

Abstract
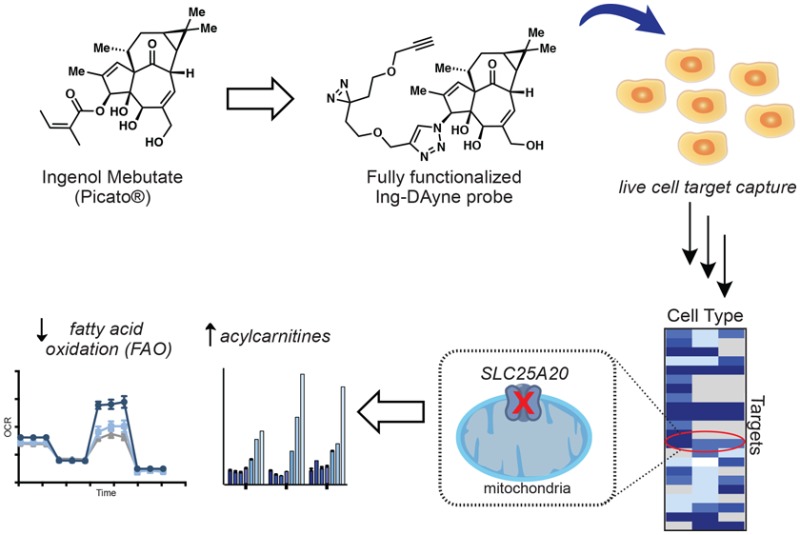
The diterpenoid ester ingenol mebutate (IngMeb) is the active ingredient in the topical drug Picato, a first-in-class treatment for the precancerous skin condition actinic keratosis. IngMeb is proposed to exert its therapeutic effects through a dual mode of action involving (i) induction of cell death that is associated with mitochondrial dysfunction followed by (ii) stimulation of a local inflammatory response, at least partially driven by protein kinase C (PKC) activation. Although this therapeutic model has been well characterized, the complete set of molecular targets responsible for mediating IngMeb activity remains ill-defined. Here, we have synthesized a photoreactive, clickable analogue of IngMeb and used this probe in quantitative proteomic experiments to map several protein targets of IngMeb in human cancer cell lines and primary human keratinocytes. Prominent among these targets was the mitochondrial carnitine-acylcarnitine translocase SLC25A20, which we show is inhibited in cells by IngMeb and the more stable analogue ingenol disoxate (IngDsx), but not by the canonical PKC agonist 12-O-tetradecanoylphorbol-13-acetate (TPA). SLC25A20 blockade by IngMeb and IngDsx leads to a buildup of cellular acylcarnitines and blockade of fatty acid oxidation (FAO), pointing to a possible mechanism for IngMeb-mediated perturbations in mitochondrial function.
Short abstract
Chemical proteomics with a photoreactive, clickable analogue of the actinic keratosis drug ingenol mebutate reveals multiple protein targets in human cells, including inhibition of the mitochondrial transporter SLC25A20.
Introduction
Natural products and natural product-inspired compounds make up roughly 40% of all drugs approved for clinical use, including a substantial fraction of anticancer drugs.1,2 Natural products, likely due to a combination of their sophisticated chemical structures and diverse biological activities refined by millions of years of evolution, have been found to access unique biological target space when compared to synthetic libraries of small molecules, underscoring the importance of natural products for the discovery of new chemical probes and therapeutic agents.3−7
Despite the venerable role played by natural products in drug discovery, the relative number of natural product-based therapeutics has been on a steady decline over the past several decades.8 This change has been attributed, in part, to barriers associated with the isolation and screening of natural products, as well as to challenges stemming from their structural complexity that impact analytical characterization and access to synthetic material and derivatives.9,10 Such problems are now being addressed by technological advances in natural product isolation and screening strategies9,11 combined with modern synthetic methods that emphasize efficient, scalable production of natural products with interceptable intermediates and opportunity for late-stage chemical derivatization.
Synthetic advances also have the potential to address another persistent bottleneck in the characterization of natural products, namely, elucidation of biological targets for these compounds. By generating derivatives of natural products with, for instance, affinity handles, as well as structurally related, inactive analogues, organic synthesis can provide key chemical probes for enriching and identifying proteins that interact with natural products in biological systems. There are many compelling examples where the synthesis of probe derivatives proved critical for elucidating the mechanism of action of natural products,12−18 although success in this area still depends on sites and routes for chemical derivatization, which may not be readily available for some natural products.
Natural products from the extracts of Euphorbia peplus have been used to treat cancer for centuries.19,20 Ingenol mebutate (IngMeb, 1), an esterified version of the parent diterpenoid ingenol (2) and a bioactive component of the Euphorbia peplus extract, was approved by the FDA in 2012 for the treatment of actinic keratosis (AK).21,22 AK is a precancerous skin condition that, if left untreated, can develop into squamous cell carcinoma (SCC).23 IngMeb is thought to exert anticancer activity through a complex mechanism involving cellular necrosis followed by a localized inflammatory immune response.24−26 Like structurally related phorbol esters, IngMeb is a potent agonist of protein kinase C (PKC), and this property is proposed to induce cytokine release27,28 followed by immune cell recruitment, cutaneous infiltration, and immune-mediated clearance of residual tumor cells.29,30 However, at higher concentrations similar to those used in topical applications, IngMeb disrupts mitochondrial structure and function and produces necrotic cell death in both cell-based and in vivo models.30,31 Although PKCs are established molecular targets of IngMeb, additional proteins are likely involved in the cellular mechanism of action of this drug. Consistent with this hypothesis, IngMeb, but not other PKC agonists, such as the phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA), blocked the growth of subcutaneous tumors in mouse models.32 The identification of additional protein targets for IngMeb that may be involved in the mode of action of the drug thus represents an important objective.
Herein, we report the development of a photoreactive, clickable ingenol-based probe and its use to identify IngMeb-binding proteins in human cancer cells and primary keratinocytes using quantitative mass spectrometry (MS)-based proteomics. Crucial to the development of these chemical probes was an understanding of the SAR for IngMeb’s biological activities, as well as efficient synthetic access to IngMeb triazole derivatives, enabling the development of a “fully functionalized” probe. Prominent among several protein targets of IngMeb identified in human cells was the mitochondrial carnitine/acylcarnitine carrier protein SLC25A20. Functional studies revealed that IngMeb blocks SLC25A20 activity in cells, leading to deregulated acylcarnitine metabolism and impairment of mitochondrial fatty acid oxidation. Similar effects were produced by a clinically relevant analogue IngDsx, but not by TPA, therefore suggesting a common PKC-independent mode of action by which IngMeb and IngDsx perturb mitochondrial metabolism in human cells.
Results and Discussion
Design and Synthesis of a Fully Functionalized IngMeb Probe
We initially set out to identify a site on IngMeb for installation of two functional groups: (1) a dialkyl diazirine33−40 photoreactive group for UV light-induced covalent trapping of proteins that bind IngMeb in cells; and (2) an alkyne group for conjugation to azide reporter tags (e.g., fluorophores, biotin) via copper catalyzed azide–alkyne cycoloaddition (CuAAC)41 for visualization and identification of IngMeb-binding proteins. We refer to such probes bearing both photoreactive and clickable groups as “fully functionalized” for enablement of the chemical proteomic discovery of small-molecule-interacting proteins in human cells.
Previous SAR studies demonstrated that ingenol itself is pharmacologically inactive42,43 and the methylation of C5- or C20-hydroxyl groups of IngMeb also eliminates biological activity.44 In addition, simultaneous removal of the C18/C19 methyl groups and dimethylcyclopropane moiety resulted in substantial loss in activity.45,46 Recent studies have further revealed that IngMeb activity requires hydroxyl groups at the C4 and C5 positions and is maintained with various esters at the C3 position.44,47 The 4-, 5-, and 20-hydroxyl groups, however, also form a framework for acyl migration of the ester, leading to subsequent loss in IngMeb potency.44 Based on these observations, we pursued installation of a photoreactive, clickable tag at the C3 position through coupling chemistry that would not suffer from the stability issues associated with ester group migration. Eying CuAAC chemistry as an attractive potential solution, we developed two routes for the synthesis of azido ingenol (6), a product that was then poised for late-stage attachment of alkyne substituents to create analogues with triazoles as a stable bioisotere for the ester linkage in IngMeb.
Our initial route leveraged one of the key intermediates (compound 3) from our 14-step total synthesis of ingenol (Figure 1A).48 Mesylation of 3 and subsequent treatment with TBACl and LiN3 afforded azide 4, which displayed the desired stereochemistry at C3. We found that TBS deprotection was best achieved using TASF. Elimination of the alcohol at C7 followed by allylic oxidation at C20 and concomitant cleavage of the carbonate using previously developed conditions48 provided crystalline azido-ingenol (6), for which we obtained a crystal structure that unambiguously confirmed the desired stereochemistry of the azide at C3. With 6 in hand, we were able to access the first triazole analogues using CuAAC chemistry. In order to provide a more scalable and robust approach to 6, we also developed an orthogonal semisynthesis of azido-ingenol that started from ingenol itself (Figure 1B). This semisynthetic route commenced with selective protection of the C5,C20-diol as the corresponding acetonide,49 followed by mesylation of the C3 alcohol. Notably, treatment of the crude mesylate with sodium azide in DMF afforded azide 8 with retention of the desired stereochemistry. All three steps could be run on decagram scales without any issues. Final deprotection of the acetonide under acidic conditions afforded azido ingenol 6. Following this semisynthetic approach, more than 1 g of azido-ingenol could be readily prepared.
Figure 1.

Structure–activity relationship studies and development of a fully functionalized ingenol-based probe. (A) Six-step synthesis of “azido-ingenol” (6) leveraging an intermediate from our ingenol total synthesis. (B) Gram-scale synthesis of azido-ingenol via a four-step semisynthetic route starting from ingenol. (C) Synthesis of ingenol-triazole analogues via CuACC along with associated biological activities. (D) Structures of a triazole-based fully functionalized probe utilizing a diazirine photoreactive group (Ing-DAyne, 9) with corresponding biological activities. Human protein kinase C (PKC) delta isoform activity was derived from measuring phosphorylation of PKC substrate peptide using 33P-ATP. Cytokine (IL-8) induction activity was measured in primary human keratinocytes (HeKa) via sandwich ELISA. The ability of compounds to induce an oxidative burst was performed in primary polymorphonuclear leukocytes (PMNs) using the superoxide indicator hydroethidine. Acute cancer cell cytotoxicity (CC50) was determined after treatment of HeLa cells with compounds and subsequent measurement of mitochondrial activity with a resazurin-based dye as a surrogate for cell viability. Further details can be found in the Supporting Information.
With azido-ingenol (6) in hand, we generated an alkynylated dialkyl diazirine fully functionalized probe—Ing-DAyne, 9—via CuAAC chemistry with the corresponding diazirine diyne S4 (Figures 1C and 1D). We next examined the activity of the Ing-DAyne probe in previously described assays that include in vitro activation of recombinant PKCδ, stimulation of IL-8 release in human primary keratinocytes, and induction of oxidative burst in human polymorphonuclear leukocytes (PMNs, i.e., neutrophils).42 The Ing-DAyne probe showed ∼10–100-fold reductions in biological activity compared to IngMb, but still maintained submicromolar potency in both in vitro and cell-based assays (Figure 1D), prompting us to move forward with the Ing-DAyne probe to identify cellular targets of IngMeb.
Gel-Based Profiling of Protein Targets of IngMeb in Human Cells
We initially assessed the protein-interaction profile of the Ing-DAyne probe by gel-based analysis of treated cells (Figure 2A). The human squamous carcinoma cell line HSC-5 was treated with Ing-DAyne (30 min), followed by exposure to UV light (10 min, 4 °C), cell lysis, coupling to tetramethylrhodamine (TAMRA)-azide via CuAAC chemistry, separation into soluble and particulate fractions, SDS–PAGE, and visualization of probe-captured proteins by in-gel fluorescence scanning. The Ing-DAyne probe showed substantial concentration-dependent labeling of proteins in both the soluble and particulate fractions of HSC5 cells, with little to no protein labeling in control cells not exposed to UV irradiation (Figure 2B). Treatment of HSC-5 cells with excess IngMeb blocked Ing-DAyne labeling of a subset of protein targets in a concentration-dependent manner (Figure 2C). The biologically inactive but structurally related analogue ingenol (2) was less effective at disrupting Ing-DAyne–protein interactions in HSC-5 cells (Figure 2C). These results prompted us to proceed forward with using the Ing-DAyne probe in quantitative MS-based proteomic experiments.
Figure 2.

Gel-based profiling of a fully functionalized ingenol probe in the human cancer cell line HSC-5. (A) Experimental workflow to visualize Ing-DAyne probe–protein interactions in cells by SDS–PAGE coupled with in-gel fluorescence scanning. (B, C) Ing-DAyne–protein interactions in cells. HSC-5 cells were treated with Ing-DAyne (10 μM) for 30 min in situ, followed by photo-cross-linking, separation of soluble and membrane fractions, and analysis. (B) Ing-DAyne (9) shows UV- and concentration-dependent labeling of protein targets. (C) Excess IngMeb, but not ingenol, blocks Ing-DAyne probe labeling of several proteins in HSC-5 cells (marked with red arrows).
Mass Spectrometry-Based Profiling of Protein Targets of IngMeb in Human Cells
We pursued IngMeb-binding proteins using two types of quantitative mass spectrometry (MS)-based proteomic experiments: (1) identification of proteins that were enriched by the Ing-DAyne probe from human cells in a UV light-dependent manner and (2) identification of proteins for which Ing-DAyne enrichment was competitively blocked by 10× IngMeb, but not 10× biologically inactive ingenol. We quantified proteins in each chemical proteomic experiment by isotopic labeling, either SILAC (stable isotope labeling with amino acids in cell culture)50 for human cancer cell lines (HSC-5 and HeLa cells) or an isotopic reductive dimethylation (ReDiMe)51,52 labeling method for primary human keratinocytes (Figure 3A). Proteins that were enriched in a UV-dependent manner (UV/no-UV ratio >5) and competed by 10× IngMeb (DMSO/IngMeb ratio >3), but not ingenol (DMSO/ingenol ratio <2), were considered as potential targets relevant to the biological activity of IngMeb (Figure 3B).
Figure 3.

MS-based profiling with a fully functionalized ingenol probe in human cells. (A) Experimental workflow to identify proteins that interact with Ing-DAyne and IngMeb in cells by quantitative MS-based proteomics. (B) Representative competition plots showing proteins for which enrichment by the Ing-DAyne probe (10 μM) was substantially blocked (>3-fold, red dotted line) by IngMeb (100 μM) in the indicated human cells. (C) Heatmap of high-occupancy protein targets of IngMeb (with comparison to ingenol). (D) Representative MS1 chromatograms of SLC25A20 tryptic peptides from Ing-DAyne enrichment and IngMeb competition experiments in corresponding cell types. (E) Concentration-dependent blockade of Ing-DAyne labeling of recombinantly expressed SLC25A20, SCCPDH, PON2, and NUCB1 in HEK293T cells by IngMeb, but not ingenol.
Isotopically light cells were treated with Ing-DAyne (10 μM, 30 min) followed by UV irradiation and compared to isotopically heavy cells also treated with Ing-DAyne, but not exposed to UV light. Following the protocol outline in Figure 3A, we identified 57, 261, and 47 proteins that were substantially enriched (>5-fold) in a UV-dependent manner by Ing-DAyne in HSC-5, HeLa, and keratinocyte cells, respectively (Figures S1A–S1C) with a total of 27 Ing-DAyne-enriched proteins that were shared across the three cell types (Table S1). We next performed competition experiments, where both heavy and light cells were treated with the Ing-DAyne probe (10 μM) along with excess competitor compound (10× IngMeb (Figure 3B) or ingenol (2, Figure S1D); heavy) or DMSO (light), respectively. These competition experiments identified 28 high-occupancy protein targets of IngMeb (light/heavy ratios >3) across HSC-5, HeLa, and keratinocyte cells (Figure 3B and Table S1) with several targets observed in all three cell types (Figure 3C,D and Table S1). Most of these proteins did not interact with ingenol (Figure 3C), which instead competitively blocked Ing-DAyne labeling of a mostly distinct subset of proteins in human cells (Figure S1D). We also performed an additional competition study in HSC-5 cells with a biologically active analogue of IngMeb—ingenol disoxate (10, IngDsx, Figure S2A)—that shows enhanced chemical stability and is currently in phase 3 clinical development.42 We observed that the majority of high-occupancy protein targets for IngMeb were also targets of IngDsx (Figure S2B, Table S1), which tended to show higher competition ratios for these shared targets.
Curiously, we did not identify members of the protein kinase C (PKC) family as IngMeb targets in our chemical proteomic experiments, despite Ing-DAyne showing submicromolar agonistic activity in PKC-δ functional assays (Figure 1D). We hypothesized that the lack of PKC enrichment could be due to low expression of PKC-δ in the cell lines examined53 and/or poor capture efficiency of this protein by the Ing-DAyne probe. In support of the latter possibility, we recombinantly expressed PKC-δ in HEK293T cells and failed to observe the labeling of this protein with the Ing-DAyne probe (Figure S3). These data underscore one of the challenges with retroactively introducing photoreactive groups into biologically active small molecules, namely, that, by placing the photoreactive group in a position that does not perturb a functional protein interaction, the photoreactive group may, in some cases, exhibit minimal physical interactions with the protein itself and correspondingly poor UV light induced labeling of the protein. Regardless, our discovery of several other IngMeb-binding proteins suggested that some of these targets may be relevant to the PKC-independent biological activities of IngMeb. We therefore set out to confirm and further characterize some of these targets.
Confirmation of IngMeb Targets Identified by Chemical Proteomics
We verified a representative subset of high-occupancy IngMeb targets by recombinant expression in HEK293T cells. The cDNAs for solute carrier family 25 member 20 (SLC25A20), paraoxonase 2 (PON2), nucleobindin-1 (NUCB1), and saccharopine dehydrogenase-like oxidoreductase (SCCPDH) were transfected into HEK293T cells, and these cells were then treated with Ing-DAyne (10 μM) and increasing concentrations of IngMeb (1), ingenol (2), or DMSO for 30 min, followed by UV cross-linking, lysis, conjugation to TAMRA-N3, and analysis by gel-based profiling. Each protein showed strong labeling by Ing-DAyne compared to mock-transfected control cells, and these labeling signals were blocked in a concentration-dependent manner by IngMeb, but not ingenol (Figure 3E).
IngMeb and IngDsx Inhibit the Activity of SLC25A20 in Human Cells
The necrotic cell death induced by IngMeb in human keratinocyte, HSC-5, and HeLa cells correlates with rupture of the mitochondrial network and parallel cytosolic calcium release, suggesting a mechanism that involves organellar dysfunction.31,54 We noted that over half (14) of the high-occupancy IngMeb targets identified in our chemical proteomic experiments were endoplasmic reticulum (ER) or mitochondria proteins (Figure 3C). One protein that stood out was SLC25A20, also known as CACT, which is a multipass integral membrane protein localized to the inner mitochondrial membrane, where it transports long-chain acylcarnitines into the mitochondrial matrix in exchange for free carnitine. These acylcarnitines are then converted to fatty acyl CoAs by carnitine palmitoyltransferase 2 (CPT-2) to provide fatty acid substrates for β-oxidation.55−57
We identified SLC25A20 as a high-occupancy target of IngMeb in all three tested cell types (Figure 3C,D), as well as a high-occupancy target of IngDsx (Figure S2B), but not ingenol (Figure S1D). In a separate study, we recently discovered a selective and cell-active inhibitor of SLC25A20—EN968724936 (or EN936, compound 21 from reference (33); Figure S4A)—using a fully functionalized fragment (FFF) platform for chemical proteomics.33 We confirmed here that EN936 blocks Ing-DAyne labeling of endogenous SLC25A20 (Figure S4B and Table S1) and, conversely, that IngMeb blocks SLC25A20 labeling with a FFF probe 12 (Figure S4C and Table S1) that is structurally related to EN936 (Figure S4A). These results indicate that IngMeb and EN936 may interact with SLC25A20 at a common site.
We next tested whether IngMeb blocks SLC25A20 function by treating HSC-5 cells with increasing concentrations of the compound (1–200 μM 3 h) and then measuring cellular acylcarnitine content by targeted LC–MS analysis. IngMeb produced a strong concentration-dependent increase in long-chain (C16, C18, C18:1) acylcarnitines in HSC-5 cells (Figure 4A). This pharmacological effect was also observed with IngDsx (Figure 4B), but not ingenol (Figure 4C). Significantly higher levels of C18 and C18:1 acylcarnitines were observed in HSC-5 cells treated with 100 μM IngDsx compared to 100 μM IngMeb, consistent with the more complete competitive blockade of Ing-DAyne labeling of SLC25A20 produced by IngDsx compared to IngMb (Table S1). Importantly, the general PKC activator TPA58 did not alter long-chain acylcarnitines in HSC-5 cells (Figure 4C), and the PKC inhibitor GF 109203X (GF)59 did not block acylcarnitine elevations induced by IngMeb (Figure 4C). Lastly, IngMeb and IngDsx did not alter the cellular amounts of short- or medium-chain (<C14) acylcarnitines (Figures 4A and 4B), which are thought to cross mitochondrial membranes by an SLC25A20-independent mechanism, a hypothesis that is supported by the selective elevations in long-chain acylcarnitines observed in the serum of patients with SLC25A20 deficiency disorders.60,61
Figure 4.

IngMeb and IngDsx block acylcarnitine metabolism and fatty acid oxidation in human cells. (A) IngMeb produces a concentration-dependent increase in long-chain (>C14) acylcarnitine content in HSC-5 cells. Cells were treated with the indicated concentration of IngMeb for 3 h prior to analysis of acylcarnitine content. (B) IngDsx (10, 100 μM 3 h) increases long-chain acylcarnitine content of HSC-5 cells. (C) IngMeb and omeprazole (13, 100 μM each, 3 h), but not ingenol (2, 100 μM) or the pan-PKC agonist 12-O-tetradecanoylphorbal-13-acetate (TPA, 100 μM), elevate long-chain acylcarnitines in HSC-5 cells. For A–C, data represent average values ± SD; n = 3 per group; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 for compound- versus DMSO-treated groups (A–C) and ##p < 0.01 and ####p < 0.0001 for IngMeb vs IngDsx-treated groups (B) or IngMeb vs omeprazole-treated groups (C) using unpaired Student’s t-tests. (D) IngMeb (1), omeprazole (13), and (E) IngDsx (100 μM each) reduce exogenous fatty acid oxidation in HSC-5 cells. Plots depict effect of compounds on spare respiratory capacity, defined as the difference between maximal respiration and basal respiration, normalized to vehicle treated cells. See Figure S5 for full oxygen consumption (OCR) plots. Data represents averaged values ± SD; n = 5–6 per group; **p < 0.01 and ****p < 0.0001 for compound- versus DMSO-treated groups and #p < 0.05 and ##p < 0.01 for IngMeb vs IngDsx-treated groups using unpaired Student’s t-tests.
Previously, we reported that blockade of SLC25A20 activity by EN936 resulted in impaired fatty acid oxidation (FAO) in cells.33 We found that IngMeb and IngDsx also blocked FAO, as HSC-5 cells treated with these compounds and palmitate (as an exogenous fatty acid source) showed substantially reduced oxygen consumption compared to control cells treated with DMSO and palmitate (Figures 4D, 4E, and S5). IngDsx produced significantly more FAO inhibition compared to IngMb, consistent with their respective effects on SLC25A20-dependent acylcarnitine content of HSC-5 cells. Tonazzi and colleagues recently reported that omeprazole, an inhibitor of K+/H+-ATPase used in the treatment of gastric secretions, blocks SLC25A20-mediated transport of acylcarnitines across proteoliposomes.62 We found that omeprazole substantially blocked Ing-DAyne labeling of endogenous SLC25A20 in HSC-5 cells (Figure S4D, Table S1), elevated long-chain acylcarnitines (Figure 4C), and disrupted fatty acid oxidation (Figure 4D). Our data, taken together, point to a highly ligandable site on SLC25A20 where tool compounds (e.g., EN936) and drugs (IngMeb, IngDsx, and omeprazole) converge to inactivate this transporter and impair mitochondrial fatty acid oxidation in cells.
We finally attempted to directly relate blockade of SLC25A20 to the potential cytotoxic action of IngMb in HeLa cells, but found that recombinantly expressing SLC25A20 (Figure S6A) did not shift the half-maximal cytotoxic effect (CC50) of the drug in this cell type (Figure S6B). We did note, however, that expression of SLC25A20 reduced the effect of IngMeb on buildup of acylcarnitines (Figure S6C), further validating SLC25A20 as a functionally perturbed target of IngMeb. While these data might suggest that IngMeb cytotoxicity occurs through primarily an SLC25A20-independent mechanism, we note that the very low potency of cell killing in HeLa cells by IngMeb may not reflect the mode of epiderimal cell death induced by this drug in vivo, where SLC25A20 disruption could still play an important role.
Conclusion
The clinical efficacy of IngMeb against AK is attributed to a dual mechanism of action involving direct cytotoxicity and immune-mediated clearance. Part of this activity appears to reflect agonism of various PKC isoforms, leading to a proinflammatory immune response as well as antiproliferative effects.27,28,63 The cell death induced by IngMeb at higher concentrations similar to those achieved in topical treatments of skin appears to reflect a distinct mechanism, bearing resemblance to necrosis and being associated with a rise in intracellular calcium as well as mitochondrial swelling and rupture.26,31 To begin to understand the molecular interactions of IngMeb that might contribute to the drug’s PKC-independent activities, we designed and prepared a fully functionalized ingenol-based probe containing photoreactive and clickable groups and used this probe to map IngMeb interactions in human cancer cells and keratinocytes by quantitative MS-based proteomics. Several protein targets of IngMeb were identified and verified in follow-up studies, including the mitochondrial acylcarnitine transporter SLC25A20, for which we found that IngMeb serves as an inhibitor to block acylcarnitine uptake into the mitochondria and subsequent mitochondrial FAO. Similar effects were also observed with the clinically relevant analogue IngDsx, but not by the PKC agonist TPA.
The discovery that IngMeb and IngDsx block SLC25A20 could provide a molecular mechanism to explain, at least in part, some of the mitochondrial defects observed in cells treated with high concentrations of this drug. Indeed, global genetic impairment of SLC25A20 alters energy metabolism and results in an inherited lethal syndrome in humans.64 Determining the extent to which such altered mitochondrial function contributes to the therapeutic mechanism of IngMeb and IngDsx represents an important topic for future research. The unchanged CC50 for IngMeb in HeLa cells that overexpress SLC25A20 may indicate that the drug’s high-μM cytotoxicity in cultured human cell lines occurs through distinct targets. Still, this finding does not exclude a role for SLC25A20 inhibition in IngMeb-induced epidermal cell death in vivo, where diverse fatty acid products contribute to keratinocyte differentiation and function, and abnormalities in fatty acid metabolism result in epidermal dysfunction.65 Further, given that several high-occupancy targets were identified for IngMeb, it is possible that functional perturbation of multiple targets (i.e., polypharmacology66) contributes to IngMeb-mediated mitochondrial and cytotoxic effects.
More generally, our findings underscore that IngMeb and IngDsx, at pharmacological concentrations likely achieved in topical application of the drug for treatment of AK, engage several proteins beyond the established interactions with PKCs. Future studies aimed at determining which of these IngMeb-binding proteins may contribute to cytotoxicity could include genetic knockdown or knockout of individual proteins in various IngMeb-sensitive cell types. That many of the detected IngMeb–protein interactions appear to be of only moderate (mid-μM) potency emphasizes an advantage of chemical proteomic methods employing fully functionalized probes, which can covalently trap lower affinity small molecule–protein binding events in cells. On the other hand, our inability to verify PKCs as targets of IngMeb serves as a cautionary reminder that photoreactive probes may also fail to detect pharmacologically relevant targets of small molecules. This challenge may be addressed by placing the photoreactive group at other locations on the bioactive drug, further emphasizing the value of efficient and flexible synthetic routes for the varied modification of structurally complex natural products.
Acknowledgments
Financial support for this work was provided by LEO Pharma and the NIH (CA132630 and S10OD016357). C.G.P. was supported by a Postdoctoral Fellowship PF-14-100-01-CDD from the American Cancer Society. We thank Martin Stahlhut for logistical support in biochemical characterization of various compounds and probes.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00420.
The authors declare the following competing financial interest(s): L.J., B.N., G.D., and J.F. are employees of LEO Pharma.
Supplementary Material
References
- Butler M. S.; Robertson A. A. B.; Cooper M. A. Natural product and natural product derived drugs in clinical trials. Nat. Prod. Rep. 2014, 31 (11), 1612–1661. 10.1039/C4NP00064A. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79 (3), 629–661. 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- Harvey A. L.; Edrada-Ebel R.; Quinn R. J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discovery 2015, 14 (2), 111–129. 10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]
- Kellenberger E.; Hofmann A.; Quinn R. J. Similar interactions of natural products with biosynthetic enzymes and therapeutic targets could explain why nature produces such a large proportion of existing drugs. Nat. Prod. Rep. 2011, 28 (9), 1483–1492. 10.1039/c1np00026h. [DOI] [PubMed] [Google Scholar]
- Lachance H.; Wetzel S.; Kumar K.; Waldmann H. Charting, Navigating, and Populating Natural Product Chemical Space for Drug Discovery. J. Med. Chem. 2012, 55 (13), 5989–6001. 10.1021/jm300288g. [DOI] [PubMed] [Google Scholar]
- Carlson E. E. Natural Products as Chemical Probes. ACS Chem. Biol. 2010, 5 (7), 639–653. 10.1021/cb100105c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feher M.; Schmidt J. M. Property distributions: Differences between drugs, natural products, and molecules from combinatorial chemistry. J. Chem. Inf. Comput. Sci. 2003, 43 (1), 218–227. 10.1021/ci0200467. [DOI] [PubMed] [Google Scholar]
- Patridge E.; Gareiss P.; Kinch M. S.; Hoyer D. An analysis of FDA-approved drugs: natural products and their derivatives. Drug Discovery Today 2016, 21 (2), 204–207. 10.1016/j.drudis.2015.01.009. [DOI] [PubMed] [Google Scholar]
- Li J. W.; Vederas J. C. Drug discovery and natural products: end of an era or an endless frontier?. Science 2009, 325 (5937), 161–165. 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- Koehn F. E.; Carter G. T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discovery 2005, 4 (3), 206–220. 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- Potterat O.; Hamburger M. Concepts and technologies for tracking bioactive compounds in natural product extracts: generation of libraries, and hyphenation of analytical processes with bioassays. Nat. Prod. Rep. 2013, 30 (4), 546–564. 10.1039/c3np20094a. [DOI] [PubMed] [Google Scholar]
- Taunton J.; Collins J. L.; Schreiber S. L. Synthesis of natural and modified trapoxins, useful reagents for exploring histone deacetylase function. J. Am. Chem. Soc. 1996, 118 (43), 10412–10422. 10.1021/ja9615841. [DOI] [Google Scholar]
- Taunton J.; Hassig C. A.; Schreiber S. L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272 (5260), 408–411. 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- Adam G. C.; Vanderwal C. D.; Sorensen E. J.; Cravatt B. F. (−)-FR182877 is a potent and selective inhibitor of carboxylesterase-1. Angew. Chem., Int. Ed. 2003, 42 (44), 5480–5484. 10.1002/anie.200352576. [DOI] [PubMed] [Google Scholar]
- Kotake Y.; Sagane K.; Owa T.; Mimori-Kiyosue Y.; Shimizu H.; Uesugi M.; Ishihama Y.; Iwata M.; Mizui Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007, 3 (9), 570–575. 10.1038/nchembio.2007.16. [DOI] [PubMed] [Google Scholar]
- Wang G. L.; Shang L. B.; Burgett A. W. G.; Harran P. G.; Wang X. D. Diazonamide toxins reveal an unexpected function for ornithine delta-amino transferase in mitotic cell division. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (7), 2068–2073. 10.1073/pnas.0610832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub I.; Sieber S. A. beta-lactams as selective chemical probes for the in vivo labeling of bacterial enzymes involved in cell wall biosynthesis, antibiotic resistance, and virulence. J. Am. Chem. Soc. 2008, 130 (40), 13400–13409. 10.1021/ja803349j. [DOI] [PubMed] [Google Scholar]
- Eirich J.; Burkhart J. L.; Ullrich A.; Rudolf G. C.; Vollmar A.; Zahler S.; Kazmaier U.; Sieber S. A. Pretubulysin derived probes as novel tools for monitoring the microtubule network via activity-based protein profiling and fluorescence microscopy. Mol. BioSyst. 2012, 8 (8), 2067–2075. 10.1039/c2mb25144b. [DOI] [PubMed] [Google Scholar]
- Hartwell J. L. Plants Used against Cancer - a Survey. Lloydia 1970, 33 (1), 97–194. [PubMed] [Google Scholar]
- Appendino G.; Szallasi A. Euphorbium: modern research on its active principle, resiniferatoxin, revives an ancient medicine. Life Sci. 1997, 60 (10), 681–696. 10.1016/S0024-3205(96)00567-X. [DOI] [PubMed] [Google Scholar]
- Ramsay J. R.; Suhrbier A.; Aylward J. H.; Ogbourne S.; Cozzi S. J.; Poulsen M. G.; Baumann K. C.; Welburn P.; Redlich G. L.; Parsons P. G. The sap from Euphorbia peplus is effective against human nonmelanoma skin cancers. Br. J. Dermatol. 2011, 164 (3), 633–636. 10.1111/j.1365-2133.2010.10184.x. [DOI] [PubMed] [Google Scholar]
- Keating G. M. Ingenol mebutate gel 0.015% and 0.05%: in actinic keratosis. Drugs 2012, 72 (18), 2397–2405. 10.2165/11470090-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Goldenberg G.; Perl M. Actinic keratosis: update on field therapy. J. Clin. Aesthetic Dermatol. 2014, 7 (10), 28–31. [PMC free article] [PubMed] [Google Scholar]
- Gillespie S. K.; Zhang X. D.; Hersey P. Ingenol 3-angelate induces dual modes of cell death and differentially regulates tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in melanoma cells. Mol. Cancer Ther. 2004, 3 (12), 1651–1658. [PubMed] [Google Scholar]
- Rosen R. H.; Gupta A. K.; Tyring S. K. Dual mechanism of action of ingenol mebutate gel for topical treatment of actinic keratoses: Rapid lesion necrosis followed by lesion-specific immune response. J. Am. Acad. Dermatol. 2012, 66 (3), 486–493. 10.1016/j.jaad.2010.12.038. [DOI] [PubMed] [Google Scholar]
- Ogbourne S. M.; Suhrbier A.; Jones B.; Cozzi S. J.; Boyle G. M.; Morris M.; McAlpine D.; Johns J.; Scott T. M.; Sutherland K. P.; et al. Antitumor activity of 3-ingenyl angelate: plasma membrane and mitochondrial disruption and necrotic cell death. Cancer Res. 2004, 64 (8), 2833–2839. 10.1158/0008-5472.CAN-03-2837. [DOI] [PubMed] [Google Scholar]
- Kedei N.; Lundberg D. J.; Toth A.; Welburn P.; Garfield S. H.; Blumberg P. M. Characterization of the interaction of ingenol 3-angelate with protein kinase C. Cancer Res. 2004, 64 (9), 3243–3255. 10.1158/0008-5472.CAN-03-3403. [DOI] [PubMed] [Google Scholar]
- Hampson P.; Chahal H.; Khanim F.; Hayden R.; Mulder A.; Assi L. K.; Bunce C. M.; Lord J. M. PEP005, a selective small-molecule activator of protein kinase C, has potent antileukemic activity mediated via the delta isoform of PKC. Blood 2005, 106 (4), 1362–1368. 10.1182/blood-2004-10-4117. [DOI] [PubMed] [Google Scholar]
- Challacombe J. M.; Suhrbier A.; Parsons P. G.; Jones B.; Hampson P.; Kavanagh D.; Rainger G. E.; Morris M.; Lord J. M.; Le T. T.; et al. Neutrophils are a key component of the antitumor efficacy of topical chemotherapy with ingenol-3-angelate. J. Immunol. 2006, 177 (11), 8123–8132. 10.4049/jimmunol.177.11.8123. [DOI] [PubMed] [Google Scholar]
- Emmert S.; Haenssle H. A.; Zibert J. R.; Schon M.; Hald A.; Hansen M. H.; Litman T.; Schon M. P. Tumor-Preferential Induction of Immune Responses and Epidermal Cell Death in Actinic Keratoses by Ingenol Mebutate. PLoS One 2016, 11 (9), e0160096. 10.1371/journal.pone.0160096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahlhut M.; Bertelsen M.; Hoyer-Hansen M.; Svendsen N.; Eriksson A. H.; Lord J. M.; Scheel-Toellner D.; Young S. P.; Zibert J. R. Ingenol mebutate: induced cell death patterns in normal and cancer epithelial cells. J. Drugs Dermatol 2012, 11 (10), 1181–1192. [PubMed] [Google Scholar]
- Li L. W.; Shukla S.; Lee A.; Garfield S. H.; Maloney D. J.; Ambudkar S. V.; Yuspa S. H. The Skin Cancer Chemotherapeutic Agent Ingenol-3-Angelate (PEP005) Is a Substrate for the Epidermal Multidrug Transporter (ABCB1) and Targets Tumor Vasculature. Cancer Res. 2010, 70 (11), 4509–4519. 10.1158/0008-5472.CAN-09-4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker C. G.; Galmozzi A.; Wang Y.; Correia B. E.; Sasaki K.; Joslyn C. M.; Kim A. S.; Cavallaro C. L.; Lawrence R. M.; Johnson S. R.; et al. Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 2017, 168 (3), 527–541.e529. 10.1016/j.cell.2016.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horning B. D.; Suciu R. M.; Ghadiri D. A.; Ulanovskaya O. A.; Matthews M. L.; Lum K. M.; Backus K. M.; Brown S. J.; Rosen H.; Cravatt B. F. Chemical Proteomic Profiling of Human Methyltransferases. J. Am. Chem. Soc. 2016, 138 (40), 13335–13343. 10.1021/jacs.6b07830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulce J. J.; Cognetta A. B.; Niphakis M. J.; Tully S. E.; Cravatt B. F. Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat. Methods 2013, 10 (3), 259–264. 10.1038/nmeth.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. Q.; Hao P. L.; Li L.; Tan C. Y. J.; Cheng X. M.; Chen G. Y. J.; Sze S. K.; Shen H. M.; Yao S. Q. Design and Synthesis of Minimalist Terminal Alkyne-Containing Diazirine Photo-Crosslinkers and Their Incorporation into Kinase Inhibitors for Cell- and Tissue-Based Proteome Profiling. Angew. Chem., Int. Ed. 2013, 52 (33), 8551–8556. 10.1002/anie.201300683. [DOI] [PubMed] [Google Scholar]
- Li Z. Q.; Wang D. Y.; Li L.; Pan S. J.; Na Z. K.; Tan C. Y. J.; Yao S. Q. ″Minimalist″ Cyclopropene-Containing Photo-Cross-Linkers Suitable for Live-Cell Imaging and Affinity-Based Protein Labeling. J. Am. Chem. Soc. 2014, 136 (28), 9990–9998. 10.1021/ja502780z. [DOI] [PubMed] [Google Scholar]
- Niphakis M. J.; Lum K. M.; Cognetta A. B.; Correia B. E.; Ichu T. A.; Olucha J.; Brown S. J.; Kundu S.; Piscitelli F.; Rosen H.; et al. A Global Map of Lipid-Binding Proteins and Their Ligandability in Cells. Cell 2015, 161 (7), 1668–1680. 10.1016/j.cell.2015.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H. B.; Zhang C. J.; Chen G. Y. J.; Yao S. Q. Cell-Based Proteome Profiling of Potential Dasatinib Targets by Use of Affinity-Based Probes. J. Am. Chem. Soc. 2012, 134 (6), 3001–3014. 10.1021/ja208518u. [DOI] [PubMed] [Google Scholar]
- Su Y.; Pan S. J.; Li Z. Q.; Li L.; Wu X. Y.; Hao P. L.; Sze S. K.; Yao S. Q. Multiplex Imaging and Cellular Target Identification of Kinase Inhibitors via an Affinity-Based Proteome Profiling Approach. Sci. Rep. 2015, 10.1038/srep07724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective ″ligation″ of azides and terminal alkynes. Angew. Chem., Int. Ed. 2002, 41 (14), 2596–2599. . [DOI] [PubMed] [Google Scholar]
- Bertelsen M.; Stahlhut M.; Grue-Sorensen G.; Liang X.; Christensen G. B.; Skak K.; Engell K. M.; Hogberg T. Ingenol Disoxate: A Novel 4-Isoxazolecarboxylate Ester of Ingenol with Improved Properties for Treatment of Actinic Keratosis and Other Non-Melanoma Skin Cancers. Dermatol Ther (Heidelb) 2016, 6 (4), 599–626. 10.1007/s13555-016-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opferkuch H. J.; Hecker E. On the Active Principles of the Spurge Family (Euphorbiaceae) 0.4. Skin Irritant and Tumor Promoting Diterpene Esters from Euphorbia Ingens E-Mey. J. Cancer Res. Clin. Oncol. 1982, 103 (3), 255–268. 10.1007/BF00409701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X.; Grue-Sorensen G.; Mansson K.; Vedso P.; Soor A.; Stahlhut M.; Bertelsen M.; Engell K. M.; Hogberg T. Syntheses, biological evaluation and SAR of ingenol mebutate analogues for treatment of actinic keratosis and non-melanoma skin cancer. Bioorg. Med. Chem. Lett. 2013, 23 (20), 5624–5629. 10.1016/j.bmcl.2013.08.038. [DOI] [PubMed] [Google Scholar]
- Winkler J. D.; Hong E. C.; Bahador A.; Kazanietz M. G.; Blumberg P. M. Synthesis of Ingenol Analogs with Affinity for Protein-Kinase-C. Bioorg. Med. Chem. Lett. 1993, 3 (4), 577–580. 10.1016/S0960-894X(01)81232-4. [DOI] [Google Scholar]
- Winkler J. D.; Kim S. H.; Harrison S.; Lewin N. E.; Blumberg P. M. Synthesis and biological evaluation of highly functionalized analogues of ingenol. J. Am. Chem. Soc. 1999, 121 (2), 296–300. 10.1021/ja9741842. [DOI] [Google Scholar]
- Grue-Sorensen G.; Liang X. F.; Mansson K.; Vedso P.; Sorensen M. D.; Soor A.; Stahlhut M.; Bertelsen M.; Engell K. M.; Hogberg T. Synthesis, biological evaluation and SAR of 3-benzoates of ingenol for treatment of actinic keratosis and non-melanoma skin cancer. Bioorg. Med. Chem. Lett. 2014, 24 (1), 54–60. 10.1016/j.bmcl.2013.11.073. [DOI] [PubMed] [Google Scholar]
- Jorgensen L.; McKerrall S. J.; Kuttruff C. A.; Ungeheuer F.; Felding J.; Baran P. S. 14-step synthesis of (+)-ingenol from (+)-3-carene. Science 2013, 341 (6148), 878–882. 10.1126/science.1241606. [DOI] [PubMed] [Google Scholar]
- Liang X. F.; Grue-Sorensen G.; Petersen A. K.; Hogberg T. Semisynthesis of Ingenol 3-Angelate (PEP005): Efficient Stereoconservative Angeloylation of Alcohols. Synlett 2012, 23, 2647–2652. 10.1055/s-0032-1317415. [DOI] [Google Scholar]
- Ong S. E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 1 (5), 376–386. 10.1074/mcp.M200025-MCP200. [DOI] [PubMed] [Google Scholar]
- Boersema P. J.; Raijmakers R.; Lemeer S.; Mohammed S.; Heck A. J. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 2009, 4 (4), 484–494. 10.1038/nprot.2009.21. [DOI] [PubMed] [Google Scholar]
- Hsu J. L.; Huang S. Y.; Chow N. H.; Chen S. H. Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 2003, 75 (24), 6843–6852. 10.1021/ac0348625. [DOI] [PubMed] [Google Scholar]
- D’Costa A. M.; Robinson J. K.; Maududi T.; Chaturvedi V.; Nickoloff B. J.; Denning M. F. The proapoptotic tumor suppressor protein kinase C-delta is lost in human squamous cell carcinomas. Oncogene 2006, 25 (3), 378–386. 10.1038/sj.onc.1209065. [DOI] [PubMed] [Google Scholar]
- Cozzi S. J.; Le T. T.; Ogbourne S. M.; James C.; Suhrbier A. Effective treatment of squamous cell carcinomas with ingenol mebutate gel in immunologically intact SKH1 mice. Arch. Dermatol. Res. 2013, 305 (1), 79–83. 10.1007/s00403-012-1270-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indiveri C.; Tonazzi A.; Palmieri F. Identification and purification of the carnitine carrier from rat liver mitochondria. Biochim. Biophys. Acta, Bioenerg. 1990, 1020 (1), 81–86. 10.1016/0005-2728(90)90096-M. [DOI] [PubMed] [Google Scholar]
- Indiveri C.; Tonazzi A.; Palmieri F. The Reconstituted Carnitine Carrier from Rat-Liver Mitochondria - Evidence for a Transport Mechanism Different from That of the Other Mitochondrial Translocators. Biochim. Biophys. Acta, Biomembr. 1994, 1189 (1), 65–73. 10.1016/0005-2736(94)90281-X. [DOI] [PubMed] [Google Scholar]
- Palmieri F. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol. Aspects Med. 2013, 34 (2–3), 465–484. 10.1016/j.mam.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Castagna M.; Takai Y.; Kaibuchi K.; Sano K.; Kikkawa U.; Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem. 1982, 257 (13), 7847–7851. [PubMed] [Google Scholar]
- Toullec D.; Pianetti P.; Coste H.; Bellevergue P.; Grandperret T.; Ajakane M.; Baudet V.; Boissin P.; Boursier E.; Loriolle F.; et al. The Bisindolylmaleimide Gf-109203x Is a Potent and Selective Inhibitor of Protein-Kinase-C. J. Biol. Chem. 1991, 266 (24), 15771–15781. [PubMed] [Google Scholar]
- Violante S.; Ijlst L.; Te Brinke H.; Tavares de Almeida I.; Wanders R. J.; Ventura F. V.; Houten S. M. Carnitine palmitoyltransferase 2 and carnitine/acylcarnitine translocase are involved in the mitochondrial synthesis and export of acylcarnitines. FASEB J. 2013, 27 (5), 2039–2044. 10.1096/fj.12-216689. [DOI] [PubMed] [Google Scholar]
- Rubio-Gozalbo M. E.; Bakker J. A.; Waterham H. R.; Wanders R. J. Carnitine-acylcarnitine translocase deficiency, clinical, biochemical and genetic aspects. Mol. Aspects Med. 2004, 25 (5–6), 521–532. 10.1016/j.mam.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Tonazzi A.; Eberini I.; Indiveri C. Molecular Mechanism of Inhibition of the Mitochondrial Carnitine/Acylcarnitine Transporter by Omeprazole Revealed by Proteoliposome Assay, Mutagenesis and Bioinformatics. PLoS One 2013, 8 (12), e82286. 10.1371/journal.pone.0082286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiberger S. N.; Cheng P. F.; Iotzova-Weiss G.; Neu J.; Liu Q.; Dziunycz P.; Zibert J. R.; Dummer R.; Skak K.; Levesque M. P.; et al. Ingenol Mebutate Signals via PKC/MEK/ERK in Keratinocytes and Induces Interleukin Decoy Receptors IL1R2 and IL13RA2. Mol. Cancer Ther. 2015, 14 (9), 2132–2142. 10.1158/1535-7163.MCT-15-0023-T. [DOI] [PubMed] [Google Scholar]
- Indiveri C.; Iacobazzi V.; Tonazzi A.; Giangregorio N.; Infantino V.; Convertini P.; Console L.; Palmieri F. The mitochondrial carnitine/acylcarnitine carrier: function, structure and physiopathology. Mol. Aspects Med. 2011, 32 (4–6), 223–233. 10.1016/j.mam.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Rizzo W. B. Fatty aldehyde and fatty alcohol metabolism: review and importance for epidermal structure and function. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2014, 1841 (3), 377–389. 10.1016/j.bbalip.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A. S.; Zhang S. Polypharmacology: drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6 (1), 41–47. 10.1586/ecp.12.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.