1998, 120, 8978–8985. 10.1021/ja9742388. [DOI] [Google Scholar]
- Han S.; Movassaghi M. J. Am. Chem. Soc. 2011, 133, 10768–10771. 10.1021/ja204597k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albeniz A. C.; Casares J. A. Adv. Organomet. Chem. 2014, 62, 1–110. 10.1016/B978-0-12-800976-5.00001-1. [DOI] [Google Scholar]
- Perez-Temprano M. H.; Gallego A. M.; Casares J. A.; Espinet P. Organometallics 2011, 30, 611–617. 10.1021/om100978w. [DOI] [Google Scholar]
- Carrera N.; Gutierrez E.; Benavente R.; Villavieja M. M.; Albeniz A. C.; Espinet P. Chem. - Eur. J. 2008, 14, 10141–10148. 10.1002/chem.200800558. [DOI] [PubMed] [Google Scholar]
- Martinez-Arranz S.; Carrera N.; Albeniz A. C.; Espinet P.; Vidal-Moya A. Adv. Synth. Catal. 2012, 354, 3551–3560. 10.1002/adsc.201200624. [DOI] [Google Scholar]
- Li Z.; Lu J.; Tse S.-C.; Zhou J.; Du X.; Tao Y.; Ding J. J. Mater. Chem. 2011, 21, 3226–3233. 10.1039/c0jm04166a. [DOI] [Google Scholar]
- Albeniz A. C.; Espinet P.; Martin-Ruiz B. Chem. - Eur. J. 2001, 7, 2481–2489. . [DOI] [PubMed] [Google Scholar]
- Wunderlich S. H.; Knochel P. Angew. Chem., Int. Ed. 2009, 48, 1501–1504. 10.1002/anie.200804966. [DOI] [PubMed] [Google Scholar]
- Mosrin M.; Knochel P. Org. Lett. 2009, 11, 1837–1840. 10.1021/ol900342a. [DOI] [PubMed] [Google Scholar]
- Lumeras W.; Caturla F.; Vidal L.; Esteve C.; Balague C.; Orellana A.; Dominguez M.; Roca R.; Huerta J. M.; Godessart N.; Vidal B. J. Med. Chem. 2009, 52, 5531–5545. 10.1021/jm9008604. [DOI] [PubMed] [Google Scholar]
- Glendenning M. E.; Goodby J. W.; Hird M.; Toyne K. J. J. Chem. Soc., Perkin Trans. 2 1999, 2, 481–491. 10.1039/a808747d. [DOI] [Google Scholar]
- Sharif M.; Zeeshan M.; Reimann S.; Villinger A.; Langer P. Tetrahedron Lett. 2010, 51, 2810–2812. 10.1016/j.tetlet.2010.03.067. [DOI] [Google Scholar]
- Sharif M.; Reimann S.; Villinger A.; Langer P. Synlett 2010, 2010, 913–916. 10.1055/s-0029-1219552. [DOI] [Google Scholar]
- Sharif M.; Maalik A.; Reimann S.; Feist H.; Iqbal J.; Patonay T.; Villinger A.; Langer P. J. Fluorine Chem. 2013, 146, 19–36. 10.1016/j.jfluchem.2012.12.003. [DOI] [Google Scholar]
- Heiss C.; Schlosser M. Eur. J. Org. Chem. 2003, 2003, 447–451. 10.1002/ejoc.200390078. [DOI] [Google Scholar]
- Feuerstein M.; Berthiol F.; Doucet H.; Santelli M. Synlett 2002, 2002, 1807–1810. 10.1055/s-2002-34896. [DOI] [Google Scholar]
- Kotsikorou E.; Song Y. C.; Chan J. M. W.; Faelens S.; Tovian Z.; Broderick E.; Bakalara N.; Docampo R.; Oldfield E. J. Med. Chem. 2005, 48, 6128–6139. 10.1021/jm058220g. [DOI] [PubMed] [Google Scholar]
- Wood W. J. L.; Patterson A. W.; Tsuruoka H.; Jain R. K.; Ellman J. A. J. Am. Chem. Soc. 2005, 127, 15521–15527. 10.1021/ja0547230. [DOI] [PubMed] [Google Scholar]
- Kylmala T.; Kuuloja N.; Xu Y. J.; Rissanen K.; Franzen R. Eur. J. Org. Chem. 2008, 2008, 4019–4024. 10.1002/ejoc.200800119. [DOI] [Google Scholar]
- Amarne H.; Baik C.; Murphy S. K.; Wang S. N. Chem. - Eur. J. 2010, 16, 4750–4761. 10.1002/chem.200903582. [DOI] [PubMed] [Google Scholar]
- Liu N.; Liu C.; Jin Z. L. J. Organomet. Chem. 2011, 696, 2641–2647. 10.1016/j.jorganchem.2011.04.007. [DOI] [Google Scholar]
- Chen W. B.; Xing C. H.; Dong J.; Hu Q. S. Adv. Synth. Catal. 2016, 358, 2072–2076. 10.1002/adsc.201600205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Li X. M. Chem. Rec. 2016, 16, 84–97. 10.1002/tcr.201500218. [DOI] [PubMed] [Google Scholar]
- Bulfield D.; Huber S. M. Chem. - Eur. J. 2016, 22, 14434–14450. 10.1002/chem.201601844. [DOI] [PubMed] [Google Scholar]
- Korenaga T.; Kosaki T.; Fukumura R.; Ema T.; Sakai T. Org. Lett. 2005, 7, 4915–4917. 10.1021/ol051866i. [DOI] [PubMed] [Google Scholar]
- Kinzel T.; Zhang Y.; Buchwald S. L. J. Am. Chem. Soc. 2010, 132, 14073–14075. 10.1021/ja1073799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohn H. J.; Adonin N. Y.; Bardin V. V.; Starichenko V. F. J. Fluorine Chem. 2003, 122, 195–199. 10.1016/S0022-1139(03)00088-5. [DOI] [Google Scholar]
- Frohn H. J.; Adonin N. Y.; Bardin V. V.; Starichenko V. F. Tetrahedron Lett. 2002, 43, 8111–8114. 10.1016/S0040-4039(02)01922-6. [DOI] [Google Scholar]
- Frohn H. J.; Adonin N. Y.; Bardin V. V.; Starichenko V. F. J. Fluorine Chem. 2002, 117, 115–120. 10.1016/S0022-1139(02)00157-4. [DOI] [Google Scholar]
- Adonin N. Y.; Babushkin D. E.; Parmon V. N.; Bardin V. V.; Kostin G. A.; Mashukov V. I.; Frohn H. J. Tetrahedron 2008, 64, 5920–5924. 10.1016/j.tet.2008.04.043. [DOI] [Google Scholar]
- Ahrens T.; Kohlmann J.; Ahrens M.; Braun T. Chem. Rev. 2015, 115, 931–972. 10.1021/cr500257c. [DOI] [PubMed] [Google Scholar]
- Gadsby J. M.; McPake C. B.; Murray C. B.; Sandford G. J. Fluorine Chem. 2016, 181, 51–55. 10.1016/j.jfluchem.2015.10.015. [DOI] [Google Scholar]
- Do H. Q.; Daugulis O. J. Am. Chem. Soc. 2007, 129, 12404–12405. 10.1021/ja075802+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafrance M.; Rowley C. N.; Woo T. K.; Fagnou K. J. Am. Chem. Soc. 2006, 128, 8754–8756. 10.1021/ja062509l. [DOI] [PubMed] [Google Scholar]
- Lafrance M.; Shore D.; Fagnou K. Org. Lett. 2006, 8, 5097–5100. 10.1021/ol0619967. [DOI] [PubMed] [Google Scholar]
- Song Y. Y.; Fu Z. J.; Xiong Q. H.; Li Z. J.; Li J.; Cai H. J. Iran. Chem. Soc. 2016, 13, 1931–1936. 10.1007/s13738-016-0909-8. [DOI] [Google Scholar]
- Zhu X.; Li F.; Su W. Tetrahedron Lett. 2013, 54, 1285–1289. 10.1016/j.tetlet.2012.12.110. [DOI] [Google Scholar]
- Chen L.; Sanchez D. R.; Zhang B.; Carrow B. P. J. Am. Chem. Soc. 2017, 139, 12418–12421. 10.1021/jacs.7b07687. [DOI] [PubMed] [Google Scholar]
- Erami R. S.; Diaz-Garcia D.; Prashar S.; Rodriguez-Dieguez A.; Fajardo M.; Amirnasr M.; Gomez-Ruiz S. Catalysts 2017, 7, 76. 10.3390/catal7030076. [DOI] [Google Scholar]
- Senaweera S.; Weaver J. D. J. Am. Chem. Soc. 2016, 138, 2520–2523. 10.1021/jacs.5b13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer A. U.; Slanina T.; Yao C. J.; Konig B. ACS Catal. 2016, 6, 369–375. 10.1021/acscatal.5b02410. [DOI] [Google Scholar]
- Lee J. C. H.; Hall D. G.. State-of-the-Art in Metal-Catalyzed Cross-Coupling Reactions of Organoboron Compounds with Organic Electrophiles. In Metal-Catalyzed Cross-Coupling Reactions and More; Wiley-VCH Verlag GmbH & Co. KGaA, 2014; pp 65–132. [Google Scholar]
- Tannaci J. F.; Noji M.; McBee J. L.; Tilley T. D. J. Org. Chem. 2008, 73, 7895–7900. 10.1021/jo8017268. [DOI] [PubMed] [Google Scholar]
- Amatore C.; Jutand A.; Le Duc G. Chem. - Eur. J. 2012, 18, 6616–6625. 10.1002/chem.201200516. [DOI] [PubMed] [Google Scholar]
- Frohn H.; Adonin N. Y.; Bardin V. V.; Starichenko V. F. Z. Anorg. Allg. Chem. 2002, 628, 2834–2838. . [DOI] [Google Scholar]
- Amatore C.; Jutand A.; Le Duc G. Angew. Chem., Int. Ed. 2012, 51, 1379–1382. 10.1002/anie.201107202. [DOI] [PubMed] [Google Scholar]
- Wong M. S.; Zhang X. L. Tetrahedron Lett. 2001, 42, 4087–4089. 10.1016/S0040-4039(01)00637-2. [DOI] [Google Scholar]
- Smith K. A.; Campi E. M.; Jackson W. R.; Marcuccio S.; Naeslund C. G. M.; Deacon G. B. Synlett 1997, 1, 131–132. 10.1055/s-1997-710. [DOI] [Google Scholar]
- Parrish J. P.; Jung Y. C.; Floyd R. J.; Jung K. W. Tetrahedron Lett. 2002, 43, 7899–7902. 10.1016/S0040-4039(02)01894-4. [DOI] [Google Scholar]
- Clavier H.; Nolan S. P. Chem. Commun. 2010, 46, 841–861. 10.1039/b922984a. [DOI] [PubMed] [Google Scholar]
- Altman R. A.; Buchwald S. L. Nat. Protoc. 2007, 2, 3115–3121. 10.1038/nprot.2007.411. [DOI] [PubMed] [Google Scholar]
- Kawatsura M.; Hartwig J. F. J. Am. Chem. Soc. 1999, 121, 1473–1478. 10.1021/ja983378u. [DOI] [Google Scholar]
- Littke A. F.; Fu G. C. J. Am. Chem. Soc. 2001, 123, 6989–7000. 10.1021/ja010988c. [DOI] [PubMed] [Google Scholar]
- Bruno N. C.; Buchwald S. L. Org. Lett. 2013, 15, 2876–2879. 10.1021/ol401208t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno N. C.; Niljianskul N.; Buchwald S. L. J. Org. Chem. 2014, 79, 4161–4166. 10.1021/jo500355k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau A.; Roche M.; Alami M.; Messaoudi S. ACS Catal. 2015, 5, 1386–1396. 10.1021/cs502011x. [DOI] [Google Scholar]
- Tolman C. A. Chem. Rev. 1977, 77, 313–348. 10.1021/cr60307a002. [DOI] [Google Scholar]
- Tolman C. A. J. Am. Chem. Soc. 1970, 92, 2956–2965. 10.1021/ja00713a007. [DOI] [Google Scholar]
- Clavier H.; Nolan S. P. Chem. Commun. 2010, 46, 9260–9261. [DOI] [PubMed] [Google Scholar]
- Gillie A.; Stille J. K. J. Am. Chem. Soc. 1980, 102, 4933–4941. 10.1021/ja00535a018. [DOI] [Google Scholar]
- Surry D. S.; Buchwald S. L. Chem. Sci. 2011, 2, 27–50. 10.1039/C0SC00331J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christmann U.; Vilar R. Angew. Chem., Int. Ed. 2005, 44, 366–374. 10.1002/anie.200461189. [DOI] [PubMed] [Google Scholar]
- Recently, Lloyd-Jones and co-workers have published an article on the mechanism of protodeboronation of polyfluorinated boronic acids and showed in detail how the fluorine substituents influence this reaction:; Cox P. A.; Reid M.; Leach A. G.; Campbell A. D.; King E. J.; Lloyd-Jones G. C. J. Am. Chem. Soc. 2017, 139, 13156–13165. 10.1021/jacs.7b07444. [DOI] [PubMed] [Google Scholar]
- Lennox A. J. J.; Lloyd-Jones G. C. Chem. Soc. Rev. 2014, 43, 412–443. 10.1039/C3CS60197H. [DOI] [PubMed] [Google Scholar]
- Gillis E. P.; Burke M. D. J. Am. Chem. Soc. 2008, 130, 14084–14085. 10.1021/ja8063759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrow B. P.; Hartwig J. F. J. Am. Chem. Soc. 2011, 133, 2116–2119. 10.1021/ja1108326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barder T. E.; Walker S. D.; Martinelli J. R.; Buchwald S. L. J. Am. Chem. Soc. 2005, 127, 4685–4696. 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- We also tried to use corresponding organotrifluoroborates in the case of the pentafluorophenyl boronic acid, which is known to be very unstable. However, in all tested cases only very small amounts of the cross-coupling product were obtained (<5%). The syntheses of other organotrifluoroborate salts were attempted (from the corresponding lithium organyl and BF3, see Ramachandran P. V.; Mitsuhashi W.. J. Fluorine Chem.; 2016, 190, pp 7–11), but the compounds were never obtained pure and were often contaminated with R2BF2K. Therefore we decided not to pursue this any further.
- Matharu A. S.; Cowling S. J.; Wright G. Liq. Cryst. 2007, 34, 489–506. 10.1080/02678290601176559. [DOI] [Google Scholar]
- Frohn H.; Adonin N. Y.; Bardin V. V.; Starichenko V. F. Z. Anorg. Allg. Chem. 2002, 628, 2827–2833. . [DOI] [Google Scholar]
- Lee S. J.; Gray K. C.; Paek J. S.; Burke M. D. J. Am. Chem. Soc. 2008, 130, 466–468. 10.1021/ja078129x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.