

Abstract
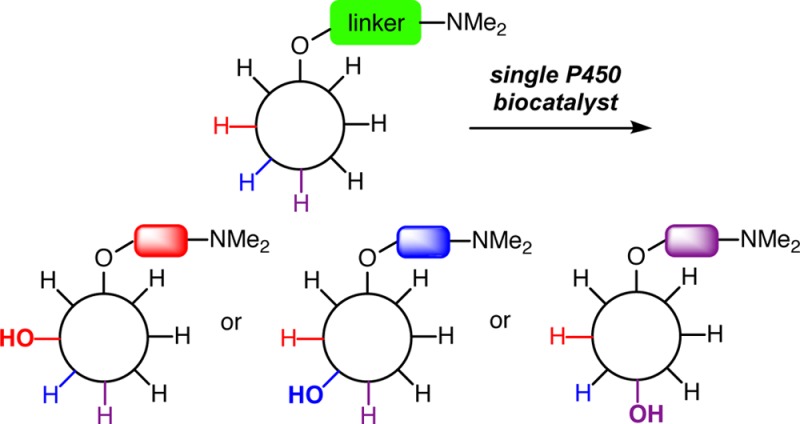
The diversification of late stage synthetic intermediates provides significant advantages in efficiency in comparison to conventional linear approaches. Despite these advantages, accessing varying ring scaffolds and functional group patterns from a common intermediate poses considerable challenges using existing methods. The combination of regiodivergent nickel-catalyzed C–C couplings and site-selective biocatalytic C–H oxidations using the cytochrome P450 enzyme PikC addresses this problem by enabling a single late-stage linear intermediate to be converted to macrolactones of differing ring size and with diverse patterns of oxidation. The approach is made possible by a novel strategy for site-selective biocatalytic oxidation using a single biocatalyst, with site selectivity being governed by a temporarily installed directing group. Site selectivities of C–H oxidation by this directed approach can overcome positional bias due to C–H bond strength, acidity, inductive influences, steric accessibility, or immediate proximity to the directing group, thus providing complementarity to existing approaches.
Short abstract
A new strategy for tuning site selectivity in biocatalytic oxidations is paired with catalytic regiodivergent macrocyclizations to provide a versatile approach for structural diversification.
Introduction
Diversifying chemical structures at a late stage of a synthetic sequence provides an efficient way to tailor molecular properties. Effective strategies typically involve either modifying the framework of a core structural scaffold or introducing new substituents through functional group interconversions and C–H functionalization methods. Core scaffold modifications are often achieved by accessing multiple cycloaddition pathways available to a single substrate or by catalyzing a thermodynamically driven rearrangement such as transesterification, sigmatropic rearrangement, or ring expansion or contraction.1−5 Alternatively, the introduction of new substituents can be accomplished through the interchange of functional groups after a fully functionalized core structure is accessed. A disadvantage of this approach is that assembly of the late-stage common intermediate often requires a lengthy linear synthesis and provides structures that are closely related with an identical core framework. Site-selective C–H functionalization methods provide greater overall efficiency,6−12 but limitations in both site selectivity and functional group compatibility (i.e., alkenes or basic amines) remain as a considerable challenge in complex environments with multiple potentially reactive centers. Biocatalytic C–H oxidations can provide impressive levels of site selectivity and functional-group compatibility, but this advantage typically requires extensive optimization of enzyme structure in order to fundamentally change the site of functionalization.
Considering the array of available diversification approaches described above, a significant gap exists in synthetic strategies that involve late-stage scaffold variation combined with a versatile site-selective C–H functionalization approach. By first converting a single substrate into two or more molecular scaffolds, and then accomplishing site-selective diversification of each of the obtained structural motifs through C–H functionalization, the rapid assembly of structural analogues differing in both the core scaffold and peripheral functionality would become possible (Figure 1). Such an approach offers the simplicity of accessing a linear substrate with minimal functionality as the point of divergence and would provide a powerful strategy for the rapid assembly of molecular diversity and complexity. In this report, we describe proof-of-concept experiments toward this objective with the demonstration of a new strategy that enables regiodivergent cyclization paired with site-selective biocatalytic oxidations. This approach provides access to a collection of 11- and 12-membered macrocycles with varying oxidation patterns.
Figure 1.

Conceptual framework for late-stage diversification. Regiodivergent scaffold assembly followed by site-selective C–H oxidation presents a versatile strategy for accessing diverse products from a single late-stage intermediate.
Results and Discussion
A key goal of this study was to develop a versatile oxidation method that utilizes a single biocatalyst to provide multiple oxidation patterns selectively, enabled by the design of directing groups temporarily installed on the substrate. To address this objective, preparation of test substrates by a nickel-catalyzed reductive macrocyclization process allows access to either regiochemical outcome (i.e., endo- or exocyclization) in macrolactone assembly by tailoring the ligand structure and reaction conditions (Figure 2A).13,14 This regiodivergent catalytic process provides an ideal approach for accessing different ring sizes of macrolactone substrates that possess multiple unactivated methylene (CH2) groups for exploration of new strategies for site-selective C–H oxidation. While impressive strides have been made in strategies that enable site-selective oxidations in multiring structures with well-defined conformations,15−18 selective access to multiple patterns of oxidation in macrocyclic compounds represents a challenge unmet by previous approaches. In order to address this limitation, the straightforward modular assembly of an enantiopure ynal precursor 1 was followed by regiodivergent and highly diastereoselective reductive macrocyclization to afford macrocycles 2 and 3. This approach, governed largely by steric properties of the N-heterocyclic carbene (NHC) ligand, enables access to a large array of linear and macrocyclic structures through predictable reversals of regiochemistry in the C–C bond-forming step.
Figure 2.

Synergy of small molecule catalysis and biocatalysis in late-stage diversification. (A) Regiodivergent reductive macrocyclization enabling access to 11- and 12-membered macrolactones. (B) Modular strategy for enabling site-selective biocatalytic oxidation with the potential to alter site of oxidation based on linker design.
Macrocyclic substrates 2 and 3 provide a challenging context for developing site-selective C–H oxidations on different scaffolds that possess a multitude of similarly reactive C–H bonds. To address this challenge, we examined the utility of an engineered fusion protein of the bacterial cytochrome P450 enzyme PikC. In prior work, engineered mutants of the PikC enzyme displayed excellent site selectivity in oxidations of amine-containing substrates closely related to the endogenous macrolide substrate YC-1719 or of smaller, structurally compact substrates such as simple terpenes (e.g., menthol).20 This strategy typically provided either a single major product or the unselective generation of numerous products. Therefore, a general strategy for changing the site of oxidation while preserving high selectivity represents a major unmet need from the prior approaches. The conformational flexibility of macrocycles such as 2 and 3 that possess six or more methylene groups poses a tremendous challenge in site-selective oxidation that has not yet been efficiently addressed by chemical or biological catalysis. With such subtle differences in the reactivity of similar methylene groups, the use of directing groups presents the best opportunity for achieving tunable site selectivity. While recent developments in enzyme engineering have demonstrated remarkable advances in organic synthesis,21−24 the use of simple directing groups to achieve site selectivity in enzymatic transformations is greatly underutilized. Structural variations in rigid directing groups have recently demonstrated enormous promise in remote C–H functionalizations using small molecule catalysts.25,26 Despite pioneering early work from Breslow in designing biomimetic site-selective functionalizations based on template design,27,28 this approach has not previously been developed as a strategy to enable multiple selective outcomes in an enzyme-catalyzed process.
Previous studies of PikC-catalyzed oxidations illustrated that substrates possessing numerous contiguous methylene groups led to unselective oxidation when substrate engineering approaches were attempted.29 Thus, although the presence of the aminosugar desosamine is essential for binding and selectivity of the PikC enzyme toward its native macrolide substrates, at least seven oxidized products were generated when using a desosaminylated, but unfunctionalized, 12-membered macrocyclic substrate. While the use of simple aromatic spacers avoids the cumbersome installation of aminosugars,19 we recognized the need for a more versatile strategy that enables an anchoring basic amine to be covalently attached through a linker. The method should provide a rapid and high-throughput synthesis, facile length and shape variations of the linker component, and efficient chemoselective removal of the linker following C–H oxidation. These criteria can all be addressed by the modular connection of hydroxyl-containing substrates, amino acid-derived azido acids, and acetylenic amines (Figure 2B). Through the appropriate choice of spacer elements S1 and S2 and access to regiochemistry reversal in azide–alkyne click cycloadditions,30,31 a large number of linker structures are readily available. Notably, click reactions are typically utilized as a simple means to connect two structures without regard to the precise structural features of the linking functionality. However, in our approach, the precise shape and geometry of the unit assembled by the azide–alkyne cycloaddition is an integral feature required for success of the strategy.
Derivatives of 11-membered substrate 2 were first examined in the click anchor strategy for site-selective biocatalytic oxidation (Table 1). The engineered PikC fusion protein PikCD50ND176QE246A-RhFRED, which was designed by molecular dynamics-guided analysis20 utilizing crystallographic insights from the endogenous substrate YC-17,32 was used in our studies. The active site residues that were predicted to lead to unproductive substrate binding orientations were mutated in order to promote formation of the closed, active form of the enzyme. A collection of 13 triazole anchors was then assembled from seven azido acids and two amines (full listing in Table S1). Triazoles examined include those derived from azido acids possessing and lacking chirality, ortho, meta, and para-spaced benzene spacers, and both 1,4- and 1,5-triazole regioisomers. Following installation of the triazole anchor to substrate 2, analytical scale biocatalytic oxidations on derivatives of 4 were performed, and promising anchors were selected from LCMS analysis based on percent conversion and selectivity for a major product. Representative cases were then conducted on a 30–60 mg preparative scale, products were isolated by preparative HPLC, and the structure and stereochemistry of major products were elucidated by NMR. Isolated monohydroxylated products were then used as internal standards in the analysis of additional analytical scale experiments in order to identify anchors that enable synthesis of a different hydroxylated product.
Table 1. Scope of Biocatalytic Macrocycle Oxidationa.
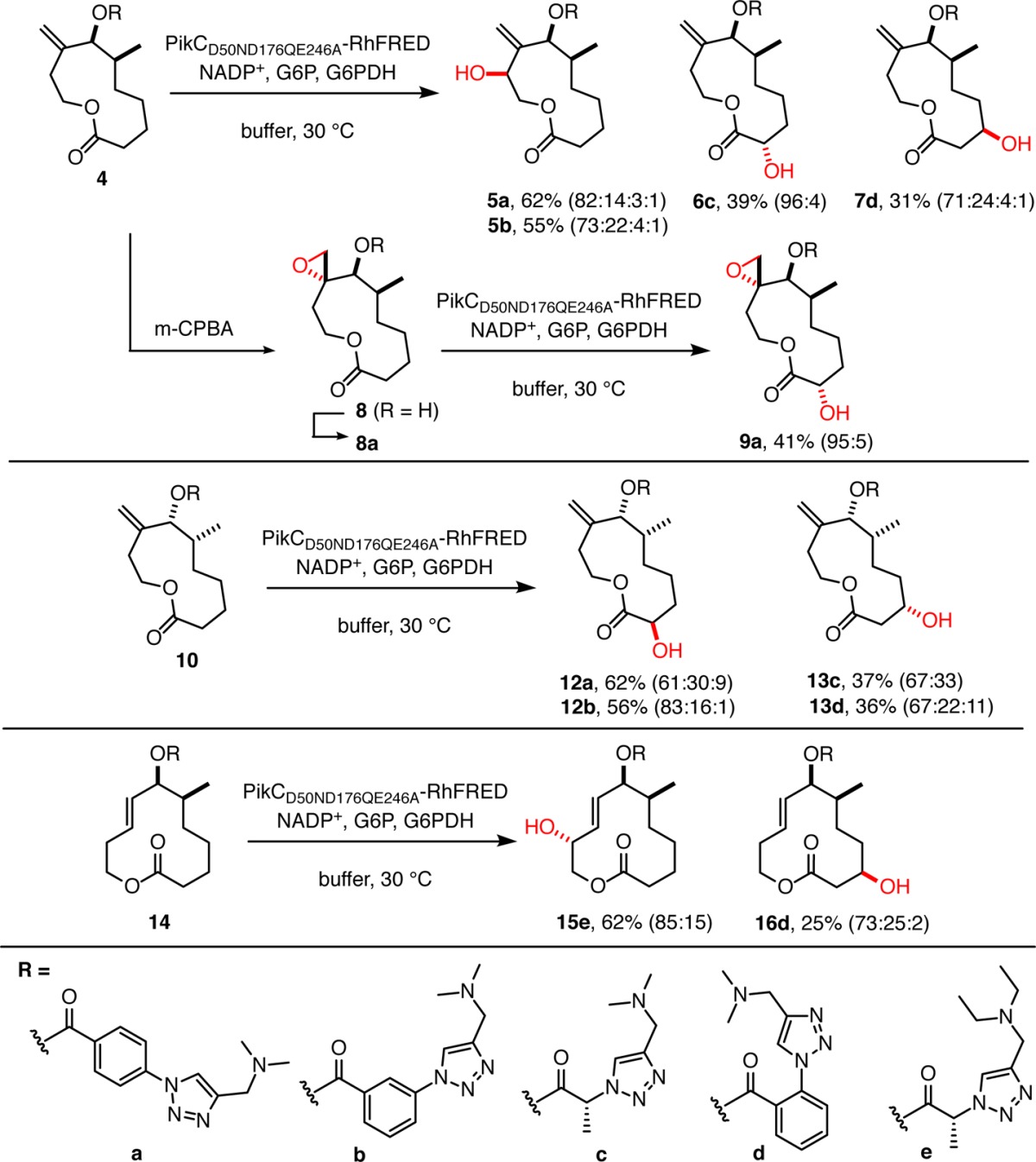
R groups are depicted by the letters a–e shown at bottom. Major product is depicted in the table. Unless otherwise noted, percent yield refers to conversion to monohydroxylated products in preparative experiments, and ratios of monohydroxylated products are given in parentheses. Products 5b and 12b were evaluated in analytical scale experiments with comparison to authentic standards. Analysis of the identity of minor products is provided in the Supporting Information.
Using this approach, three different monohydroxylated products (5, 6, and 7) were obtained from substrate 4. Notably, triazoles possessing para- or meta-substituted benzene spacers a or b enable oxidation of allylic protons proximal to the point of anchor connection to provide product 5. Alternatively, selection of anchors that possess a shorter linker motif enables oxidation of the distal region of the substrate. For example, using alanine-derived anchor c leads to the production of product 6c with oxidation α- to the carbonyl. In contrast, anchor d with an ortho-substituted benzene spacer leads to product 7d with oxidation β to the carbonyl. No evidence for epoxidation or amine oxidation was observed in PikC-catalyzed oxidations, demonstrating desirable chemoselectivity features of C–H oxidation. Epoxide 8 is cleanly obtained (when R = H) either in reactions with m-CPBA as the electrophilic oxidant or by using the White C–H oxidation catalyst,18 demonstrating orthogonal chemoselectivity of the PikC oxidation method. By installing anchor a, biocatalytic oxidation of 8a then cleanly affords epoxy alcohol 9a. The site-selective oxidation to provide 5, 6, 7, or 9 demonstrates that this method is useful for oxidizing C–H bonds that are either proximal or distal to the directing group. No conversion was noted when the dimethylamino group of 4a was replaced by an isopropyl or imidazole functionality, thus validating the directing capability of the dimethylamino group. The versatile directing capability of the triazole linkers paired with PikC catalysis thus does not directly correlate with C–H bond strength, acidity, inductive influences, steric accessibility, or immediate proximity to the directing group. Instead, the enzyme active site conformation matched with the structure, stereochemistry, and conformation of both the triazole anchor and the substrate overrides these influences.
To probe the interrelationship of the stereochemical features of the active site residues, triazole anchor, and macrocyclic substrate, the enantiomeric form of the macrocycle (substrate 10) was examined with the same biocatalyst and triazole anchors a–d. By using the meta- and para-substituted anchors a and b that enabled oxidation of the proximal allylic methylene in substrate 4, these same anchors instead led to oxidation of distal protons α to the carbonyl to provide products 12a and 12b. Use of an alanine-derived anchor c or the ortho-substituted anchor d afforded 13c and 13d as the major product via oxidation β to the carbonyl. These results demonstrate that optimal anchor structure will vary from substrate to substrate, even within an enantiomeric series, thus adding appeal to the simple and potentially high throughput access to linkers in the approach described.
In taking advantage of the regiodivergent access to macrocycles described, the 12-membered structure derived from endocyclization was also examined in biocatalytic oxidations. In this case, two different oxidized products were obtained from substrate 14. The alanine anchor e provided access to allylic oxidation product 15e, and a diethylamino anchor was employed in this case to minimize N-demethylation, which was observed as a minor byproduct in some instances. Alternatively, using the ortho-linked benzene spacer d, major product 16d was obtained via oxidation β to the carbonyl. These results collectively illustrate that a variety of monohydroxylated compounds of macrocycles varying in ring size may be efficiently obtained starting from a single, easily accessible ynal substrate.
As described above, the structure, stereochemistry, and conformation of the substrate, linker, and enzyme active site all play important roles in determining site selectivity of the C–H oxidations. In order to understand these influences and to add a predictive component to the strategy, a combined density functional theory (DFT) and molecular dynamics (MD) computational study was undertaken. DFT was used to compute the intrinsic reaction barriers to C–H abstraction at C3 allylic and C10 alpha positions of 4. The R group shown in 4 was truncated to a methyl, and conformations of the macrocycle were explored with molecular mechanics.33 These conformers were then optimized, and single point energy calculations were performed with DFT. The lowest energy conformer was identified along with a second low energy conformer only 0.9 kcal/mol higher in energy. All other conformers were >2 kcal/mol higher in energy than conformer 1. The barriers to C–H abstraction at C3 and C10 were computed for these two low energy conformers and are shown in Figure 3A. The barriers to abstraction of the hydrogens shown in purple and green are the lowest for each site of both conformers, and correspond to the hydrogens that are abstracted to form 5 and 6. Figures 3B and 3C show the transition structures for abstraction of the C3 purple (equatorial) and C3 yellow (axial) hydrogens, respectively, from conformer 1. It is clear from close inspection of the transition structures that the abstraction of the equatorial hydrogen (purple) benefits from developing conjugation with the neighboring exocyclic alkene. The axial hydrogen (yellow) is perpendicular to the π-system and cannot benefit from the allylic stabilization. A similar conjugation effect is seen with the C–H abstractions at C10. The green hydrogen is in conjugation with the carbonyl group (Figure 3D), but the ring must distort for the blue hydrogen to benefit from the same stabilization. The substrate prefers a dihedral of 16.1° between the exocyclic alkene and the carbonyl. In Figure 3E, this dihedral (shown with blue atoms) expanded to 55.6° in the transition structure for the abstraction of the blue hydrogen.
Figure 3.

(A) Lowest energy conformers of a model of structure 4, with DFT barriers (kcal/mol) to C–H abstraction at C3 and C10. (B) Transition structure of C3 (purple) hydrogen abstraction. (C) Transition structure of C3 (yellow) hydrogen. (D) Transition structure of C10 (green) hydrogen abstraction. (E) Transition structure of C10 (blue) hydrogen abstraction.
Conformer 1 was used for molecular dynamics simulations. Linker a and linker c were each attached to the macrocycle and docked into the active site of the PikCD50N/D176Q/E246A, and 500 ns trajectories were run on each of these complexes using Amber. Over the course of the simulations, both conformer 1 and conformer 2 are observed. The salt bridges between the terminal amines of the substrates and residues E84 and E95 are critical for substrate binding and reactions as studied in previous work by our groups.20 The same salt bridges are also observed in MD simulations of 4a and 4c. Though the salt bridges are similar for both linkers, different linkers lead to different substrate binding modes and oxidation patterns. The goal of our molecular dynamics studies here is to determine how the length, flexibility, and stereochemistry of these linkers allow for the different oxidation patterns. As shown in the snapshot of Figures 4A and 4B, linker a places C3 closest to the active iron–oxo. C2 is also placed close to the iron–oxo (3.7 Å averaged over the 500 ns simulation), but the computed barriers to abstraction at C2 are +5 kcal/mol higher in energy than at C3 (Figure S10). Alternatively, linker c places C10 closest to the iron–oxo species. Figures 4C and 4E show the average distance of each hydrogen to the iron–oxo oxygen over the course of the 500 ns simulations. Figures 4D and 4F are plots of the Hsubstrate–Oiron distances vs C–H–O angles over the course of the trajectories with linkers a and c respectively. Figures 4C and 4D reveal that the purple hydrogen is closest to the DFT-computed transition state geometry. Collectively these data show that the purple, equatorial hydrogen of C3 is the most accessible to the iron–oxo in the enzyme and intrinsically more reactive. Figures 4E and 4F show that the blue hydrogen is closest to the DFT-computed transition state geometry, but abstraction of this hydrogen is disfavored by up to 1.1 kcal/mol. While linker c places C10 closest to the iron–oxo, the intrinsic reactivity of the equatorial hydrogen (green) overrides the proximity of the axial hydrogen (blue). MD simulations of 4d and 14e were also able to reproduce the stereo preference of their corresponding major products, 7d and 15e respectively (Figures S11–S13). The combined DFT and MD study provides an explanation for the observed regiochemistry and stereoselectivity of these PikC-catalyzed hydroxylations.
Figure 4.

(A) Snapshot of MD trajectory of 4 with linker a overlaid with a snapshot of 4 with linker c. (B) Closeup of Figure 3A snapshot with average C–OFe distances shown. (C) Snapshot of 4 with linker a with average H–O distances shown. (D) Plot of hydrogen (of substrate C3) to oxygen (of iron–oxo) distances vs C–H–O angles throughout the MD trajectory, with transition state (TS) geometry shown in red. (E) Snapshot of 4 with linker c with average H–O distances shown. (F) Plot of hydrogen (of substrate C10) to oxygen (of iron–oxo) distances and C–H–O angles throughout the MD trajectory, with TS geometry shown in red.
Conclusions
In summary, this study describes a versatile strategy for the rapid generation of a collection of macrocyclic compounds that differ in ring size and oxidation pattern. Starting from a single linear substrate, catalyst control in a regiodivergent macrocyclization is paired with site-selective C–H oxidations enabled by the synergy of a computationally designed engineered cytochrome P450 catalyst and a tailored amine-containing directing group. This new approach for controlling selectivities of biocatalytic oxidations enables a single P450 biocatalyst to be utilized for accessing multiple oxidation patterns on flexible macrocyclic motifs containing an array of oxidizable bonds. Computational analysis uncovers a complex synergy of macrocycle conformation, inherent reactivity of various C–H bonds, linker structure, and substrate-active site interactions in controlling site selectivity of C–H oxidation. This work provides a general strategy and new opportunities for diversification through late-stage functionalization of a broad array of substrate classes.
Methods
General Procedure for Reductive Macrocyclization
In a glovebox, an oven-dried flask equipped with stir bar was charged with 33 mol % of Ni(COD)2 and 30 mol % of IPrCl. The flask was capped with a septum, removed from the glovebox, and attached to a nitrogen line. The contents of the flask were suspended in toluene (6.3 mL) and stirred for 20 min. The contents were diluted with an additional 31.4 mL of toluene and heated to 60 °C in an oil bath for 20 min. Triisopropylsilane (5 equiv) was then added in a single portion. Ynal (1 equiv) was then added by syringe drive over 1 h as a 4.15 mL solution in toluene. The reaction mixture was stirred overnight at 60 °C. The reaction mixture was cooled to room temperature, and the volatiles were removed under vacuum. The crude residue was filtered through a plug of silica with EtOAc/hexanes (1:1) and then purified by column chromatography.
General Procedure for Biocatalytic Oxidation
To an Erlenmeyer flask containing reaction buffer (50 mM NaH2PO4, 1 mM EDTA, 0.2 mM DTT, 10% (v/v) glycerol, pH 7.3) were added the following components sequentially: substrate (20 mM stock in DMSO, 1 mM final concentration), glucose-6-phosphate (100 mM stock in reaction buffer, 5 mM final concentration), glucose-6-phosphate dehydrogenase (100 U/mL stock in water, 1 U/mL final concentration), PikC-RhFRED (varied stock concentrations, 5 μM final concentration), and NADP+ (20 mM stock in reaction buffer, 1 mM final concentration). The reaction mixture was capped with a milk filter and incubated at 30 °C overnight (14–16 h) with gentle shaking (100 rpm). Reactions were typically conducted on ∼40–60 mg of each substrate (∼90–130 mL total reaction volume) and performed in 500 mL Erlenmeyer flasks. After overnight incubation, the reaction was quenched by addition of acetone (2× total reaction volume) and incubated at 4 °C for 2 h. The details of the workup and purification are provided in the Supporting Information.
Acknowledgments
The authors thank the NSF Center for Chemical Innovation: Center for Selective C–H Functionalization (Grant CHE-1700982, D.H.S., K.N.H., and J.M.) for support of the design and evaluation of biocatalysts for C–H oxidations and the National Institutes of Health (Grant R35GM118133, J.M., and R35GM118101, D.H.S.) for support of the design of synthetic strategies for macrolide analogue preparation and site-selective derivatization. S.W. thanks the Institute of Chemical Research of Catalonia (ICIQ) for summer fellowship support. The authors acknowledge assistance from Emma Gendre (ENS de Lyon), Summer Baker-Dockrey (University of Michigan), and Devin Ferguson (University of Michigan).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00450.
Methods and materials, supplementary graphics, and references (PDF)
Author Present Address
⊥ J.M.G.: Bridge Institute, University of Southern California, Los Angeles, CA, 90089, USA. S.W.: Institute of Chemical Research of Catalonia, 43007, Tarragona, Spain. H.W.: Department of Chemistry, Emory University, Atlanta, GA 30322, USA.
Author Contributions
Synthetic chemistry was conducted by M.M.G., S.W., and H.W.; biocatalytic oxidations and product analysis were conducted by M.D.D., M.M.G., and A.R.H.N.; computational studies were conducted by S.Y. and J.M.G.; and the project was supervised by D.H.S., K.N.H, and J.M.
The authors declare no competing financial interest.
Supplementary Material
References
- Burke M. D.; Schreiber S. L. A planning strategy for diversity-oriented synthesis. Angew. Chem., Int. Ed. 2004, 43, 46–58. 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- Bos P. H.; Antalek M. T.; Porco J. A.; Stephenson C. R. J. Tandem dienone photorearrangement-cycloaddition for the rapid generation of molecular complexity. J. Am. Chem. Soc. 2013, 135, 17978–17982. 10.1021/ja409992m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros M. R.; Narayan R. S.; McDougal N. T.; Schaus S. E.; Porco J. A. Skeletal diversity via cationic rearrangements of substituted dihydropyrans. Org. Lett. 2010, 12, 3222–3225. 10.1021/ol101144k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou K. C.; Petasis N. A.; Zipkin R. E. The endiandric acid cascade. Electrocyclizations in organic synthesis. 4. Biomimetic approach to endiandric acids A-G. Total synthesis and thermal studies. J. Am. Chem. Soc. 1982, 104, 5560–5562. 10.1021/ja00384a080. [DOI] [Google Scholar]
- Zacuto M. J.; Leighton J. L. Divergent synthesis of complex polyketide-like macrolides from a simple polyol fragment. Org. Lett. 2005, 7, 5525–5527. 10.1021/ol052373g. [DOI] [PubMed] [Google Scholar]
- Gutekunst W. R.; Baran P. S. C-H functionalization logic in total synthesis. Chem. Soc. Rev. 2011, 40, 1976–1991. 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]
- Newhouse T.; Baran P. S. If C-H bonds could talk: Selective C-H bond oxidation. Angew. Chem., Int. Ed. 2011, 50, 3362–3374. 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topczewski J. J.; Cabrera P. J.; Saper N. I.; Sanford M. S. Palladium-catalysed transannular C-H functionalization of alicyclic amines. Nature 2016, 531, 220–224. 10.1038/nature16957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wencel-Delord J.; Glorius F. C-H bond activation enables the rapid construction and late-stage diversification of functional molecules. Nat. Chem. 2013, 5, 369–375. 10.1038/nchem.1607. [DOI] [PubMed] [Google Scholar]
- Cernak T.; Dykstra K. D.; Tyagarajan S.; Vachal P.; Krska S. W. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. 10.1039/C5CS00628G. [DOI] [PubMed] [Google Scholar]
- Durak L. J.; Payne J. T.; Lewis J. C. Late-stage diversification of biologically active molecules via chemoenzymatic C-H functionalization. ACS Catal. 2016, 6, 1451–1454. 10.1021/acscatal.5b02558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolev J. N.; O’Dwyer K. M.; Jordan C. T.; Fasan R. Discovery of potent parthenolide-based antileukemic agents enabled by late-stage P450-mediated C-H functionalization. ACS Chem. Biol. 2014, 9, 164–173. 10.1021/cb400626w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Negretti S.; Knauff A. R.; Montgomery J. Exo-selective reductive macrocyclization of ynals. Org. Lett. 2015, 17, 1493–1496. 10.1021/acs.orglett.5b00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson E. P.; Malik H. A.; Sormunen G. J.; Baxter R. D.; Liu P.; Wang H.; Shareef A.-R.; Montgomery J. Mechanistic basis for regioselection and regiodivergence in nickel-catalyzed reductive couplings. Acc. Chem. Res. 2015, 48, 1736–1745. 10.1021/acs.accounts.5b00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaudel Q.; Journot G.; Regueiro-Ren A.; Goswami A.; Guo Z. W.; Tully T. P.; Zou L. F.; Ramabhadran R. O.; Houk K. N.; Baran P. S. Improving physical properties via C-H oxidation: Chemical and enzymatic approaches. Angew. Chem., Int. Ed. 2014, 53, 12091–12096. 10.1002/anie.201407016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde N. C.; Isomura M.; Mendoza A.; Baran P. S. TWo-phase synthesis of (−)-taxuyunnanine D. J. Am. Chem. Soc. 2014, 136, 4909–4912. 10.1021/ja501782r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gormisky P. E.; White M. C. Catalyst-controlled aliphatic C-H oxidations with a predictive model for site-selectivity. J. Am. Chem. Soc. 2013, 135, 14052–14055. 10.1021/ja407388y. [DOI] [PubMed] [Google Scholar]
- Chen M. S.; White M. C. Combined effects on selectivity in Fe-catalyzed methylene oxidation. Science 2010, 327, 566–571. 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]
- Negretti S.; Narayan A. R. H.; Chiou K. C.; Kells P. M.; Stachowski J. L.; Hansen D. A.; Podust L. M.; Montgomery J.; Sherman D. H. Directing group-controlled regioselectivity in an enzymatic C-H bond oxygenation. J. Am. Chem. Soc. 2014, 136, 4901–4904. 10.1021/ja5016052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan A. R. H.; Jimenez-Oses G.; Liu P.; Negretti S.; Zhao W. X.; Gilbert M. M.; Ramabhadran R. O.; Yang Y. F.; Furan L. R.; Li Z.; Podust L. M.; Montgomery J.; Houk K. N.; Sherman D. H. Enzymatic hydroxylation of an unactivated methylene C-H bond guided by molecular dynamics simulations. Nat. Chem. 2015, 7, 653–660. 10.1038/nchem.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho P. S.; Brustad E. M.; Kannan A.; Arnold F. H. Olefin cyclopropanation via carbene transfer catalyzed by engineered cytochrome P450 enzymes. Science 2013, 339, 307–310. 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]
- Hyster T. K.; Knorr L.; Ward T. R.; Rovis T. Biotinylated Rh(III) complexes in engineered streptavidin for accelerated asymmetric C-H activation. Science 2012, 338, 500–503. 10.1126/science.1226132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dydio P.; Key H. M.; Nazarenko A.; Rha J. Y. E.; Seyedkazemi V.; Clark D. S.; Hartwig J. F. An artificial metalloenzyme with the kinetics of native enzymes. Science 2016, 354, 102–106. 10.1126/science.aah4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanuel M. A.; Greenberg N. R.; Oblinsky D. G.; Hyster T. K. Accessing non-natural reactivity by irradiating nicotinamide-dependent enzymes with light. Nature 2016, 540, 414–417. 10.1038/nature20569. [DOI] [PubMed] [Google Scholar]
- Das S.; Incarvito C. D.; Crabtree R. H.; Brudvig G. W. Molecular recognition in the selective oxygenation of saturated C-H bonds by a dimanganese catalyst. Science 2006, 312, 1941–1943. 10.1126/science.1127899. [DOI] [PubMed] [Google Scholar]
- Tang R. Y.; Li G.; Yu J. Q. Conformation-induced remote meta-C-H activation of amines. Nature 2014, 507, 215–220. 10.1038/nature12963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow R. Biomimetic control of chemical selectivity. Acc. Chem. Res. 1980, 13, 170–177. 10.1021/ar50150a002. [DOI] [Google Scholar]
- Breslow R.; Baldwin S.; Flechtner T.; Kalicky P.; Liu S.; Washburn W. Remote oxidation of steroids by photolysis of attached benzophenone groups. J. Am. Chem. Soc. 1973, 95, 3251–3262. 10.1021/ja00791a031. [DOI] [PubMed] [Google Scholar]
- Li S.; Chaulagain M. R.; Knauff A. R.; Podust L. M.; Montgomery J.; Sherman D. H. Selective oxidation of carbolide C-H bonds by an engineered macrolide P450 mono-oxygenase. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18463–18468. 10.1073/pnas.0907203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein J. E.; Fokin V. V. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. 10.1039/b904091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boren B. C.; Narayan S.; Rasmussen L. K.; Zhang L.; Zhao H. T.; Lin Z. Y.; Jia G. C.; Fokin V. V. Ruthenium-catalyzed azide-alkyne cycloaddition: Scope and mechanism. J. Am. Chem. Soc. 2008, 130, 8923–8930. 10.1021/ja0749993. [DOI] [PubMed] [Google Scholar]
- Li S.; Ouellet H.; Sherman D. H.; Podust L. M. Analysis of transient and catalytic desosamine-binding pockets in cytochrome P-450 pikC from streptomyces venezuelae. J. Biol. Chem. 2009, 284, 5723–5730. 10.1074/jbc.M807592200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Details of computational methods are included in refs 34–50 in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.