

Abstract
Glioblastoma (GBM) is the most aggressive primary brain tumor in adults and is virtually incurable with conventional therapies. Immunotherapy with T cells expressing GBM-specific chimeric antigen receptors (CAR) is an attractive approach to improve outcomes. Although CAR T cells targeting GBM antigens, such as IL13 receptor subunit α2 (IL13Rα2), HER2, and EGFR variant III (EGFRvIII), have had antitumor activity in preclinical models, early-phase clinical testing has demonstrated limited antiglioma activity. Transgenic expression of IL15 is an appealing strategy to enhance CAR T-cell effector function. We tested this approach in our IL13Rα2-positive glioma model in which limited IL13Rα2-CAR T-cell persistence results in recurrence of antigen-positive gliomas. T cells were genetically modified with retroviral vectors encoding IL13Rα2-CARs or IL15 (IL13Rα2-CAR.IL15 T cells). IL13Rα2-CAR.IL15 T cells recognized glioma cells in an antigen-dependent fashion, had greater proliferative capacity, and produced more cytokines after repeated stimulations in comparison with IL13Rα2-CAR T cells. No autonomous IL13Rα2-CAR. IL15 T-cell proliferation was observed; however, IL15 expression increased IL13Rα2-CAR T-cell viability in the absence of exogenous cytokines or antigen. In vivo, IL13Rα2-CAR.IL15 T cells persisted longer and had greater antiglioma activity than IL13Rα2-CAR T cells, resulting in a survival advantage. Gliomas recurring after 40 days after T-cell injection had downregulated IL13Rα2 expression, indicating that antigen loss variants occur in the setting of improved T-cell persistence. Thus, CAR T cells for GBM should not only be genetically modified to improve their proliferation and persistence, but also to target multiple antigens.
Introduction
Glioblastoma (GBM) represents the most common and aggressive primary brain tumor in humans (1). Despite a multimodal therapeutic approach including surgery, chemotherapy, and radiation, outcomes for pediatric and adult GBM patients remain poor (2–4). Novel therapeutics could improve outcomes. Immunotherapy promises to meet this need as it does not rely on the cytotoxic pathways employed by the aforementioned standard therapies.
T cells, genetically engineered to be GBM specific, can be a foundation for adoptive immunotherapy of GBMs (5, 6). Several groups, including ours, have developed GBM-specific chimeric antigen receptors (CAR) targeting tumor-associated antigens (TAA), including IL13Rα2, HER2, EphA2, and EGFRvIII (7–11). On the basis of preclinical studies that demonstrated GBM activity of CAR T cells in xenograft models, phase I testing of IL13Rα2-, HER2-, and EGFRvIII-specific CAR T cells in humans is currently in progress (12–15). These clinical studies demonstrated safety, but the anti-GBM activity of CAR T cells was limited (12–15). CAR T-cell therapy studies showed similar results for solid tumors (16, 17). In contrast, CD19-specific CAR T cells showed potent antitumor activity for B-cell–derived hematologic malignancies (18–20).
Lack of efficacy of CAR T cells for brain and solid tumors is likely multifactorial, including limited proliferation and expansion of T cells at tumor sites (21, 22). Genetic modification strategies being pursued to overcome this obstacle include expression of cytokines (e.g., IL12, IL15), or manipulation of cytokine receptors (e.g., TGFβ, IL4, or PD-1) to block inhibitory signals in the tumor microenvironment or convert inhibitory into stimulatory signals (23–30).
The effectiveness of secondary genetic modifications to enhance the effector function of CAR T cells for GBM has not been explored. T cells expressing an IL13Rα2-CAR with a CD28.ζ domain (IL13Rα2-CAR T cells) have potent antitumor activity in a glioma xenograft model (8). However, antigen-positive tumors in mice treated with IL13Rα2-CAR T cells recurred due to limited T-cell persistence (8). Here, we demonstrate that transgenic expression of IL15 enhances the effector function and antiglioma activity of IL13Rα2-CAR T cells in vitro and in vivo. Gliomas in mice treated with IL13Rα2-CAR.IL15 T cells still recurred albeit at later time points in comparison with mice treated with IL13Rα2-CAR T cells. Mechanistic studies revealed that “late recurring tumors” had downregulated the expression of IL13Rα2, highlighting the need not only to optimize T-cell persistence and expansion, but also to target multiple TAAs expressed in GBM.
Materials and Methods
Cell lines
U373 (GBM), 293T (human embryonic kidney), and Raji (Burkitt lymphoma) cell lines were purchased from the ATCC). GBM6 is a primary adult GBM cell line (kindly provided by Dr. C. David James, Northwestern University, Chicago, IL; ref. 31). The generation of U373 cells expressing enhanced GFP and firefly luciferase (U373.eGFP.ffLuc), 293T cells expressing GFP (293T. GFP) or IL13Rα2 and GFP (293T.IL13Rα2.GFP) was reported previously (11). Cell lines were grown in RPMI or DMEM (GE Healthcare Life Sciences HyClone Laboratories) with 10% FBS (GE Healthcare Life Sciences HyClone) and 2 mmol/L GlutaMAX-I (Invitrogen). Cell lines were purchased between 2008 and 2011. The “Characterized Cell Line Core Facility” at MD Anderson Cancer Center (Houston, TX) performed cell line validation (2011). Once thawed, cell lines were kept in culture for a maximum of 3 months before new reference vials were thawed. All cell lines were tested on a regular basis for mycoplasma and were negative.
Generation of retroviral vectors encoding IL13Rα2-specific CAR and IL15
The generation of the IL13Rα2-specific CAR with a short spacer region, a CD28 transmembrane domain, and a CD28.ζ endodomain was described previously (8). The IL15-encoding retroviral vector was generated by replacing the ΔCD34 gene in pSFG.iC9-2A-ΔCD34-2A-IL15 (28) with a cytoplasmic domain-truncated nerve growth factor receptor gene (ΔNGFR; pSFG.iC9-2A-ΔNGFR-2A-IL15). RD114-pseudotyped retroviral particles were generated by transient transfection of 293T cells as described previously (11) using GeneJuice transfection reagent (EMD Biosciences). Super-natants containing retroviral particles were collected 48 hours after transfection for T-cell transduction.
Generation of CAR T cells
Human peripheral blood mononuclear cells (PBMC) from healthy donors were obtained under a Baylor College of Medicine Institutional Review Board–approved protocol, after informed consent was obtained in accordance with the Declaration of Helsinki. To generate IL13Rα2-CAR T cells expressing IL15, PBMCs were isolated by Lymphoprep (Greiner Bio-One) gradient centrifugation and then stimulated on nontissue culture-treated 24-well plates, which were precoated with OKT3 (CRL-8001, ATCC) and CD28 (BD Biosciences) antibodies. Recombinant human IL7 and IL15 (IL7, 10 ng/mL; IL15, 5 ng/mL; PeproTech) were added to cultures on day 2 (32). On day 3, OKT3/CD28 stimulated T cells (2.5 × 105 cells/well) were doubly transduced on RetroNectin (Clontech)-coated plates in the presence of IL7 and IL15. On day 3 or 4, T cells were transferred into new wells and subsequently expanded with IL7 and IL15. Nontransduced T cells were activated with OKT3/CD28 and expanded in parallel with IL7 and IL15. IL13Rα2-CAR and IL15 expression was determined 4 to 5 days posttransduction and at later time points (day 12 and day 20).
Flow cytometry
FACSCanto II (BD Biosciences) or BC Gallios (Beckman Coulter, Inc.) instruments were used to acquire immunofluorescence data, which were analyzed with FACSDiva (BD Biosciences) or BC Gallios (Beckman Coulter, Inc.), respectively. FlowJo v.9 (FlowJo, LLC) or Kaluza v1.2 (Beckman Coulter, Inc.) was used for final data analysis and graphic representation. Isotype controls were immunoglobulin G1–FITC (IgG1-FITC; BD Biosciences) and IgG1–PE (IgG1-PE; BD Biosciences). IL13Rα2-CAR expression was detected by staining T cells with human IL13Rα2 chimera (R&D Systems, Inc.) followed by Fc-FITC (Millipore) or Fc-PE (SouthernBiotech). U373 cells and recurrent tumors were analyzed for IL13Rα2, HER2, and EphA2 expression using anti-IL13Rα2, HER2-APC (BD Biosciences), and anti-EphA2 antibodies (R&D Systems, Inc.), respectively. Secondary antibodies anti-goat IgG-AF647 and anti-mouse IgG-AF647 were used for IL13Rα2 and EphA2, respectively (both from Invitrogen). Cell phenotype was evaluated by staining T cells with CD4-Pacific Blue, CD8-PerCP, CCR7-FITC, and CD45RA-AF750 (BD Biosciences). Forward and side scatter gating were used to distinguish normal lymphocytes. Cells were collected and washed once with PBS (Sigma) containing 1% FBS (GE Healthcare Life Sciences HyClone Laboratories; FACS buffer) prior to the addition of antibodies. Cell were incubated for 30 minutes on ice in the dark, washed once, and fixed in FACS buffer with 0.5% paraformaldehyde (BD Biosciences) prior to analysis.
Western blot analysis
Cells were dissociated with PBS + 3 mmol/L EDTA and lysed in a buffer containing 50 mmol/L Tris, 150 mmol/L NaCl, 5 mmol/L EDTA, 1% Triton X-100 (all from Sigma), and protease inhibitors (Thermo Fisher Scientific). Protein concentrations were determined using a Bio-Rad protein assay (Bio-Rad) with BSA as the standard. Samples were denatured in Laemmli buffer (Bio-Rad) at 95°C for 5 minutes. Cell lysates (5–10 μg/lane) were run on a 10% SDS polyacrylamide gel and transferred to nitrocellulose membranes (Bio-Rad). Membranes were blocked with 5% milk powder in TBS + 0.1% Tween-20 (all from Sigma) and then probed with anti-CD3.ζ (sc-1239, Santa Cruz Biotechnology, Inc.) or GAPDH (sc-47724, Santa Cruz Biotechnology, Inc.) mouse mAbs or anti-caspase-9 rabbit antibody (9502, Cell Signaling Technology), followed by a horseradish peroxidase–conjugated goat anti-mouse IgG antibody (Santa Cruz Biotechnology, Inc.) or goat anti-rabbit IgG antibody (Jackson ImmunoResearch, Inc.). Blots were developed using SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific) and exposed to GeneMate Blue Basic Autoradiography Film (BioExpress).
Coculture assays
Recombinant protein coculture assay
Nontissue culture-treated 24-well plates were precoated with recombinant human IL13Rα1, IL13Rα2 (R&D Systems, Inc.), or OKT3 (CRL-8001, ATCC) proteins at a final concentration of 500 ng/well. Plates were washed once using RPMI, and CAR, CAR.15 or NT T cells were plated. After 24 hours, supernatants were harvested, and IL15 release was measured by ELISA as per the manufacturer’s instructions (R&D Systems, Inc.).
Cell coculture assay
CAR T cells were cocultured with target cells at a 2:1 effector-to-target (E:T) ratio in a 24-well plate. Non-transduced T cells served as controls. After 24 hours, culture supernatants were harvested, and the presence of cytokines was determined by Multiplex assay (HSTCMAG28SPMX13, EMD Millipore).
Repeated stimulation assay
CAR T cells were cocultured with target cells at a 2:1 E:T ratio in a 24-well plate. CAR T cells were restimulated with fresh target cells every 7 days. Culture supernatants were harvested 24 hours after each stimulation, and the presence of cytokines was determined by Multiplex assay. CAR T cells were counted every 7 days, before the addition of fresh U373 cells.
Cytotoxicity assay
Standard chromium (51Cr) release assays were performed as described previously (11). Briefly, 1 × 106 target cells were labeled with 0.1 mCi (3.7 MBq) 51Cr and mixed with decreasing numbers of effector cells to give E:T ratios of 40:1, 20:1, 10:1, and 5:1. Target cells incubated in complete medium alone or in 1% Triton X-100 were used to determine spontaneous and maximum 51Cr release, respectively. After 4 hours, supernatants were collected, and radioactivity was measured in a gamma counter (Cobra Quantum; PerkinElmer). The mean percentage of specific lysis of triplicate wells was calculated according to the following formula:
Orthotopic xenograft SCID mouse model
All animal experiments followed a protocol approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. Experiments were performed as described previously (8). Briefly, 7- to 9-week-old ICR-SCID mice were anesthetized. The head was shaved and the mice were immobilized in a Cunningham Mouse/Neonatal Rat Adaptor (Stoelting) stereotactic apparatus and then scrubbed with 1% povidoneiodine. A 10-mm skin incision was made along the midline. A 1-mm burr hole was drilled into the skull 1 mm anterior and 2 mm to the right of the bregma. U373.eGFP.ffLuc cells (1 × 105) in 2.0 μL were injected 3 mm deep to the bregma over 5 minutes. The needle was left in place for 3 minutes, to avoid tumor cell extrusion, and then withdrawn over 5 minutes. Seven days after tumor cell injection, animals were treated with 2 × 106 effector cells in 2 μL to the same tumor coordinates. The incision was closed with 2 to 3 interrupted 7.0 Ethilon sutures (Ethicon, Inc.). A subcutaneous injection of 0.03 to 0.1 mg/kg buprenorphine (Buprenex RBH) was given for pain control.
Recurrent tumor cell isolation
Recurrent tumors were microdis-sected under sterile conditions from CAR or CAR.IL15-treated xenograft mouse models. The isolates were manually dissociated and filtered through a 40-μm mesh filter. The cell suspension was washed with PBS and cultured in RPMI + 10% FBS.
Bioluminescence imaging
Isoflurane-anesthetized animals were imaged using the IVIS system (IVIS, Xenogen Corp.) 10 to 15 minutes after 150 mg/kg D-luciferin (Xenogen) was injected per mouse intraperitoneally. The photons emitted from the luciferase-expressing tumor cells were quantified using Living Image software (Caliper Life Sciences). A pseudo-color image representing light intensity (blue least intense and red most intense) was generated and superimposed over the grayscale reference image. Mice were euthanized when the tumor radiance was greater than 1 × 109 on two occasions or when they met euthanasia criteria (neurologic deficits, weight loss, signs of distress) in accordance with the Center for Comparative Medicine at Baylor College of Medicine (Houston, TX).
Statistical analysis
All in vitro experiments were performed at least in triplicate. Data were summarized using descriptive statistics. Comparisons were made between groups using Wilcoxon rank sum test or t test, whichever is appropriate, for continuous variables. Changes from baseline to follow-up measures were compared using paired t test. Linear regression analysis was performed to evaluate the trend in cytokine secretion relationship between CAR and CAR.IL15. Survival time from the time of tumor cell injection was estimated by the Kaplan–Meier method, and differences in survival between groups were compared by the Wilcoxon test. GraphPad Prism 5 software (GraphPad software, Inc.), SAS 9.4, and R 3.3.2 were used for statistical analysis. P values <0.05 were considered statistically significant.
Results
Generation of IL13Rα2-specific CAR T cells releasing transgenic IL15
To generate IL13Rα2-specific CAR T cells that secrete transgenic IL15 (IL13Rα2-CAR.IL15 T cells), we genetically modified T cells with a retroviral vector encoding an IL13Rα2-specific scFv (scFv47) with a CD28.ζ endodomain (IL13Rα2-CAR; ref. 8), and a retroviral vector encoding inducible caspase-9 (iC9), NGFR with a truncated cytoplasmic domain (ΔNGFR), and IL15 separated by 2A sequences (iC9-2A-ΔNGFR-2AIL15; Fig. 1A). CD3/CD28–activated T cells from healthy donors were transduced with RD114-pseudotyped retroviral particles, and 4 to 5 days posttransduction, CAR expression was determined by FACS analysis. As controls, we generated T cells that only expressed IL13Rα2-CARs, IL15, or IL13Rα2-CARs in which the endodomain was deleted (IL13Rα2-CAR.Δ). Transduction efficiency was determined by FACS analysis for CAR and ΔNGFR expression (Fig. 1B and C). Single transduction with retroviral vectors encoding CAR, CAR.Δ, or iC9-2A-ΔNGFR-2A-IL15 yielded mean transduction efficiencies of 66.5% (SD ± 12.1%), 66.1% (SD ± 13.6%), and 56.3% (SD ± 14.5%), respectively. In IL13Rα2-CAR.IL15 T-cell lines, on average, 35.4% (SD ± 7.4%) of T cells were genetically modified with both vectors and in IL13Rα2-CAR.Δ.IL15 T-cell lines 39.5% (SD ± 9.6%). CAR expression was confirmed by Western blot for CD3.ζ (Fig. 1D). Phenotypic analysis of transduced cells revealed a mixture of CD4- and CD8-positive T cells and the presence of naïve (CD45RA+/CCR7+), central memory (CD45RA−/CCR7+), effector memory (CD45RA+/CCR7−), and terminally differentiated effector memory (CD45RA−/CCR7−) T cells (Supplementary Fig. S1). Genetic modification with CAR, CAR.Δ, and/or iC9-2A-ΔNGFR-2A-IL15 did not change the CD4:CD8 ratio or T-cell subset composition in comparison with nontransduced T cells. Cytotoxicity assays were performed with all T-cell populations using IL13Rα2-negative (239T-GFP) and IL13Rα2-positive (239T-GFP. IL13Rα2, U373, GBM6) target cells (Supplementary Fig. S2). Only IL13Rα2-CAR T cells and IL13Rα2.IL15-CAR T cells killed IL13Rα2-positive target cells in contrast to IL13Rα2-CAR.Δ. IL15, IL15, and nontransduced T cells, demonstrating that genetic modification of T cells with iC9-2A-ΔNGFR-2A-IL15 did not induce unspecific T-cell killing or influence the lytic activity of IL13Rα2-CAR T cells (Fig. 1E).
Figure 1.
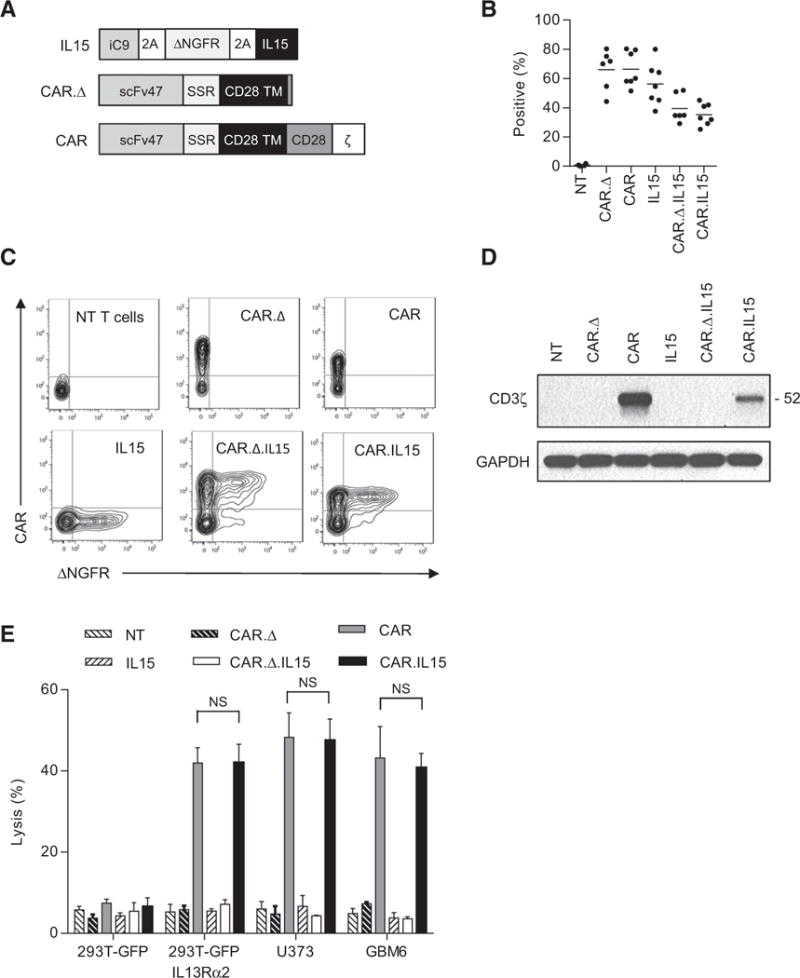
Generation of IL13Rα2-specific CAR T cells expressing transgenic IL15. A, Scheme of IL13Rα2-specific CAR and IL15 retroviral vectors. scFv47 is the name of the scFv that specifically recognize IL13Rα2. B and C, CAR and IL15 expression was confirmed using FACS analysis. Summary data [B; range, 37.7%–80.4% for single transduction, 30%–51.1% for double transduction (CAR.Δ.IL15 and CAR.IL15), n = 6–7 per construct; 4–5 independent experiments] and representative plots (C) are shown. D, Expression of full-length CAR by Western blot analysis using a CD3-ζ antibody. E, Four-hour cytotoxicity assay at an E:T ratio of 10:1 (n = 4; two independent experiments); CAR versus CAR.IL15: NS; two-way ANOVA; error bars, SEM). Targets: 293T-GFP, 293T-GFP-IL13Rα2, U373, and GBM6 cells (all positive for IL13Rα2 except 293T-GFP).
IL13Rα2-CAR.IL15 T cells displayed activation-dependent IL15 production
To determine IL15 production by IL15, IL13Rα2-CAR.Δ.IL15, IL13Rα2-CAR.IL15, or nontransduced T cells at baseline and after activation, we cultured T cells on tissue culture plates that were coated with recombinant IL13Rα1 or IL13Rα2 proteins. Non-coated plates or plates coated with OKT3 served as controls. After 24 hours, IL15 concentration in culture media was measured by ELISA. At baseline IL15, IL13Rα2-CAR.Δ.IL15, and IL13Rα2-CAR. IL15 T cells produced similar amounts of IL15 as did nontransduced T cells. However, in the absence of exogenous cytokines, viability of IL15-expressing T cells was improved in comparison with their unmodified counterpart after 14 days of culture (Supplementary Fig. S3). After OKT3 activation, IL15 (mean, 159.5 pg/mL ±70.8), IL13Rα2-CAR.Δ.IL15 (mean, 137.9 pg/mL ±73.5), and IL13Rα2-CAR.IL15 (mean, 152.2 pg/mL ±72.4) T cells produced significantly (P < 0.001) more IL15 in comparison with nontransduced T cells (mean, 16.3 pg/mL ±2.6; Fig. 2A). IL13Rα2-CAR.IL15 T cells produced significantly (P < 0.001) more IL15 when stimulated with recombinant IL13Rα2 protein (mean, 176.7 pg/mL; ±55.4), whereas no increase in IL15 production was observed for all T-cell populations in the presence of recombinant IL13Rα1 protein. Thus, IL15 production by IL15, IL13Rα2-CAR.Δ.IL15, and IL13Rα2-CAR.IL15 T cells was activation dependent. This was not specific for IL15, because we observed an activation-dependent increase in expression also for a second transgene (Supplementary Fig. S4).
Figure 2.
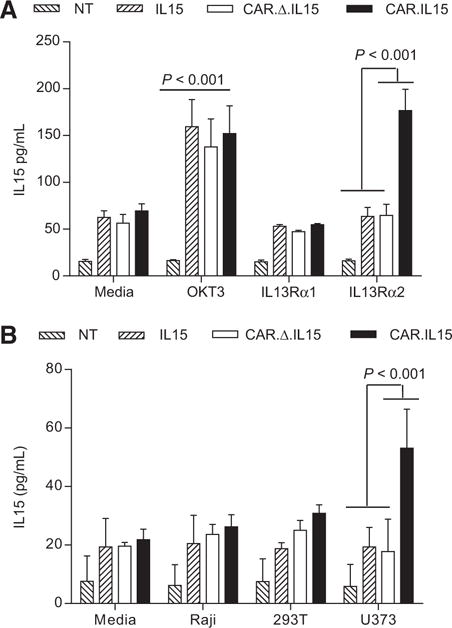
IL13Rα2-CAR.IL15 T cells display activation-dependent IL15 production. A, IL13Rα2-CAR or nontransduced (NT) T cells were stimulated with recombinant IL13Rα1 or IL13Rα2 protein, or OKT3. After 24 hours, IL15 was measured by ELISA. T cells expressing IL15 and CAR.IL15 constructs, but not controls, expressed significant concentrations of IL15 when stimulated with recombinant IL13Rα2 protein and OKT3 in comparison with IL13Rα1 stimulated T cells (n = 6; three independent experiments); poststimulation with OKT3: NT versus IL15/CAR/CAR.IL15 P < 0.001; poststimulation with IL13Rα2: NT/IL15/CAR.Δ.IL15 versus CAR.IL15 P < 0.001; two-way ANOVA; error bars, SEM). B, IL13Rα2-CAR T cells were cocultured with Raji, 293T, and U373 cells at a 2:1 E:T ratio. NT, IL15, and CAR.D.IL15 T cells served as controls. After 24 hours IL15 was measured by ELISA (n = 4; two independent experiments); poststimulation with U373: NT/IL15/CAR versus CAR.IL15 P < 0.001; two-way ANOVA; error bars, SEM).
We next determined whether IL13Ra2 expression on glioma cells is sufficient to induce IL15 production of IL13Rα2-CAR.IL15 T cells. IL15, IL13Rα2-CAR.Δ.IL15, IL13Rα2-CAR.IL15, or non-transduced T cells were cultured with Raji (IL13Rα1-/IL13Rα2-), 293T (IL13Rα1+/IL13Rα2−), or U373 (IL13Rα1+/IL13Rα2+) cells, or media alone. After 24 hours, IL15 concentration in culture media was determined by ELISA. Only IL13Rα2−CAR.IL15 T cells produced significant amounts of IL15 in the presence of IL13Rα2-positive cells (U373) compared with IL15, IL13Rα2-CAR.Δ.IL15, or nontransduced T cells (Fig. 2B). These data were confirmed using 293T cells genetically modified to express IL13Rα2 (239T-GFP.IL13Rα2; Supplementary Fig. S5). Target cells that were only positive for IL13Rα1 (293T cells) or negative for IL13Rα1 and IL13Rα2 (Raji) did not induce significant IL15 production in any of the tested T-cell populations. Thus, as for the experiments with recombinant protein, antigen-specific CAR activation induces IL15 production by IL13Rα2-CAR.IL15 T cells. As the IL15 vector also encodes iC9, we wanted to determine its presence and functionality. Western blot analysis for C9 at baseline revealed the presence of iC9 in IL15 and IL13Rα2-CAR.IL15 T-cell lines. However, 24 hours postexposure to 10 nmol/L CID (Chemical Inducible Dimerizer; A/C Heterodimerizer, Clontech), only native C9 was present, indicating the killing of iC9-positive T cells (Supplementary Fig. S6A). Cell killing was confirmed by FACS analysis for Annexin V and 7-AAD staining (Supplementary Fig. S6B).
IL13Rα2-CAR.IL15 T cells proliferate more than IL13Rα2-CAR T cells
To determine the benefits of transgenic IL15 expression on the effector function of CAR T cells, IL13Rα2-CAR and IL13Rα2-CAR.IL15 T cells were stimulated every 7 days with U373 or 293T cells without exogenous cytokines. Prior to each stimulation T cells were counted and CAR and ΔNGFR expression was determined by FACS analysis. IL15 and non-transduced T cells served as controls. Repeat stimulations with U373 cells resulted in a significant (P < 0.05) expansion of IL13Rα2-CAR and IL13Rα2-CAR.IL15 T cells in comparison with IL15 and NT T cells (Fig. 3A). There was no significant difference between IL13Rα2-CAR and IL13Rα2-CAR.IL15 T-cell expansion after the first and second simulation. With subsequent stimulations, only IL13Rα2-CAR.IL15 T cells continued to expand. Addition of exogenous IL15 rescued IL13Rα2-CAR T cells, indicating that the observed effect was specific to IL15 (Supplementary Fig. S7). 293T cells did not induce T-cell expansion, confirming antigen specificity. Successive stimulations resulted in enrichment of IL13Rα2-CAR.IL15 T cells as judged by FACS analysis for each single transgene (before the first stimulation compared with after the sixth stimulation: for CAR: P < 0.05; for IL15: P < 0.01) in contrast to controls (nontransduced, IL15, IL13Rα2-CAR T cells; Fig. 3B).
Figure 3.
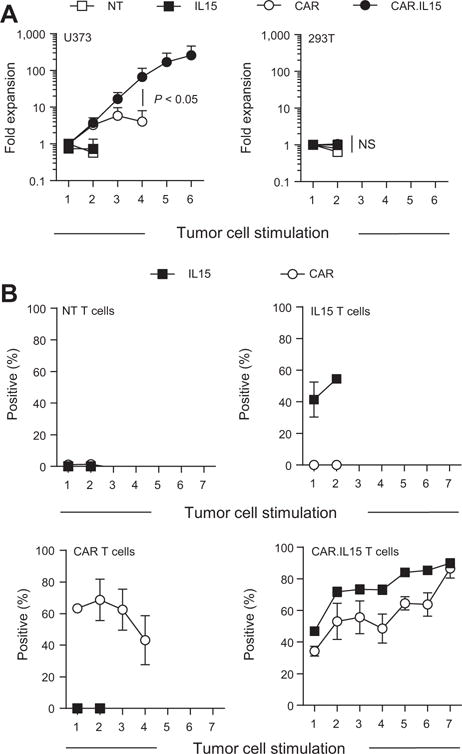
IL13Rα2-CAR.IL15 T cells have greater proliferative capacity compared with IL13Rα2-CAR T cells. CAR and CAR.IL15 T cells were cocultured with U373 cells at a 2:1 E:T ratio. T cells were stimulated weekly with fresh U373 cells, and T cells were counted before addition of fresh target cells. A, Cumulative data of CAR T-cell expansion (n = 4; two independent experiments); post third stimulation with U373: CAR versus CAR.IL15 P < 0.05; t test; error bars, SEM). B, Transgene expression after each co-culture with U373 tumor cells as determined before each stimulation by FACS analysis. Bottom right, percent of CAR or IL15-positive cells in CAR.IL15 T-cell lines (n = 4; two independent experiments); pre first versus post sixth stimulation: for CAR: P < 0.05; for IL15: P < 0.01; paired t test; error bars, SEM).
Transgenic IL15 expression maintains cytokine production
Having established that IL13Rα2-CAR.IL15 T cells expand better than IL13Rα2-CAR T cells after repeated antigen-specific stimulations, we next determined the benefits of transgenic expression of IL15 on cytokine production. NT, IL15, IL13Rα2-CAR, or IL13Rα2-CAR.IL15 T cells were stimulated weekly with U373 cells (up to 5 stimulations), and 24 hours after each stimulation, an aliquot of media was removed to determine the concentrations of cytokines by Multiplex analysis. After the first stimulation, only IL13Rα2-CAR and IL13Rα2-CAR.IL15 T cells produced TH1 (IFNγ, TNFα, GM-CSF, IL2), TH1/2 cytokine (IL6), and TH2 (IL4, IL5, IL10) cytokines (Fig. 4). There was no difference in cytokine production between both T-cell populations. IL15 and nontransduced T cells produced negligible amounts of cytokines, confirming antigen specificity. With subsequent stimulations, there was a decline in cytokine production by IL13Rα2-CAR T and IL13Rα2-CAR.IL15 T cells. However, the decline in IFNγ, TNFα, and IL5 production by IL13Rα2-CAR T cells was significantly (P = 0.0008, P = 0.0253, P = 0.0291, respectively) greater in comparison with IL13Rα2-CAR.IL15 T cells as evaluated by the trend in cytokine secretion relationship between CAR and CAR.IL15 T cells. In addition, we measured IL15 secretion in long-term cultures and confirmed continuous production of IL15 as judged by IL15 ELISA (Fig 4; bottom right).
Figure 4.
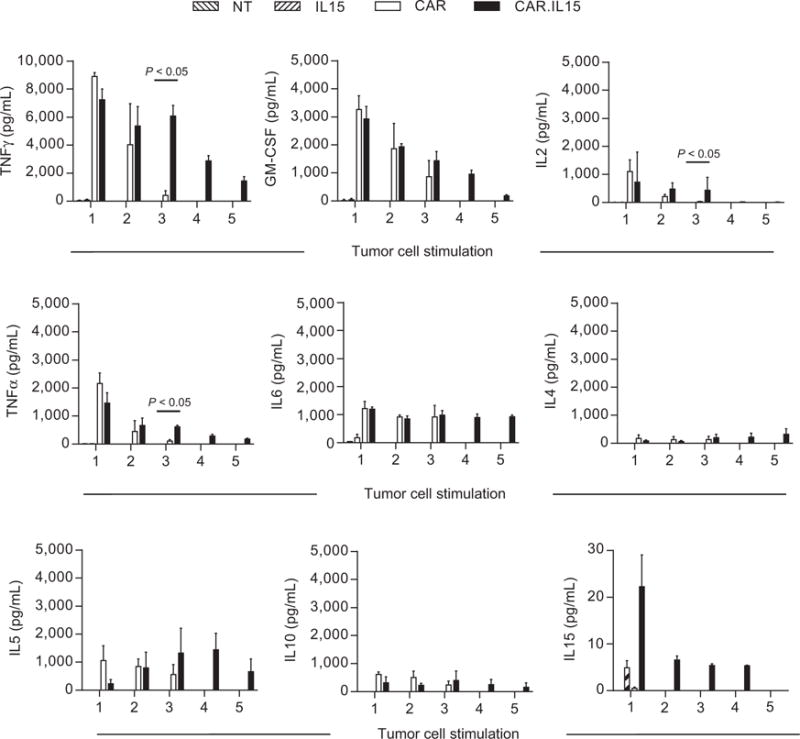
Transgenic IL15 expression improves cytokine production after repeated stimulations of IL13Rα2-CAR.IL15 T cells with glioma cells. CAR and CAR.IL15 T cells were cocultured with U373 cells at a 2:1 E:T ratio. T cells were stimulated weekly with U373 cells, and 24 hours after each stimulation, a small aliquot of media was removed to determine the concentrations of cytokines by Multiplex assay (HSTCMAG28SPMX13, EMD Millipore) and IL15 ELISA Kit (R&D Systems, Inc.). Nontransduced and IL15 T cells served as controls (n = 4; two independent experiments); post third stimulation CAR versus CAR.IL15: for IFNγ, IL2, and TNFα: P < 0.05; Wilcoxon rank sum test; error bars, SEM).
IL13Rα2-CAR.IL15 T cells have improved antitumor activity
Finally, we evaluated the impact of IL15 on the antitumor activity of IL13Rα2-CAR T-cell in the U373 human glioblastoma orthotopic xenograft mouse model. On day 0, U373.eGFP.ffLuc cells were injected into the brains of SCID mice followed by the intratumoral injection of IL13Rα2-CAR and IL13Rα2-CAR.IL15 T cells on day 7. Mice injected with IL13Rα2-CAR.Δ.IL15 T cells served as controls. Mice treated with IL13Rα2-CAR.Δ.IL15 T cells showed continuous tumor growth within 4 days of T-cell injection, whereas mice treated with IL13Rα2-CAR ± IL15 T cells did not (Fig. 5A and B). Comparison of bioluminescence imaging results revealed no difference between IL13Rα2-CAR T cells and the IL13Rα2-CAR.IL15 T-cell groups up to 28 days after T-cell injection. However, mice treated with IL13Rα2-CAR.IL15 had lower tumor signals starting 35 day posttreatment (Supplementary Table 1). This resulted in greater progression-free survival (PFS) and overall survival (OS) of mice treated with IL13Rα2-CAR.IL15 T cells than mice treated with IL13Rα2-CAR T cells (CAR vs. CAR.IL15: PFS, P = 0.0021; OS, P = 0.0019; Fig. 5C) without inducing GVHD from nonspecific T-cell activation. To investigate the mechanism of improved PFS and OS of IL13Rα2-CAR.IL15 T cells, we compared the in vivo persistence of IL13Rα2-CAR and IL13Rα2-CAR.IL15 T cells. U373 glioma-bearing mice were injected on day 7 with IL13Rα2-CAR, IL13Rα2-CAR.IL15, or CAR.Δ.IL15 T cells that were also genetically modified to express eGFP.ffLuc. T cells modified only with eGFP.ffLuc served as controls. IL13Rα2-CAR.IL15 persisted longer than nontransduced (P < 0.05) and IL13Rα2-CAR T cells (P < 0.01). However, there was no difference between IL13Rα2-CAR.IL15 and CAR.Δ.IL15 T cells (Fig. 5D). No T cells were detected outside the brain as judged by bioluminescence imaging.
Figure 5.
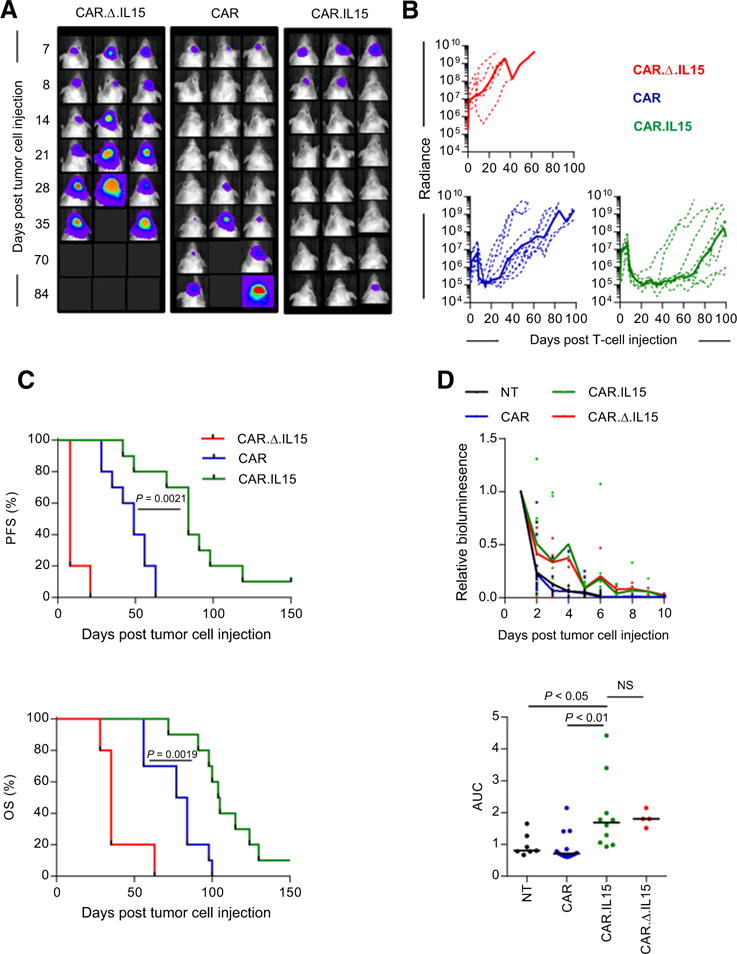
Treatment of glioma xenografts with IL13Rα2-CAR.IL15 T cells resulted in tumor regression and improved OS compared with IL13Rα2-CAR T cells. U373 glioma-bearing mice were treated on day 7 with CAR (n = 10), CAR.IL15 (n = 10; two independent experiments) T cells. CAR.Δ.IL15 T cells (n = 5; one independent experiment) served as a control. A and B, Representative images for each group (A) and quantitative bioluminescence (B; radiance = photons/sec/cm2/sr) imaging data for all mice are shown (dotted lines: individual mice; solid lines: median). C, Kaplan–Meier survival analysis (CAR versus CAR. IL15, P = 0.0019) and PFS (CAR vs. CAR.IL1, P = 0.0021). No GVHD from nonspecific activation was observed. D, Unmodified U373 cells (1 × 105) were injected intracranially into mice. On day 7, mice received 2 × 106 CAR. eGFP.ffLuc or CAR.IL15.eGFP.ffLuc CAR T cells intracranially using the same tumor coordinates. Top, bioluminescence measurement was used to monitor T-cell persistence; bottom, AUC of bioluminescence data [nontransduced (NT; n = 7; two independent experiments) vs. CAR. IL15 (n = 10; two independent experiments), P < 0.05; CAR (n = 13; three independent experiments) vs. CAR.IL15 (n = 10; two independent experiments), P < 0.01; CAR.IL15 vs. CAR.Δ.IL15 (n = 4; one independent experiment), ns; t test].
Therapy failure is due to antigen loss variants
Although IL13Rα2-CAR.IL15 T cells persisted longer than IL13α2-CAR T cells in vivo, IL13Rα2-CAR.IL15 T cells only persisted for 10 days after T-cell injection as judged by bioluminescence imaging. To differentiate between lack of long-term T-cell persistence and antigen escape as an etiology of therapy failure, recurrent gliomas from IL13Rα2-CAR and IL13Rα2-CAR.IL15 T-cell–treated mice were harvested, and FACS analysis was performed for IL13Rα2 and two other glioma-associated antigens (HER2, EphA2) after short-term culture. Tumors that recurred within 20 days after T-cell injection were IL13Rα2 positive, whereas more than 80% of cells in tumors that recurred after day 40 were IL13Rα2 negative (Fig. 6A and B). Tumors recurring between day 20 and 40 exhibited variable levels of IL13Rα2 expression. In contrast, HER2 and EphA2 expression was preserved in all tumors except in one late-recurring tumor (day 63) that was HER2 negative (Fig. 6B, middle). Cytotoxicity assays with CAR T cells were used to confirm loss of IL13Rα2 and HER2 expression (Supplementary Fig. S8). Thus, antigen negative immune escape is the most likely explanation for late (>day 40 after T-cell injection) tumor recurrence.
Figure 6.
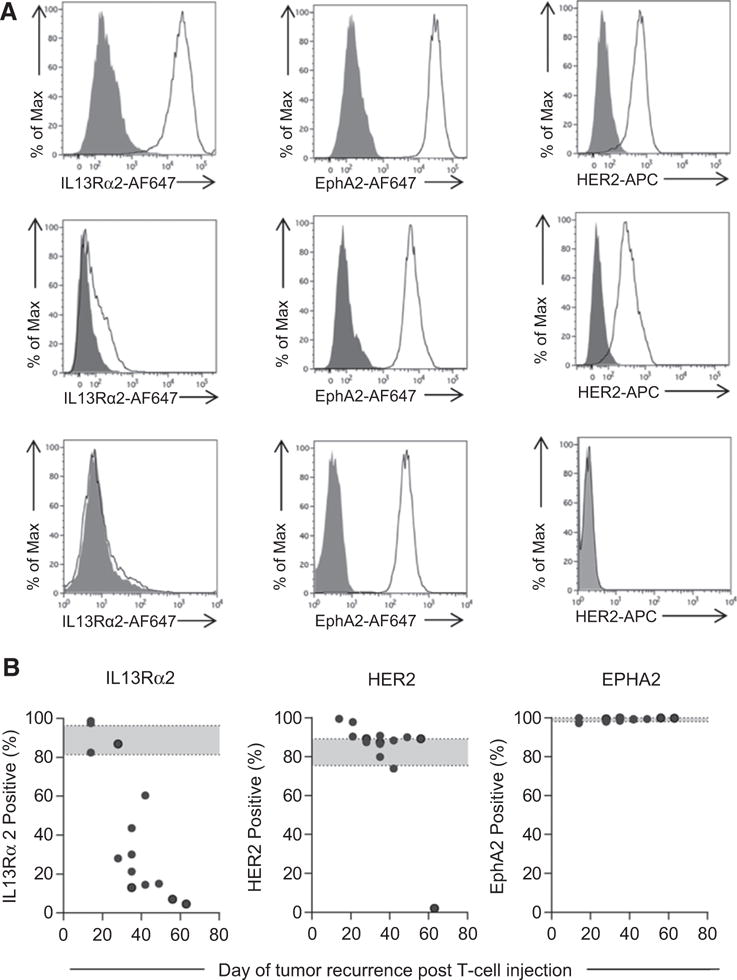
Late-recurring U373 gliomas did not express IL13Rα2. Cells isolated from recurrent tumors were analyzed for IL13Rα2, EphA2, and HER2 expression using primary goat anti-IL13Rα2 (AF146, R&D Systems), mouse anti-EphA2 (MAB3035, R&D Systems), and HER2-APC (340554, BD Biosciences) followed by secondary (except for HER2) rabbit anti-goat or goat anti-mouse IgG Alexa647 (Life Technologies). A, Representative FACS plots. Top row, IL13Rα2-positive recurrent tumors; middle row, IL13Rα2 low-expressing recurrent tumors; bottom row, IL13Rα2-negative recurrent tumors; gray, isotype control; and white, antigen-specific antibody. B, Summary data of GBM-associated antigen (IL13Rα2, HER2, EphA2) expression; each dot represents a recurring tumor (n = 13; one independent experiment per one recurrence, total 13).
Discussion
Here, we described the generation and characterization of T cells expressing an IL13Rα2-CAR with a CD28.ζ signaling domain and IL15. We showed that transgenic expression of IL15 in IL13Rα2-CAR T cells enhanced their effector function as judged by cytokine production and T-cell expansion after repeated stimulations, resulting in selection of T cells that expressed CAR and IL15. In vivo, IL13Rα2-CAR.IL15 T cells had greater antitumor activity in comparison with IL13Rα2-CAR T cells. Recurring tumors presented as antigen loss variants, highlighting the need to target multiple TAAs once the effector function of monospecific T cells is optimized.
We genetically modified T cells with our SFG retroviral vector encoding iC9-2A-ΔNGFR-2A-IL15. Modification was confirmed by FACS analysis for ΔNGFR and Western blot for iC9. At baseline, there was no increase in IL15 production in comparison with unmodified T cells. Upon T-cell activation, IL15 production increased in comparison with IL15 gene-unmodified T cells, which was confirmed for a second transgene. The expression of all transgenes in SFG retroviral vectors, including IL15 and GFP. ffLuc, is driven by the Molony Murine Leukemia Virus (MoMuLV) long terminal repeat (LTR) promoter/enhancer. The MoMuLV LTR enhancer is located within the U3 region of the LTR and contains binding sites for at least six distinct transcription factors (33). T-cell activation results in a broad upregulation of multiple transcription factors, including NFAT and NF-κB (34), which likely enhance the expression of LTR-driven genes including IL15 and GFP.ffLuc. Our findings are consistent with previous findings that demonstrated activation-dependent transgenic expression of IL15 in Epstein–Barr virus-specific or CD19-CAR T cells that were genetically modified with a SFG retroviral vector encoding IL15 (27, 28).
We did not observe autonomous growth of IL13Rα2-CAR.IL15, IL13Rα2-CAR.Δ.IL15, or IL15 T cells, which is in agreement with previous studies (27, 28); however, baseline IL15 expression enhanced T-cell survival in the absence of antigen. This should be a favorable attribute for T-cell therapy of brain and solid tumors in which TAAs are not readily available to activate infused T cells, as in the setting of leukemia. Repeat stimulation resulted in an enrichment of T cells expressing IL15 and IL13Rα2-CARs with approximately 100% of T cells expressing both transgenes after six stimulations. Selection of IL13Rα2-CAR.IL15 T cells after repeat stimulations argues for an autocrine loop in which IL15 is only secreted in sufficient amounts to enhance the function of IL15 gene-modified T cells. As we observed enrichment of IL13Rα2-CAR.IL15 T cells after repeat stimulations, we did not sort double-positive cells for the in vivo experiments.
Transgenic expression of IL15 did not change the T-cell memory subsets as defined by CD45RA and CCR7 expression in comparison with nontransduced, CAR, and CAR.Δ T cells. Transgenic expression of membrane-bound or “tethered” IL15, and the generation of CAR T cells in IL7 and IL15 have been reported to increase the percentage of T cells with memory stem cell–like (CD45RA+, CCR7+) phenotype (32, 35, 36). We expected no phenotypic differences between IL15 gene–modified and unmodified T cells, as we routinely generated T cells in the presence of IL7 and IL15.
Transgenic expression of IL15 did not improve IL13Rα2-CAR T-cell proliferation or cytokine secretion after the first antigen-specific stimulation; however, starting with the third stimulations, there were significant (proliferation, P < 0.05; IFNγ, IL2, TNFα secretion, P < 0.05) differences between IL13Rα2-CAR and IL13Rα2-CAR.IL15 T cells. After the third stimulation, no viable IL13Rα2-CAR T cells remained, whereas IL13Rα2-CAR.IL15 T cells continued to expand and produce cytokines for at least two additional stimulations. Cytokine production with subsequent stimulations declined in IL13Rα2-CAR.IL15 T cells. This finding is consistent with an exhausted T-cell phenotype (37), which is expected after T-cell culture for more than 40 days. Studies are in progress to evaluate this in detail for IL13Rα2-CAR.IL15 T cells.
As in our previous study, IL13Rα2-CAR T cells did not expand in vivo after local injection (8). IL13Rα2-CAR.IL15 T cells also did not expand, but persisted longer than IL13Rα2-CAR T cells. These findings are in contrast to previous studies in which T cells, expressing only a CAR with a CD28.ζ signaling domain, initially expand in an antigen-dependent manner after intravenous or intraperitoneal administration (38, 39).
The benefits of transgenic expression of cytokines, such as IL12 and IL15, have been explored by several groups of investigators for solid tumors (23, 24, 40, 41). For example, transgenic expression of IL12 enhanced effector function and antitumor activity of MUC-16-CAR T cells, as judged by increased T-cell persistence and prolonged survival of tumor-bearing mice (40). In addition, transgenic expression of IL12 in VEGF-CAR T cells enhanced their antitumor activity by reversing the immunosuppressive tumor microenvironment (41). Finally, transgenic expression of IL15 in GD2-CAR NKT cells rendered NKT resistant to hypoxia and tumor-associated macrophages resulting in improved activity against neuroblastoma (24). Our study extends these findings to glioma and highlights that improving T-cell persistence by transgenic expression of IL15 leaves T cells vulnerable to other mechanisms of immune escape.
Antigen-negative immune escape was the major mode of CAR T-cell therapy failure in tumors recurring more than 40 days after T-cell injection. Antigen escape was specific for the targeted antigens, as all tested recurring tumors continued to express the glioma-associated antigen EphA2, and all but one expressed HER2. Finding an HER2-negative, recurring, tumor highlights the need to profile TAA expression in recurring tumors to determine the best second antigen to target. Investigators have targeted two TAAs either by mixing two CAR T-cell populations, expressing two CARs within the same T-cell, or developing CARs that contain two antigen-binding domains (42–44). On the basis of our findings, we favor targeting IL13Rα2 and EphA2 with CAR T cells to prevent immune escape. How to best target both antigens needs to be determined experimentally, given the growing literature that there is an intricate structural/functional relationship between the targeted antigen and the CAR (8, 45). In addition, CARs can multimerize, resulting in baseline or tonic signaling (45–47), which can have detrimental effects on T-cell function (46, 47). Expressing single-specificity CARs in T cells that recognize TAAs through their endogenous αβ TCR, or bispecific T-cell engagers (BiTEs) in CAR T cells present alternative strategies to generate bi- or multispecific T-cell products for glioma, which should mitigate the risk of heterodimerization of multiple CARs expressed in a single T cell.
A limitation of our study and the majority of published CAR T-cell therapy studies is the use of xenograft models, which do not recapitulate the immunosuppressive tumor microenvironment created by tumors, including glioma (48–50). Others have reported the development of an immunocompetent glioma model to evaluate CAR T-cell therapies; however, this model does not recapitulate physiologic TAA expression, as glioma cells were genetically modified to express the targeted TAA, EGFRvIII (48). Our xenograft model allowed for the study of human tumor and human T cells in vivo, which enabled us to delineate attributes of effective CAR T-cell products for glioma. We are planning to establish an immunocompetent glioma model to evaluate the effects of transgenic expression of IL15 on resident immune cell.
In conclusion, we showed that transgenic expression of IL15 in IL13Rα2-CAR T cells enhanced their effector function, resulting in improved antiglioma activity in vitro and in vivo. Recurring gliomas had downregulated the expression of IL13Rα2, highlighting the need to target multiple TAAs once the effector function of monospecific T cells is optimized. Hence, optimal CAR T-cell products for glioma should be engineered to increase their effector function and to target multiple TAAs.
Supplementary Material
Summary.
Glioblastoma responds imperfectly to immunotherapy. Transgenic expression of IL15 in T cells expressing CARs improved their proliferative capacity, persistence, and cytokine production. The emergence of antigen loss variants highlights the need to target multiple tumor antigens.
Acknowledgments
This work was supported by NIH Grants 1R01CA173750-01 and 1R21NS089802-01, NCI Cancer Center Support grant P30CA125123, American Brain Tumor Association Basic Research Fellowship in the honor of Joel A. Gingras Jr. (BRF160004), Alex Lemonade Stand Foundation, Curing Kids Cancer, Cookies for Kids Cancer, and the James S. McDonnell Foundation.
Footnotes
Note: Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
G. Dotti reports receiving other commercial research support from Bluebird Bio and is a consultant/advisory board member for Cell Medica. I.V. Balyasnikova has ownership interest in a submitted patent application. S. Gottschalk has ownership interest in a patent and is a consultant/advisory board member for Merrimack. No potential conflicts of interest were disclosed by the other authors.
Authors’ Contributions
Conception and design: G. Krenciute, G. Dotti, I.V. Balyasnikova, S. Gottschalk
Development of methodology: G. Krenciute, S. Gottschalk
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): G. Krenciute, B.L. Prinzing, Z. Yi, I.V. Balyasnikova, S. Gottschalk
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): G. Krenciute, Z. Yi, M.-F. Wu, H. Liu, S. Gottschalk
Writing, review, and/or revision of the manuscript: G. Krenciute, B.L. Prinzing, M.-F. Wu, H. Liu, G. Dotti, I.V. Balyasnikova, S. Gottschalk
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): G. Krenciute, G. Dotti, S. Gottschalk
Study supervision: G. Krenciute, S. Gottschalk
Other (designed and developed anti-IL13Ra2 scFv47): I.V. Balyasnikova
References
- 1.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310:1842–50. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 2.Thakkar JP, Dolecek TA, Horbinski C, Ostrom QT, Lightner DD, Barnholtz-Sloan JS, et al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol Biomarkers Prev. 2014;23:1985–96. doi: 10.1158/1055-9965.EPI-14-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arvold ND, Reardon DA. Treatment options and outcomes for glioblastoma in the elderly patient. Clin Interv Aging. 2014;9:357–67. doi: 10.2147/CIA.S44259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Konar SK, Bir SC, Maiti TK, Nanda A. A systematic review of overall survival in pediatric primary glioblastoma multiforme of the spinal cord. J Neurosurg Pediatr. 2017;19:239–48. doi: 10.3171/2016.8.PEDS1631. [DOI] [PubMed] [Google Scholar]
- 5.Suryadevara CM, Verla T, Sanchez-Perez L, Reap EA, Choi BD, Fecci PE, et al. Immunotherapy for malignant glioma. Surg Neurol Int. 2015;6:S68–S77. doi: 10.4103/2152-7806.151341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krebs S, Rodriguez-Cruz TG, Derenzo C, Gottschalk S. Genetically modified T cells to target glioblastoma. Front Oncol. 2013;3:322. doi: 10.3389/fonc.2013.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64:9160–6. doi: 10.1158/0008-5472.CAN-04-0454. [DOI] [PubMed] [Google Scholar]
- 8.Krenciute G, Krebs S, Torres D, Wu MF, Liu H, Dotti G, et al. Characterization and functional analysis of scFv-based chimeric antigen receptors to redirect T cells to IL13Ralpha2-positive Glioma. Mol Ther. 2016;24:354–63. doi: 10.1038/mt.2015.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16:474–85. doi: 10.1158/1078-0432.CCR-09-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015;7:275ra22. doi: 10.1126/scitranslmed.aaa4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21:629–37. doi: 10.1038/mt.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Rourke D, Desai A, Morrissette J, Martinez-Lage M, Nasrallah M, Brem S, et al. Pilot study of T cell redirected to EGFRvIII with a chimeric antigen receptor in patients with EGFRvIII+ glioblastoma. Neuro Oncol. 2015;17:v110–1. [Google Scholar]
- 13.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375:2561–9. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and Safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21:4062–72. doi: 10.1158/1078-0432.CCR-15-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017 Apr 20; doi: 10.1001/jamaoncol.2017.0184. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. 2015;33:1688–96. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–70. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park JH, Geyer MB, Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. 2016;127:3312–20. doi: 10.1182/blood-2016-02-629063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruella M, June CH. Chimeric antigen receptor T cells for B cell neoplasms: choose the right CAR for you. Curr Hematol Malig Rep. 2016;11:368–84. doi: 10.1007/s11899-016-0336-z. [DOI] [PubMed] [Google Scholar]
- 20.Turtle CJ, Riddell SR, Maloney DG. CD19-Targeted chimeric antigen receptor-modified T-cell immunotherapy for B-cell malignancies. Clin Pharmacol Ther. 2016;100:252–8. doi: 10.1002/cpt.392. [DOI] [PubMed] [Google Scholar]
- 21.Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257:107–26. doi: 10.1111/imr.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newick K, O’Brien S, Moon E, Albelda SM. CAR T Cell Therapy for Solid Tumors. Ann Rev Med. 2016;68:139–52. doi: 10.1146/annurev-med-062315-120245. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res. 2015;21:2278–88. doi: 10.1158/1078-0432.CCR-14-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu D, Song L, Wei J, Courtney AN, Gao X, Marinova E, et al. IL-15 protects NKT cells from inhibition by tumor-associated macrophages and enhances antimetastatic activity. J Clin Invest. 2012;122:2221–33. doi: 10.1172/JCI59535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilkie S, Burbridge SE, Chiapero-Stanke L, Pereira AC, Cleary S, van der Stegen SJ, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem. 2010;285:25538–44. doi: 10.1074/jbc.M110.127951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foster AE, Dotti G, Lu A, Khalil M, Brenner MK, Heslop HE, et al. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J Immunother. 2008;31:500–5. doi: 10.1097/CJI.0b013e318177092b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24:1160–70. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quintarelli C, Vera JF, Savoldo B, Giordano Attianese GM, Pule M, Foster AE, et al. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood. 2007;110:2793–802. doi: 10.1182/blood-2007-02-072843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5:215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leen AM, Sukumaran S, Watanabe N, Mohammed S, Keirnan J, Yanagisawa R, et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol Ther. 2014;22:1211–20. doi: 10.1038/mt.2014.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarkaria JN, Yang L, Grogan PT, Kitange GJ, Carlson BL, Schroeder MA, et al. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Mol Cancer Ther. 2007;6:1167–74. doi: 10.1158/1535-7163.MCT-06-0691. [DOI] [PubMed] [Google Scholar]
- 32.Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123:3750–9. doi: 10.1182/blood-2014-01-552174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Speck NA, Baltimore D. Six distinct nuclear factors interact with the 75-base-pair repeat of the Moloney murine leukemia virus enhancer. Mol Cell Biol. 1987;7:1101–10. doi: 10.1128/mcb.7.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121:573–84. doi: 10.1182/blood-2012-05-431718. [DOI] [PubMed] [Google Scholar]
- 36.Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci USA. 2016;113:E7788–E97. doi: 10.1073/pnas.1610544113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–99. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vera J, Savoldo B, Vigouroux S, Biagi E, Pule M, Rossig C, et al. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood. 2006;108:3890–7. doi: 10.1182/blood-2006-04-017061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santos EB, Yeh R, Lee J, Nikhamin Y, Punzalan B, Punzalan B, et al. Sensitive in vivo imaging of T cells using a membrane-bound Gaussia princeps luciferase. Nat Med. 2009;15:338–44. doi: 10.1038/nm.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. 2015;4:e994446. doi: 10.4161/2162402X.2014.994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res. 2012;18:1672–83. doi: 10.1158/1078-0432.CCR-11-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. 2013;21:2087–101. doi: 10.1038/mt.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, et al. TanCAR: A novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids. 2013;2:e105. doi: 10.1038/mtna.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32:1059–70. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 45.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21:581–90. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O’Neill A, et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother. 2005;28:203–11. doi: 10.1097/01.cji.0000161397.96582.59. [DOI] [PubMed] [Google Scholar]
- 47.Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3:125–35. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. 2014;20:972–84. doi: 10.1158/1078-0432.CCR-13-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010;116:3875–86. doi: 10.1182/blood-2010-01-265041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morgan RA. Human tumor xenografts: the good, the bad, and the ugly. Mol Ther. 2012;20:882–4. doi: 10.1038/mt.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.